Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases


Abstract
1. Introduction
2. HDACs in the Brain
3. HDAC Inhibitors
4. Radioligands for HDACs
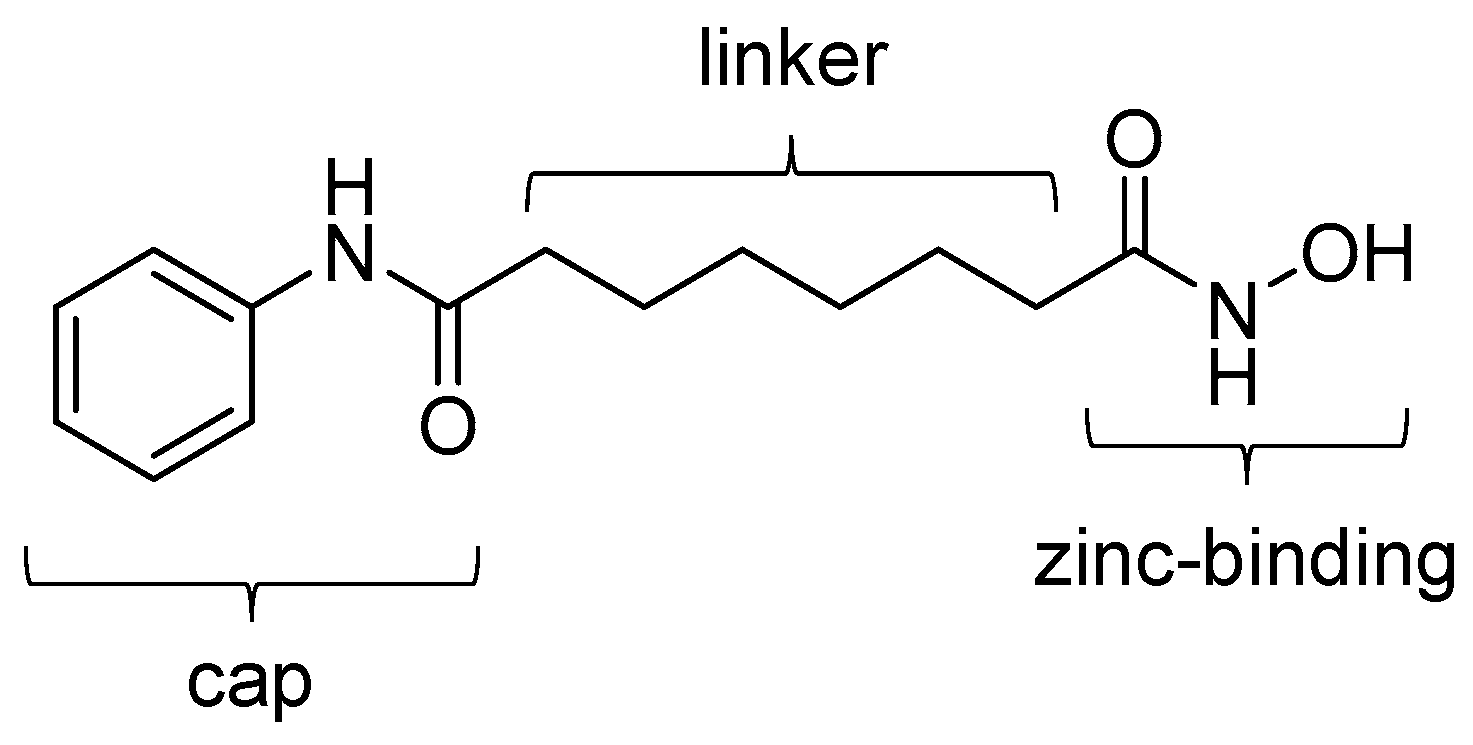
4.1. SAHA-Based Ligands
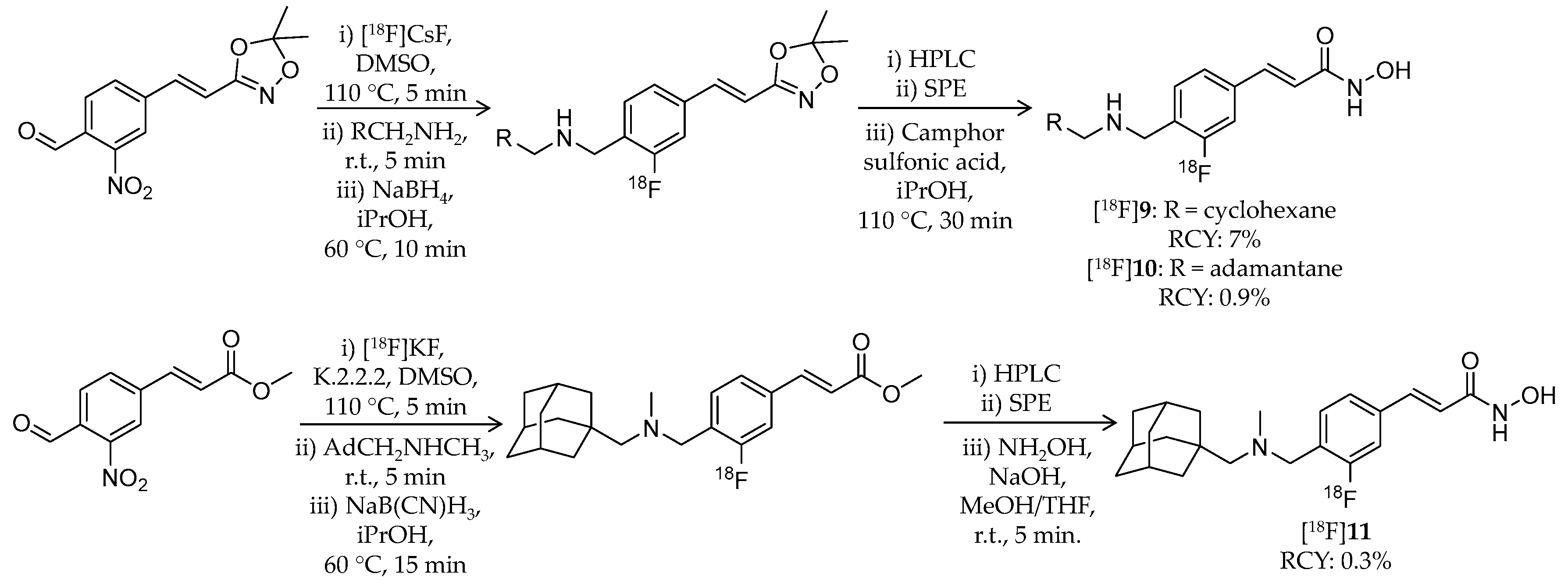
4.2. Adamantane-Conjugated Ligands
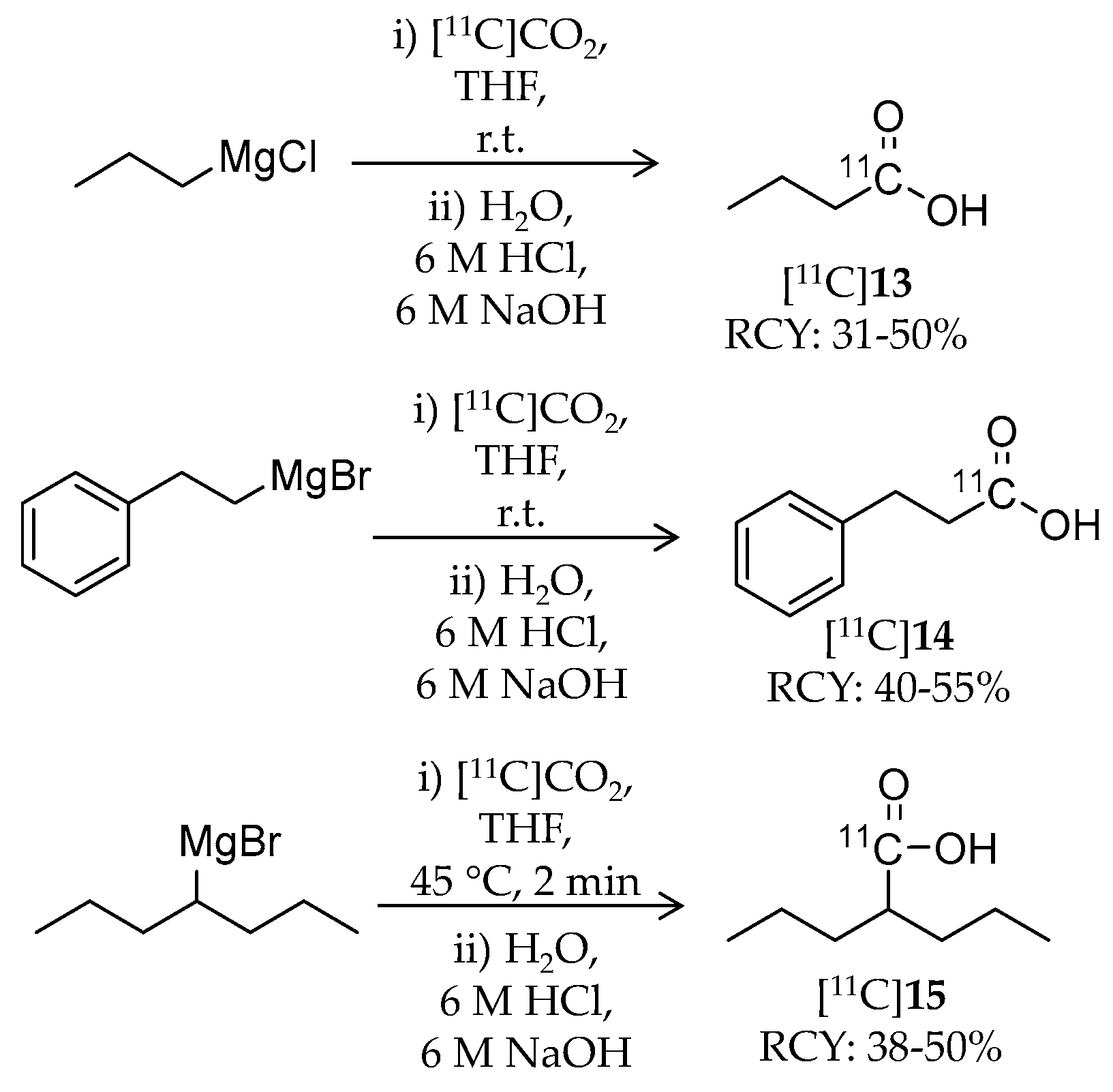
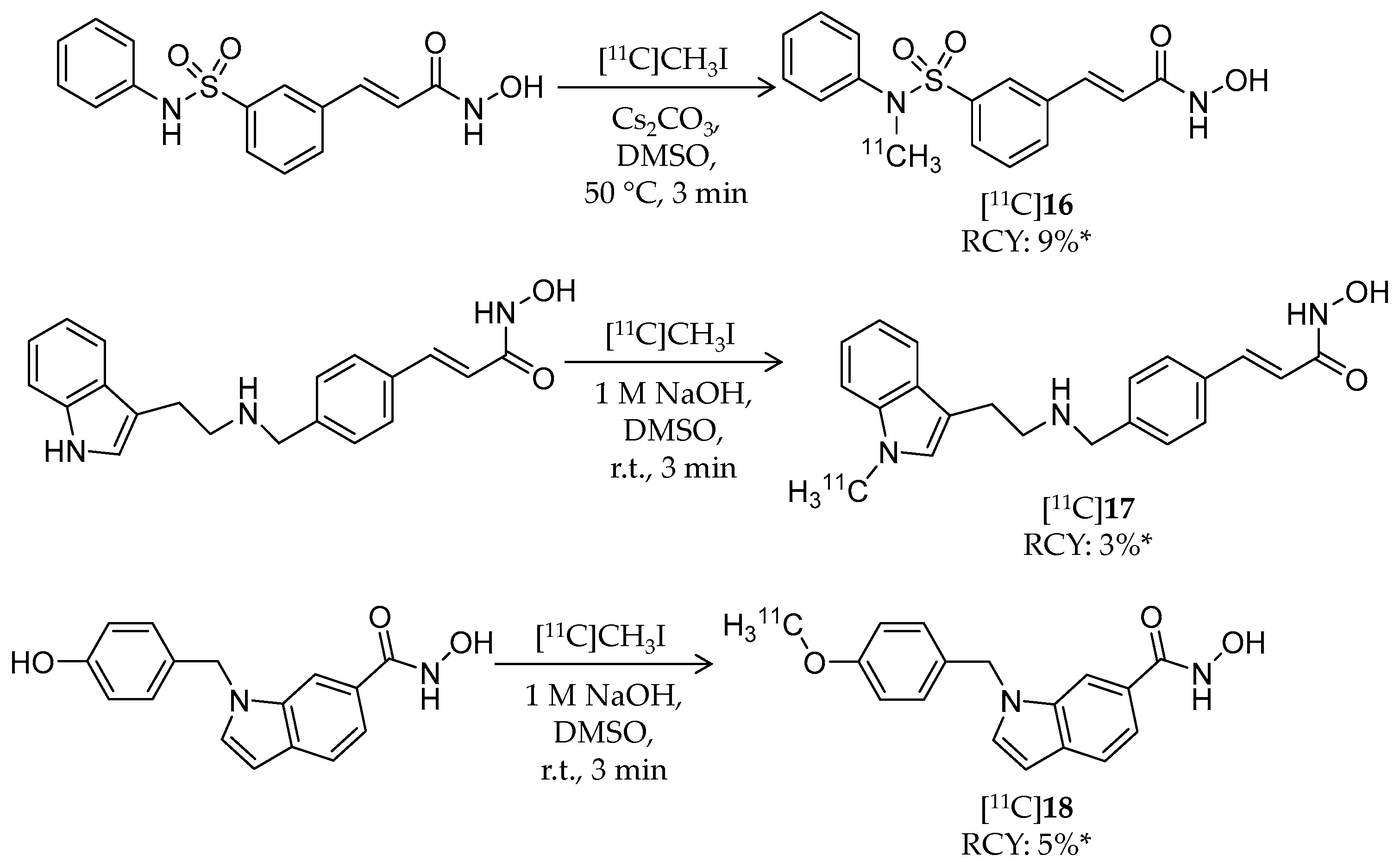
4.3. Other Carboxylic Acid- and Hydroxamic Acid-Based Ligands
4.4. Ortho-Aminoanilide-Based Ligands
4.5. First-in-Human PET Study
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Landgrave-Gomez, J.; Mercado-Gomez, O.; Guevara-Guzman, R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar] [PubMed]
- Lovrečić, L.; Maver, A.; Zadel, M.; Peterlin, B. The Role of Epigenetics in Neurodegenerative Diseases. In Neurodegenerative Diseases; InTech: London, UK, 2013; pp. 345–365. ISBN 978-953-51-1088-0. [Google Scholar]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A. On the Enzymatic Properties of Dnmt1: Specificity, Processivity, Mechanism of Linear Diffusion and Allosteric Regulation of the Enzyme. Epigenetics 2014, 1, 63–66. [Google Scholar] [CrossRef]
- Turek-Plewa, J.; Jagodzinski, P.P. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell. Mol. Biol. Lett. 2005, 10, 631–647. [Google Scholar] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.M.; Wood, M.A. Beyond transcription factors: The role of chromatin modifying enzymes in regulating transcription required for memory. Learn. Mem. 2008, 15, 460–467. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Volpe, T.A.; Kidner, C.; Hall, I.M.; Teng, G.; Grewal, S.I.; Martienssen, R.A. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 2002, 297, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Joseph, N.F.; Horn, M.E.; Samiei, A.; Meng, J.; Seo, J.; Rei, D.; Bero, A.W.; Phan, T.X.; Wagner, F.; et al. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell 2014, 156, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Renthal, W.; Nestler, E.J. Epigenetic mechanisms in drug addiction. Trends Mol. Med. 2008, 14, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Langley, B.; Lubin, F.D.; Renthal, W.; Wood, M.A.; Yasui, D.H.; Kumar, A.; Nestler, E.J.; Akbarian, S.; Beckel-Mitchener, A.C. Epigenetics in the nervous system. J. Neurosci. 2008, 28, 11753–11759. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; McKee, A.; Grover, A.; Rogers, J.; Coleman, P.D. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE 2009, 4, e6617. [Google Scholar] [CrossRef] [PubMed]
- Politis, M. Neuroimaging in Parkinson disease: From research setting to clinical practice. Nat. Rev. Neurol. 2014, 10, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.P.; Liang, S.H.; Rotstein, B.H.; Collier, T.L.; Stephenson, N.A.; Greguric, I.; Vasdev, N. Alternative approaches for PET radiotracer development in Alzheimer’s disease: Imaging beyond plaque. J. Label. Comp. Radiopharm. 2014, 57, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Wesmall yi, U.M.; Lewis, M.C.; Holson, E.B. Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics 2013, 10, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.L.; Yang, W.M. Beyond histone and deacetylase: An overview of cytoplasmic histone deacetylases and their nonhistone substrates. J. Biomed. Biotechnol. 2011, 2011, 146493. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed]
