Effect of Melatonin on the Renin-Angiotensin-Aldosterone System in l-NAME-Induced Hypertension

 ,
, 
Abstract
:1. Introduction
2. Results
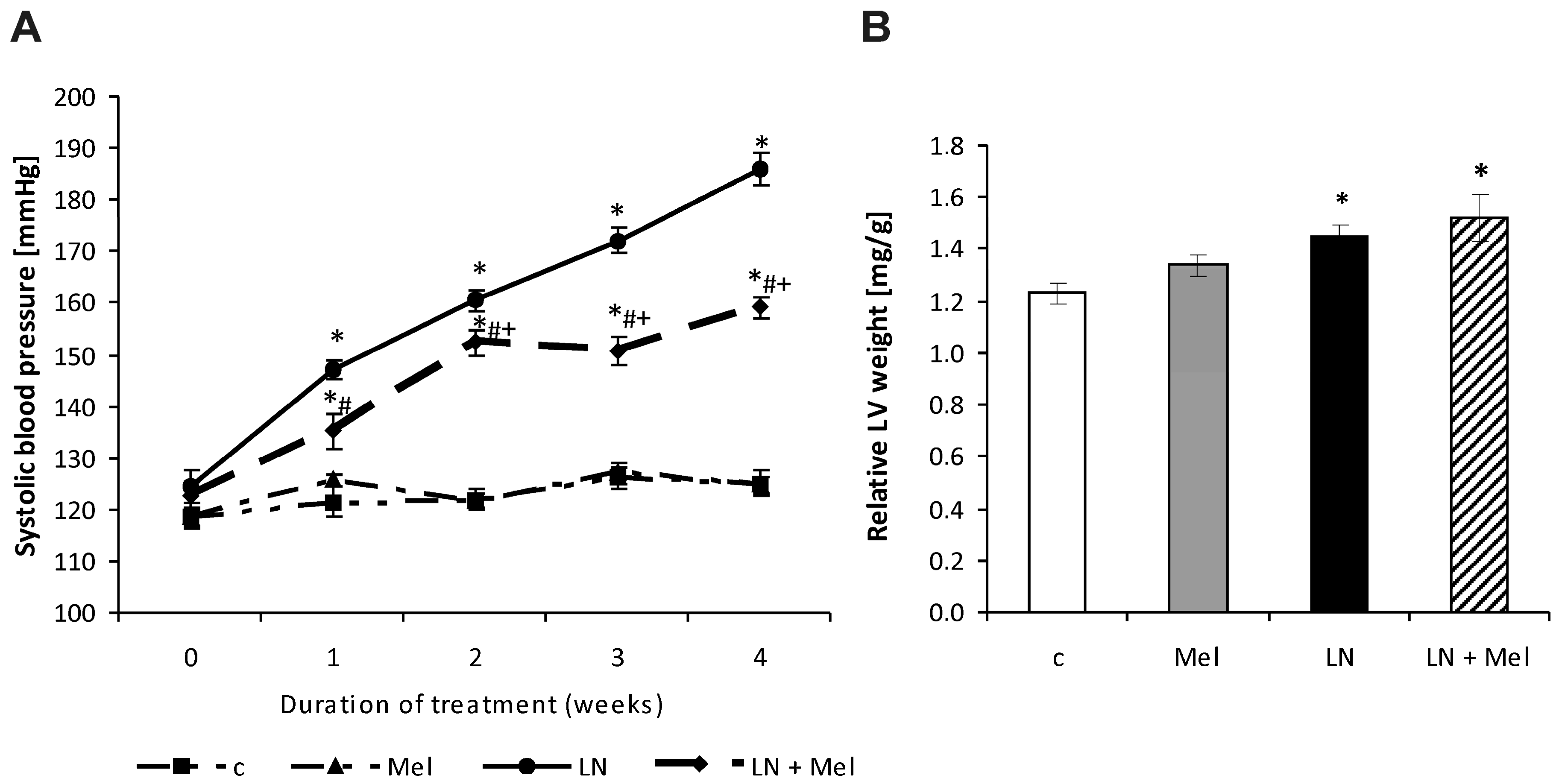
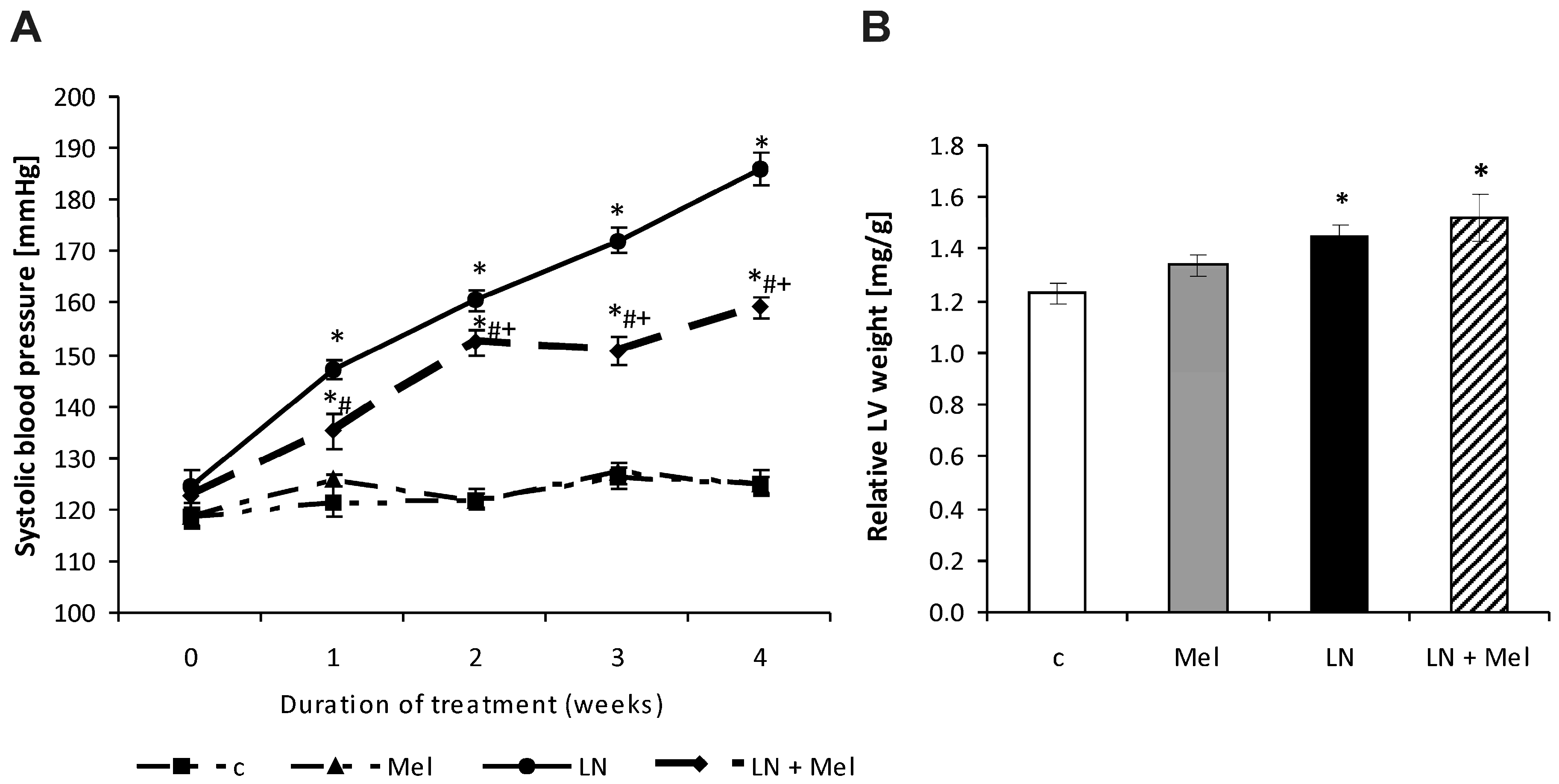
