A Systematic Review of Antiamyloidogenic and Metal-Chelating Peptoids: Two Structural Motifs for the Treatment of Alzheimer’s Disease

Abstract
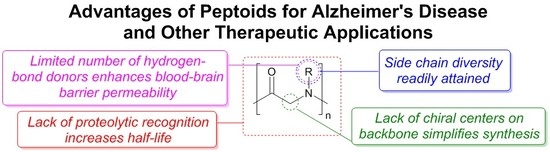
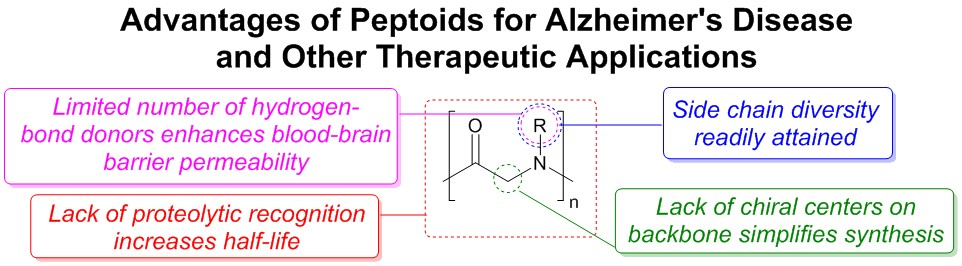
:
1. Introduction
2. Amyloid-Targeting Peptoids
2.1. Overview
2.2. Peptoid-Based Aβ1–42-Aggregation Inhibitors
2.3. Peptoid-Based Aβ1–40-Aggregation Inhibitors
2.4. Peptoids Targeting the Apolipoprotein E4-Aβ Interaction
2.5. Comparative Structure-Activity Relationship for Amyloid-Targeting Peptoids
3. Metal-Chelating Peptoids
3.1. Connections between Metal Dysregulation, the Amyloid Pathway, and AD Pathology
3.2. Cu(II)-Binding Peptoids
3.3. Zn(II)-Binding Peptoids
3.4. Fe(II)-Binding Peptoids
3.5. Screening Approaches for the Discovery of Metal-Binding Peptoids
3.6. Comparative Structure-Activity Relationship for Metal-Chelating Peptoids
3.7. Advantages of Metal-Chelating Peptoids
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.G.; Francis, P.T.; Schwam, E.; Payne-Parrish, J. Cholinesterase inhibitors used in the treatment of Alzheimer’s disease: The relationship between pharmacological effects and clinical efficacy. Drugs Aging 2004, 21, 453–478. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [PubMed]
- Robert, A.; Liu, Y.; Nguyen, M.; Meunier, B. Regulation of Copper and Iron Homeostasis by Metal Chelators: A Possible Chemotherapy for Alzheimer’s Disease. Acc. Chem. Res. 2015, 48, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Weiner, M.W.; Veitch, D.P.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Green, R.C.; Harvey, D.; Jack, C.R.; Jagust, W.; Morris, J.C.; et al. The Alzheimer’s Disease Neuroimaging Initiative 3: Continued innovation for clinical trial improvement. Alzheimer’s Dement. 2017, 13, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, M.C.; Galvin, J.E. Using biomarkers to improve detection of Alzheimer’s disease. Neurodegener. Dis. Manag. 2011, 1, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Scarano, S.; Lisi, S.; Ravelet, C.; Peyrin, E.; Minunni, M. Detecting Alzheimer’s disease biomarkers: From antibodies to new bio-mimetic receptors and their application to established and emerging bioanalytical platforms—A critical review. Anal. Chim. Acta 2016, 940, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Banks, W.A. Developing drugs that can cross the blood-brain barrier: Applications to Alzheimer’s disease. BMC Neurosci. 2008, 9, S2. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; et al. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar] [CrossRef] [PubMed]