- Broide, R.S.; Redwine, J.M.; Aftahi, N.; Young, W.; Bloom, F.E.; Winrow, C.J. Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 2007, 31, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Anna, G.D.S.S.; Elsner, V.R.; Moyses, F.; Cechinel, R.L.; Lovatel, G.A.; Siqueira, I.R. Histone deacetylase activity is altered in brain areas from aged rats. Neurosci. Lett. 2013, 556, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.W.; Chen, J.; Wang, M.; Mast, N.; Pikuleva, I.A.; Turko, I.V. Quantification of histone deacetylase isoforms in human frontal cortex, human retina, and mouse brain. PLoS ONE 2015, 10, e0126592. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Tian, M.; Hinz, R.; Young, D.; Shavrin, A.; Mukhapadhyay, U.; Flores, L.G.; Balatoni, J.; Soghomonyan, S.; Jeong, H.J.; et al. Imaging epigenetic regulation by histone deacetylases in the brain using PET/MRI with 18F-FAHA. Neuroimage 2013, 64, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Lucio-Eterovic, A.K.; Cortez, M.A.; Valera, E.T.; Motta, F.J.; Queiroz, R.G.; Machado, H.R.; Carlotti, C.G., Jr.; Neder, L.; Scrideli, C.A.; Tone, L.G. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: Class II and IV are hypoexpressed in glioblastomas. BMC Cancer 2008, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Wey, H.Y.; Gilbert, T.M.; Zurcher, N.R.; She, A.; Bhanot, A.; Taillon, B.D.; Schroeder, F.A.; Wang, C.; Haggarty, S.J.; Hooker, J.M. Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci. Transl. Med. 2016, 8, 351ra106. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, A.; Doherty, K.; Yeh, H.H.; Robinson, A.C.; Rollinson, S.; Pickering-Brown, S.; Snowden, J.; Thompson, J.C.; Davidson, Y.S.; Mann, D.M. Histone deacetylases (HDACs) in frontotemporal lobar degeneration. Neuropathol. Appl. Neurobiol. 2015, 41, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Dietz, K.C.; Casaccia, P. HDAC inhibitors and neurodegeneration: At the edge between protection and damage. Pharmacol. Res. 2010, 62, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Webb, Y.; Merger, R.; Sheppard, T.; Jursic, B.; Ngo, L.; Civoli, F.; Breslow, R.; Rifkind, R.A.; Marks, P.A. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 5705–5708. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A. Discovery and development of SAHA as an anticancer agent. Oncogene 2007, 26, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef] [PubMed]
- Laubach, J.P.; Moreau, P.; San-Miguel, J.F.; Richardson, P.G. Panobinostat for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4767–4773. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A.; Opal, P. The promise and perils of HDAC inhibitors in neurodegeneration. Ann. Clin. Transl. Neurol. 2015, 2, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Perez-Mediavilla, A.; Frechilla, D.; Del Rio, J.; Garcia-Osta, A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology 2009, 34, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Gardian, G.; Yang, L.; Cleren, C.; Calingasan, N.Y.; Klivenyi, P.; Beal, M.F. Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. Neuromol. Med. 2004, 5, 235–241. [Google Scholar] [CrossRef]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.; Tong, W.P.; Gelovani, J.G.; Alauddin, M.M. Radiosynthesis of 6-([18F]fluoroacetamido)-1-hexanoicanilide ([18F]FAHA) for PET imaging of histone deacetylase (HDAC). J. Label. Compd. Radiopharm. 2006, 49, 997–1006. [Google Scholar] [CrossRef]
- Nishii, R.; Mukhapadhyay, U.; Yeh, H.; Soghomonyan, S.; Volgin, A.; Alauddin, M.; Tong, W.; Gelovani, J. PET imaging of histone deacetylase activity in a rat brain using 6-([18F]-fluoroacetamide)-1-hexanoicanilide ([18F]-FAHA). J. Nucl. Med. 2007, 48 (Suppl. 2), 336. [Google Scholar]
- Nishii, R.; Mukhapadhyay, U.; Yeh, H.; Soghomonyan, S.; Volgin, A.; Alauddin, M.; Tong, W.; Gelovani, J. Non-invasive imaging of histone deacetylase activity in human breast carcinoma xenografts in rats using positron emission tomography (PET) with [18F]-FAHA. J. Nucl. Med. 2007, 48 (Suppl. 2), 34. [Google Scholar]
