A Novel Flavonoid Glucoside from the Fruits of Lycium ruthenicun

 , and
, and
Abstract
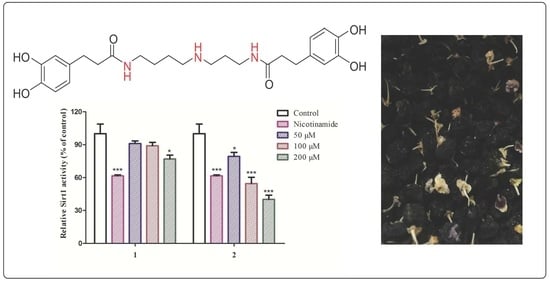
:
1. Introduction
2. Results and Discussion
2.1. Structure Elucidation of the Compounds
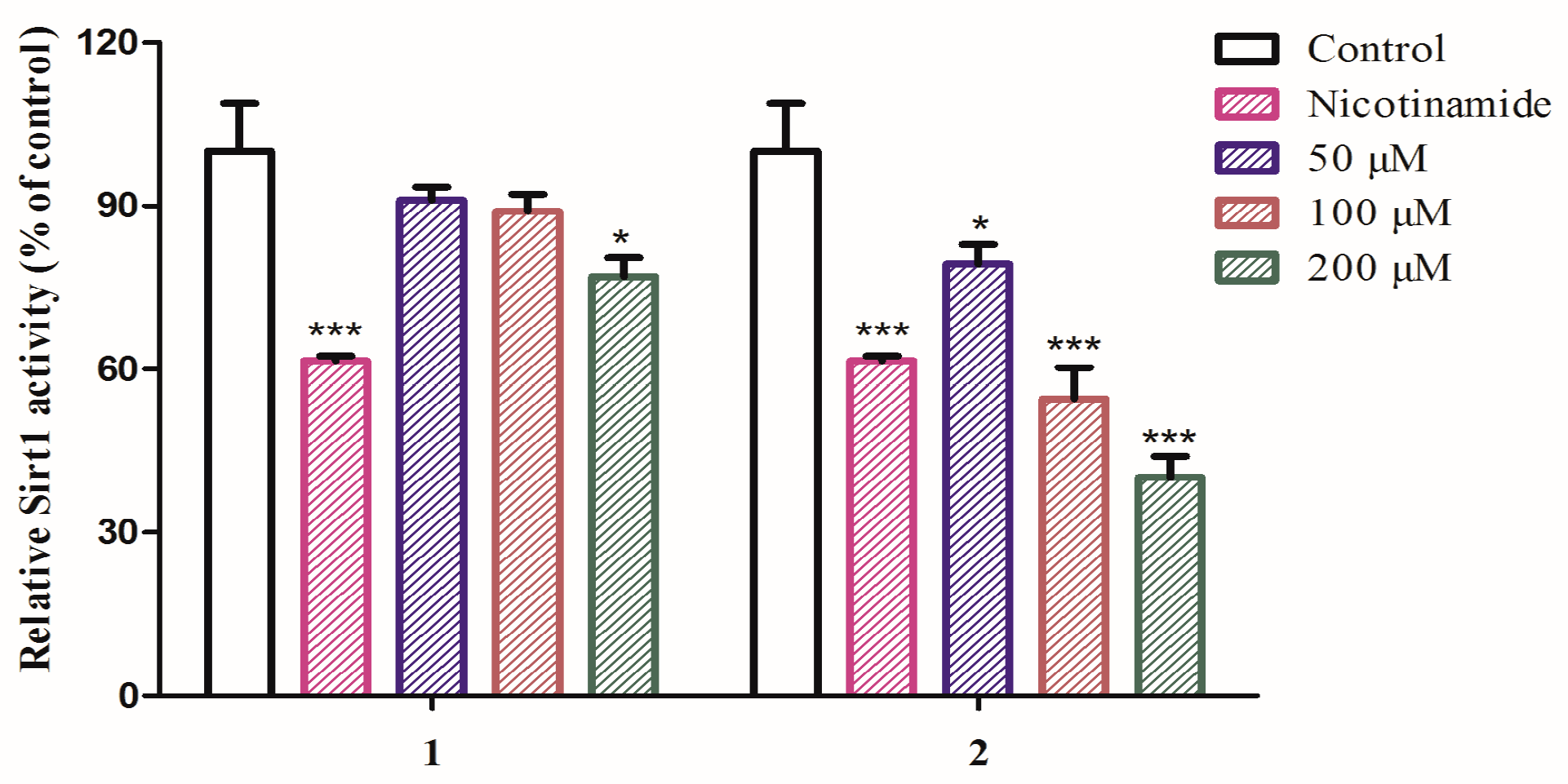
2.2. Biological Evaluation
3. Experimental Section
3.1. General Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Compound Characterization Data
3.5. Acid Hydrolysis and Sugar Analysis
3.6. SIRT1 Inhibition
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhao, J.; Xu, F.; Ji, T.F.; Li, J. A new spermidine from the fruits of Lycium ruthenicum. Chem. Nat. Compd. 2014, 50, 880–883. [Google Scholar] [CrossRef]
- Rao, A.V.; Snyde, D.M. Raspberries and human health: A review. J. Agric. Food Chem. 2010, 58, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Zilic, S.; Serpen, A.; Akillioglu, G.; Gokmen, V.; Vancetovic, J. Phenolic compounds, carotenoids, anthocyanins, and antioxidant capacity of colored maize (Zea mays L.) Kernels. J. Agric. Food Chem. 2012, 60, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qu, W.J.; Zhang, S.J.; Lv, H.Y. Study on antioxidant activity of pigment of Lycium ruthenicum. Chin. J. Chin. Mater. Med. 2006, 31, 1179–1183. [Google Scholar] [CrossRef]
- Zheng, J.; Ding, C.X.; Wang, L.S.; Li, G.L.; Shi, J.Y.; Li, H.; Wang, H.L.; Suo, Y.R. Anthocyanins composition and antioxidant activity of wild Lycium ruthenicum Murr. from Qinghai-Tibet Plateau. Food Chem. 2011, 126, 859–865. [Google Scholar] [CrossRef]
- Li, J.; Qu, W.J.; Lv, H.Y.; Yuan, H. Study on extracting and refining of the pigments from Lycium ruthenicum Murr. Nat. Prod. Res. Dev. 2006, 18, 650–654. [Google Scholar] [CrossRef]
- Li, S.Z.; Li, J.; Yang, Z.J.; Yuan, H. Study on separation and purifeation of total flavonoids from Lycium ruthenicum Murr. with macroreticular resin. Food Sci. 2009, 30, 19–24. [Google Scholar] [CrossRef]