- Guarna, A.; Trabocchi, A. Peptidomimetics in Organic and Medicinal Chemistry: The Art of Transforming Peptides in Drugs; John Wiley & Sons: Chichester, UK, 2014; pp. 123–135. ISBN 9781119950608. [Google Scholar]
- Udugamasooriya, G. Peptoids: An Emerging Class of Peptidomimetics for Cancer Therapy and Diagnostics. J. Biomol. Res. Ther. 2014, 3. [Google Scholar] [CrossRef]
- Horne, W.S. Peptide and peptoid foldamers in medicinal chemistry. Expert Opin. Drug Discov. 2011, 6, 1247–1262. [Google Scholar] [CrossRef] [PubMed]
- Mándity, I.M.; Fülöp, F. An overview of peptide and peptoid foldamers in medicinal chemistry. Expert Opin. Drug Discov. 2015, 10, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, R.N.; Kerr, J.M.; Kent, S.B.H.; Moos, W.H. Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar] [CrossRef]
- Olivos, H.J.; Alluri, P.G.; Reddy, M.M.; Salony, D.; Kodadek, T. Microwave-Assisted Solid-Phase Synthesis of Peptoids. Org. Lett. 2002, 4, 4057–4059. [Google Scholar] [CrossRef] [PubMed]
- Roy, O.; Caumes, C.; Esvan, Y.; Didierjean, C.; Faure, S.; Taillefumier, C. The tert-Butyl Side Chain: A Powerful Means to Lock Peptoid Amide Bonds in the Cis Conformation. Org. Lett. 2013, 15, 2246–2249. [Google Scholar] [CrossRef] [PubMed]
- Astle, J.M.; Udugamasooriya, D.G.; Smallshaw, J.E.; Kodadek, T. A VEGFR2 Antagonist and Other Peptoids Evade Immune Recognition. Int. J. Pept. Res. Ther. 2008, 14, 223–227. [Google Scholar] [CrossRef]
- Zuckermann, R.N.; Kodadek, T. Peptoids as potential therapeutics. Curr. Opin. Mol. Ther. 2009, 11, 299–307. [Google Scholar] [PubMed]
- Kwon, Y.-U.; Kodadek, T. Quantitative Evaluation of the Relative Cell Permeability of Peptoids and Peptides. J. Am. Chem. Soc. 2007, 129, 1508–1509. [Google Scholar] [CrossRef] [PubMed]
- Schwochert, J.; Turner, R.; Thang, M.; Berkeley, R.F.; Ponkey, A.R.; Rodriguez, K.M.; Leung, S.S.F.; Khunte, B.; Goetz, G.; Limberakis, C.; et al. Peptide to Peptoid Substitutions Increase Cell Permeability in Cyclic Hexapeptides. Org. Lett. 2015, 17, 2928–2931. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Townsend, C.E.; Schwochert, J.; Pye, C.R.; Bednarek, M.A.; Lokey, R.S. Passive Membrane Permeability in Cyclic Peptomer Scaffolds Is Robust to Extensive Variation in Side Chain Functionality and Backbone Geometry. J. Med. Chem. 2016, 59, 9503–9512. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.C.; Yu, P.; Kwon, Y.