- Reid, A.E.; Hooker, J.; Shumay, E.; Logan, J.; Shea, C.; Kim, S.W.; Collins, S.; Xu, Y.; Volkow, N.; Fowler, J.S. Evaluation of 6-([18F]fluoroacetamido)-1-hexanoicanilide for PET imaging of histone deacetylase in the baboon brain. Nucl. Med. Biol. 2009, 36, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.L.; Ackermann, R.F. Evaluation of radiolabeled acetate and fluoroacetate as potential tracers of cerebral oxidative metabolism. Metab. Brain Dis. 1990, 5, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Pourghiasian, M.; Hundal, N.; Lau, J.; Benard, F.; Dedhar, S.; Lin, K.S. 2-[18F]fluoroethanol and 3-[18F]fluoropropanol: Facile preparation, biodistribution in mice, and their application as nucleophiles in the synthesis of [18F]fluoroalkyl aryl ester and ether PET tracers. Nucl. Med. Biol. 2013, 40, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Luurtsema, G.; Schuit, R.C.; Takkenkamp, K.; Lubberink, M.; Hendrikse, N.H.; Windhorst, A.D.; Molthoff, C.F.; Tolboom, N.; van Berckel, B.N.; Lammertsma, A.A. Peripheral metabolism of [18F]FDDNP and cerebral uptake of its labelled metabolites. Nucl. Med. Biol. 2008, 35, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, S.S.; Shetty, H.U.; Ichise, M.; Fujita, M.; Imaizumi, M.; Liow, J.S.; Shah, J.; Musachio, J.L.; Pike, V.W.; Innis, R.B. PET imaging of the dopamine transporter with 18F-FECNT: A polar radiometabolite confounds brain radioligand measurements. J. Nucl. Med. 2006, 47, 520–527. [Google Scholar] [PubMed]
- Ponde, D.E.; Dence, C.S.; Oyama, N.; Kim, J.; Tai, Y.C.; Laforest, R.; Siegel, B.A.; Welch, M.J. 18F-fluoroacetate: A potential acetate analog for prostate tumor imaging—In vivo evaluation of 18F-fluoroacetate versus 11C-acetate. J. Nucl. Med. 2007, 48, 420–428. [Google Scholar] [PubMed]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfi, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Kuruvilla, S.A.; Galitovskiy, V.; Pan, M.L.; Grando, S.A.; Mukherjee, J. Targeting histone deacetylase in lung cancer for early diagnosis: 18F-FAHA PET/CT imaging of NNK-treated A/J mice model. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 324–332. [Google Scholar] [PubMed]
- Neal, J.W.; Sequist, L.V. Exciting new targets in lung cancer therapy: ALK, IGF-1R, HDAC, and Hh. Curr. Treat. Opt. Oncol. 2010, 11, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Gordon, W.; Galitovskiy, V.; Edwards, R.; Andersen, B.; Grando, S.A. The tobacco carcinogen nitrosamine induces a differential gene expression response in tumour susceptible A/J and resistant C3H mouse lungs. Eur. J. Cancer 2013, 49, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Galitovskiy, V.; Kuruvilla, S.A.; Sevriokov, E.; Corches, A.; Pan, M.L.; Kalantari-Dehaghi, M.; Chernyavsky, A.I.; Mukherjee, J.; Grando, S.A. Development of novel approach to diagnostic imaging of lung cancer with 18F-Nifene PET/CT using A/J mice treated with NNK. J. Cancer Res. Ther. 2013, 1, 128–137. [Google Scholar]
- Bonomi, R.; Mukhopadhyay, U.; Shavrin, A.; Yeh, H.H.; Majhi, A.; Dewage, S.W.; Najjar, A.; Lu, X.; Cisneros, G.A.; Tong, W.P.; et al. Novel Histone Deacetylase Class IIa Selective Substrate Radiotracers for PET Imaging of Epigenetic Regulation in the Brain. PLoS ONE 2015, 10, e0133512. [Google Scholar] [CrossRef] [PubMed]