- Luo, H.; Jin, L.; Gao, S.F.; Chen, H.G. Determination of Anthocyanin in Lycium ruthenicum Murr. from different producing areas in Hei River basin by UV. Chin. Med. J. Res. Pract. 2015, 29, 24–27. [Google Scholar] [CrossRef]
- Lopes, P.; Richard, T.; Saucier, C.; Teissedre, P.; Monti, J.; Glories, Y. Anthocyanone A: A quinone methide derivative resulting from malvidin 3-O-glucoside degradation. J. Agric. Food Chem. 2007, 55, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, H.; Yanase, E.; Nakatsuka, S. Novel oxidation products of cyanidin 3-O-glucoside with 2,2′-azobis-(2,4-dimethyl)valeronitrile and evaluation of anthocyanin content and its oxidation in black rice. Food Chem. 2014, 155, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.J.; Yan, Y.M.; Wang, C.X.; Cheng, Y.X. Compounds from Lycium ruthenicum. Nat. Prod. Res. Dev. 2017. Available online: http://kns.cnki.net/kcms/detail/51.1335.Q.20170904.1418.016.html (accessed on 4 September 2017).
- Yingyongnarongkul, B.; Apiratikul, N.; Aroonrerk, N.; Suksamrarn, A. Synthesis of bis, tris and tetra (dihydrocaffeoyl) polyamine conjugates as antibacterial agents against VRSA. Arch. Pharm. Res. 2008, 31, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.X.; Hui, Y.X.; Rupprecht, J.K.; Mclaughlin, J.L.; Wood, K.V. Additional bioactive compounds and trilobacin, a novel highly cytotoxic acetogenin, from the bark of Asimina triloba. J. Nat. Prod. 1992, 55, 347–356. [Google Scholar] [CrossRef] [PubMed]
- King, R.R.; Calhoun, L.A. Characterization of cross-linked hydroxycinnamic acid amides isolated from potato common scab lesions. Phytochemistry 2005, 66, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Cutillo, F.; Dabrosca, B.; Dellagreca, M.; Marino, C.D.; Golino, A.; Previtera, L.; Zarrelli, A. Cinnamic acid amides from Chenopodium album: Effects on seeds germination and plant growth. Phytochemistry 2003, 64, 1381–1387. [Google Scholar] [CrossRef]