2.1. Cardiovascular Parameters
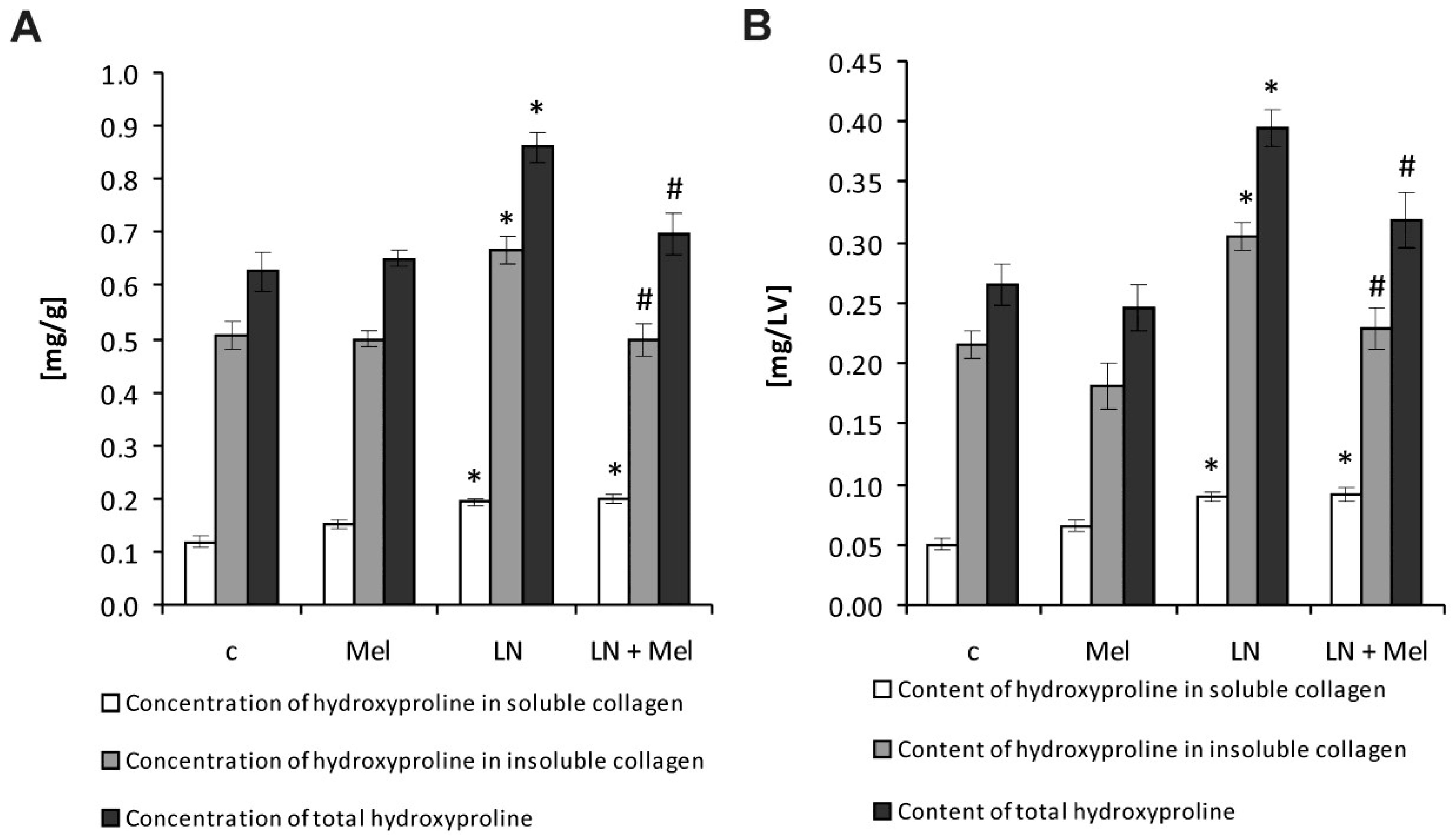
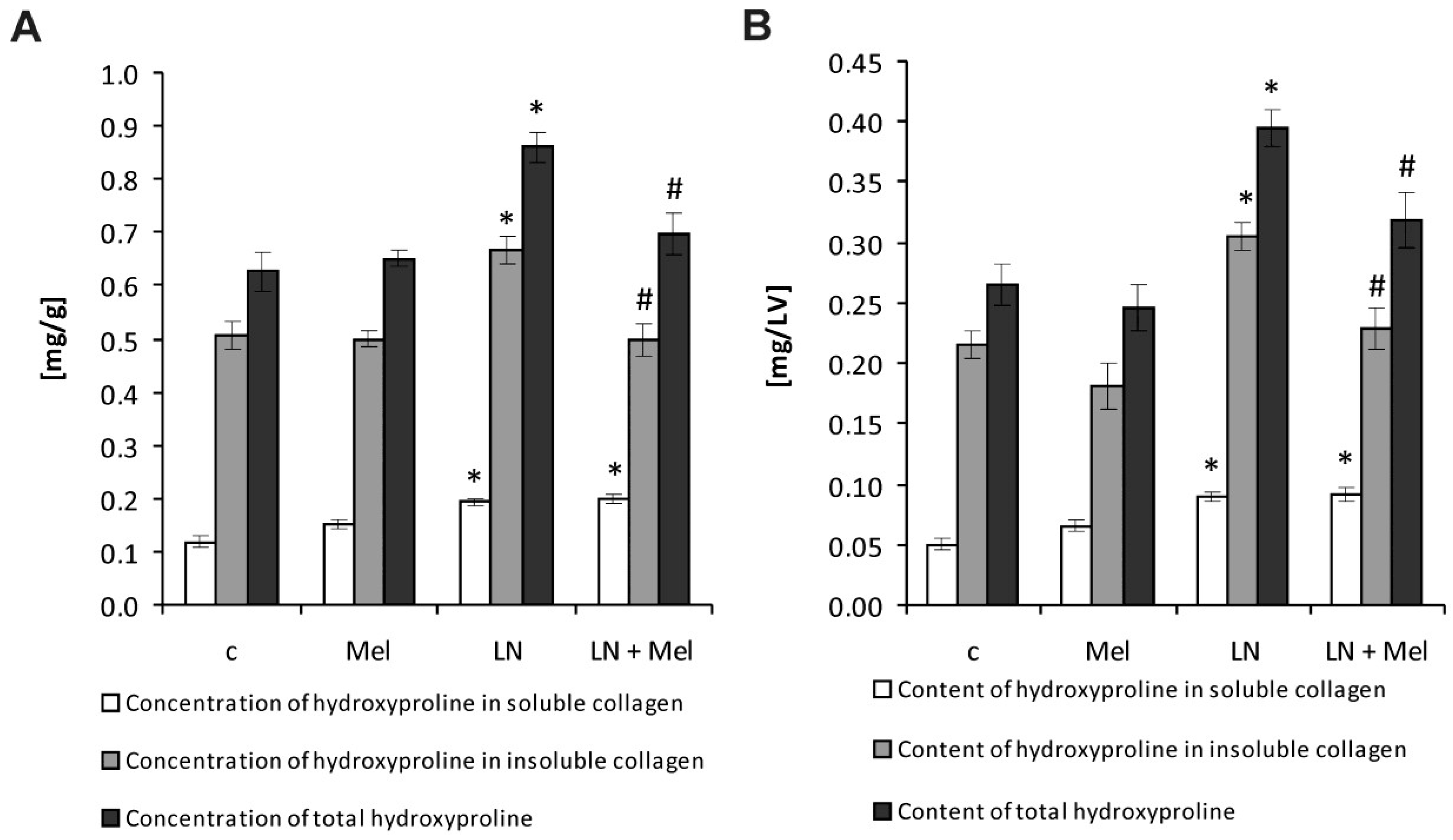