- Zeglis, B.M.; Pillarsetty, N.; Divilov, V.; Blasberg, R.A.; Lewis, J.S. The synthesis and evaluation of N1-(4-(2-[18F]-fluoroethyl)phenyl)-N8-hydroxyoctanediamide ([18F]-FESAHA), a PET radiotracer designed for the delineation of histone deacetylase expression in cancer. Nucl. Med. Biol. 2011, 38, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, J.A.; Keliher, E.J.; Marinelli, B.; Reiner, T.; Weissleder, R.; Mazitschek, R. In vivo PET imaging of histone deacetylases by 18F-suberoylanilide hydroxamic acid (18F-SAHA). J. Med. Chem. 2011, 54, 5576–5582. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Muench, L.; Reid, A.; Chen, J.; Kang, Y.; Hooker, J.M.; Volkow, N.D.; Fowler, J.S.; Kim, S.W. Radionuclide labeling and evaluation of candidate radioligands for PET imaging of histone deacetylase in the brain. Bioorg. Med. Chem. Lett. 2013, 23, 6700–6705. [Google Scholar] [CrossRef] [PubMed]
- Majdzadeh, N.; Morrison, B.E.; D’Mello, S.R. Class IIA HDACs in the regulation of neurodegeneration. Front. Biosci. 2008, 13, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Riester, D.; Hildmann, C.; Grunewald, S.; Beckers, T.; Schwienhorst, A. Factors affecting the substrate specificity of histone deacetylases. Biochem. Biophys. Res. Commun. 2007, 357, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zou, X.; Berger, A.D.; Twiss, C.; Peng, Y.; Li, Y.; Chiu, J.; Guo, H.; Satagopan, J.; Wilton, A.; et al. Increased expression of histone deacetylaces (HDACs) and inhibition of prostate cancer growth and invasion by HDAC inhibitor SAHA. Am. J. Transl. Res. 2009, 1, 62–71. [Google Scholar] [PubMed]
- Gediya, L.K.; Chopra, P.; Purushottamachar, P.; Maheshwari, N.; Njar, V.C. A new simple and high-yield synthesis of suberoylanilide hydroxamic acid and its inhibitory effect alone or in combination with retinoids on proliferation of human prostate cancer cells. J. Med. Chem. 2005, 48, 5047–5051. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Strobel, E.; Ralbovsky, J.; Galemmo, R.A., Jr. Improved solution- and solid-phase preparation of hydroxamic acids from esters. J. Org. Chem. 2005, 70, 4873–4875. [Google Scholar] [CrossRef] [PubMed]
- Hooker, J.M.; Xu, Y.; Schiffer, W.; Shea, C.; Carter, P.; Fowler, J.S. Pharmacokinetics of the potent hallucinogen, salvinorin A in primates parallels the rapid onset and short duration of effects in humans. Neuroimage 2008, 41, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Schroeder, F.A.; Wey, H.Y.; Borra, R.; Wagner, F.F.; Reis, S.; Kim, S.W.; Holson, E.B.; Haggarty, S.J.; Hooker, J.M. In vivo imaging of histone deacetylases (HDACs) in the central nervous system and major peripheral organs. J. Med. Chem. 2014, 57, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Banister, S.D.; Wilkinson, S.M.; Longworth, M.; Stuart, J.; Apetz, N.; English, K.; Brooker, L.; Goebel, C.; Hibbs, D.E.; Glass, M.; et al. The synthesis and pharmacological evaluation of adamantane-derived indoles: Cannabimimetic drugs of abuse. ACS Chem. Neurosci. 2013, 4, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, N.; Hama, T.; Kawada, M.; Hasui, A.; Konishi, R.; Shiwa, S.; Ochi, Y.; Futaki, S.; Kitagawa, K. Adamantane as a brain-directed drug carrier for poorly absorbed drug. 2. AZT derivatives conjugated with the 1-adamantane moiety. J. Pharm. Sci. 1994, 83, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Wanka, L.; Iqbal, K.; Schreiner, P.R. The lipophilic bullet hits the targets: Medicinal chemistry of adamantane derivatives. Chem. Rev. 2013, 113, 3516–3604. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, B.; Ponpandian, T.; Kachhadia, V.; Bharathimohan, K.; Vignesh, R.; Sivasudar, V.; Narayanan, S.; Mandar, B.; Praveen, R.; Saranya, N.; et al. Discovery of adamantane based highly potent HDAC inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2532–2537. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Oda, K.; Ishiwata, K. Age-related changes of the [11C]CFT binding to the striatal dopamine transporters in the Fischer 344 rats: A PET study. Ann. Nucl. Med. 2003, 17, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, F.A.; Wang, C.; Van de Bittner, G.C.; Neelamegam, R.; Takakura, W.R.; Karunakaran, A.; Wey, H.Y.; Reis, S.A.; Gale, J.; Zhang, Y.L.; et al. PET imaging demonstrates histone deacetylase target engagement and clarifies brain penetrance of known and novel small molecule inhibitors in rat. ACS Chem. Neurosci. 2014, 5, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [PubMed]
