Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors

 ,
, 
Abstract
1. Introduction
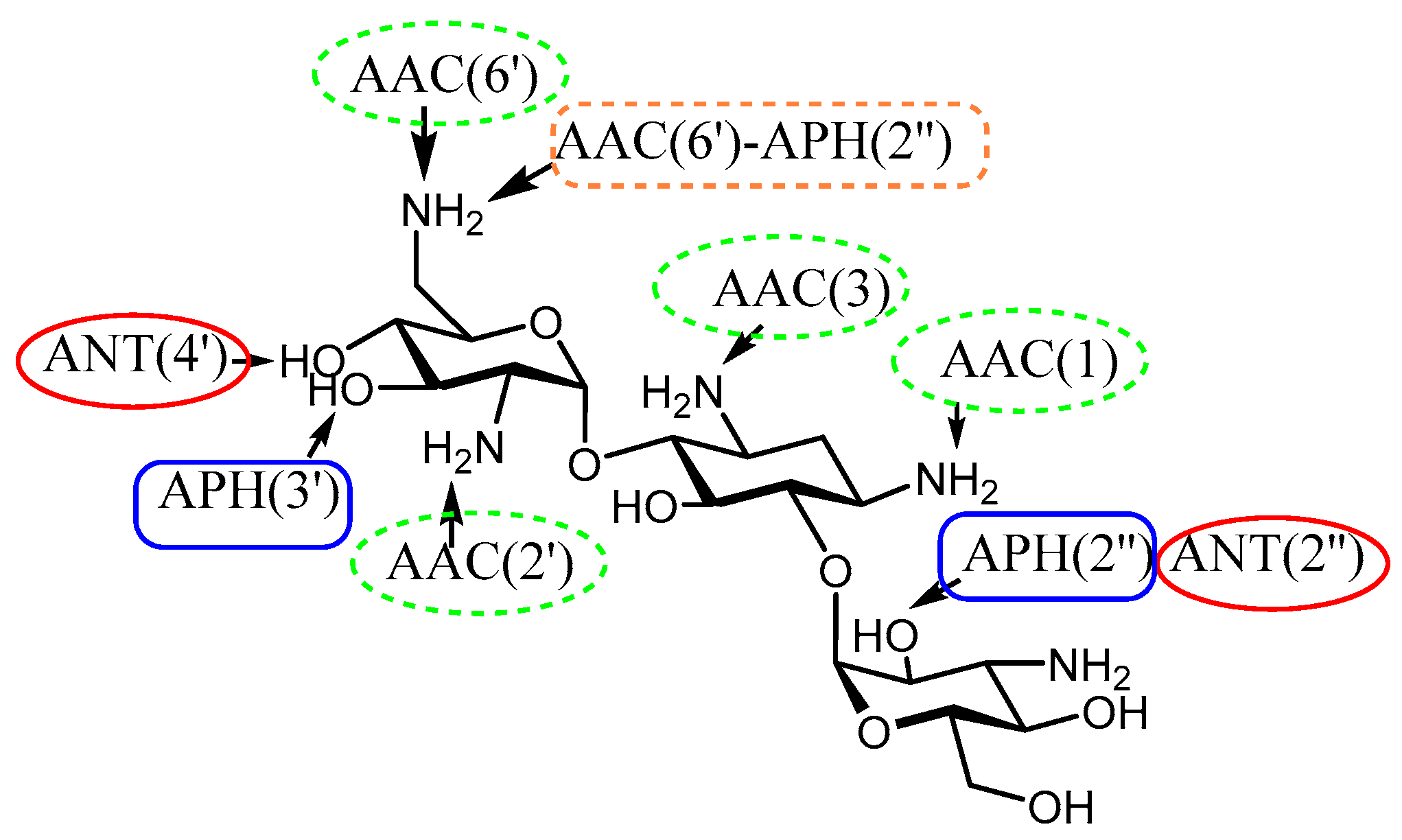
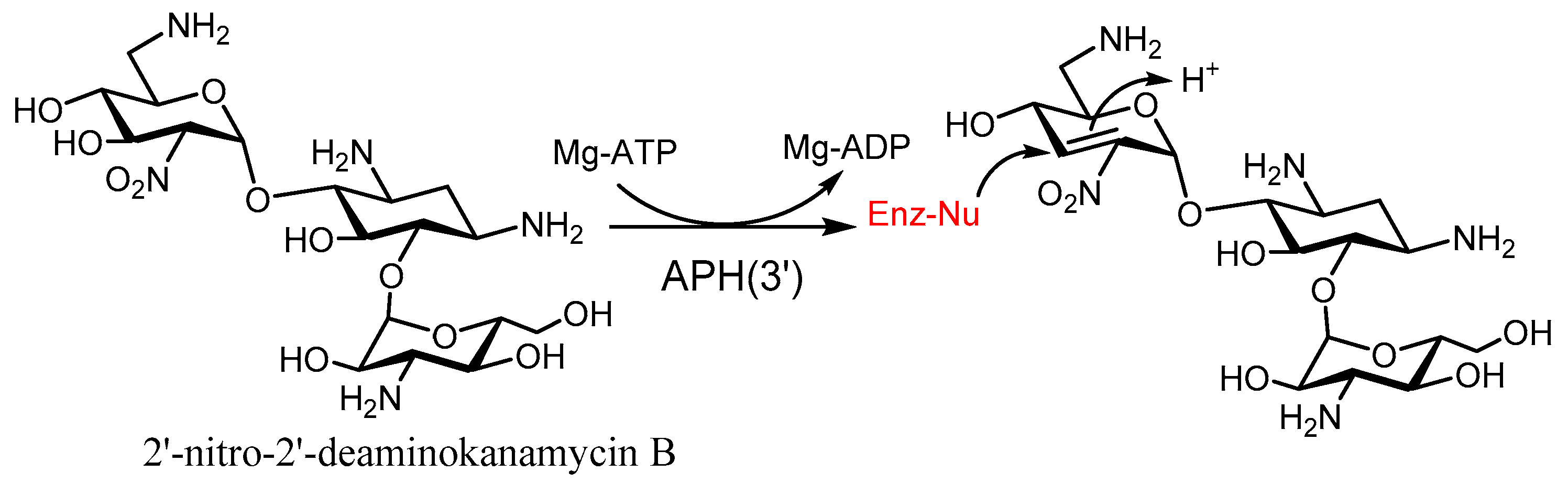
2. Understanding the Modifying Enzymes
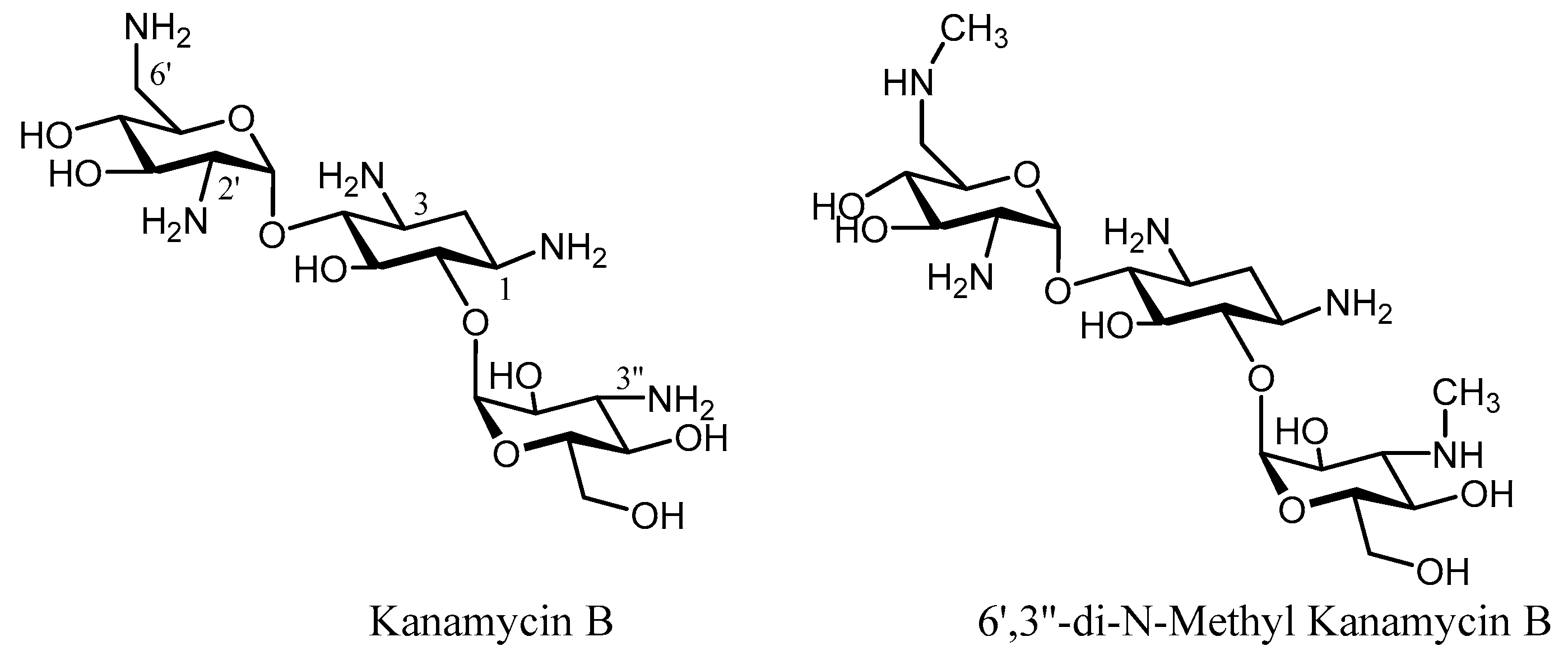
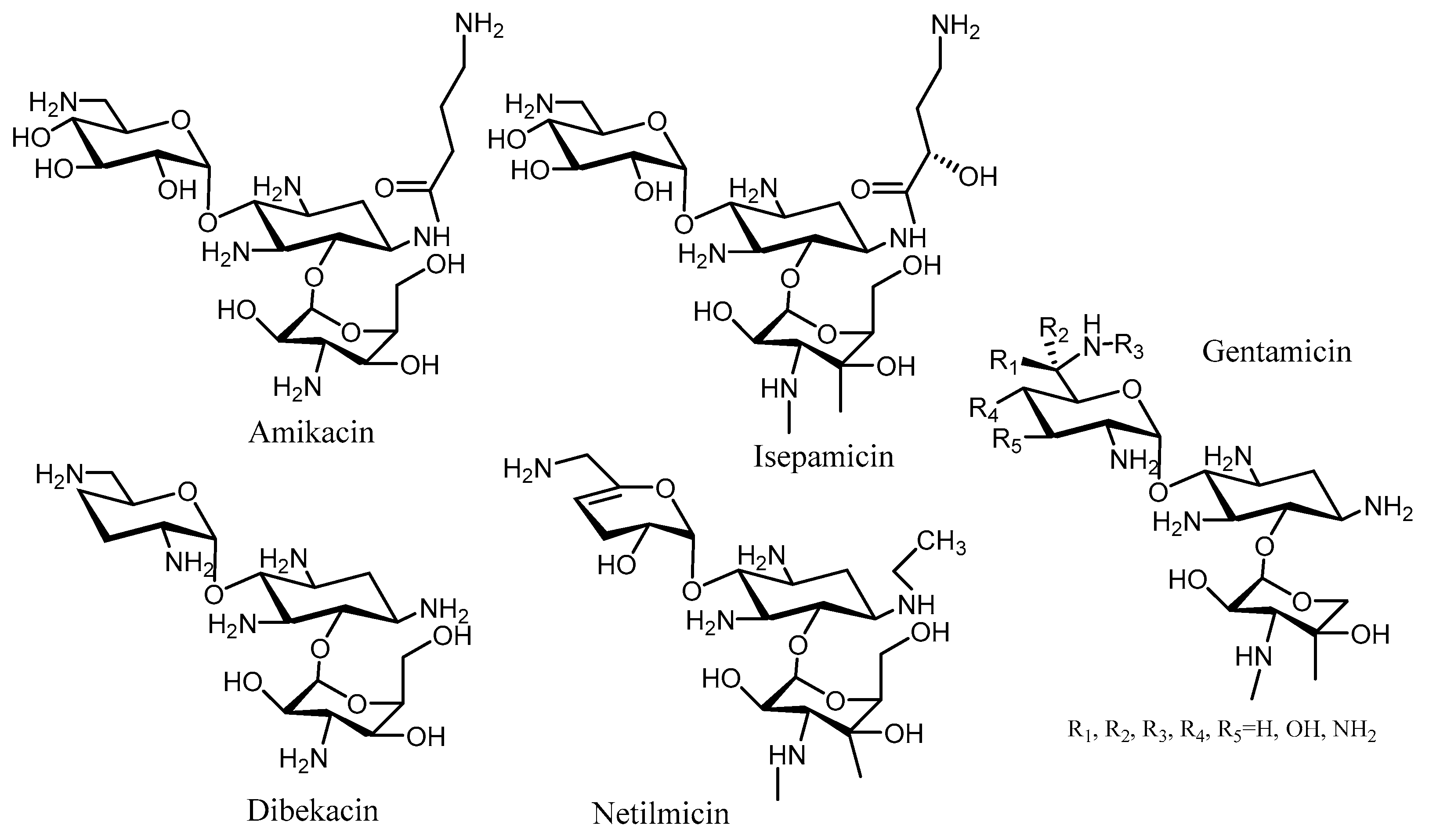
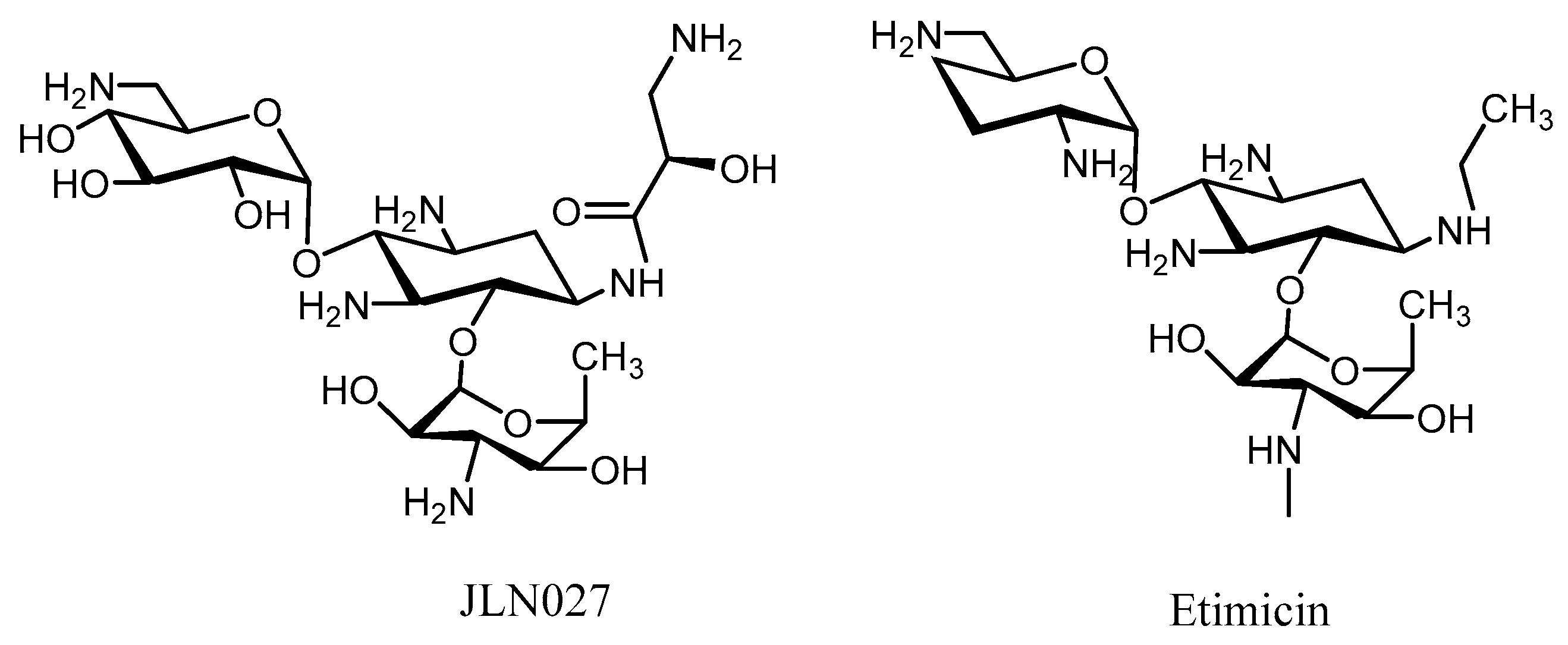
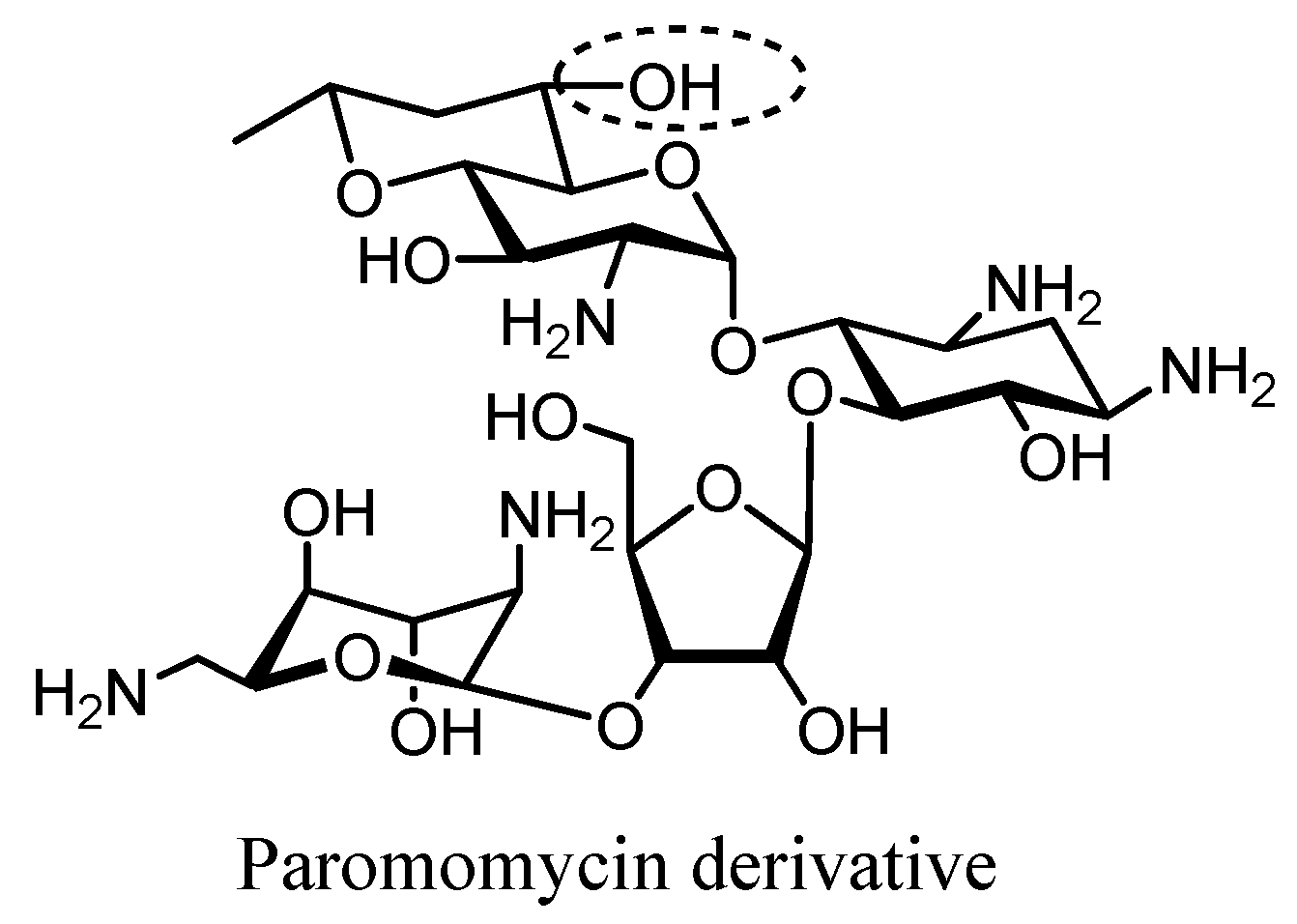
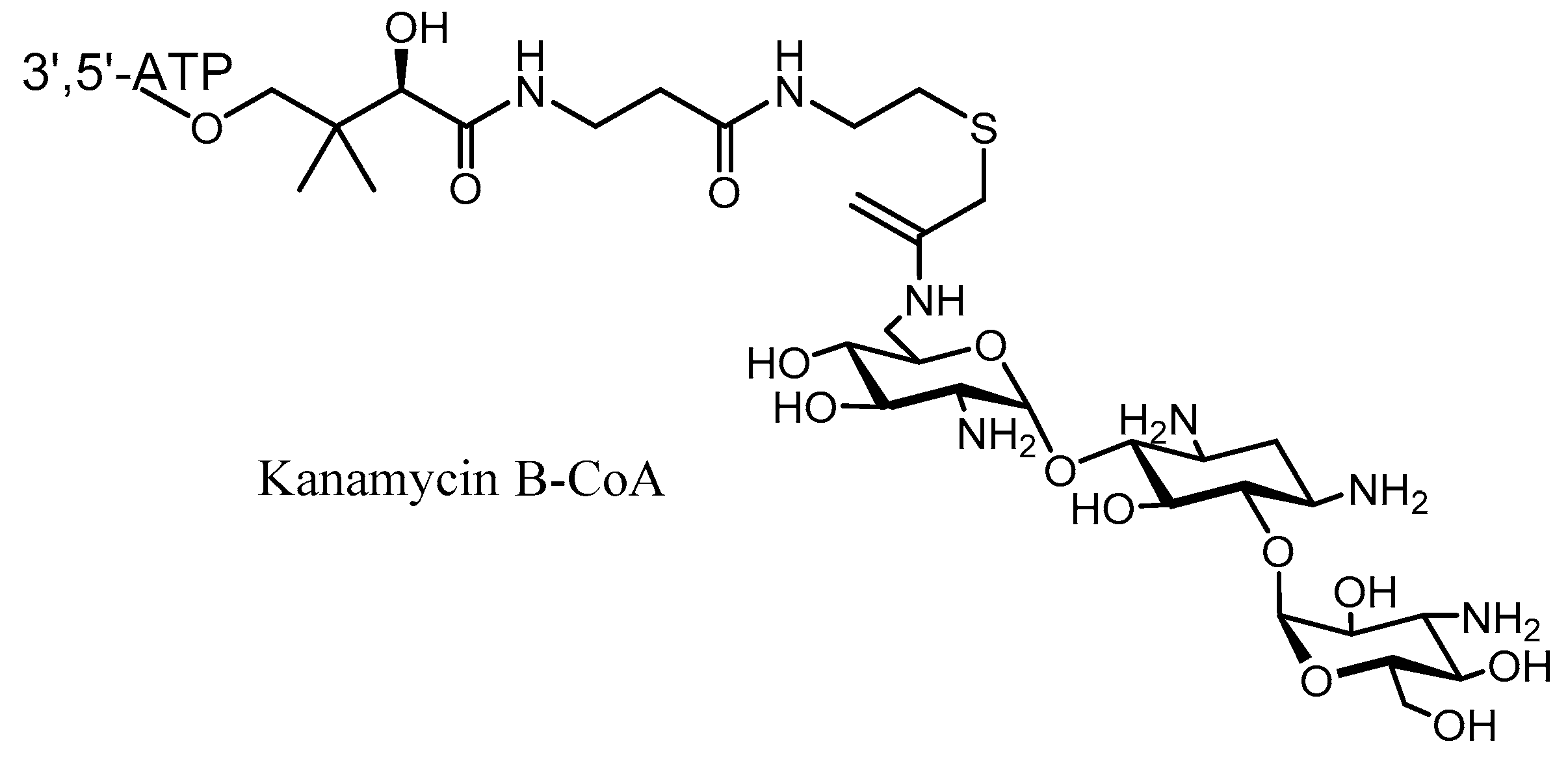
3. Semi-Synthetic Aminoglycoside Derivatives
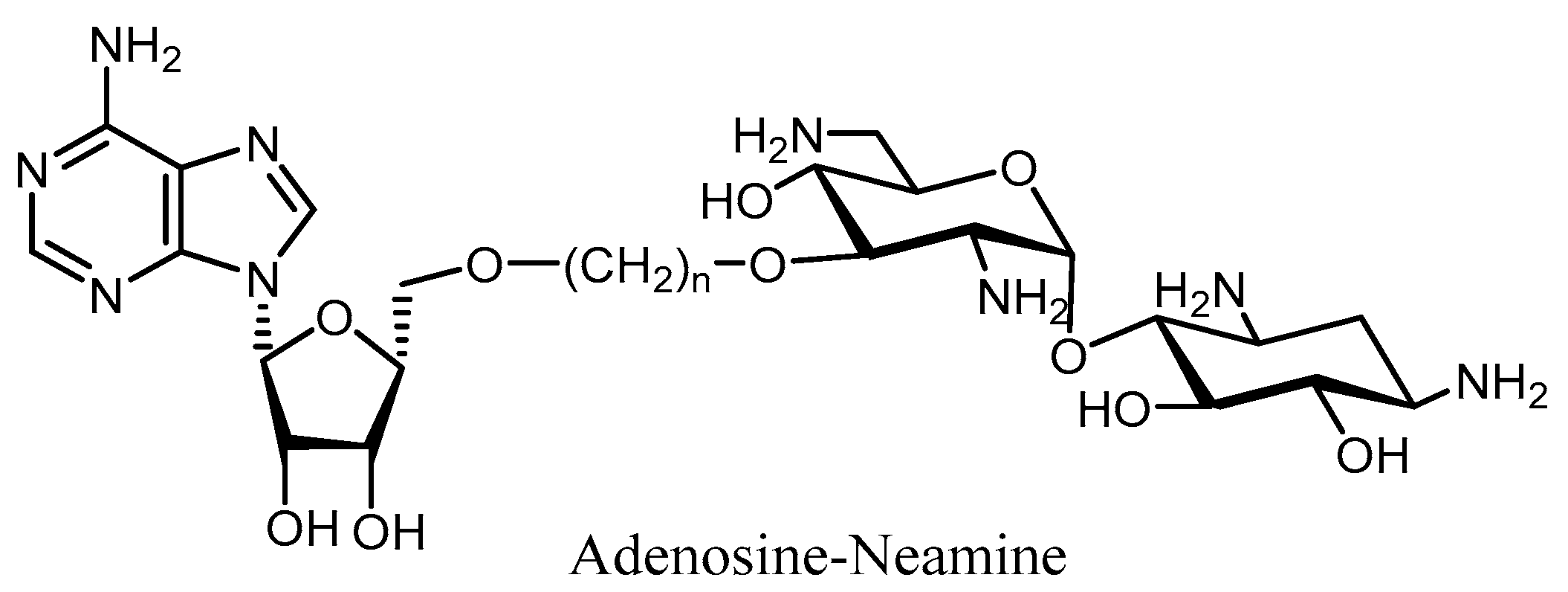
4. Combination Therapy
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Fourteen years in resistance. Int. J. Antimicrob. Agents 2012, 39, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.A.; Murray, B.E. Antibiotic-resistant bugs in the 21st century—A clinical super-challenge. N. Engl. J. Med. 2009, 360, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, D.; Finch, R.; Davey, P.; Wilcox, M. Antimicrobial Chemotherapy, 4th ed.; Oxford University Press: Oxford, UK, 2000; pp. 32–48. ISBN 9780192631954. [Google Scholar]
- Chen, G.-H.; Pan, P.; Yao, Y.; Cheng, Y.; Meng, X.-B.; Li, Z.-J. Regioselective modification of amino groups in aminoglycosides based on cyclic carbamate formation. Tetrahedron 2008, 64, 9078–9087. [Google Scholar] [CrossRef]
- Chen, G.-H.; Pan, P.; Yao, Y.; Chen, Y.; Meng, X.-B.; Li, Z.-J. Selective deprotection of the Cbz amine protecting group for the facile synthesis of kanamycin A dimers linked at N-3″ position Original. Tetrahedron 2009, 65, 5922–5927. [Google Scholar] [CrossRef]
- Umezawa, H.; Okanishi, M.; Kondo, S.; Hamana, K.; Utahara, R.; Maeda, K.; Mitsuhashi, S. Phosphorylative inactivation of aminoglycosidic antibiotics by Escherichia coli carrying R factor. Science 1967, 157, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Doi, O.; Miyamoto, M.; Tanaka, N.; Umezawa, H. Inactivation and phosphorylation of kanamycin by drug-resistant Staphylococcus aureus. Appl. Microbiol. 1968, 16, 1282–1284. [Google Scholar] [PubMed]
- Miller, G.H.; Sabatelli, F.J.; Naples, L.; Hare, R.S.; Shaw, K.J. The changing nature of aminoglycoside resistance mechanisms and the role of isepamicin—A new broad-spectrum aminoglycoside. The Aminoglycoside Resistance Study Groups. J. Chemother. 1995, 7, 31–44. [Google Scholar] [PubMed]
- Mitscher, L.A. Antibiotics and Antimicrobial Agents; Lippincott Williams & Wilkins: Baltimore, PA, USA, 2002; pp. 788–791. [Google Scholar]
- Weinstein, M.J.; Luedemann, G.M.; Oden, E.M.; Wagman, G.H. Gentamicin, a new broad-spectrum antibiotic complex. Antimicrob. Agents Chemother. 1963, 161, 1–7. [Google Scholar] [PubMed]
- Martin, C.M.; Ikari, N.S.; Zimmerman, J.; Waitz, J.A. A virulent nosocomial Klebsiella with a transferable R factor for gentamicin: Emergence and suppression. J. Infect. Dis. 1971, 124, 24–29. [Google Scholar] [CrossRef]
- Benveniste, R.; Davies, J. R-factor mediated gentamicin resistance: A new enzyme which modifies aminoglycoside antibiotics. FEBS Lett. 1971, 14, 293–296. [Google Scholar] [CrossRef]
- Woo, P.; Dion, H.; Bartz, Q. Butirosins A and B, aminoglycoside antibiotics. III. structures. Tetrahedron Lett. 1971, 12, 2625–2628. [Google Scholar] [CrossRef]
- Hayashi, S.F.; Norcia, L.J.; Seibel, S.B.; Silvia, A.M. Structure activity relationships of hygromycin A and its analogs: Protein synthesis inhibition activity in a cell free system. J. Antibiot. 1997, 50, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.J.; Rather, P.N.; Hare, R.S.; Miller, G.H. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138–163. [Google Scholar] [PubMed]
- Hotta, K.; Zhu, C.B.; Ogata, T.; Sunada, A.; Ishikawa, J.; Mizuno, S.; Kondo, S. Enzymatic 2′-N-acetylation of arbekacin and antibiotic activity of its product. J. Antibiot. 1996, 49, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Labby, K.J.; Garneau-Tsodikova, S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med. Chem. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.R.; Paulsen, I.T.; Tetu, S.G. Genomics and the evolution of antibiotic resistance. Ann. N. Y. Acad. Sci. 2017, 1388, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Vassilev, A.; Makino, Y.; Sali, A.; Nakatani, Y.; Burley, S.K. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell 1998, 94, 439–449. [Google Scholar] [CrossRef]