2.2. Hydroxyproline Concentration and Content in the Soluble, Insoluble and Total Collagenous Proteins
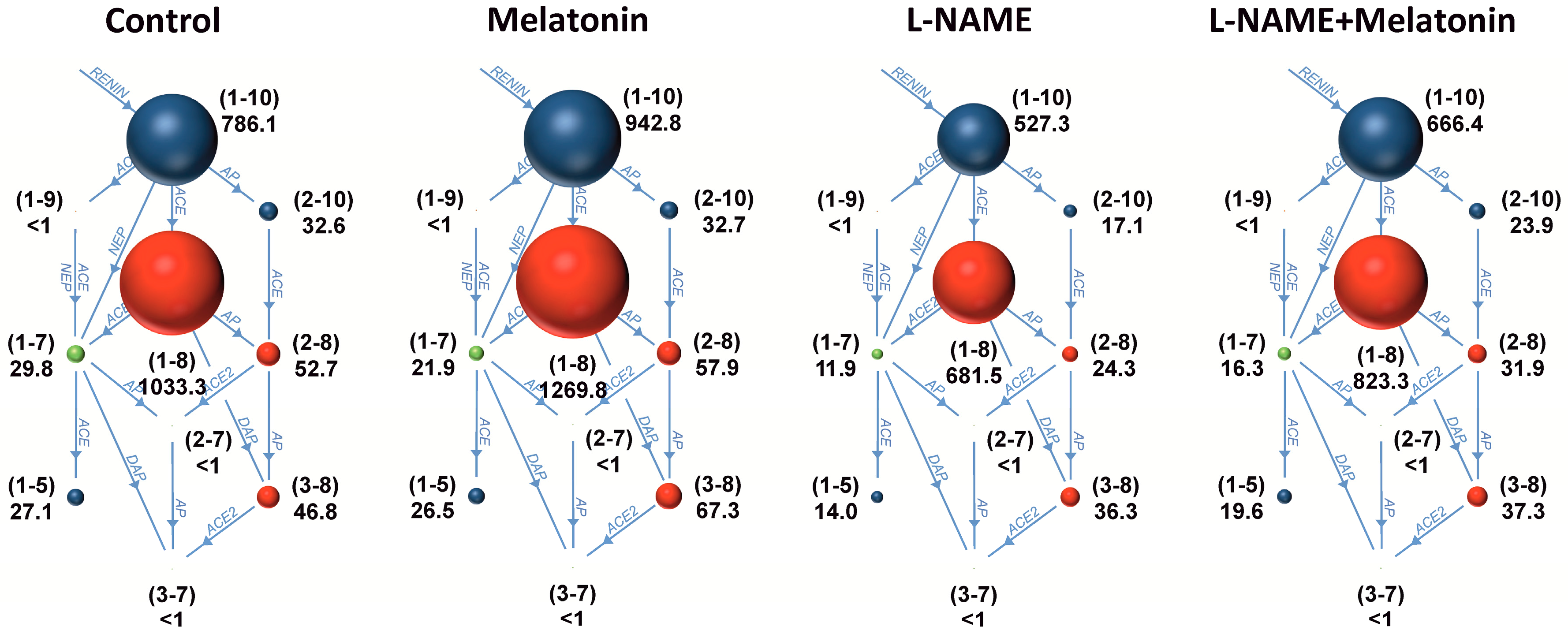
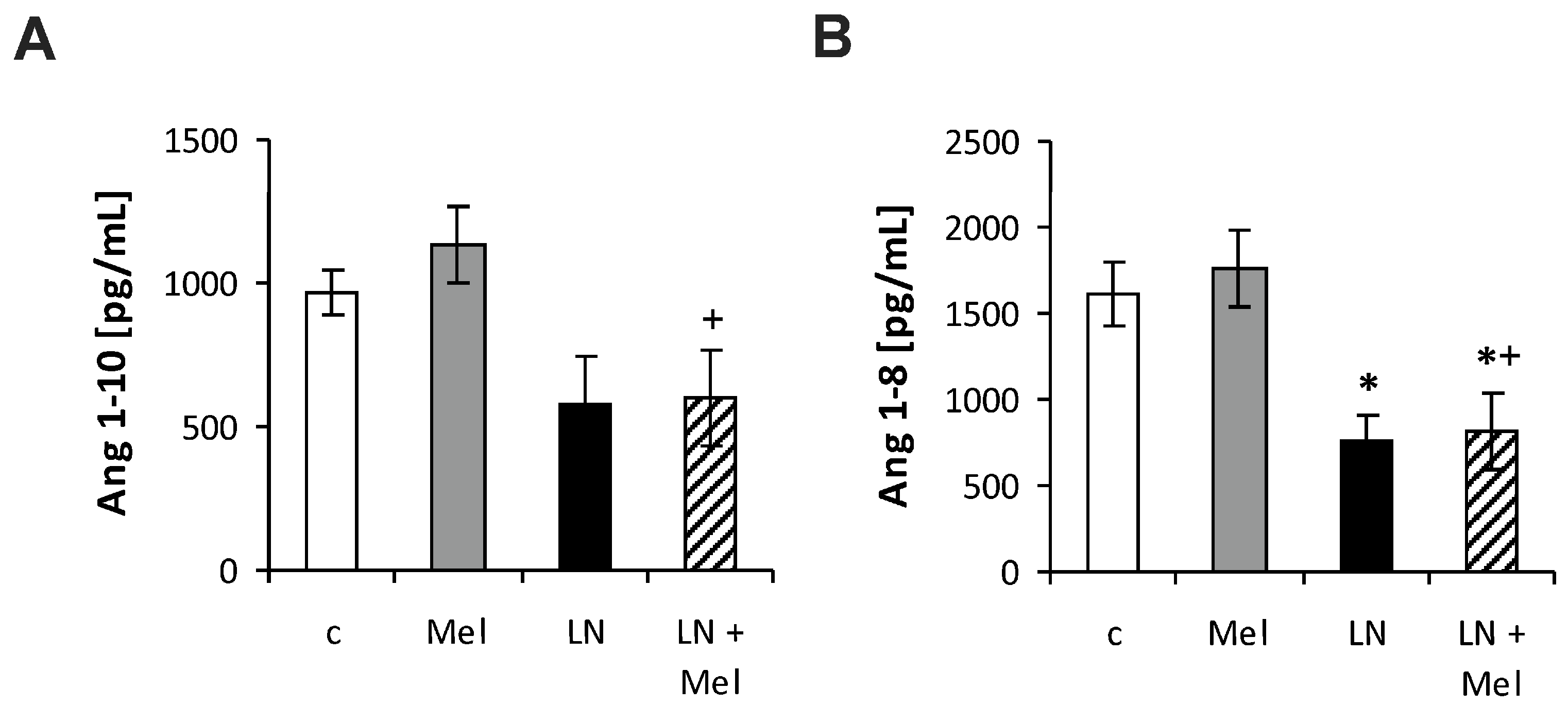
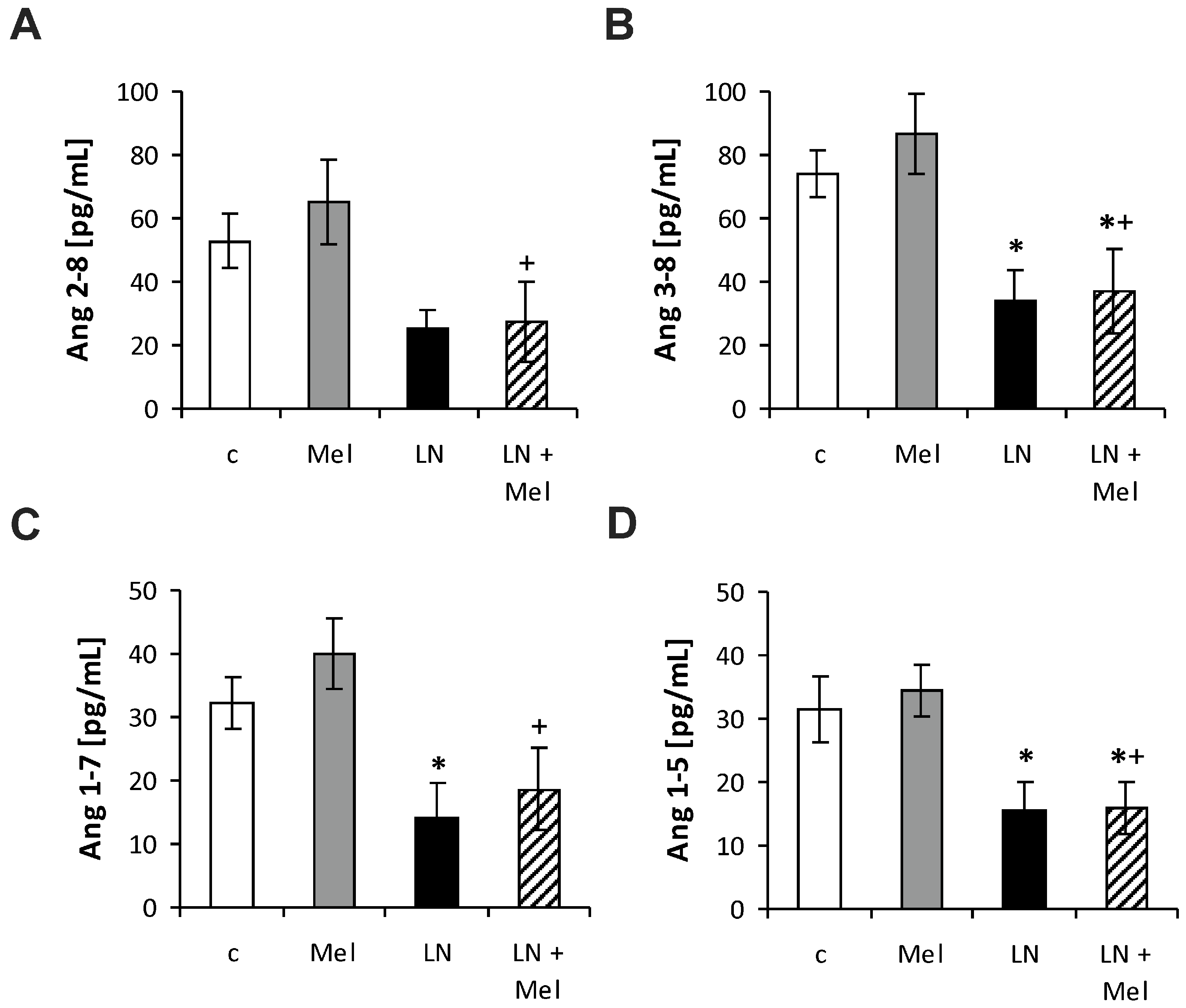