- Binaschi, M.; Boldetti, A.; Gianni, M.; Maggi, C.A.; Gensini, M.; Bigioni, M.; Parlani, M.; Giolitti, A.; Fratelli, M.; Valli, C.; et al. Antiproliferative and differentiating activities of a novel series of histone deacetylase inhibitors. ACS Med. Chem. Lett. 2010, 1, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, F.A.; Lewis, M.C.; Fass, D.M.; Wagner, F.F.; Zhang, Y.L.; Hennig, K.M.; Gale, J.; Zhao, W.N.; Reis, S.; Barker, D.D.; et al. A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests. PLoS ONE 2013, 8, e71323. [Google Scholar] [CrossRef] [PubMed]
- Wey, H.Y.; Wang, C.; Schroeder, F.A.; Logan, J.; Price, J.C.; Hooker, J.M. Kinetic Analysis and Quantification of [11C]Martinostat for In Vivo HDAC Imaging of the Brain. ACS Chem. Neurosci. 2015, 6, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Strebl, M.G.; Wang, C.; Schroeder, F.A.; Placzek, M.S.; Wey, H.Y.; Van de Bittner, G.C.; Neelamegam, R.; Hooker, J.M. Development of a Fluorinated Class-I HDAC Radiotracer Reveals Key Chemical Determinants of Brain Penetrance. ACS Chem. Neurosci. 2016, 7, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [18F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Beyzavi, M.H.; Mandal, D.; Strebl, M.G.; Neumann, C.N.; D’Amato, E.M.; Chen, J.; Hooker, J.M.; Ritter, T. 18F-Deoxyfluorination of Phenols via Ru pi-Complexes. ACS Cent. Sci. 2017, 3, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.N.; Hooker, J.M.; Ritter, T. Concerted nucleophilic aromatic substitution with 19F− and 18F−. Nature 2016, 534, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Ritter, T. PhenoFluorMix: Practical chemoselective deoxyfluorination of phenols. Org. Lett. 2015, 17, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Hooker, J.M.; Otto, N.; Win, K.; Muench, L.; Shea, C.; Carter, P.; King, P.; Reid, A.E.; Volkow, N.D.; et al. Whole-body pharmacokinetics of HDAC inhibitor drugs, butyric acid, valproic acid and 4-phenylbutyric acid measured with carbon-11 labeled analogs by PET. Nucl. Med. Biol. 2013, 40, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Fass, D.M.; Shah, R.; Ghosh, B.; Hennig, K.; Norton, S.; Zhao, W.N.; Reis, S.A.; Klein, P.S.; Mazitschek, R.; Maglathlin, R.L.; et al. Short-Chain HDAC Inhibitors Differentially Affect Vertebrate Development and Neuronal Chromatin. ACS Med. Chem. Lett. 2010, 2, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, P.; Poddar, B.; Parmar, V. Fatal cardiac malformation in fetal valproate syndrome. Indian J. Pediatr. 2001, 68, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Eessalu, T.E.; Barth, V.N.; Mitch, C.H.; Wagner, F.F.; Hong, Y.; Neelamegam, R.; Schroeder, F.A.; Holson, E.B.; Haggarty, S.J.; et al. Design, synthesis, and evaluation of hydroxamic acid-based molecular probes for in vivo imaging of histone deacetylase (HDAC) in brain. Am. J. Nucl. Med. Mol. Imaging 2013, 4, 29–38. [Google Scholar] [PubMed]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; La Thangue, N.B.; Brown, R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2003, 2, 721–728. [Google Scholar] [PubMed]