- Vetting, M.W.; Hegde, S.; Javid-Majd, F.; Blanchard, J.S.; Roderick, S.L. Aminoglycoside 2′-N-acetyltrasferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat. Struct. Biol. 2002, 9, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Lovering, A.M.; White, L.O.; Reeves, D.S. AAC-(1): A new aminoglycoside-acetylating enzyme modifying the Cl aminogroup of apramycin. J. Antimicrob. Chemother. 1987, 20, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Sunada, A.; Nakajima, M.; Ikeda, Y.; Kondo, S.; Hotta, K. Enzymatic 1-N-acetylation of paromomycin by an actinomycete strain 8 with multiple aminoglycoside resistance and paromomycin sensitivity. J. Antibiot. 1999, 52, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. Phosphotransferase sequence homology. Nature 1987, 329, 6134. [Google Scholar] [CrossRef] [PubMed]
- Burk, D.L.; Hon, W.C.; Leung, A.K.; Berghuis, A.M. Structural analysis of nucleotide binding to an aminoglycoside phosphotransferase. Biochemistry 2001, 40, 8756–8764. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.H.; Berghuis, A.M. Substrate promiscuity of an aminoglycoside antibiotic resistance enzyme via target mimicry. EMBO J. 2002, 21, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.L.; Serpersu, E.H. Ligand promiscuity through the eyes of the aminoglycoside N3 acetyltransferase IIa. Protein Sci. 2013, 22, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.R.; Hughes, D.W.; Wright, G.D. Regiospecificity of aminoglycoside phosphotransferase from Enterococci and Staphylococci (APH(3’)-IIIa). Biochemistry 1996, 35, 8686–8695. [Google Scholar] [CrossRef] [PubMed]
- McKay, G.A.; Wright, G.D. Kinetic mechanism of aminoglycoside phosphotransferase type IIIa. Evidence for a Theorell-Chance mechanism. J. Biol. Chem. 1995, 270, 24686–24692. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; Stogios, P.J.; Savchenko, A.; Wright, G.D. Structural and Molecular Basis for Resistance to Aminoglycoside Antibiotics by the Adenylyltransferase ANT(2’’)-Ia. Mbio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Bassenden, A.V.; Rodionov, D.; Shi, K.; Berghuis, A.M. Structural Analysis of the Tobramycin and Gentamicin Clinical Resistome Reveals Limitations for Next-generation Aminoglycoside Design. ACS Chem. Biol. 2016, 11, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Nasvall, J.; Wu, S.; Andersson, D.I.; Selmer, M. Crystal structure of AadA an aminoglycoside adenyltransferase. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 2267. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.C.; Benning, M.M.; Holden, H.M. Structural investigation of the antibiotic and ATP-binding sites in kanamycin nucleotidyltransferase. Biochemistry 1995, 34, 13305–13311. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Wawrzak, Z.; Minasov, G.A.; Evdokimova, E.; Egorova, O.; Yim, V.; Kudritska, M.; Courvalin, P.; Savchenko, A.; Anderson, W.F. Crystal structure of aminoglycoside 4′-O-adenylyltransferase ANT(4’)-IIb, apo. Cent. Struct. Genom. Infect. Dis. (CSGID) 2012. [Google Scholar] [CrossRef]
- Tyagi, R.; Eswaramoorthy, S.; Burley, S.K.; Swaminathan, S. New York SGX Research Center for Structural Genomics. Crystal structure of an aminoglycoside 6-adenyltransferase from Bacillus subtilis. Unpublished work. 2007. [Google Scholar]
- Murphy, E. Nucleotide sequence of a spectinomycin adenyltransferase AAD(9) determinant from Staphylococcus aureus and its relationship to AAD(3’)(9). Mol. Gen. Genet. 1985, 200, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Matesanz, R.; Díaz, J.F.; Corzana, F.; Santana, A.G.; Bastida, A.; Asensio, J.L. Multiple keys for a single lock: The unusual structural plasticity of the nucleotidyltransferase (4’)/kanamycin complex. Chemistry 2012, 18, 2875–2889. [Google Scholar] [CrossRef] [PubMed]