-U.; Kodadek, T. High-throughput evaluation of relative cell permeability between peptoids and peptides. Bioorg. Med. Chem. 2008, 16, 5853–5861. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. Iron dysregulation in Alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog. Neurobiol. 2007, 82, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Vanderstichele, H.; Kodadek, T. Roadblocks for integration of novel biomarker concepts into clinical routine: The peptoid approach. Alzheimers Res. Ther. 2014, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid -protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.A.; Willbold, D. Peptides for therapy and diagnosis of Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Adessi, C.; Frossard, M.-J.; Boissard, C.; Fraga, S.; Bieler, S.; Ruckle, T.; Vilbois, F.; Robinson, S.M.; Mutter, M.; Banks, W.A.; et al. Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer’s disease. J. Biol. Chem. 2003, 278, 13905–13911. [Google Scholar] [CrossRef] [PubMed]
- Penninkilampi, R.; Brothers, H.M.; Eslick, G.D. Safety and Efficacy of Anti-Amyloid-β Immunotherapy in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Neuroimmune Pharmacol. 2017, 12, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhu, L.; Li, H.; Cheng, P.; Peng, J.; Yin, Y.; Yang, Y.; Wang, C.; Hu, Z.; Yang, Y. Antiamyloidogenic Activity of Aβ42-Binding Peptoid in Modulating Amyloid Oligomerization. Small 2017, 13. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Luo, Y.; Chen, X.; Kodadek, T. Peptoid Compositions for the Treatment of Alzheimer’s Disease and Polyglutamine Expansion Disorder. WO 2,013,043,669 A1, 28 March 2013. [Google Scholar]
- Luo, Y.; Vali, S.; Sun, S.; Chen, X.; Liang, X.; Drozhzhina, T.; Popugaeva, E.; Bezprozvanny, I. Aβ42-binding peptoids as amyloid aggregation inhibitors and detection ligands. ACS Chem. Neurosci. 2013, 4, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Servoss, S.; Moss, M. Peptoids and Methods for Treating Alzheimer’s Disease. U.S. Patent 20,130,102,539, 25 April 2013. [Google Scholar]
- Turner, J.P.; Lutz-Rechtin, T.; Moore, K.A.; Rogers, L.; Bhave, O.; Moss, M.A.; Servoss, S.L. Rationally Designed Peptoids Modulate Aggregation of Amyloid-Beta 40. ACS Chem. Neurosci. 2014, 5, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Näslund, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of -Amyloid Fibril Formation by a Pentapeptide Ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Lilliehöök, C.; Callaway, D.J.; Näslund, J.; Hahne, S.; Thyberg, J.; Terenius, L.; Nordstedt, C. Controlling amyloid beta-peptide fibril formation with protease-stable ligands. J. Biol. Chem. 1997, 272, 12601–12605. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.P.; Chastain, S.E.; Park, D.; Moss, M.A.; Servoss, S.L. Modulating amyloid-β aggregation: The effects of peptoid side chain placement and chirality. Bioorg. Med. Chem. 2017, 25, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z. CNS Physicochemical Property Space Shaped by a Diverse Set of Molecules with Experimentally Determined Exposure in the Mouse Brain: Miniperspective. J. Med. Chem. 2017, 60, 5943–5954. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.J.; Kirshenbaum, K. Peptoid and Synthetic Oligomers, Pharmaceutical Compositions and Methods of Using Same. U.S. Patent 9,364,449 B2, 14 June 2016. [Google Scholar]
- Liu, S.; Park, S.; Allington, G.; Prelli, F.; Sun, Y.; Martá-Ariza, M.; Scholtzova, H.; Biswas, G.; Brown, B.; Verghese, P.B.; et al. Targeting Apolipoprotein E/Amyloid β Binding by Peptoid CPO_Aβ17-21 P Ameliorates Alzheimer’s Disease Related Pathology and Cognitive Decline. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, C. Aβ42 is more rigid than Abeta40 at the C terminus: Implications for Aβ aggregation and toxicity. J. Mol. Biol. 2006, 364, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25, reprinted in Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Zabrodski, T.; Baskin, M.; Kaniraj, P.; Maayan, G. Click To Bind: Microwave-Assisted Solid-Phase Synthesis of Peptoids Incorporating Pyridine–Triazole Ligands and Their Copper(II) Complexes. Synlett 2015, 26, 461–466. [Google Scholar] [CrossRef]
- Maayan, G.; Yoo, B.; Kirshenbaum, K. Heterocyclic amines for the construction of peptoid oligomers bearing multi-dentate ligands. Tetrahedron Lett. 2008, 49, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Baskin, M.; Maayan, G. Water-soluble chiral metallopeptoids: Water-Soluble Chiral Metallopeptoids. Biopolymers 2015, 104, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Maayan, G.; Ward, M.D.; Kirshenbaum, K. Metallopeptoids. Chem. Commun. 2009, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Zborovsky, L.; Smolyakova, A.; Baskin, M.; Maayan, G. A Pure Polyproline Type I-like Peptoid Helix by Metal Coordination. Chem. A Eur. J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Baskin, M.; Maayan, G. A rationally designed metal-binding helical peptoid for selective recognition processes. Chem. Sci. 2016, 7, 2809–2820. [Google Scholar] [CrossRef] [PubMed]