- Giles, F.; Fischer, T.; Cortes, J.; Garcia-Manero, G.; Beck, J.; Ravandi, F.; Masson, E.; Rae, P.; Laird, G.; Sharma, S.; et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin. Cancer Res. 2006, 12, 4628–4635. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, L.; Kalin, J.; Liow, J.S.; Gladding, R.L.; Innis, R.B.; Kozikowski, A.P.; Pike, V.W. Synthesis and evaluation of [methyl-11C]KB631—A candidate radioligand for histone deacetylase isozyme 6 (HDAC6). J. Label. Comp. Radiopharm. 2013, 56, S319. [Google Scholar]
- Kalin, J.H.; Butler, K.V.; Akimova, T.; Hancock, W.W.; Kozikowski, A.P. Second-generation histone deacetylase 6 inhibitors enhance the immunosuppressive effects of Foxp3+ T-regulatory cells. J. Med. Chem. 2012, 55, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, Y.; Kalin, J.H.; Cai, L.; Kozikowski, A.P.; Pike, V.W. Exploration of the labeling of [11C]tubastatin A at the hydroxamic acid site with [11C]carbon monoxide. J. Label. Comp. Radiopharm. 2016, 59, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Li, F.; Jiang, S.; Li, Z. Novel 64Cu-Labeled CUDC-101 for In Vivo PET Imaging of Histone Deacetylases. ACS Med. Chem. Lett. 2013, 4, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew. Chem. Int. Ed. Engl. 2008, 47, 8998–9033. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhai, H.X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDc-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Lu, Z.; Luo, R.Z.; Zhang, X.; Seto, E.; Liao, W.S.; Yu, Y. Multiple histone deacetylases repress tumor suppressor gene ARHI in breast cancer. Int. J. Cancer 2007, 120, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Hooker, J.M.; Kim, S.W.; Alexoff, D.; Xu, Y.; Shea, C.; Reid, A.; Volkow, N.; Fowler, J.S. Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [11C]MS-275 using Positron Emission Tomography. ACS Chem. Neurosci. 2010, 1, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Dul, E.; Sung, C.M.; Chen, Z.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Liu, R.; Lago, A.; Hofmann, G.; et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J. Pharmacol. Exp. Ther. 2003, 307, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Simonini, M.V.; Camargo, L.M.; Dong, E.; Maloku, E.; Veldic, M.; Costa, E.; Guidotti, A. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc. Natl. Acad. Sci. USA 2006, 103, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Hooker, J.M.; Reibel, A.T.; Hill, S.M.; Schueller, M.J.; Fowler, J.S. One-pot, direct incorporation of [11C]CO2 into carbamates. Angew. Chem. Int. Ed. Engl. 2009, 48, 3482–3485. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Kang, Y.; Muench, L.; Reid, A.; Caesar, S.; Jean, L.; Wagner, F.; Holson, E.; Haggarty, S.J.; Weiss, P.; et al. Image-guided synthesis reveals potent blood-brain barrier permeable histone deacetylase inhibitors. ACS Chem. Neurosci. 2014, 5, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.L.; Hennig, K.; Gale, J.P.; Hong, Y.; Cha, A.; Riley, M.; Wagner, F.; Haggarty, S.J.; Holson, E.; et al. Class I HDAC imaging using [3H]CI-994 autoradiography. Epigenetics 2013, 8, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. PET radiotracers: Crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
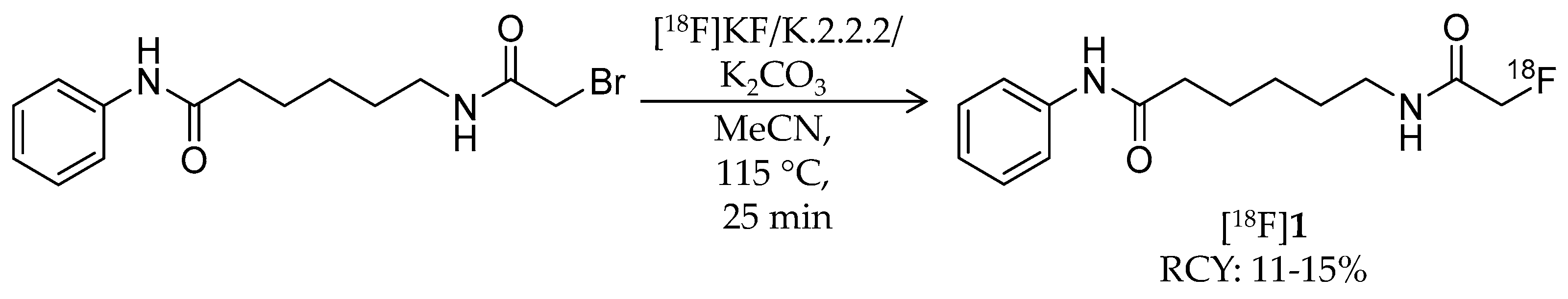{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
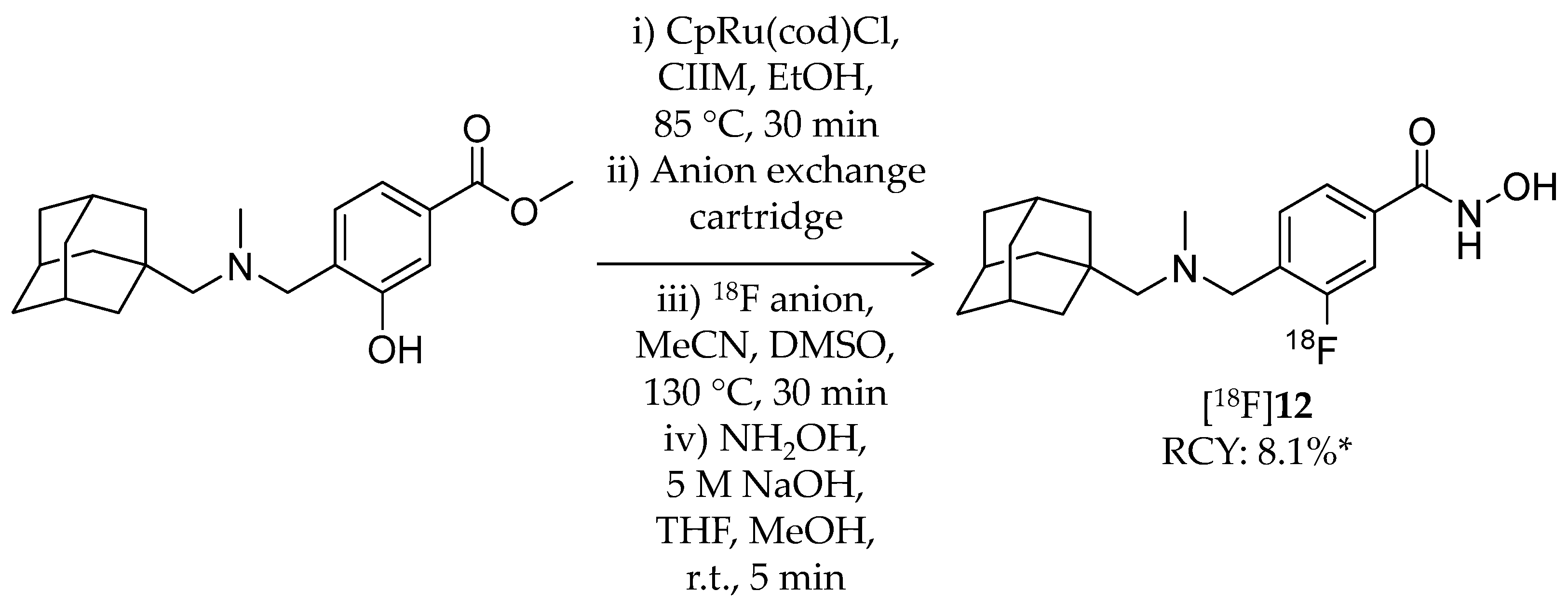{kind=link}
{kind=link}
{kind=link}
{kind=link}
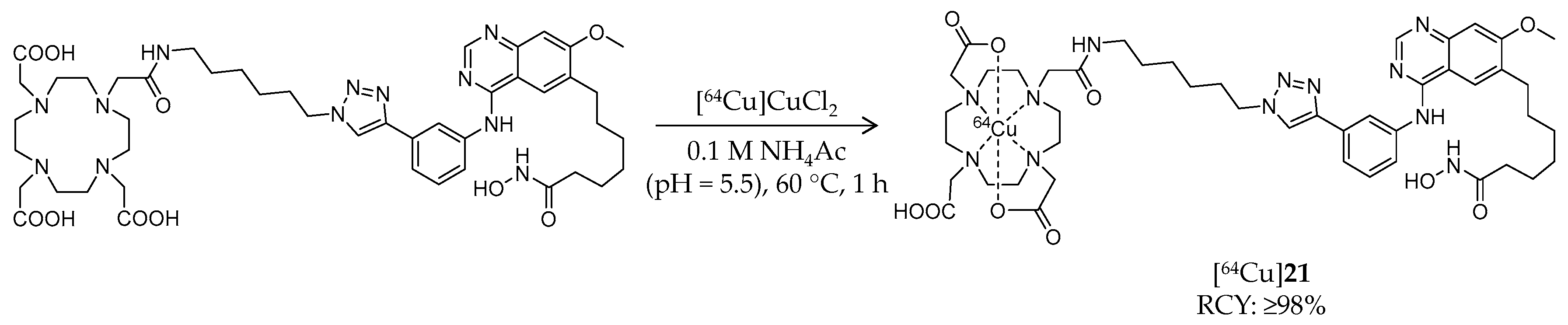{kind=link}
{kind=link}
{kind=link}
| Compound | MW | Log D | RCY | Target | IC50 for HDACs | In Vivo Properties in Rodents | Reference |
|---|---|---|---|---|---|---|---|
| [18F]1 | 265.32 | 1.39 | 11–15% | Class IIa | – | Brain uptake: 0.44 and 0.40% ID/g at 5 and 60 min, respectively (rat) | [26,43,44,46] |