- Revuelta, J.; Corzana, F.; Bastida, A.; Asensio, J.L. The unusual nucleotide recognition properties of the resistance enzyme ANT(4’): Inorganic tri/polyphosphate as a substrate for aminoglycoside inactivation. Chem. A Eur. J. 2010, 16, 8635–8640. [Google Scholar] [CrossRef] [PubMed]
- Chen-Goodspeed, M.; Vanhooke, J.L.; Holden, H.M.; Raushel, F.M. Kinetic mechanism of kanamycin nucleotidyltransferase from Staphylococcus aureus. Bioorg. Chem. 1999, 27, 395–408. [Google Scholar] [CrossRef]
- Magnet, S.; Blanchard, J.S. Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev. 2005, 105, 477–497. [Google Scholar] [CrossRef] [PubMed]
- Van Pelt, J.E.; Northrop, D.B. Purification and properties of gentamicin nucleotidyltransferase from Escherichia coli: Nucleotide specificity, pH optimum, and the separation of two electrophoretic variants. Arch. Biochem. Biophys. 1984, 230, 250–263. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside Modifying Enzymes. Drug Resist. Update 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Gates, C.A.; Northrop, D.B. Alternative substrate and inhibition kinetics of aminoglycoside nucleotidyltransferase 2”-I in support of a Theorell-Chance kinetic mechanism. Biochemistry 1988, 27, 3826–3833. [Google Scholar] [CrossRef] [PubMed]
- Bacot-D, V.R.; Bassenden, A.V.; Sprules, T.; Berghuis, A.M. Effect of solvent and protein dynamics in ligand recognition and inhibition of aminoglycoside adenyltransferase 2″-Ia. Protein Sci. 2017, 26, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Corzana, F.; Cuesta, I.; Bastida, A.; Hidalgo, A.; Latorre, M.; González, C.; García-Junceda, E.; Jiménez-Barbero, J.; Asensio, J.L. Molecular recognition of aminoglycoside antibiotics by bacterial defense proteins: NMR analysis of Streptomycin inactivation by Bacillus subtilis Aminoglycoside-6-adenyl Transferas. Chem. Eur. J. 2005, 11, 5102–5113. [Google Scholar] [CrossRef] [PubMed]
- Latorre, M.; Revuelta, J.; García-Junceda, E.; Bastida, A. 6-O-Nucleotidyltransferase: An aminoglycoside-modifying enzyme specific for streptomycin/streptidine. MedChemComm 2016, 7, 177–183. [Google Scholar] [CrossRef]
- Ferretti, J.J.; Gilmore, K.S.; Courvalin, P. Nucleotide-sequence analysis of the gene specifying the bifunctional 6’-aminoglycoside acetyltransferase 2’’-aminoglycoside phosphotransferase enzyme in Streptococcus faecalis and identification and cloning of gene regions specifying the two activities. J. Bacteriol. 1986, 167, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Yıldız, Ö.; Çoban, A.Y.; Şener, A.G.; Coşkuner, S.A.; Bayramoğlu, G.; Güdücüoğlu, H.; Özyurt, M.; Tatman-Otkun, M.; Karabiber, N.; Özkütük, N.; et al. Antimicrobial susceptibility and resistance mechanisms of methicillin resistant Staphylococcus aureus isolated from 12 Hospitals in Turkey. Ann. Clin. Microbiol. Antimicrob. 2014, 16, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Centron, D.; Roy, P.H. Presence of a group II intron in a multiresistant Serratia marcescens strain that harbors three integrons and a novel gene fusion. Antimicrob. Agents Chemother. 2002, 46, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Dubois, W.; Poirel, L.; Marie, C.; Arpin, C.; Nordmann, P.; Quentin, C. Molecular characterization of a novel class 1 integron containing blaGES-1 and a fused product of aac(3)-Ib/aac(6’)-Ib’ gene cassettes in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2002, 46, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Hesek, D.; Zajícek, J.; Vakulenko, S.B.; Mobashery, S. Characterization of the bifunctional aminoglycoside-modifying enzyme ANT(3’’)-Ii/AAC(6’)-IId from Serratia marcescens. Biochemistry 2006, 45, 8368–8377. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Villegas-Estrada, A.; Hesek, D.; Mobashery, S. Mechanistic characterization of the bifunctional aminoglycoside-modifying enzyme AAC(3)-Ib/AAC(6’)-Ib’ from Pseudomonas aeruginosa. Biochemistry 2007, 46, 5270–5282. [Google Scholar] [CrossRef] [PubMed]