- Savithri, A.; Thulasi, S.; Varma, R.L. Narrow-Rim Functionalization of Calix[4]arene through Ugi-4CR: Synthesis of a Series of Calix[4]arene Peptoids. J. Org. Chem. 2014, 79, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-C.; Chu, T.K.; Dill, K.A.; Zuckermann, R.N. Biomimetic Nanostructures: Creating a High-Affinity Zinc-Binding Site in a Folded Nonbiological Polymer. J. Am. Chem. Soc. 2008, 130, 8847–8855. [Google Scholar] [CrossRef] [PubMed]
- Elchinger, P.-H.; Delattre, C.; Faure, S.; Roy, O.; Badel, S.; Bernardi, T.; Michaud, P.; Taillefumier, C. Antioxidant Activities of Peptoid-Grafted Chitosan Films. Appl. Biochem. Biotechnol. 2017, 181, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Nalband, D.M.; Warner, B.P.; Zahler, N.H.; Kirshenbaum, K. Rapid identification of metal-binding peptoid oligomers by on-resin X-ray fluorescence screening. Biopolymers 2014, 102, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Pirrung, M.C.; Park, K. Discovery of selective metal-binding peptoids using 19F encoded combinatorial libraries. Bioorg. Med. Chem. Lett. 2000, 10, 2115–2118. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Park, K.; Tumey, L.N. 19F-encoded combinatorial libraries: Discovery of selective metal binding and catalytic peptoids. J. Comb. Chem. 2002, 4, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.H.K.; Simon, C.G. Fast setting calcium phosphate–chitosan scaffold: Mechanical properties and biocompatibility. Biomaterials 2005, 26, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ni, M.; Zhang, M.; Ratner, B. Calcium Phosphate—Chitosan Composite Scaffolds for Bone Tissue Engineering. Tissue Eng. 2003, 9, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Prior, M.; Chiruta, C.; Currais, A.; Goldberg, J.; Ramsey, J.; Dargusch, R.; Maher, P.A.; Schubert, D. Back to the Future with Phenotypic Screening. ACS Chem. Neurosci. 2014, 5, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2017. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Zuckermann, R.N. Peptoid origins. Biopolymers 2011, 96, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. (Ed.) Pseudo-Peptides in Drug Discovery; Wiley-VCH: Weinheim, Germany, 2004; ISBN 978-3-527-30633-6. [Google Scholar]
- Hecht, S.; Huc, I. Foldamers: Structure, Properties, and Applications; Wiley InterScience (Online service); Wiley-VCH: Weinheim, Germany, 2007; ISBN 978-3-527-61147-8. [Google Scholar]