| [18F]2 | 283.31 | – | 25% | Class IIa | – | Brain uptake: SUVs were around 1 in various brain regions during the 60-min scan (rat) | [57] |
| [18F]3 | 301.30 | – | 22% | Class IIa | – | Brain uptake: SUVs were around 1 in various brain regions during the 60-min scan (rat) | [57] |
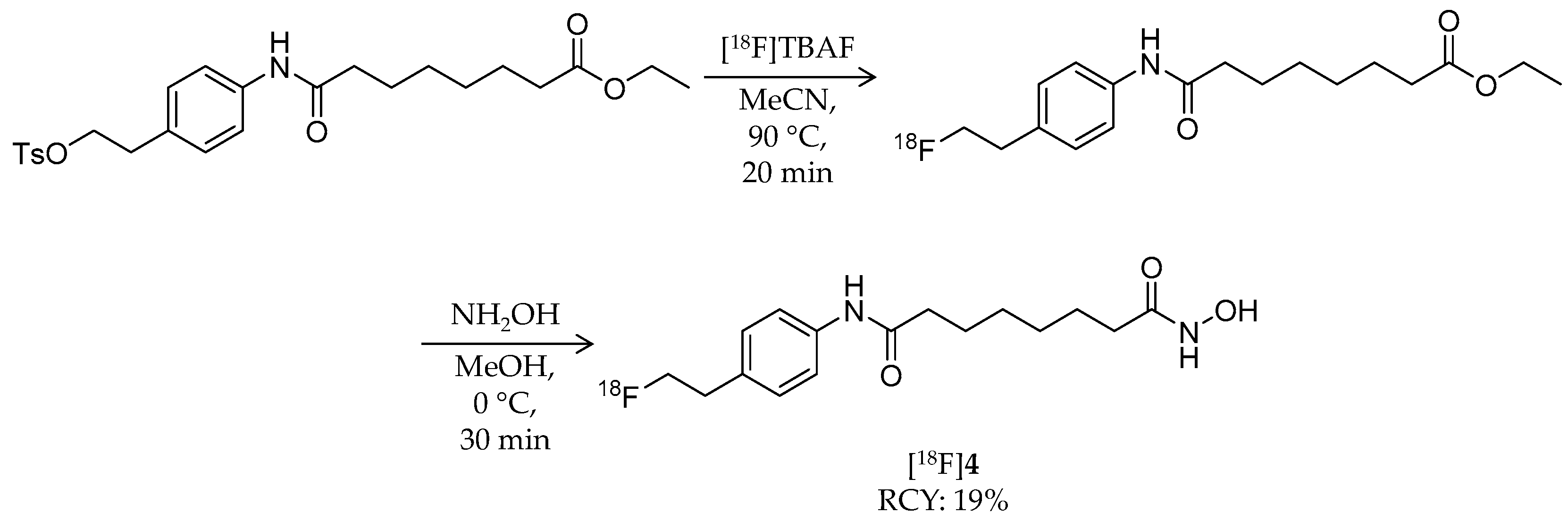
| [18F]4 | 309.37 | 1.01 | 19% | Non-selective | HDAC1: 56 nM HDAC5: 67 nM HDAC6: 3 nM HDAC7: 85 nM | Brain uptake: 1.0, 0.7, and 0.4% ID/g at 30, 60, and 120 min, respectively Defluorination: 9.6, 11.1, and 13.4% ID/g bone uptake at 30, 60, and 120 min, respectively (tumor-bearing mouse) | [58] |
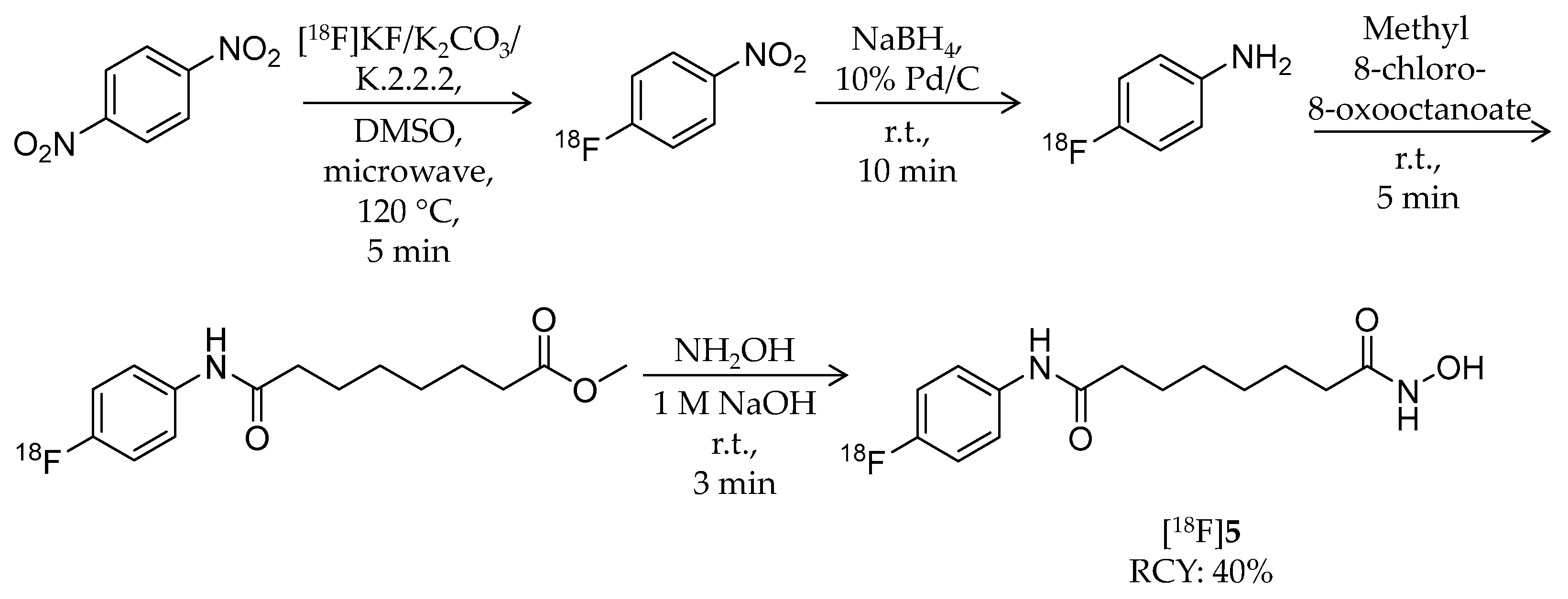
| [18F]5 | 281.32 | – | 40% | HDAC1–3/6 | HDAC1: 9.0 nM HDAC2: 13 nM HDAC3: 24 nM HDAC6: 50 nM | Brain uptake: Very limited in biodistribution study (mouse/tumor-bearing mouse) | [59] |
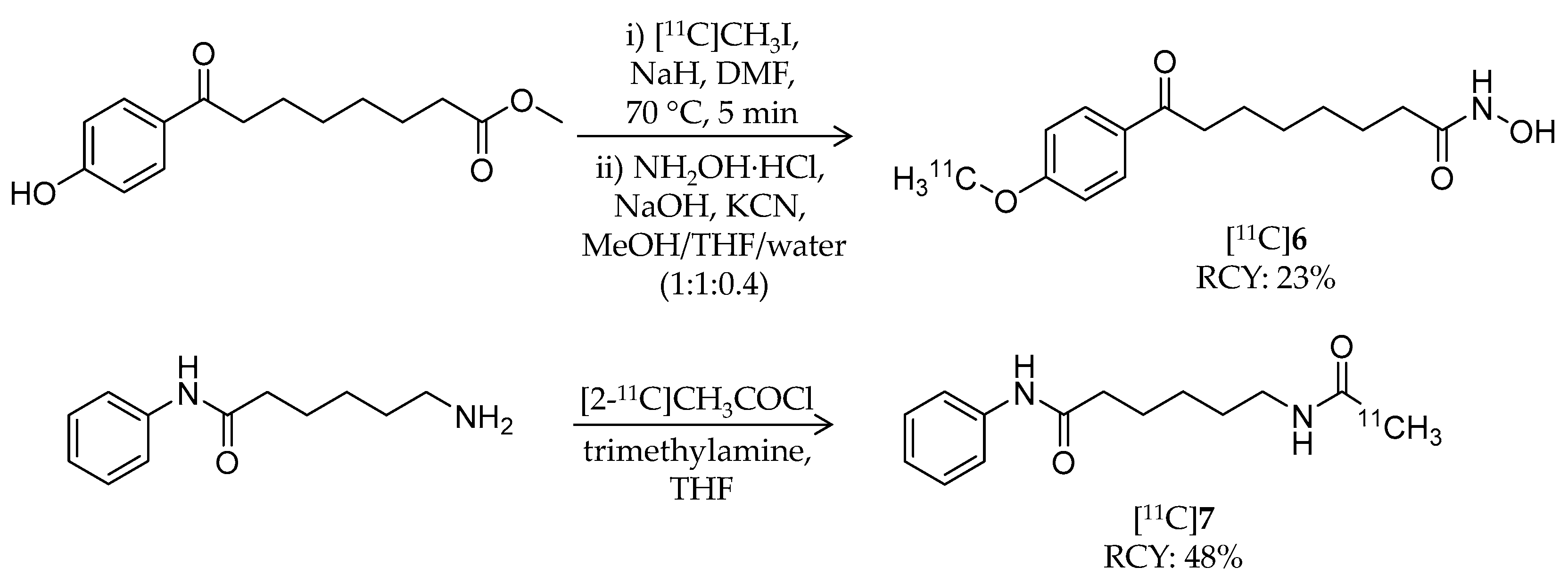
| [11C]6 | 278.34 | 0.5 | 23% | – | – | – | [60] |
| [11C]7 | 247.33 | 1.7 1 | 48% | – | – | – | [60] |
| Compound | MW | Log D | RCY | Target | IC50 for HDACs | In Vivo Properties in Rodents | Reference |
|---|---|---|---|---|---|---|---|
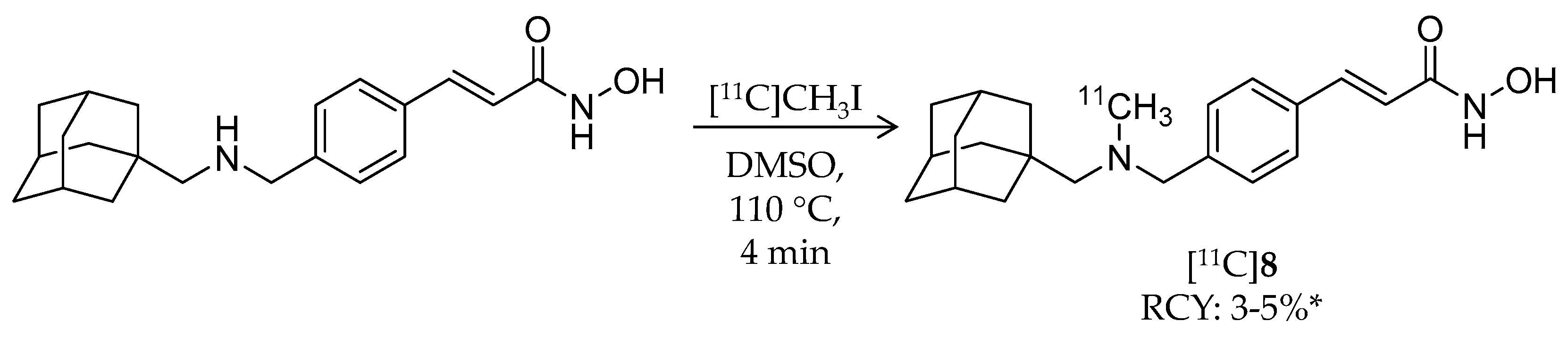
| [11C]8 | 353.49 | 2.03 | 3–5% 2 | Class I/IIb | HDAC1: 0.3 nM HDAC2: 2.0 nM HDAC3: 0.6 nM HDAC6: 4.1 nM | Brain uptake: Around 0.5% ID/cc during the 60-min scan Metabolism: Radiometabolites in the brain were limited (rat) | [28,68,74,78] |
| [18F]9 | 305.38 | 3.07 1 | 7% | Class I/IIb | HDAC1: 1.6 nM HDAC2: 14 nM HDAC3: 0.5 nM HDAC6: 12 nM | Brain uptake: Rapid brain uptake and limited washout were observed Specific binding in the brain was confirmed in a blocking study (rat) | [79] |
| [18F]10 | 357.46 | 3.77 1 | 0.9% | Class I/IIb | HDAC1: 0.8 nM HDAC2: 6.4 nM HDAC3: 0.5 nM HDAC6: 9.5 nM | Brain uptake: Rapid brain uptake and limited washout were observed Specific binding in the brain was confirmed in a blocking study (rat) | [79] |
| [18F]11 | 371.49 | 4.17 1 | 0.3% | Class I/IIb | HDAC1: 0.8 nM HDAC2: 7.0 nM HDAC3: 0.8 nM HDAC6: 12 nM | Brain uptake: Rapid brain uptake and limited washout were observed Specific binding in the brain was confirmed in a blocking study (rat) | [79] |
| [18F]12 | 345.45 | – | 8.1% 2 | HDAC6 | HDAC6: 60 nM Others: ≥1 μM | Brain uptake: Rapid brain uptake and limited washout were observed Specific binding in the brain was confirmed in a blocking study (rat) | [80] |
| Compound | MW | Log D | RCY | Target | IC50 for HDACs | In Vivo Properties in Rodents | Reference |
|---|---|---|---|---|---|---|---|
| [11C]13 | 87.11 | 1.02 | 31–50% | Class I | HDAC1: 16 μM HDAC2: 12 μM HDAC3: 9 μM HDAC8: 15 μM | – | [86,87] |
| [11C]14 | 149.18 | −0.20 | 40–55% | Class I | HDAC1: 64 μM HDAC2: 65 μM HDAC3: 260 μM HDAC8: 93 μM | – | [86,87] |
| [11C]15 | 143.21 | 0.26 | 38–50% | Class I | HDAC1: 39 μM HDAC2: 62 μM HDAC3: 161 μM HDAC8: 1103 μM | – | [86,87] |
| [11C]16 | 331.37 | 2.03 1 | 9% 3 | HDAC1–3/6 | HDAC1: 5 nM HDAC2: 32 nM HDAC3: 3.4 nM HDAC6: 5 nM | Brain uptake: Around 0.1% ID/cc immediately after i.v. injection (rat) | [89] |
| [11C]17 | 348.43 | 2.66 1 | 3% 3 | HDAC1–3/6 | HDAC1: 0.2 nM HDAC2: 1.2 nM HDAC3: 0.4 nM HDAC6: 1.6 nM | Brain uptake: Around 0.15% ID/cc immediately after i.v. injection (rat) | [89] |
| [11C]18 | 295.33 | 2.61 1 | 5% 3 | HDAC8 | HDAC8: 18 nM | Brain uptake: Around 0.2% ID/cc immediately after i.v. injection (rat) | [89] |
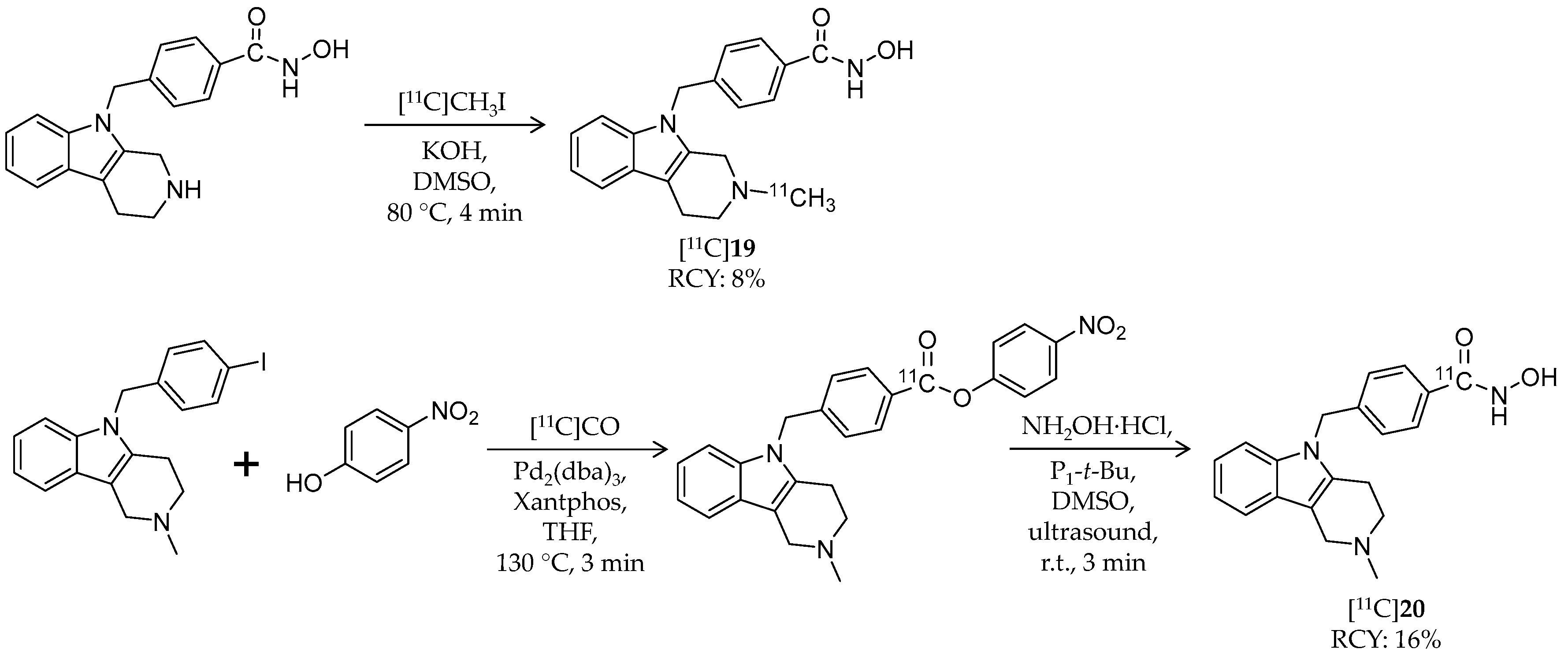
| [11C]19 | 334.41 | 1.33 2 | 8% | HDAC6 | HDAC1: 5.2 μM HDAC6: 1.4 nM | Brain uptake: Radioactivity peaked in forebrain at 0.44 SUV and in cerebellum at 0.48 SUV (rat) | [93,94] |
| [11C]20 | 334.41 | 1.33 2 | 16% | HDAC6 | HDAC6: 4 nM Others: ≥1.3 μM | – | [82,95] |