- Shaul, P.; Green, K.D.; Rutenberg, R.; Kramer, M.; Berkov-Zrihen, Y.; Breiner-Goldstein, E.; Garneau-Tsodikova, S.; Fridman, M. Assessment of 6’- and 6””-N-acylation of aminoglycosides as a strategy to overcome bacterial resistance. Org. Biomol. Chem. 2011, 9, 4057–4063. [Google Scholar] [CrossRef] [PubMed]
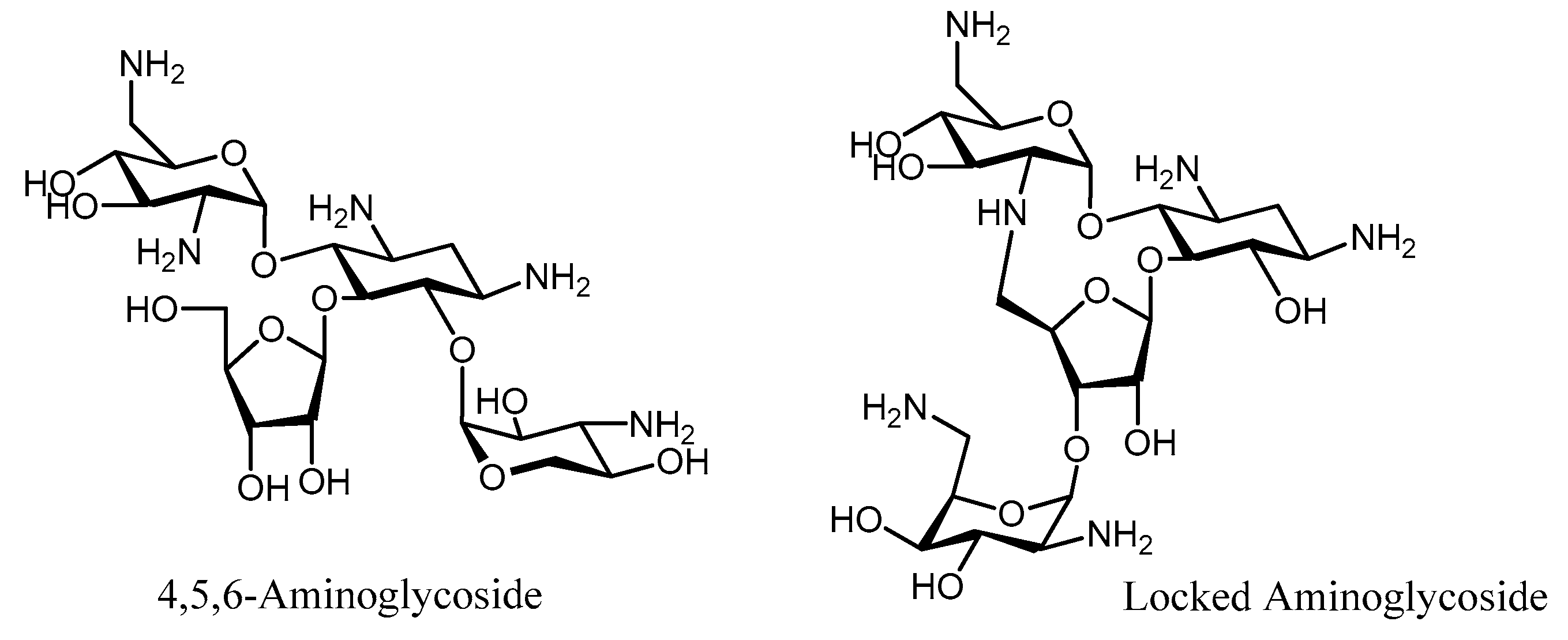
- Bastida, A.; Hidalgo, A.; Chiara, J.L.; Torrado, M.; Corzana, F.; Pérez-Cañadillas, J.M.; Groves, P.; García-Junceda, E.; González, C.; Jiménez-Barbero, J.; et al. Exploring the use of conformationally locked aminoglycosides as a new strategy to o.vercome bacterial resistance. J. Am. Chem. Soc. 2006, 128, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Fair, R.J.; McCoy, L.S.; Hensler, M.E.; Aguilar, B.; Nizet, V.; Tor, Y. Singly modified amikacin and tobramycin derivatives show increased rRNA A-site binding and higher potency against resistant bacteria. ChemMedChem 2014, 9, 2164–2171. [Google Scholar] [CrossRef] [PubMed]
- Fair, R.J.; Hensler, M.E.; Thienphrapa, W.; Dam, Q.N.; Nizet, V.; Tor, Y. Selectively guanidinylated aminoglycosides as antibiotics. ChemMedChem 2012, 7, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ye, X-S. Development of Aminoglycoside Antibiotics Effective Against Resistant Bacterial Strains. Curr. Top. Med. Chem. 2010, 10, 1898–1926. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.-B.; Yuan, M.; Wu, Y.; You, X.; Ye, X.-S. Rational design and synthesis of potent aminoglycoside antibiotics against resistant bacterial strains. Bioorg. Med. Chem. 2011, 19, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, H.; Umezawa, S.; Tsuchiya, T.; Okazaki, I. 3’,4’-Dideoxykanamycin B active against kanamycin-resistant Escherichia coli and Pseudomonas aeruginosa. J. Antibiot. 1971, 24, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.R.; Nucken, E.J.; Henschke, R.B. Nucleotide sequence analysis of 2’’-aminoglycoside nucleotidyl-transferase ANT(2’’) from Tn4000: Its relationship with AAD(3’) and impact on Tn21 evolution. Mol. Microbiol. 1988, 2, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Daigle, D.M.; Hughes, D.W.; Wright, G.D. Prodigious substrate specificity of AAC(6’)-APH(2”), an aminoglycoside antibiotic resistance determinant in enterococci and staphylococci. Chem. Biol. 1999, 6, 99–110. [Google Scholar] [CrossRef]
- Woo, P.W.K.; Haskell, T.H. Deoxy derivatives of butirosin A and 5”-amino-5”-deoxybutirosin A, aminoglycoside antibiotics resistant to bacterial 3’-phosphorylative enzymatic inactivation. Synthesis and NMR studies. J. Antibiot. 1982, 35, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Fujii, F.; Kondo, S.; Umezawa, H. Chemical derivation of antibiotics active against resistant bacteria. Jpn. J. Med. Sci. Biol. 1967, 21, 224–227. [Google Scholar] [PubMed]
- Kagawuchi, H.; Naito, T.; Nakagawa, S.; Fujisawa, K.I. B-K8, a new semisynthetic aminoglycoside antibiotic. J. Antibiot. 1972, 25, 695–708. [Google Scholar] [CrossRef]
- Kim, Y.A.; Park, Y.S.; Youk, T.; Lee, H.; Lee, K. Correlation of Aminoglycoside Consumption and Amikacin- or Gentamicin-Resistant Pseudomonas aeruginosa in Long-Term Nationwide Analysis: Is Antibiotic Cycling an Effective Policy for Reducing Antimicrobial Resistance? Ann. Lab. Med. 2018, 38, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Iinuma, K.; Yamamoto, H.; Maeda, K.; Umezawa, H. Letter: Synthesis of 1-N-{(S)-4-amino-2-hydroxybutyryl}-kanamycin B and -3’,4’-dideoxykanamycin B active against kanamycin resistant bacteria. J. Antibiot. 1973, 26, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Ubutaka, K.; Yamashita, N.; Gotoh, A.; Konno, M. Purification and characterization of aminoglycoside-modifying enzymes from Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob. Agents Chemother. 1984, 25, 754–759. [Google Scholar] [CrossRef]