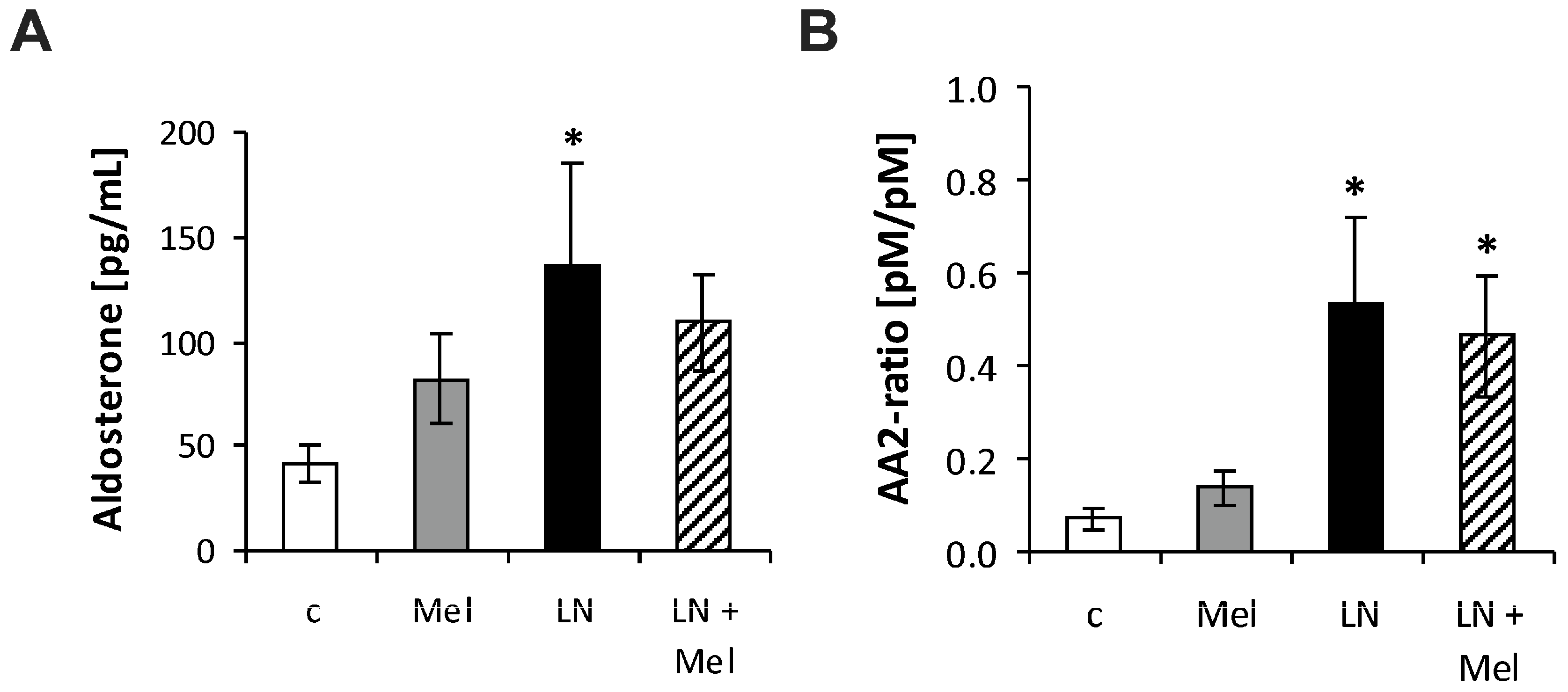
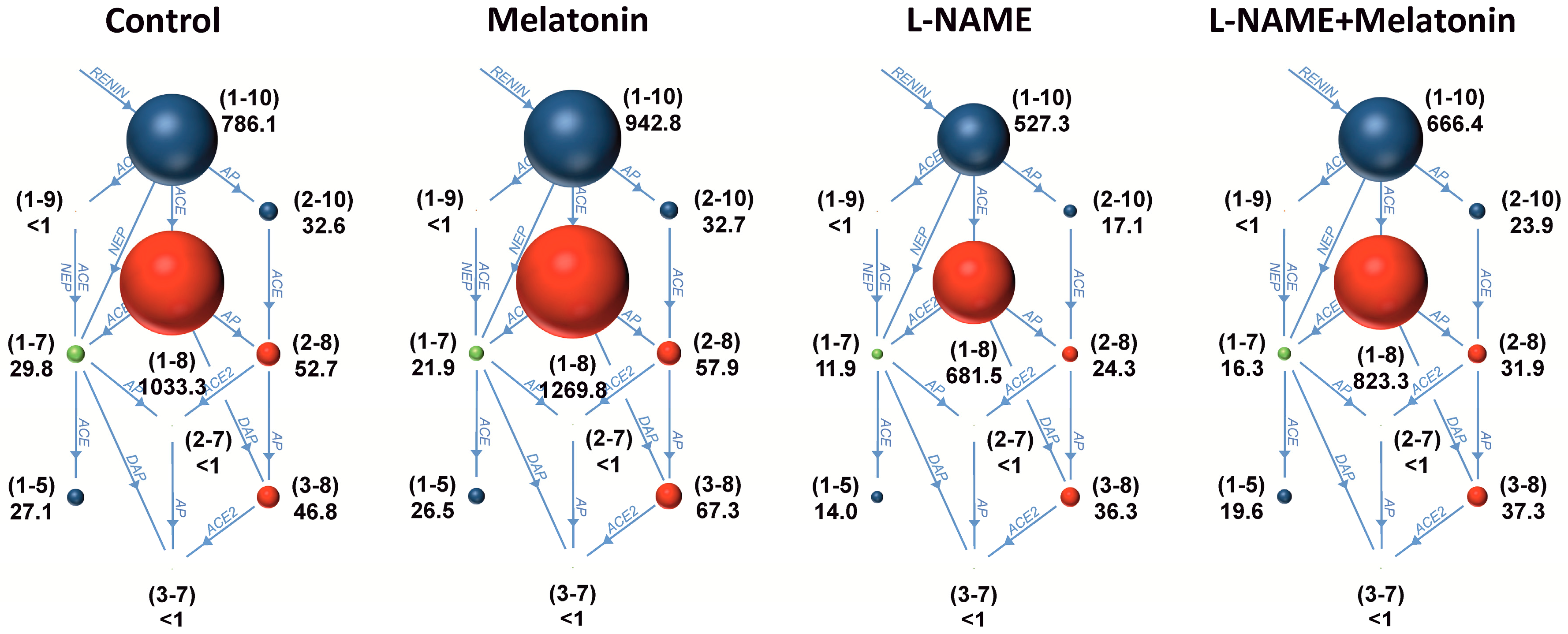