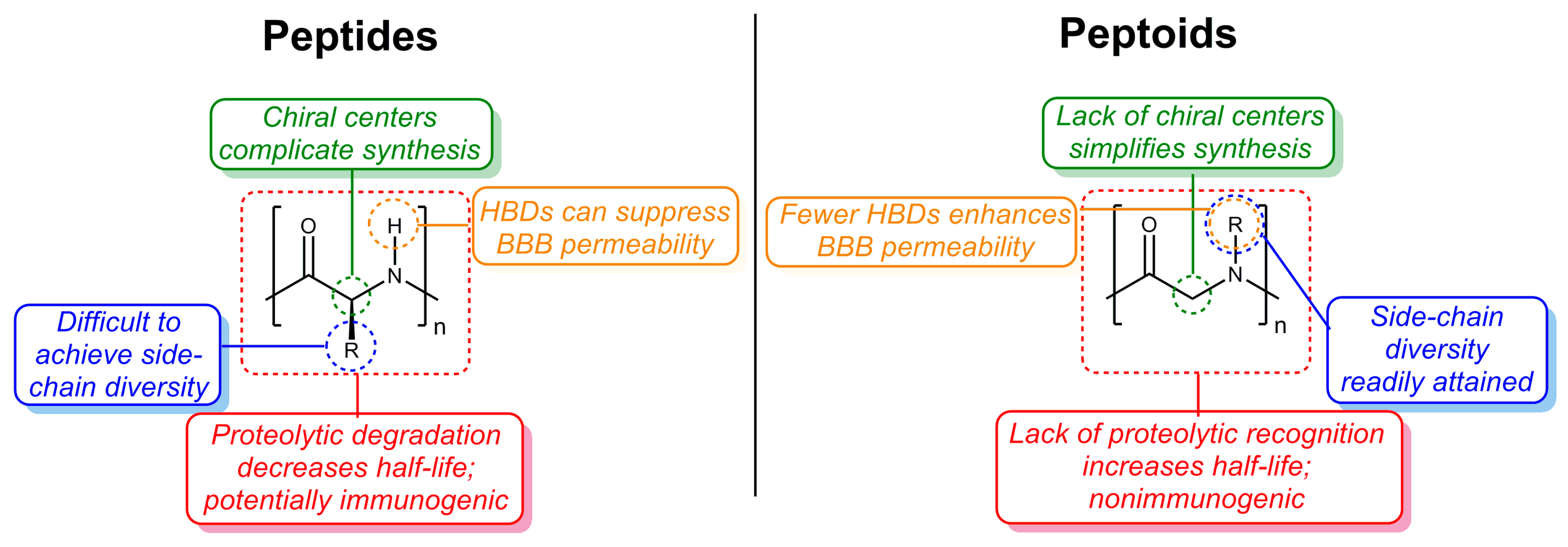



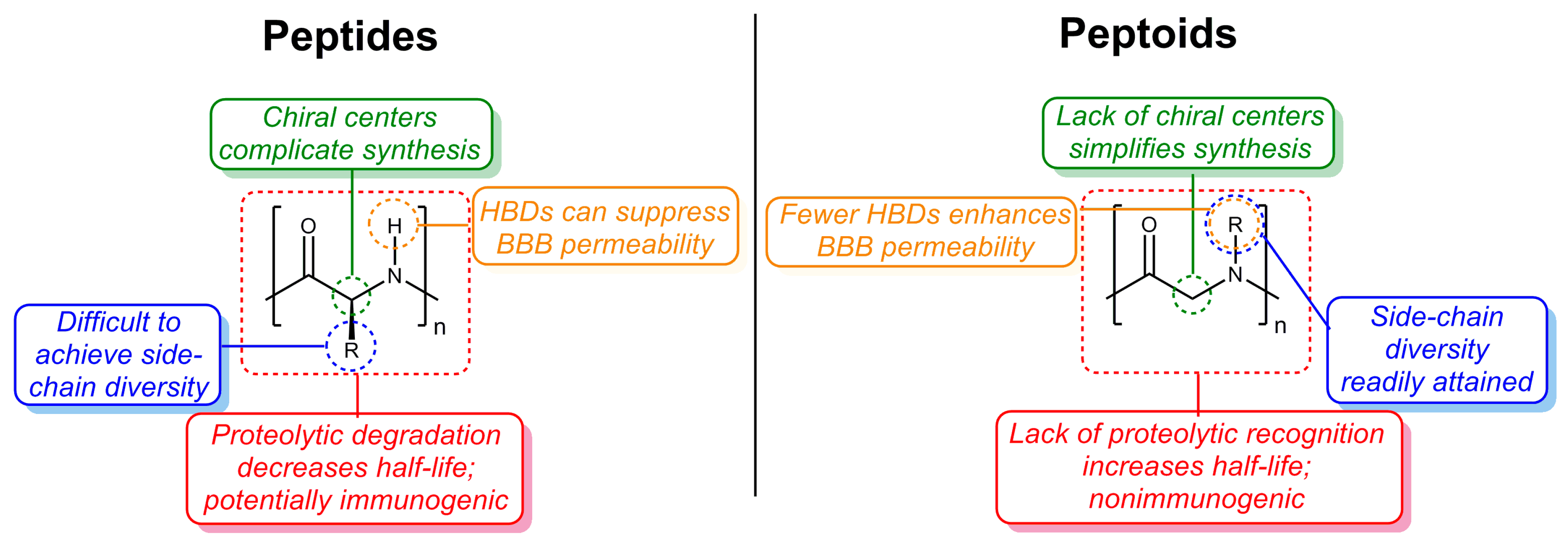
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name/Description | Methods Used | Bioactivities | References |
|---|---|---|---|
| AIP1 | Combinatorial chemistry was used to synthesize a library of over 4000 peptoids; surface plasmon resonance imaging (SPRi) used to screen peptoid library; thioflavin T (ThT) assay, atomic force microscopy (AFM), ELISA, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, docking, and circular dichroism (CD) spectroscopy used to investigate antiamyloidogenic activity; in vitro brain microvascular endothelial cell (BMVEC) model used to assess BBB permeability. | KD of binding to Aβ1–42 is 19.9 nM. Lag time to form critical nucleus increased from 1.78 to 7.18 h with AIP1. Transport ratio of 3.7 ± 0.2% (12 h) measured in BMVEC model. | [40] |
| IAM1 and (IAM1)2 | SPS used to generate an on-bead library containing over 38,000 peptoid analogs; bead-based screening methods, a ThT assay, and an amyloid toxicity assay used to test for Aβ1–42-aggregation inhibitory activities. | The Aβ1–42 binding affinities for IAM1 and (IAM1)2 are 0.43 ± 0.05 and 0.06 ± 0.04 µM, respectively. IAM1 has higher Aβ1–42:Aβ1–40 selectivity (9.6-fold vs. 2.1-fold); (IAM1)2 restored viability of neurons to 87% at 100 nM. | [41,42] |
| JPT1 | ThT fluorescence and dot blot analyses used to test antiamyloidogenic properties; fibril morphology investigated using transmission electron microscopy (TEM) and Nile Red spectroscopy; peptoid helicity investigated via CD spectroscopy. | Dose-dependent inhibition of Aβ1–40 aggregation was reported (81.2 ± 4.4% at 100 µM of JPT1); fewer Aβ fibrils were formed and the lag time was decreased. | [43,44,47] |
| Peptoid/peptide hybrids | MTT assay, a ThT aggregation assay, and an oligomerization assay used to assess impact on Aβ1–40 oligomerization. | Hybrids suppress Aβ oligomerization; one analog reduced the amount of Aβ1–40 oligomers by 61.3%. | [49] |
| CPO_Aβ17-21P | Linear and cyclic peptoid library synthesized via SPS; SPR, a ThT assay, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) cell viability assay, behavioral testing, immunohistochemistry, western blot, and ELISA used to study binding to apoE4-Aβ. | ApoE4-Aβ binding inhibited with a half-maximal inhibitory concentration (IC50) of 1.02 nM; transgenic mice exhibited significant cognitive improvement. | [50] |