- Kondo, S.; Hotta, K. Semisynthetic aminoglycoside antibiotics: Development and enzymatic modifications. J. Infect. Chemother. 1999, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.E.; Dolin, R.; Blaser, M. Principles and Practice of Infectious Diseases, 8th ed.; Douglas & Bennett’s, Churchill, Livingston: New York, NY, USA, 1995; pp. 279–301. ISBN 978-1-4557-4801-3. [Google Scholar]
- Montie, T.; Patamasucon, P. Aminoglycosides: The complex problem of antibiotic mechanisms and clinical applications. Eur. J. Clin. Microbiol. Infect. Dis. 1995, 14, 85–87. [Google Scholar] [CrossRef]
- Edson, R.S.; Terrell, C.L. The aminoglycosides. Mayo Clin. Proc. 1991, 66, 1158–1164. [Google Scholar] [CrossRef]
- Zhao, C.; Li, J.; Hou, J.; Guo, M.; Zhang, Y.; Chen, Y. A randomized controlled clinical trial, on etimicin, a new aminoglycoside antibiotic, versus netilmicin in the treatment of bacterial infections. Clin. Med. J. 2000, 113, 1026–1030. [Google Scholar] [PubMed]
- Lortholary, O.; Tod, M.; Cohen, Y.; Petitjean, O. Aminoglycosides. Med. Clin. N. Am. 1995, 79, 761–787. [Google Scholar] [CrossRef] [PubMed]
- Kaelowsky, J.A.; Zelenitsky, S.A.; Zhanel, G.G. Aminoglycoside adaptive resistance. Pharmacotherapy 1997, 17, 549–555. [Google Scholar] [CrossRef]
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
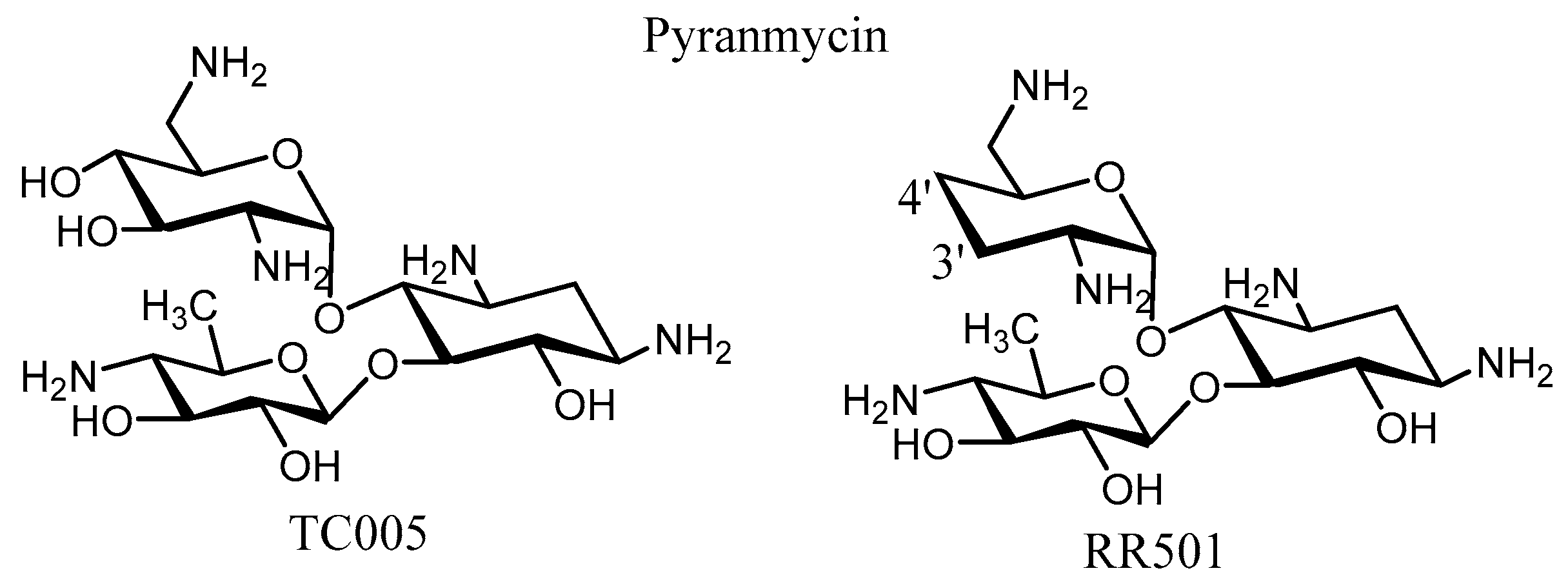
- Chang, C.-W.T.; Hui, Y.; Elchert, B.; Wang, J.; Li, J.; Rai, R. Pyranmycins, a novel class of aminoglycosides with improved acid stability: The SAR of D-pyranoses on ring III of pyranmycin. Org. Lett. 2002, 4, 4603–4606. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Chang, H.; Chang, C.-W.T. Novel Method for the Synthesis of 3’,4’-Dideoxygenated Pyranmycin and Kanamycin Compounds, and Studies of Their Antibacterial Activity against Aminoglycoside Resistant Bacteria. J. Carbohydr. Chem. 2005, 24, 131–143. [Google Scholar] [CrossRef]
- Rai, R.; Chen, H.; Czyryca, P.G.; Li, J.; Chang, C.-W.T. Design and Synthesis of Pyrankacin: A Pyranmycin Class Broad Spectrum Aminoglycoside Antibiotic. Org. Lett. 2006, 8, 887–889. [Google Scholar] [CrossRef] [PubMed]
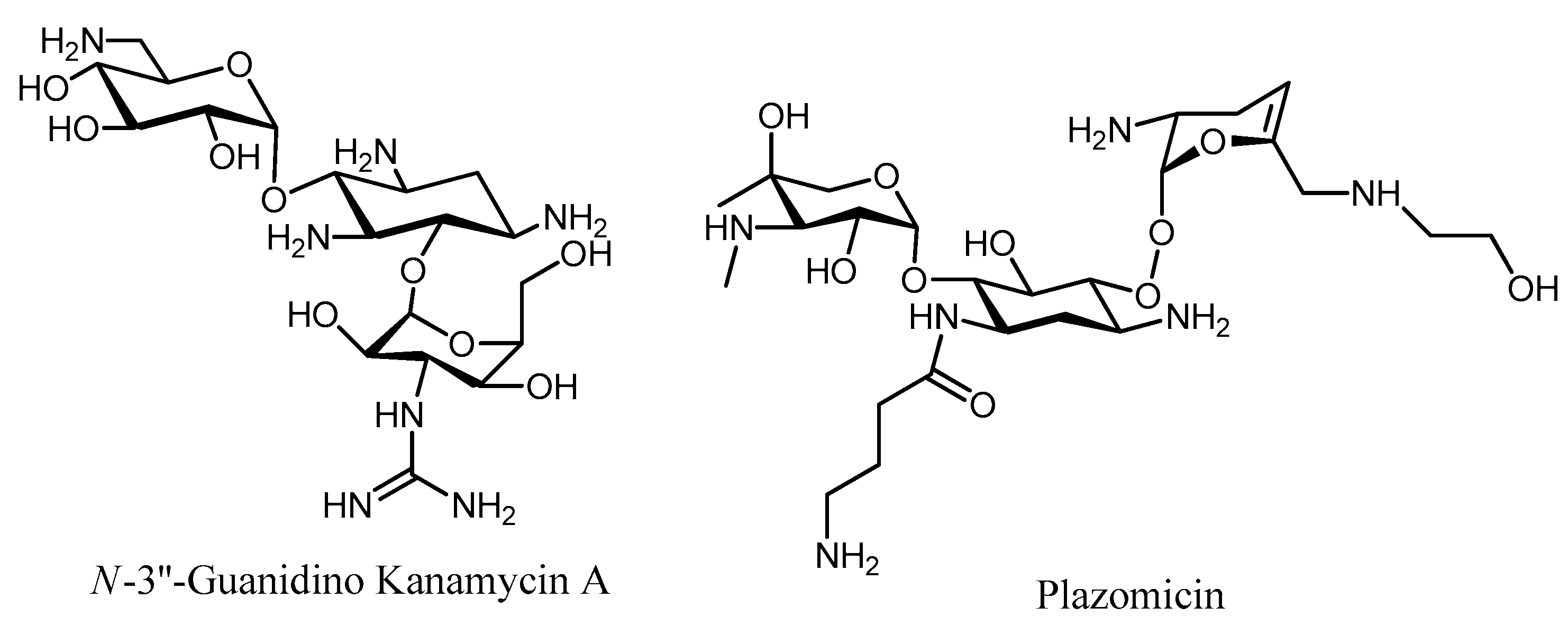
- Santana, A.G.; Zárate, S.G.; Asensio, J.L.; Revuelta, J.; Bastida, A. Selective modification of the 3′′-amino group of kanamycin prevents significant loss of activity in resistant bacterial strains. Org. Biomol. Chem. 2016, 14, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.; Souli, M.; Daikos, G.L.; Chrysouli, Z.; Poulakou, G.; Psichogiou, M.; Panagea, T.; Argyropoulou, Stefanou, I.; Plakias, G.; Giamarellou, H.; et al. Activity of Plazomicin (ACHN-490) against MDR clinical isolates of Klebsiella pneumoniae, Escherichia coli, and Enterobacter spp. J. Chemother. 2012, 24, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Mega, W.M.; Doyle-Eisele, M.; Cass, R.T.; Kostrub, C.F.; Sherwood, R.L.; Metz, M.A.; Cirz, R.T. Plazomicin is effective in a non-human primate pneumonic plague model. Bioorg. Med. Chem. 2016, 24, 6429–6439. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Mushtaq, S.; Warner, M.; Zhang, J.C.; Maharjan, S.; Doumith, M. Activity of aminoglycosides, including ACHN-490, against carbapenem-resistant Enterobacteriaceae isolates. J. Antimicrob. Chemother. 2011, 66, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Francois, B.; Russell, R.J.M.; Murray, J.B.; Aboul-ela, F.; Masquida, B.; Vicens, Q.; Westhof, E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: Role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res. 2005, 33, 5677–5690. [Google Scholar] [CrossRef] [PubMed]