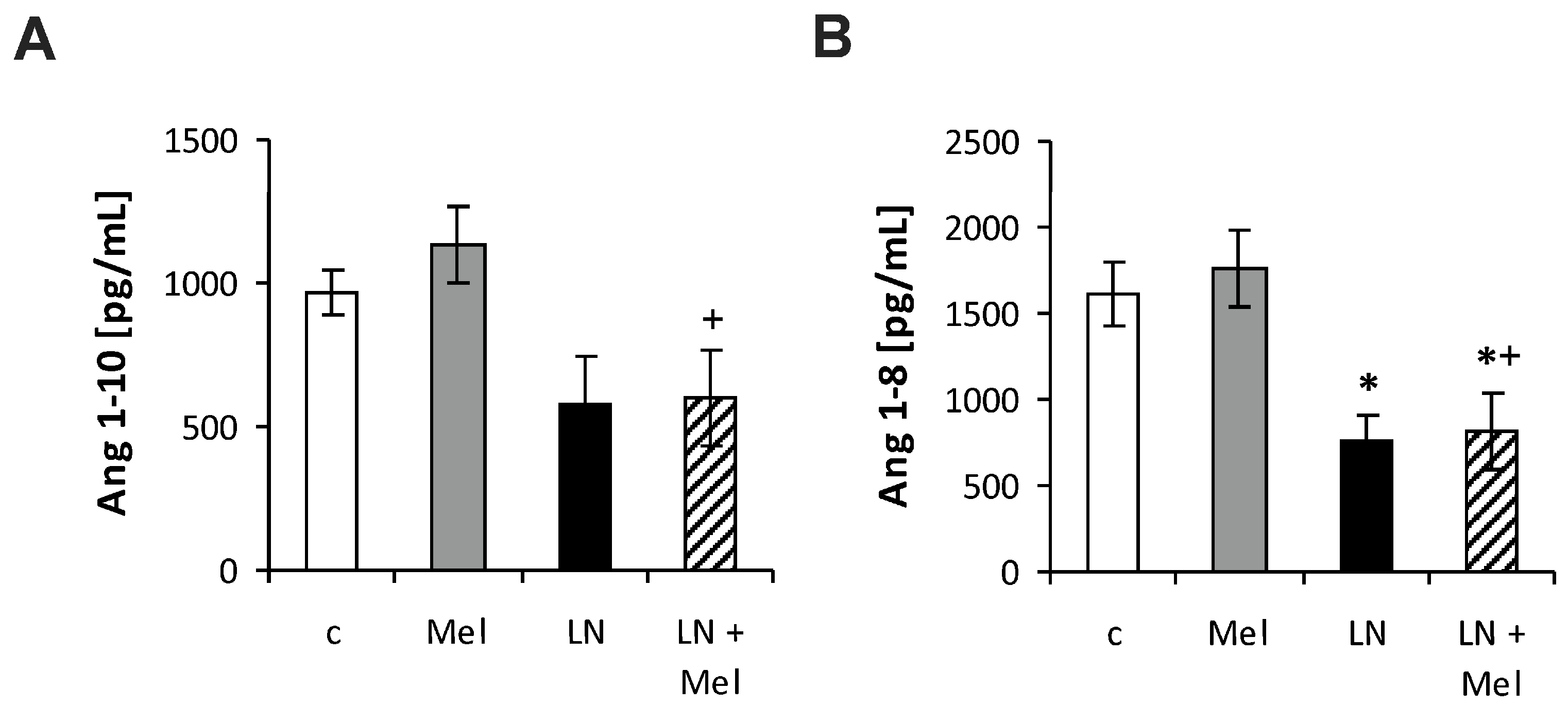
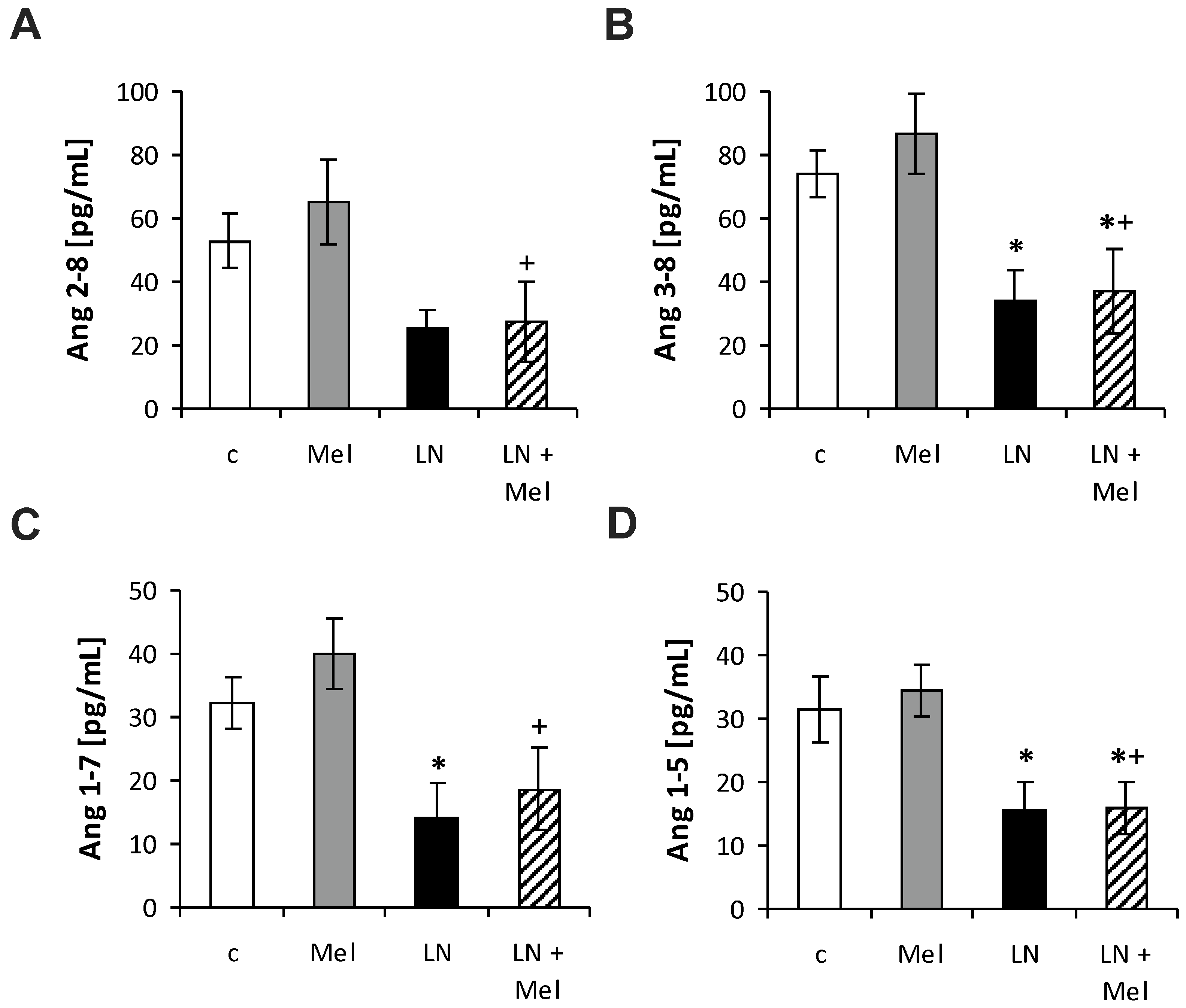
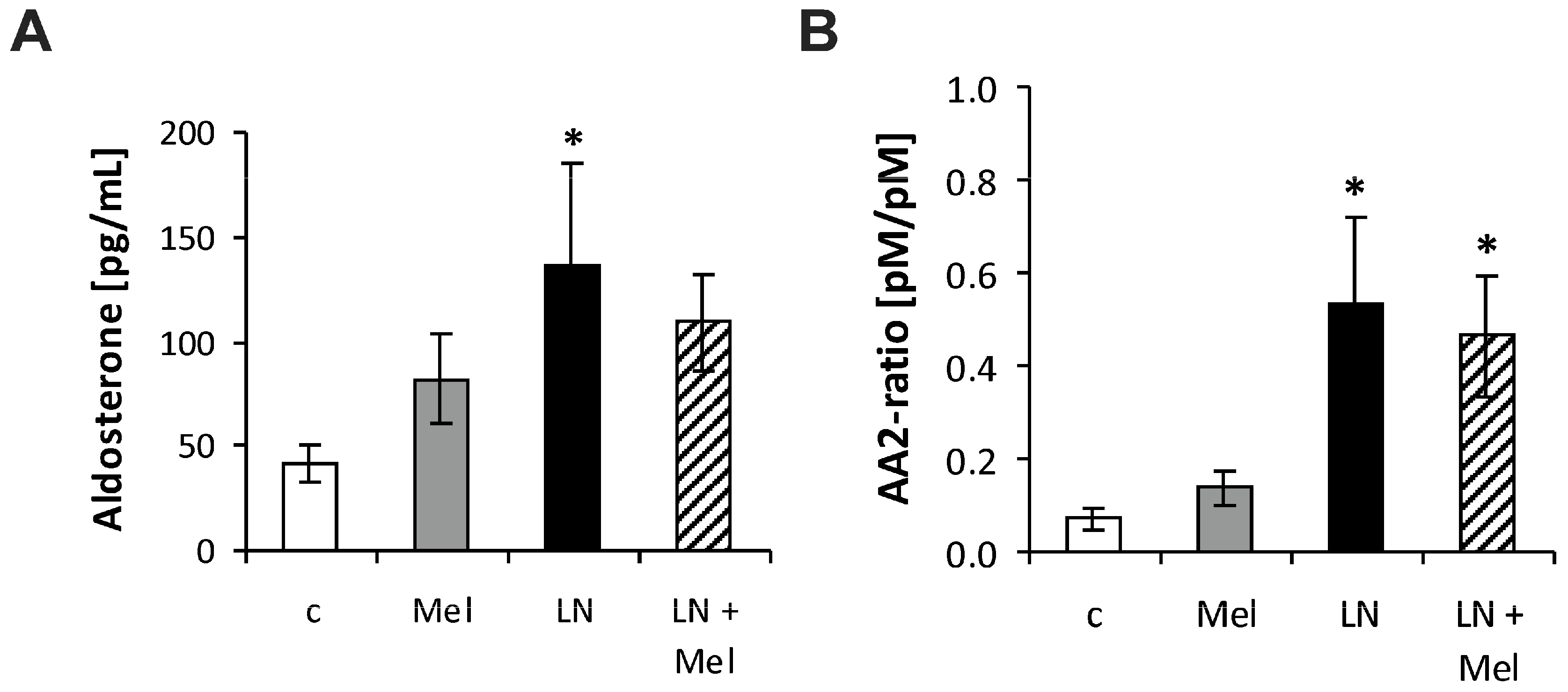