| Name | Molecular Weight (Da) | HBDs | HBAs 1 | References |
|---|---|---|---|---|
| AIP1 | 970.25 | 10 | 20 | [40] |
| IAM1 | 1037.34 | 9 | 18 | [41,42] |
| (IAM1)2 | 2368.30 2 | 20 | 42 | [41,42] |
| JPT1 | 1130.51 | 6 | 18 | [43,44,47] |
| CPO_Aβ17-21P | 703.37 3 | 2 | 14 | [50] |
| Name/Description | Methods Used | Bioactivities | References |
|---|---|---|---|
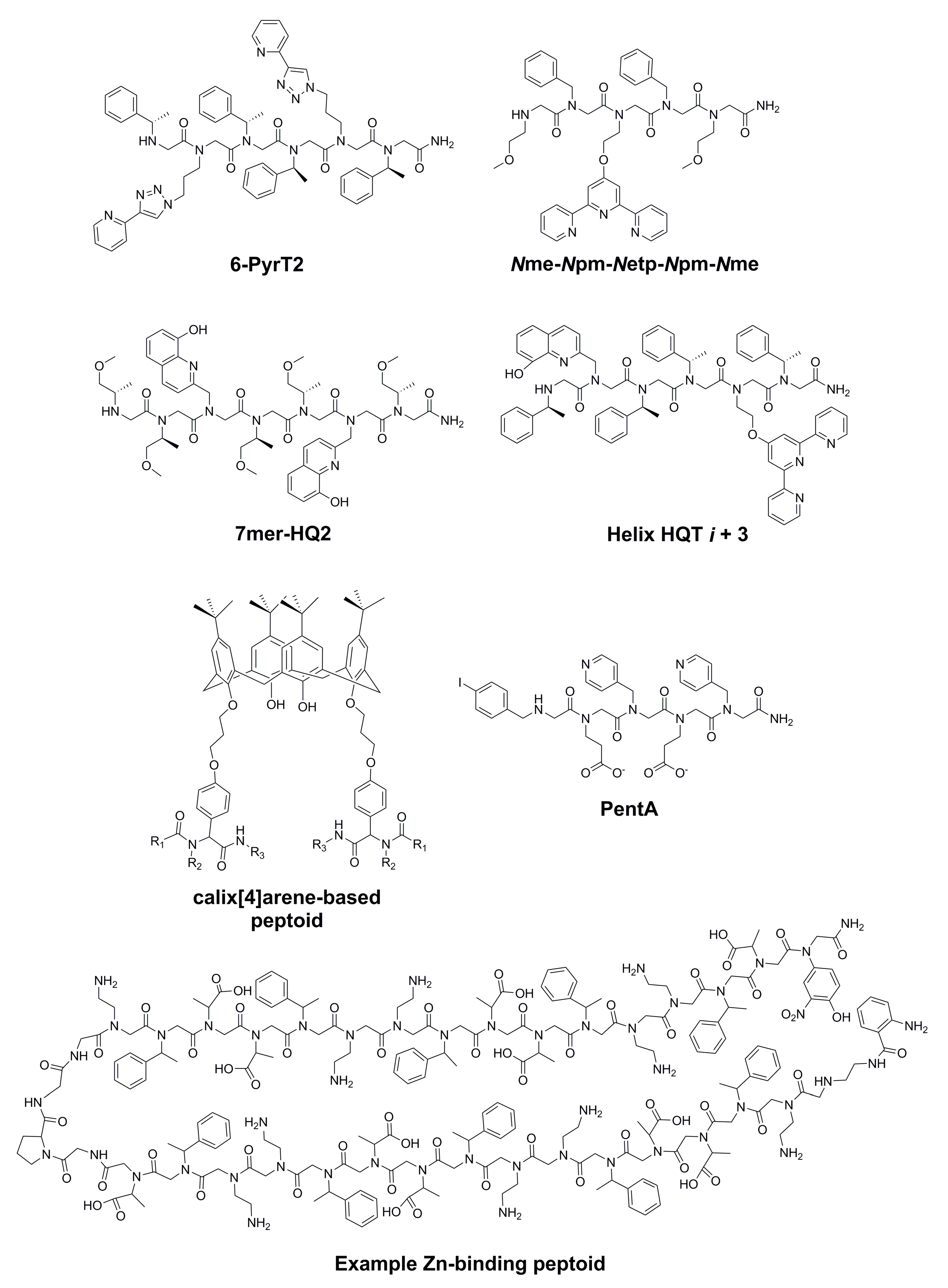
| 6-PyrT2 and other Cu(II)-binding peptoids | Peptoids synthesized via microwave-accelerated solid-phase click chemistry; CD spectroscopy and isothermal titration calorimetry (ITC) used to investigate peptoid-Cu(II) binding. | Association constants (KA) for 6-PyrT2-Cu(II) complex are 3.318 × 106 in methanol and 3.071 × 106 in acetonitrile. | [53] |
| Nme-Npm-Netp-Npm-Nme and other heterocyclic-amine- based peptoids | Peptoid 3-, 4-, and 5-mers containing metal-chelating terpyridine, phenathroline, and HQ side chains synthesized via SPS. | Metal-binding properties were not explored. | [54] |
| 7mer-HQ2 and other Cu(II)- and Co(II)-binding peptoids | Peptoids synthesized via SPS; near-UV CD spectroscopy, UV titration, and EPR used to investigate peptoid-metal binding. | 7mer-HQ2 formed a 1:1 complex with Cu(II). | [55] |
| Helix HQT i + 3 and other selective Cu(II)-binding peptoids | Peptoids synthesized via SPS; metal-binding properties studied using UV titration, CD spectroscopy, electron paramagnetic resonance (EPR), and inductively coupled plasma mass spectrometry (ICP-MS) experiments. | K for formation of peptoid-Cu complex is 1.03 ± 0.49 × 1013 M−1; Cu(II) selectivity observed in the presence of over 800× and 670× higher concentrations of Mn(II) and Ni(II), respectively. | [58] |
| Selective-Cu(II)- binding calix[4]arene peptoid tetramers | Peptoids synthesized using an isocyanide based multi-component reaction (MCR); UV-vis titration used to investigate metal binding. | One of the peptoids binds to Cu(II) selectively in the presence of various metal cations (as perchlorates). | [59] |
| Selective and tight Zn(II)-binding peptoids | Peptoids synthesized via automated SPS; ethylene glycol bis(2-aminoethyl ether)-N,N,N,N-tetraacetic acid (EGTA) competition assay, Förster resonance energy transfer (FRET), CD spectroscopy, and UV-vis spectroscopy used to investigate metal binding. | Some peptoids exhibited Zn-binding affinity at least one order of magnitude higher than that of various metal ions. | [60] |