- Nuthanakanti, A.; Boerneke, M.A.; Hermann, T.; Srivatsan, S.G. Structure of the Ribosomal RNA Decoding Site Containing a Selenium-Modified Responsive Fluorescent Ribonucleoside Probe. Angew. Chem. Int. Ed. Engl. 2017, 56, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.B.; Meroueh, S.O.; Russell, R.J.; Lentzen, G.; Haddad, J.; Mobashery, S. Interactions of designer antibiotics and the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 2006, 13, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Revuelta, J.; Vacas, T.; Corzana, F.; González, C.; Bastida, A.; Asensio, J.L. Structure-Based Design of Highly Crowded Ribostamycin/Kanamycin Hybrids as a new Family of Antibiotics. Chem. A Eur. J. 2010, 16, 2986–2991. [Google Scholar] [CrossRef] [PubMed]
- Asensio, J.L.; Hidalgo, A.; Bastida, A.; Torrado, M.; Corzana, F.; García-Junceda, E.; Cañada, J.; Chiara, J.L.; Jimenez-Barbero, J. A simple structural-based approach to prevent aminoglycoside inactivation by bacterial defense proteins. Conformational restriction provides effective protection against neomycin-B nucleotidylation by ANT4”. J. Am. Chem. Soc. 2005, 127, 8278–8279. [Google Scholar] [CrossRef] [PubMed]
- Blount, K.F.; Zhao, F.; Hermann, T.; Tor, Y. Conformational constraint as a means for understanding RNA-aminoglycoside specificity. J. Am. Chem. Soc. 2005, 127, 9818–9829. [Google Scholar] [CrossRef] [PubMed]
- Santana, A.G.; Bastida, A.; Martínez del Campo, T.; Asensio, J.L.; Revuelta, J. An efficient and general route to the synthesis of novel aminoglycosides for RNA binding. Synlett 2011, 219–222. [Google Scholar] [CrossRef]
- Sucheck, S.J.; Wong, A.L.; Koeller, K.M.; Boehr, D.D.; Draker, K.; Sears, P.; Wright, G.D. Design of bifunctional antibiotics that target bacterial rRNA and inhibit resistance-causing enzymes. J. Am. Chem. Soc. 2000, 122, 5230–5231. [Google Scholar] [CrossRef]
- Vacas, T.; Corzana, F.; Jiménez-Osés, G.; González, C.; Gómez, A.M.; Bastida, A.; Revuelta, J.; Asensio, J.L. Role of aromatic rings in the molecular recognition of aminoglycoside antibiotics: Implications for drug design. J. Am. Chem. Soc. 2010, 132, 12074–12090. [Google Scholar] [CrossRef] [PubMed]
- Mandhapati, A.R.; Yang, G.; Kato, T.; Shcherbakov, D.; Hobbie, S.N.; Vasella, A.; Boöttger, E.C.; Crich, D. Structure-Based Design and Synthesis of Apramycin−Paromomycin Analogues: Importance of the Configuration at the 6′-Position and Differences between the 6′-Amino and Hydroxy Series. J. Am. Chem. Soc. 2017, 139, 14611–14619. [Google Scholar] [CrossRef] [PubMed]
- Leban, N.; Kaplan, E.; Chaloin, L.; Godreuil, S.; Lionne, C. Kinetic characterization and molecular docking of novel allosteric inhibitors of aminoglycoside phosphotransferases. Biochim. Biophys. Acta 2017, 1861, 3464–3473. [Google Scholar] [CrossRef] [PubMed]
- Kotra, L.P.; Haddad, J.; Mobashery, S. Aminoglycosides: Perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob. Agents Chemother. 2000, 44, 3249–3256. [Google Scholar] [CrossRef] [PubMed]
- Boehr, D.D.; Draker, K.A.; Koteva, K.; Bains, M.; Hancock, R.E.; Wright, G.D. Broad-spectrum peptide inhibitors of aminoglycoside antibiotic resistance enzymes. Chem. Biol. 2003, 10, 189–196. [Google Scholar] [CrossRef]
- Gao, F.; Yan, X.; Baetting, O.M.; Berghuis, A.M.; Auclair, K. Regio- and chemoselective 6’-N-derivatization of aminoglycosides: Bisubstrate inhibitors as probes to study aminoglycoside 6’-N-acetyltransferases. Angew. Chem. Int. Ed. 2005, 44, 6859–6862. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Northrop, D.B. Synthesis of a tight-binding, multisubstrate analog inhibitor of gentamicin acetyltransferase I. J. Antibiot. (Tokyo) 1979, 32, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Haddad, J.; Azucena, E.; Kotra, L.P.; Kirzhner, M.; Mobashery, S. Tethered bisubstrate derivatives as probes for mechanism and as inhibitors of aminoglycoside 3’-phosphotransferases. J. Org. Chem. 2000, 65, 7422–7431. [Google Scholar] [CrossRef] [PubMed]
- Roestamadji, J.; Grapsas, I.; Mobashery, S. Mechanism-based inactivation of bacterial aminoglycoside 3’-phosphotransferases. J. Am. Chem. Soc. 1995, 117, 80–84. [Google Scholar] [CrossRef]