- Kim, D.K.; Lim, J.P.; Kim, J.W.; Park, H.W.; Eun, J.S. Antitumor and antiinflammatory constituents from Celtis sinensis. Arch. Pharm. Res. 2005, 28, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, Y. SIRT1: Role in cardiovascular biology. Clin. Chim. Acta 2015, 440, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.Y.; Qu, S.L.; Tang, Z.H.; Zhang, Y.; Liu, M.H.; Peng, J.; Tang, H.; Yu, K.L.; Zhang, C.; Ren, Z.; et al. SIRT1 in cardiovascular aging. Clin. Chim. Acta 2014, 437, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Clark-Knowles, K.V.; He, X.H.; Jardine, K.; Coulombe, J.; Dewar-Darch, D.; Caron, A.Z.; Gray, D.A.; McBurney, M.W. Reversible modulation of SIRT1 activity in a mouse strain. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.W.; Buer, B.C.; Marshab, E.N.G.; Kennedy, R.T. A label-free Sirtuin 1 assay based on dropletelectrospray ionization mass spectrometry. Anal. Methods 2016, 8, 3458–3465. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chauhan, S. How much successful are the medicinal chemists in modulation of SIRT1: A critical review. Eur. J. Med. Chem. 2016, 119, 45–69. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.N.; Tu, Z.C.; Wang, X.L.; Yan, Y.M.; Fang, P.; Zuo, Z.L.; Hou, B.; Yang, T.H.; Cheng, Y.X. Bioactive compounds from the insect Aspongopus chinensis. Bioorg. Med. Chem. Lett. 2014, 24, 5164–5169. [Google Scholar] [CrossRef] [PubMed]
- Jordan, D.S.; Daubenspeck, J.M.; Dybvig, K. Rhamnose biosynthesis in mycoplasmas requires precursor glycans larger than monosaccharide. Mol. Microbiol. 2013, 89, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Karbasforooshan, H.; Karimi, G. The role of SIRT1 in diabetic cardiomyopathy. Biomed. Pharmacother. 2017, 90, 386–392. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Sample of the compound 1 is available from the authors. |

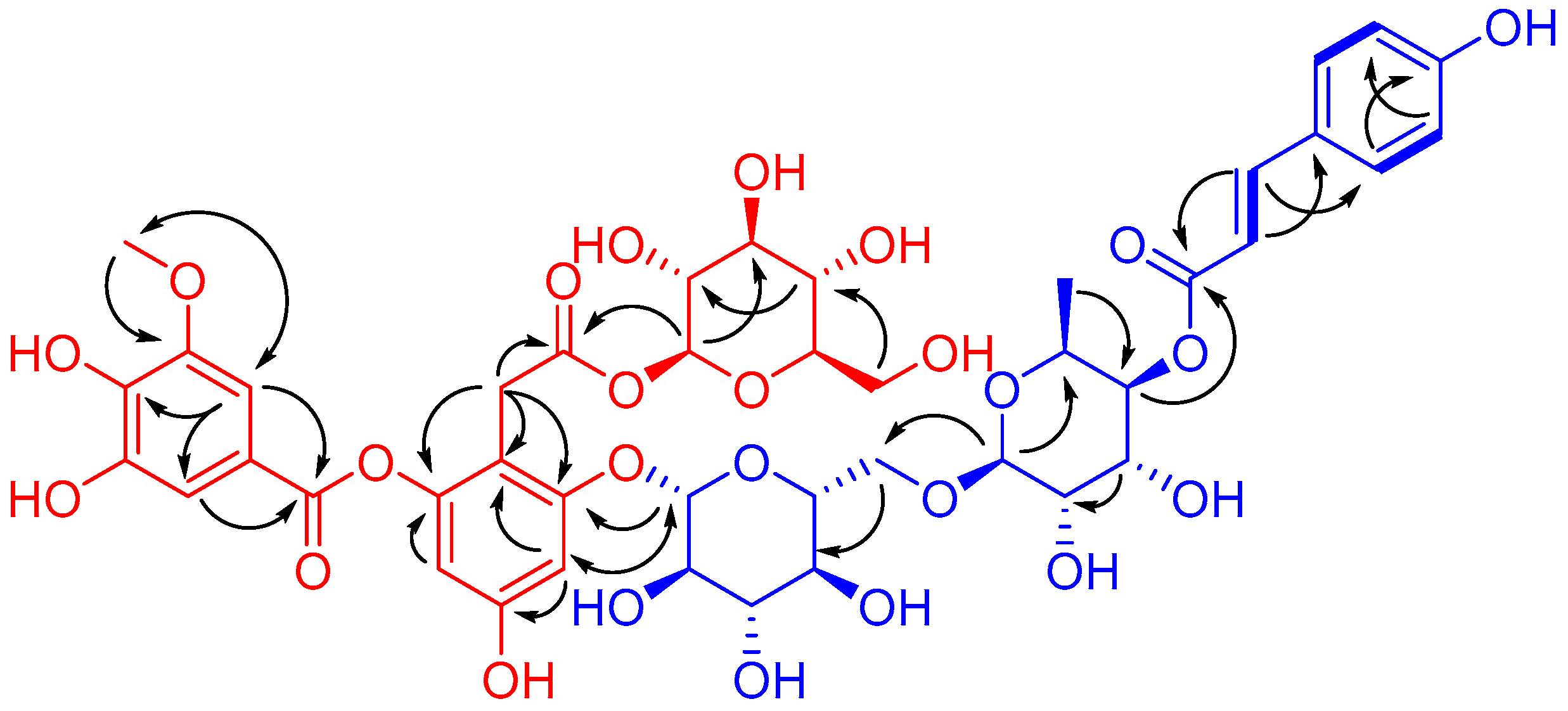
 ) and key HMBC (
) and key HMBC (  ) and ROESY (
) and ROESY (  ) correlations of 1.