| Benzyloxyethyl-based peptoids free and immobilized on a chitosan film with antioxidant and Fe-chelating properties | 2,2-Diphenyl-1-picrylhydrazyl (DPPH) and hydroxyl radical procedures were used to assess antioxidant activities; Fe-chelating properties investigated using an EDTA-competition assay. | Two of the free peptoids exhibited concentration- dependent radical scavenging effect of up to 80% at 5 g/L. | [61] |
| Screening for selective Ni(II)-binding peptoids | SPS used to synthesize peptoids; high-throughput, bench-top X-ray fluorescence screening (with ICP-MS and a colorimetric assay for validation) used to screen peptoids for Ni(II)-binding. | Two of the peptoids bind Ni(II) in the presence of other metal ions. | [62] |
| Screening for iron- and copper-binding peptoids | Split-pool SPS used to synthesize peptoid library; 19F NMR spectroscopy used to screen for metal-binding properties. Results were validated using UV titration. | A 12 nmol detection limit was achieved using a conventional NMR spectrometer; KD values of ~27–44 mM measured. | [63,64] |
| Name | Molecular Weight (Da) | HBDs | HBAs 1 | References |
|---|---|---|---|---|
| 6-PyrT2 | 1148.39 | 3 | 21 | [53] |
| Nme-Npm-Netp-Npm-Nme | 874.01 | 3 | 17 | [54] |
| 7mer-HQ2 | 1091.27 | 5 | 24 | [55] |
| Helix HQT i + 3 | 1208.43 | 4 | 19 | [58] |
| Calix[4]arene-based peptoids | 1613.94–1978.15 2 | 4–6 | 14–18 | [59] |
| Zn(II)-binding peptoids | 4267.8–4681.2 2 | ~36–40 | ~89–93 | [60] |
| Benzyloxyethyl-based peptoids | 414.22–512.29 | 1–2 | 7–8 | [61] |
| PentA (Ni(II)-binding peptoid) | 844.2 3 | 3 | 17 | [62] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Young, S.C. A Systematic Review of Antiamyloidogenic and Metal-Chelating Peptoids: Two Structural Motifs for the Treatment of Alzheimer’s Disease. Molecules 2018, 23, 296. https://doi.org/10.3390/molecules23020296
Young SC. A Systematic Review of Antiamyloidogenic and Metal-Chelating Peptoids: Two Structural Motifs for the Treatment of Alzheimer’s Disease. Molecules. 2018; 23(2):296. https://doi.org/10.3390/molecules23020296
Chicago/Turabian StyleYoung, Sherri C. 2018. "A Systematic Review of Antiamyloidogenic and Metal-Chelating Peptoids: Two Structural Motifs for the Treatment of Alzheimer’s Disease" Molecules 23, no. 2: 296. https://doi.org/10.3390/molecules23020296
APA StyleYoung, S. C. (2018). A Systematic Review of Antiamyloidogenic and Metal-Chelating Peptoids: Two Structural Motifs for the Treatment of Alzheimer’s Disease. Molecules, 23(2), 296. https://doi.org/10.3390/molecules23020296