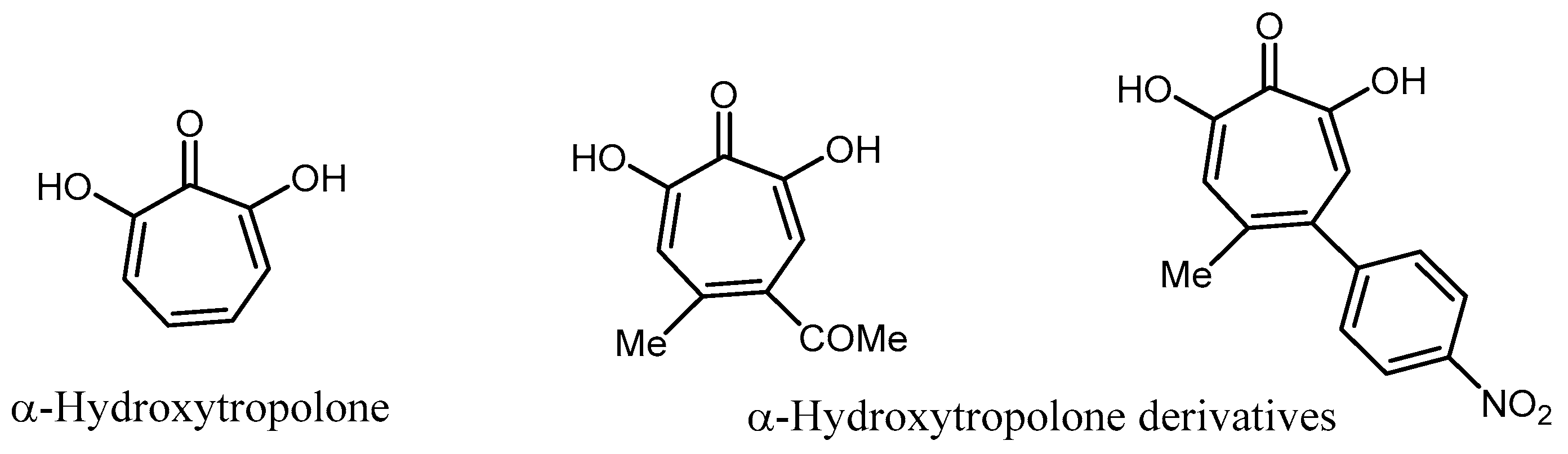
- Allen, N.E.; Alborn, W.E., Jr.; Hobbs, J.N., Jr.; Kirst, H.A. 7´hydroxytropolone: An inhibitor of aminoglycoside-2’’-O-adenyltrasfersae. Antimicrob. Agents Chemother. 1982, 22, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, D.R.; Cox, G.; D’Erasmo, M.P.; Shakya, T.; Meck, C.; Mohda, N.; Wright, G.D.; Murelli, R.P. Inhibition of the ANT(2’’)-Ia resistance enzyme and rescue of aminoglycoside antibiotic activity by synthetic α-hydroxytropolones. Bioorg. Med. Chem. Lett. 2014, 24, 4943–4947. [Google Scholar] [CrossRef] [PubMed]
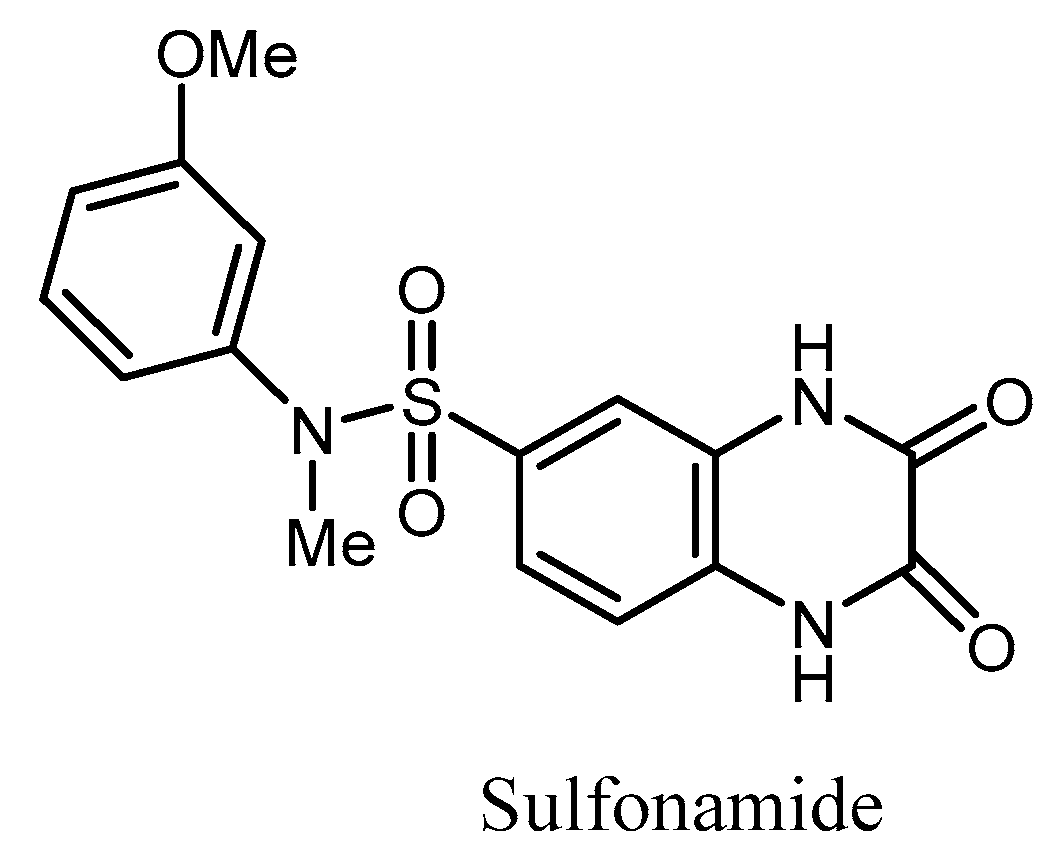
- Garzan, A.; Willby, M.J.; Green, K.D.; Gajadeera, C.S.; Hou, C.; Tsodikov, O.V.; Posey, J.E.; Garneau-Tsodikova, S. Sulfonamide-Based Inhibitors of Aminoglycoside Acetyltransferase Eis Abolish Resistance to Kanamycin in Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 10619–10628. [Google Scholar] [CrossRef] [PubMed]
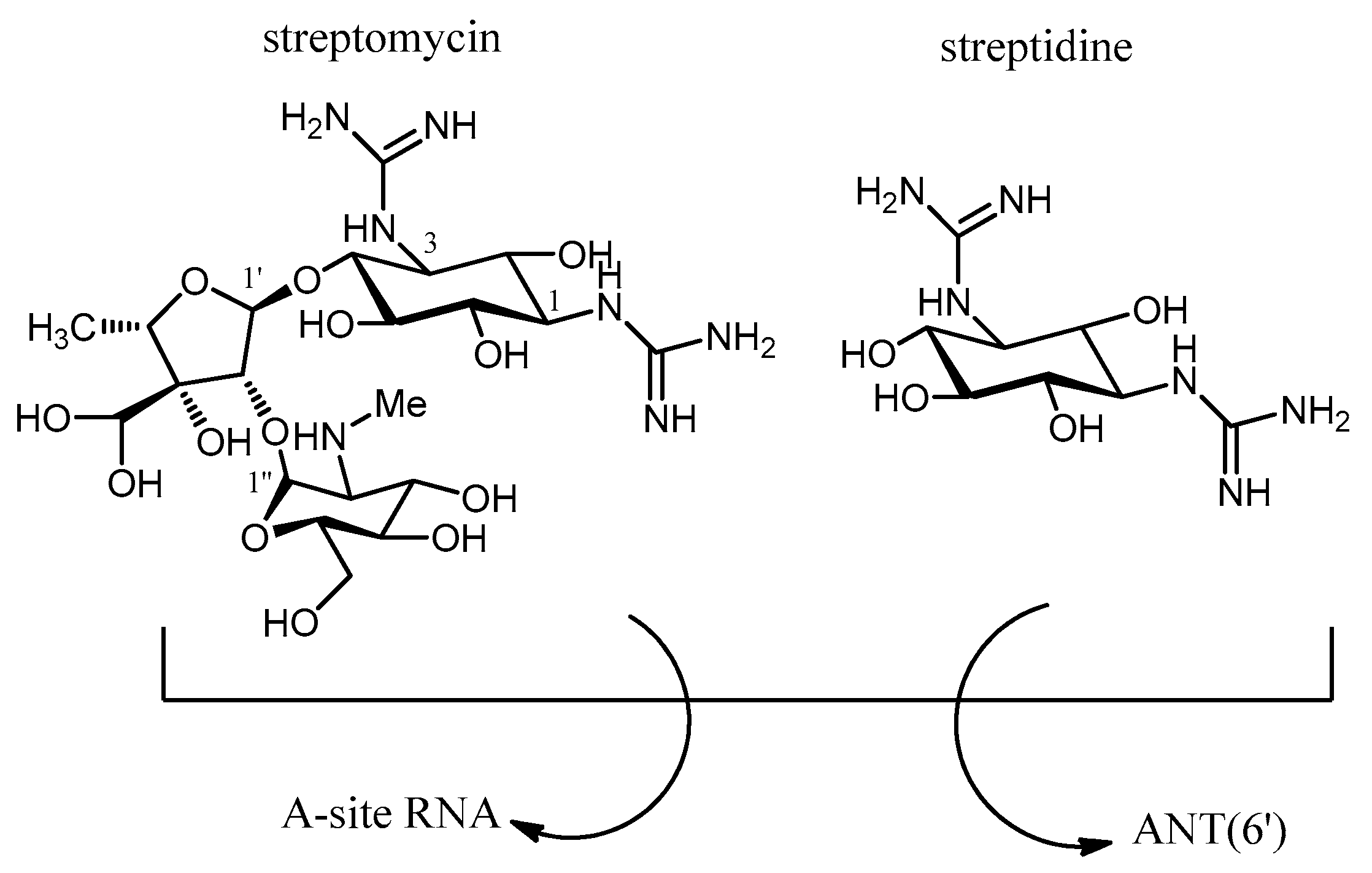
- Latorre, M.; Peñalver, P.; Revuelta, J.; Luis Asensio, J.L.; García-Junceda, E.; Bastida, A. Rescue of the streptomycin antibiotic activity by using streptidine as a “decoy acceptor” for the aminoglycoside-inactivating enzyme adenyl transferase. Chem. Commun. 2007, 2829–2831. [Google Scholar] [CrossRef] [PubMed]
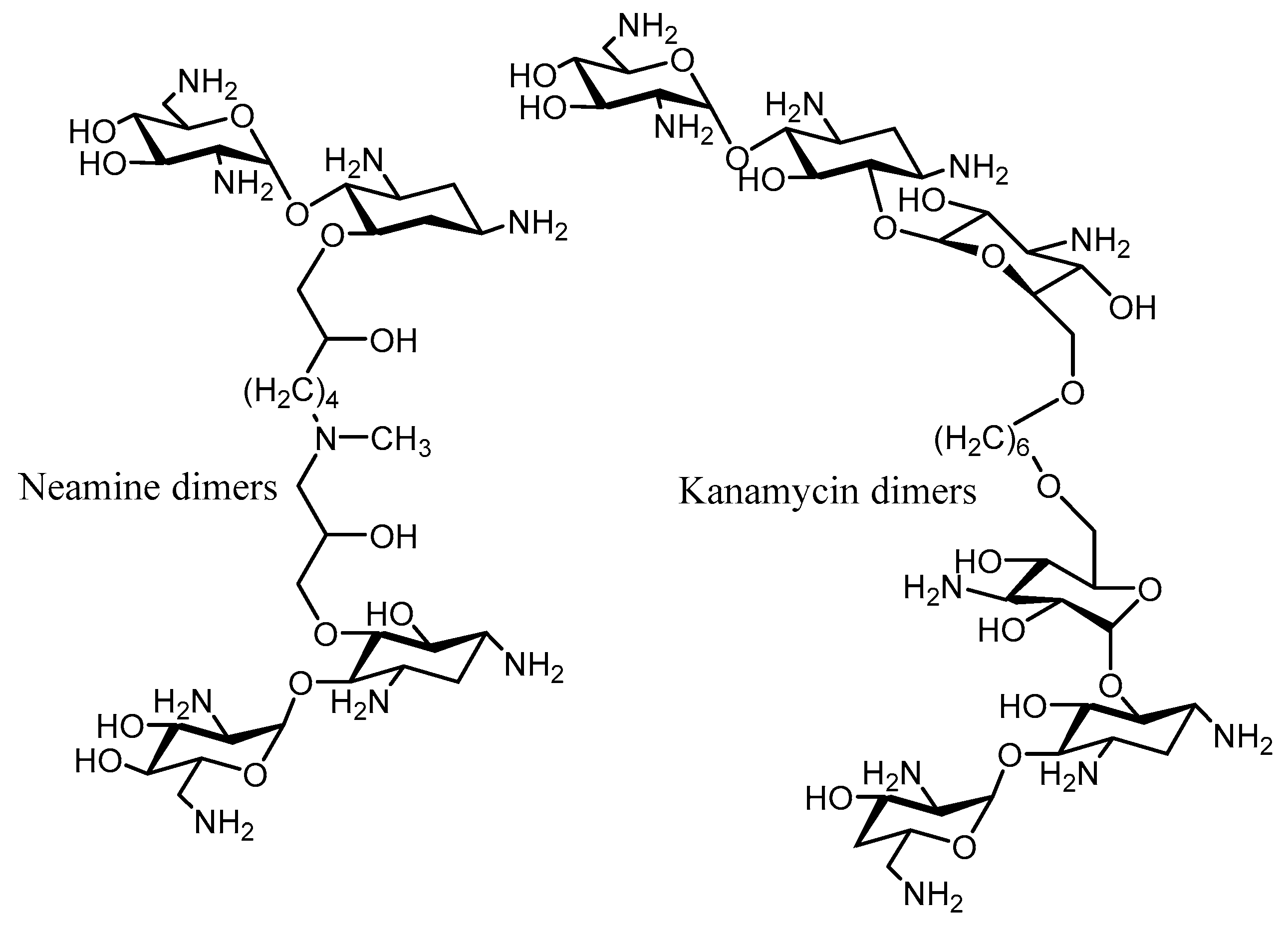
- Pokrovskaya, V.; Baasov, T. Dual-acting hybrid antibiotics: A promising strategy to combat bacterial resistance. Expert Opin. Drug Discov. 2010, 5, 883–902. [Google Scholar] [CrossRef] [PubMed]
- Berkov-Zrihen, Y.; Green, K.D.; Labby, K.J.; Feldman, M.; Garneau-Tsodikova, S.; Fridman, M. Synthesis and evaluation of hetero- and homodimers of ribosome-targeting antibiotics: Antimicrobial activity, in vitro inhibition of translation, and drug resistance. J. Med. Chem. 2013, 56, 5613–5625. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.P.; Wang, X.D.; Wang, P.F. Design, synthesis, and evaluation of novel fluoroquinolone-flavonoid hybrids as potent antibiotics against drug-resistant microorganisms. Eur. J. Med. Chem. 2014, 80, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T.; Chan, K.C.; So, P.K. Increased structural flexibility at the active site of a fluorophore-conjugated beta-lactamase distinctively impacts its binding toward diverse cephalosporin antibiotics. J. Biol. Chem. 2011, 286, 31771–31780. [Google Scholar] [CrossRef] [PubMed]
- Bremner, J.B.; Ambrus, J.I.; Samosorn, S. Dual action-based approaches to antibacterial agents. Curr. Med. Chem. 2007, 14, 1459–1477. [Google Scholar] [CrossRef] [PubMed]
- Hainrichson, M.; Pokrovskaya, V.; Shallom-Shezifi, D. Branched aminoglycosides: Biochemical studies and antibacterial activity of neomycin B derivatives. Bioorg. Med. Chem. 2005, 13, 5797–5807. [Google Scholar] [CrossRef] [PubMed]
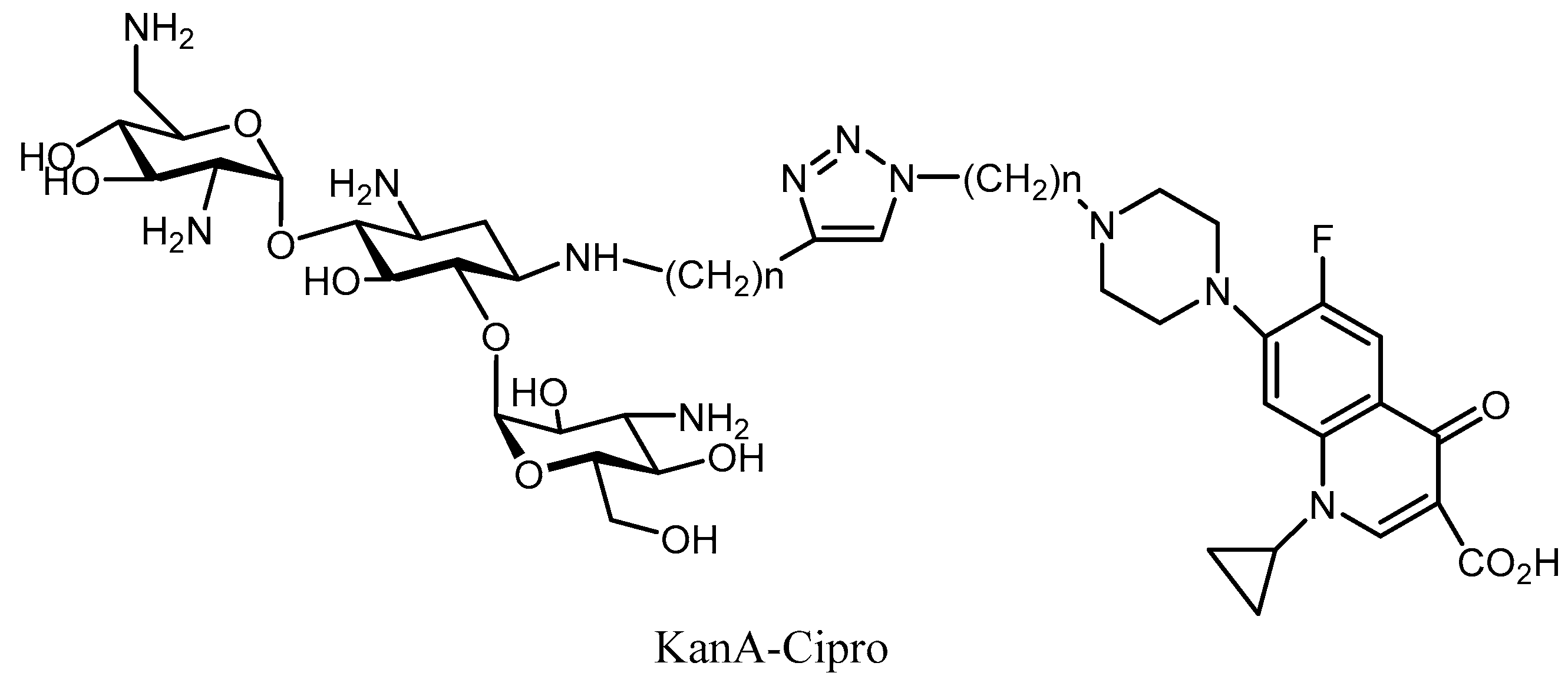
- Shavit, M.; Pokrovskaya, V.; Belakhov, V.; Baasov, T. Covalently linked kanamycin–Ciprofloxacin hybrid antibiotics as a toolto fight bacterial resistance. Bioorg. Med. Chem. 2017, 25, 2917–2925. [Google Scholar] [CrossRef] [PubMed]
- Pokrovskaya, V.; Nudelman, I.; Kandasamy, J.; Baasov, T. Aminoglycosides: Redesign Strategies for Improved Antibiotics and Compounds for Treatment of Human Genetic Diseases. In Methods in Enzymology, 1st ed.; Fukuda, M., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2010; Charper 21 Glycomics; Volume 478, pp. 437–462. ISSN 0076-6879. [Google Scholar] [CrossRef]
- Serpersu, E.H.; Norris, A.L. Effect of protein dynamics and solvent in ligand recognition by promiscuous aminoglycoside-modifying enzymes. In Advances in Carbohydrate Chemistry and Biochemistry; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 67, pp. 222–243. ISBN 978-0-12-396527-1. [Google Scholar] [CrossRef]
- Herzog, I.M.; Zada, S.L.; Fridman, M. Effects of 5-O-Ribosylation of Aminoglycosides on Antimicrobial Activity and Selective Perturbation of Bacterial Translation. J. Med. Chem. 2016, 59, 8008–8018. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Resistance Profile | Bacterial Source | Pdb Number | |