| [64Cu]21 | 1009.02 | – | ≥98% | Class I/IIb/III | Class I, IIb, III: 94 nM | Brain uptake: Biodistribution study was performed but brain uptake was not assessed (tumor-bearing mouse) | [96] |
| Compound | MW | Log D | RCY | Target | IC50 for HDACs | In Vivo Properties in Rodents | Reference |
|---|---|---|---|---|---|---|---|
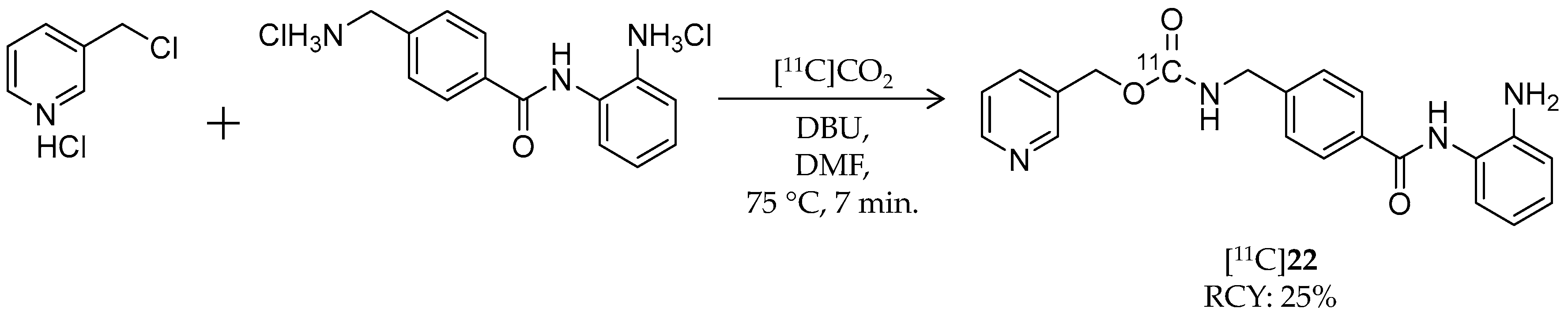
| [11C]22 | 375.42 | 1.8 | 25% | Class I | HDAC1: 60 nM HDAC2: 153 nM | Brain uptake: <0.10% ID/cm3 after 3 min Metabolism: 80% of radioactivity in the brain was unchanged [11C]22 (rat) | [101,105] |
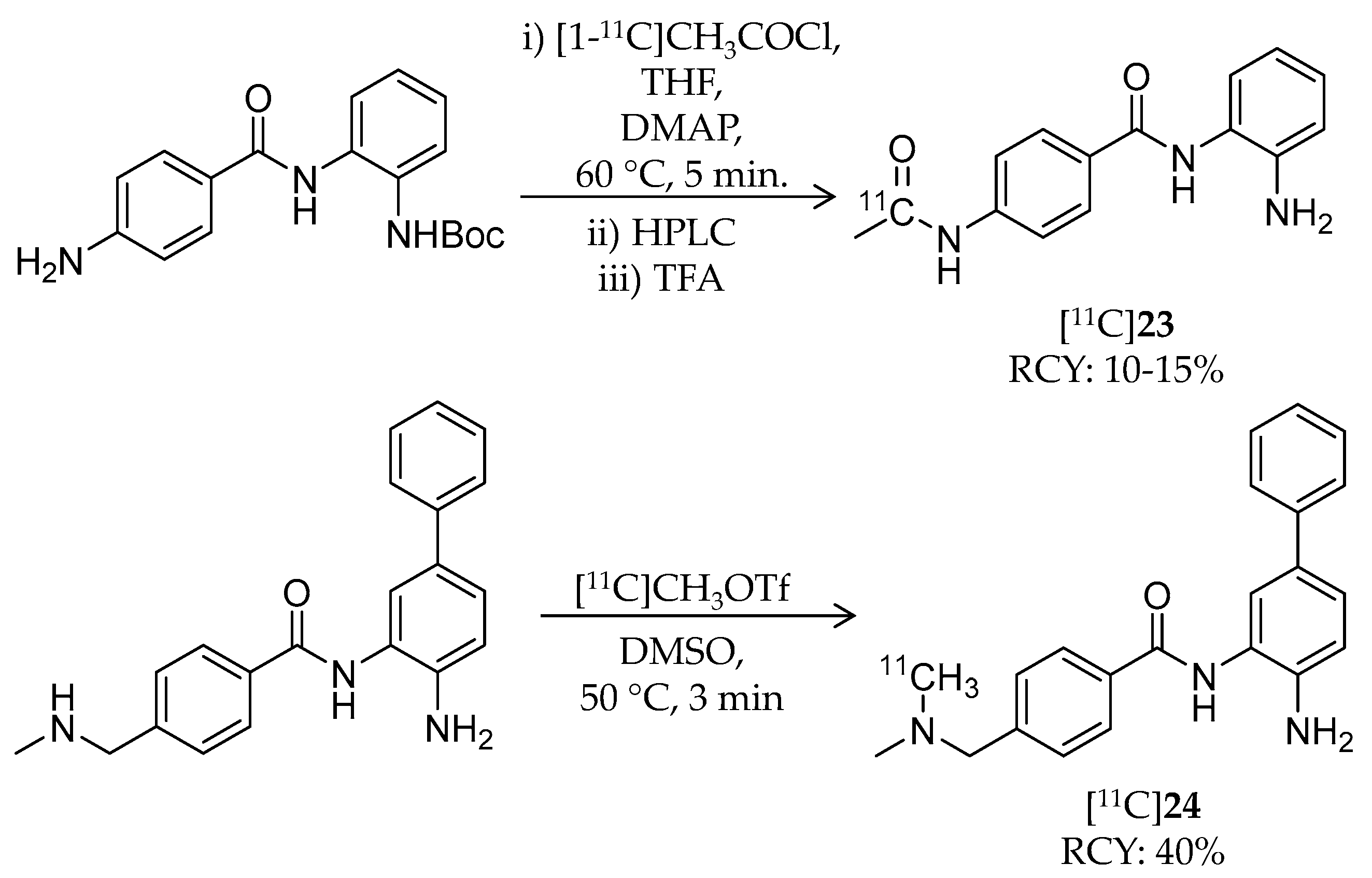
| [11C]23 | 268.30 | 1.0 | 10–15% | Class I | HDAC1: 45 nM HDAC2: 31 nM HDAC3: 20 nM | – | [105] |
| [11C]24 | 344.45 | 2.1 | 40% | Class I | HDAC1: 10 nM HDAC2: 20 nM | – | [105] |
| Compound | Target | Findings in PET Studies in NHPs | Reference |
|---|---|---|---|
| [18F]1 | Class IIa | Radioactivity accumulation was brain region specific in rhesus macaques (~0.03% ID/g) By 30 min p.i., almost all [18F]1 was metabolized to [18F]FACE Radioactivity in the brain was decreased by pre-treatment with SAHA in a dose-dependent manner | [26,46] |
| [11C]6 [11C]7 | – | Brain uptake of both ligands was very low in baboons (~0.004% ID/cc) The unchanged fraction of both ligands in baboon plasma was less than 20% at 30 min p.i. | [60] |
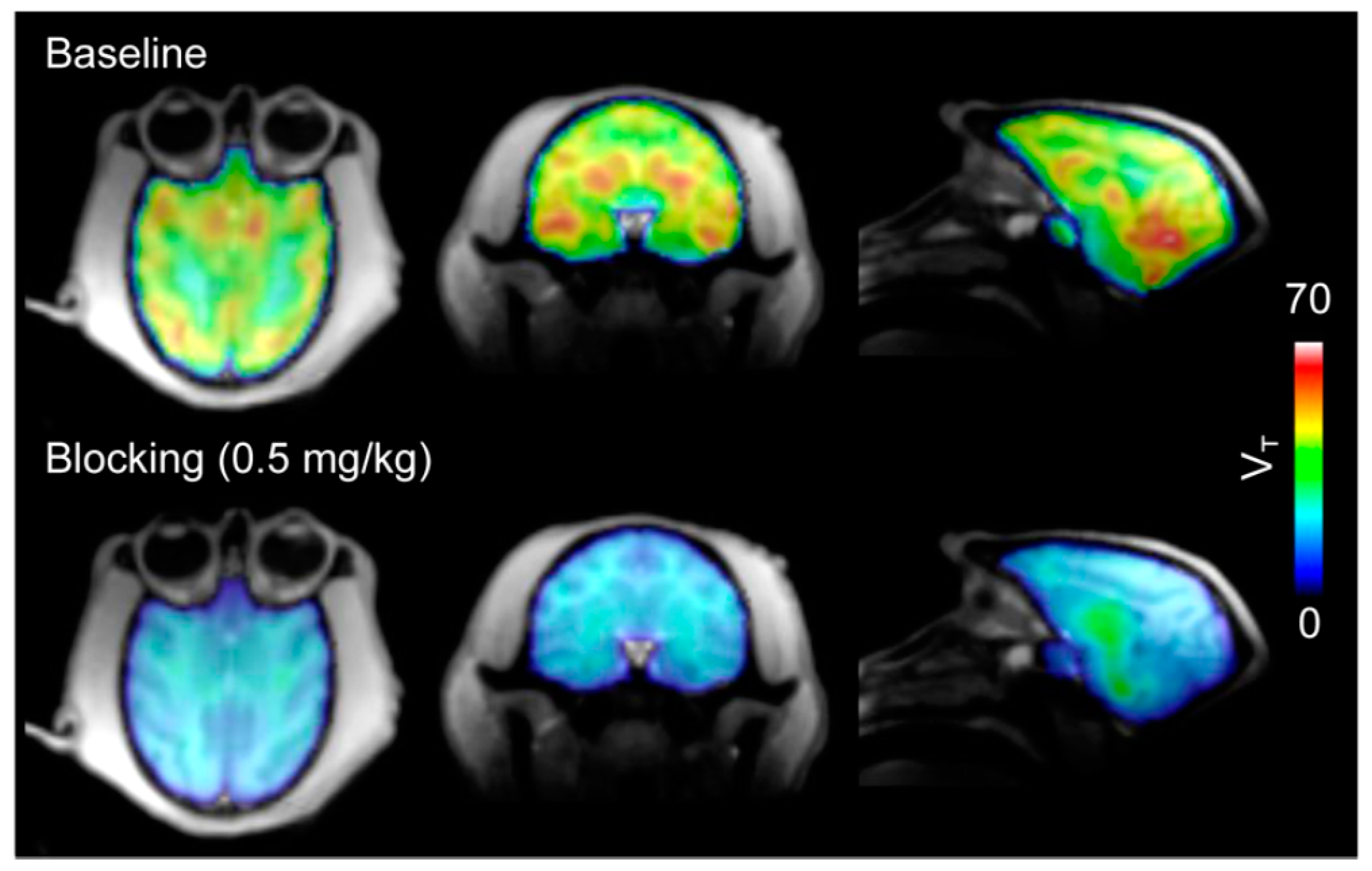
| [11C]8 | Class I/IIb | Regional VT (90-min scan) in the baboon brain ranged from 29.9 to 54.4 mL/cm3 Parent fraction in plasma decreased gradually (50% at 30 min p.i. and 40% at 60 min p.i.) The mean VT in the brain decreased by 82.3 ± 5.5% with a 1-mg/kg blocking dose | [28,68,78] |
| [18F]9 [18F]10 [18F]11 | Class I/IIb | Whole brain SUV30–60 min of [18F]9, [18F]10, and [18F]11 in baboons were 0.57, 1.2, and 1.8, respectively (2.3 for [11C]8) [18F]11 showed the highest correlation in regional brain distribution with [11C]8 | [79] |
| [18F]12 | HDAC6 | Excellent brain uptake (SUV ≈ 3 around 30 min p.i.) was observed in baboons Nonspecific binding in the brain determined with 1 mg/kg unlabeled 12 was low (<1 SUV) | [80] |
| [11C]13 [11C]14 [11C]15 | Class I | In the baboon brain, uptake of the three ligands was low (~0.006% ID/cc) | [86] |
| [11C]16 [11C]17 | HDAC1–3/6 | Brain uptake in baboons was very low over the 80-min scan time | [89] |
| [11C]18 | HDAC8 | Brain uptake in baboons was very low over the 80-min scan time | [89] |
| [11C]22 | Class I | Brain uptake in baboons was very low (<0.001% ID/cc) over the 90-min scan time Approximately 60% of the plasma radioactivity was unchanged ligand at 40 min p.i. | [101] |
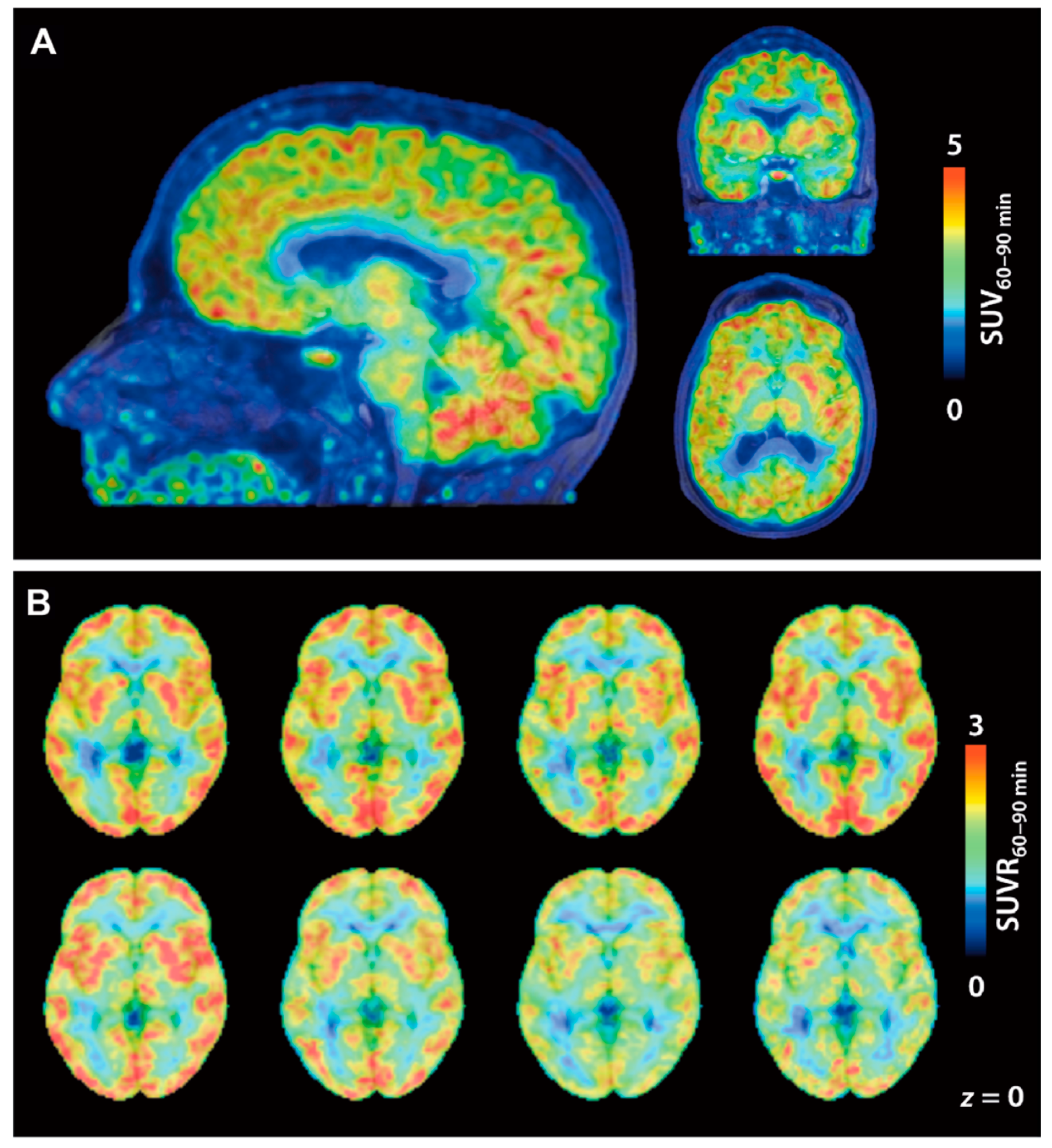
| [11C]23 [11C]24 | Class I | Total VT values of [11C]23 and [11C]24 in the baboon brain were 0.41 and 12 mL/cm3, respectively The degree of VT reduction of [11C]24 by unlabeled 24 or SAHA (1 mg/kg) ranged from 8–24% in various brain regions | [105] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tago, T.; Toyohara, J. Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases. Molecules 2018, 23, 300. https://doi.org/10.3390/molecules23020300
Tago T, Toyohara J. Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases. Molecules. 2018; 23(2):300. https://doi.org/10.3390/molecules23020300
Chicago/Turabian StyleTago, Tetsuro, and Jun Toyohara. 2018. "Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases" Molecules 23, no. 2: 300. https://doi.org/10.3390/molecules23020300
APA StyleTago, T., & Toyohara, J. (2018). Advances in the Development of PET Ligands Targeting Histone Deacetylases for the Assessment of Neurodegenerative Diseases. Molecules, 23(2), 300. https://doi.org/10.3390/molecules23020300