) correlations of 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | |||||
|---|---|---|---|---|---|
| No. | δH | δC | No. | δH | δC |
| 1 | 109.2 | 1′′′ | 4.87, brs | 103.2 | |
| 2 | 152.1 | 2′′′ | 3.51, m | 74.8 | |
| 3 | 6.42, d, 1.8 | 105.2 | 3′′′ | 3.47, m | 77.8 |
| 4 | 159.1 | 4′′′ | 3.32, overlap | 71.0 | |
| 5 | 6.67, d, 1.8 | 102.4 | 5′′′ | 3.32, overlap | 77.7 |
| 6 | 158.6 | 6′′′ | 3.96, m | 67.9 | |
| 7 | 3.73, m | 30.4 | 3.62, m | ||
| 3.66, m | 1′′′′ | 4.76, brs | 102.2 | ||
| 8 | 172.2 | 2′′′′ | 3.43, m | 78.2 | |
| 1′ | 120.1 | 3′′′′ | 3.86, m | 70.4 | |
| 2′ | 7.30, d, 1.8 | 106.9 | 4′′′′ | 5.00, m | 75.3 |
| 3′ | 149.3 | 5′′′′ | 3.79, m | 67.9 | |
| 4′ | 141.6 | 6′′′′ | 1.04, d, 6.2 | 17.8 | |
| 5′ | 146.5 | 1′′′′′ | 127.2 | ||
| 6′ | 7.35, d, 1.8 | 112.8 | 2′′′′′ | 7.48, d, 8.5 | 131.3 |
| 7′ | 166.5 | 3′′′′′ | 6.81, d, 8.5 | 116.8 | |
| 1′′ | 5.45, d, 8.2 | 96.0 | 4′′′′′ | 161.2 | |
| 2′′ | 3.89, m | 72.1 | 5′′′′′ | 6.81, d, 8.5 | 116.8 |
| 3′′ | 3.30, m | 73.8 | 6′′′′′ | 7.48, d, 8.5 | 131.3 |
| 4′′ | 3.42, m | 71.2 | 7′′′′′ | 7.63, d, 15.9 | 146.9 |
| 5′′ | 3.50, m | 77.7 | 8′′′′′ | 6.37, d, 15.9 | 115.2 |
| 6′′ | 3.92, m | 62.5 | 9′′′′′ | 169.1 | |
| 3.74, m | -OCH3 | 3.88, s | 56.9 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, J.-J.; Yan, Y.-M.; Cheng, L.-Z.; Liu, B.-H.; Qin, F.-Y.; Cheng, Y.-X. A Novel Flavonoid Glucoside from the Fruits of Lycium ruthenicun. Molecules 2018, 23, 325. https://doi.org/10.3390/molecules23020325
Qi J-J, Yan Y-M, Cheng L-Z, Liu B-H, Qin F-Y, Cheng Y-X. A Novel Flavonoid Glucoside from the Fruits of Lycium ruthenicun. Molecules. 2018; 23(2):325. https://doi.org/10.3390/molecules23020325
Chicago/Turabian StyleQi, Jing-Jing, Yong-Ming Yan, Li-Zhi Cheng, Bao-Hua Liu, Fu-Ying Qin, and Yong-Xian Cheng. 2018. "A Novel Flavonoid Glucoside from the Fruits of Lycium ruthenicun" Molecules 23, no. 2: 325. https://doi.org/10.3390/molecules23020325
APA StyleQi, J.-J., Yan, Y.-M., Cheng, L.-Z., Liu, B.-H., Qin, F.-Y., & Cheng, Y.-X. (2018). A Novel Flavonoid Glucoside from the Fruits of Lycium ruthenicun. Molecules, 23(2), 325. https://doi.org/10.3390/molecules23020325