|---|---|---|---|---|
| AAC(6′) | I (a–d,e,f–z) | T, A, N, D, S, K, I | Salmonella enterica | 1S60, 2VBQ, 1S3Z, 1S5K, 2QIR |
| II | T, G, N, D, S, K | Enterococcus faecium | 2A4N, 5E96 | |
| Acinetobacter haemolyticus | 4F0Y, 4EVY, 4F0Y | |||
| Acinetobacter baumannii | 4E80 | |||
| Escherichia coli | 6BFF, 6BFH, 1V0C, 2BUE, 2VQY | |||
| Staphylococcus warneri | 4QC6 | |||
| AAC(3) | I (a–b) | G, S, F | Serratia marcesans | 1B04 |
| II (a–c) | T, G, N, D, S | Pseudomonas aeruginosa | 4YFJ | |
| III (a–c) | T, G, D, S, K, N, P, L | Klebsiella pneumoniae, | ||
| IV | T, S, N, D, S, A | Campylobacter jejuni | ||
| VII | G | Actinomycetes | ||
| AAC(2′) | I (a–c) | T, S, N, D, Ne | Providencia stuartii | 5US1 |
| Mycobacterium tuberculosis | 1M44, 1M4D, 1M4G, 1M41 | |||
| AAC(1) | Ia | P, L, R, AP | E. coli | |
| Campylobacter spp. |
| Enzyme | Resistance Profile | Bacterial Source | Pdb | |
|---|---|---|---|---|
| APH(3′) | I (a–d) | K, Ne, R, L, P | Acinetobacter baumannii | 4FEV |
| II | K, Ne, B, P, R | Stenotrophomonas maltophilia | ||
| III (a–b) | K, Ne, P, B, L, R, B, A, I | |||
| IV | K, Ne, B, P, R | |||
| V | Ne, P, R | |||
| VI | K, Ne, P, R, B, A, I | Bacillus circulans | ||
| APH(2″) | I-a | K, G, T, S, D | ||
| I-(b,d) | K, G, T, N, D | Escherichia coli | 4DCA | |
| II-(a–b) | K, G, T | Enterococcus faecium | 3HAM, 3HAV | |
| IVa | G, K, S | Enterococcus cassaliflavus | 5C4K, 5C4L, 4N57, 4DT8, 4DT9, 4DTA, 4DTB, 3SG8, 3SG9 | |
| APH(3″) | I (a–b) | St | Acinetobacter baumannii | 4EJ7, 4FEU, 4FEV, 4FEX, 4FEW |
| III a | St | Enterococcus faecalis | 2BKK | |
| APH(7) | I a | H | Streptomyces hygroscopicus | |
| APH(4) | I-(a–b) | H | Escherichia coli | 3W0O, 3TYK, 3W0M, 3W0N |
| APH(6) | I-(a–d) | St | Streptomyces griseus | |
| APH(9) | I-(a–b) | Sp | Legionella pneumophila | 3I0O, 3I0Q, 3I1A, 3Q2M |
| Enzyme | Resistance Profile | Bacterial Host | Pdb Number |
|---|---|---|---|
| ANT(2″) | K, T, G, D, S | Pseudomonas aeruginosa | 4XJE, 5CFT, 5CFS, 5CFU |
| Klebsiella pneumoniae | 4WQK, 4WQL, 5KQJ | ||
| ANT(3″) | St, Sp | Salmonella enterica | 4CS6, 5G4A |
| ANT(4′) | K, Ne, T, A, D, I | Pseudomonas aeruginosa | 4EBJ, 4EBK |
| Staphylococcus aureus | 1KNY | ||
| ANT(6) | St | Bacillus subtilis | 2PBE, 1B87 |
| ANT(9) | Sp | Enterococcus avium |
| Enzyme | Resistance Profile | Bacterial Source | Pdb Number |
|---|---|---|---|
| Enterococcus faecalis | |||
| AAC(6′)-Ie-APH(2″)-IVa | G, K, T, A | Staphylococcus aureus | 4ORQ |
| APH(2″)-Id-APH(2″)-IVa | K, G, T, S, D | Enterococcus casseliflavus | 4DBX, 4DE4, 4DFB |
| APH(2″)-Ia-APH(6′)-Ie | K, G, T, S, D, St | Staphylococcus aureus | 5IQF |
| ANT(3)-Ib-AAC(6′)-IId | T, A, N, D, S, K, St, Sp | Serratia marcescens | |
| AAC(3)-Ib-AAC(6′)-Ib | G, S, F, T, A, N, D, K, I | Pseudomonas aeruginosa |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zárate, S.G.; De la Cruz Claure, M.L.; Benito-Arenas, R.; Revuelta, J.; Santana, A.G.; Bastida, A. Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors. Molecules 2018, 23, 284. https://doi.org/10.3390/molecules23020284
Zárate SG, De la Cruz Claure ML, Benito-Arenas R, Revuelta J, Santana AG, Bastida A. Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors. Molecules. 2018; 23(2):284. https://doi.org/10.3390/molecules23020284
Chicago/Turabian StyleZárate, Sandra G., M. Luisa De la Cruz Claure, Raúl Benito-Arenas, Julia Revuelta, Andrés G. Santana, and Agatha Bastida. 2018. "Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors" Molecules 23, no. 2: 284. https://doi.org/10.3390/molecules23020284
APA StyleZárate, S. G., De la Cruz Claure, M. L., Benito-Arenas, R., Revuelta, J., Santana, A. G., & Bastida, A. (2018). Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors. Molecules, 23(2), 284. https://doi.org/10.3390/molecules23020284