2.3. The Serum Concentrations of Angiotensins and Aldosterone
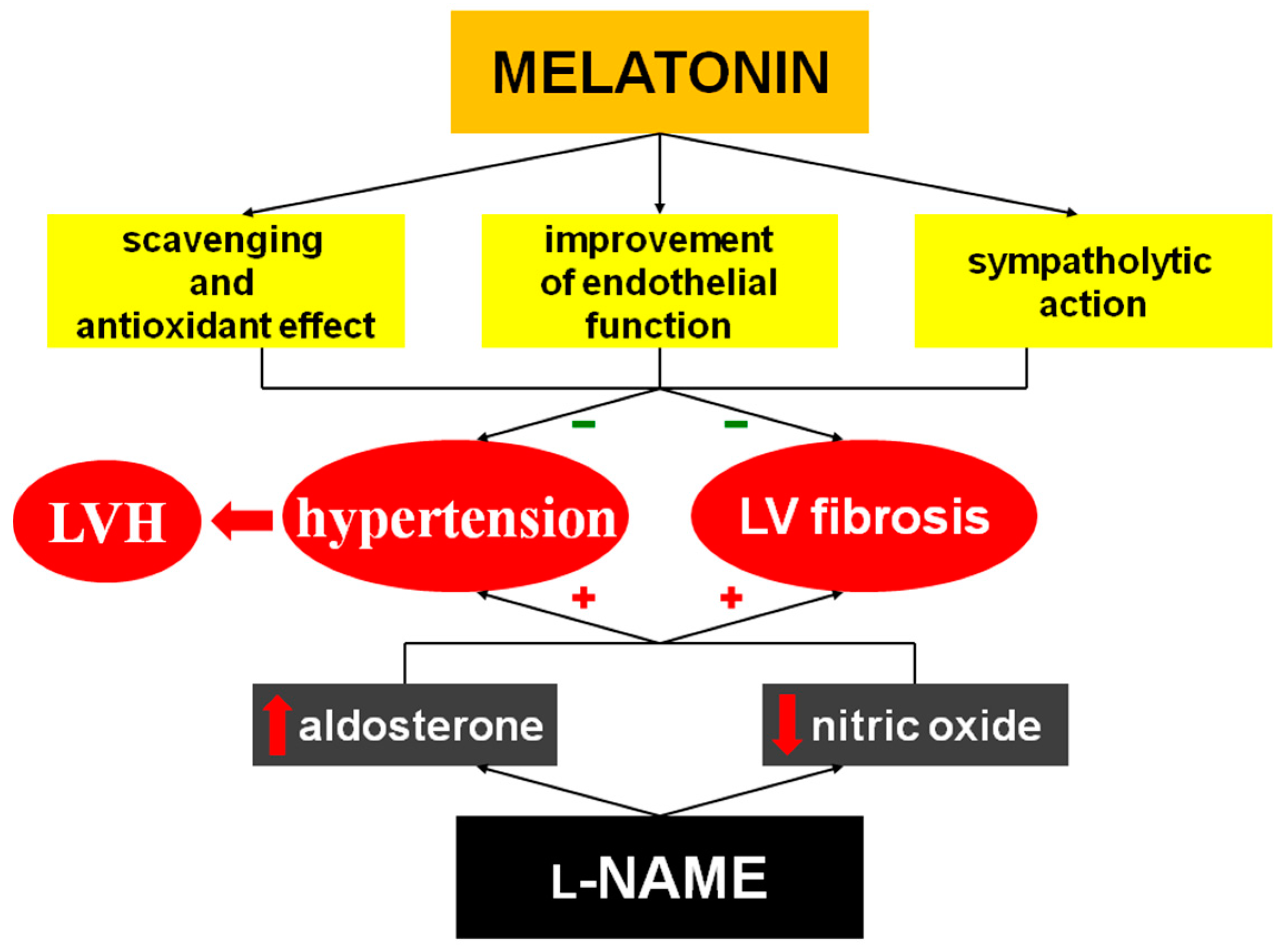
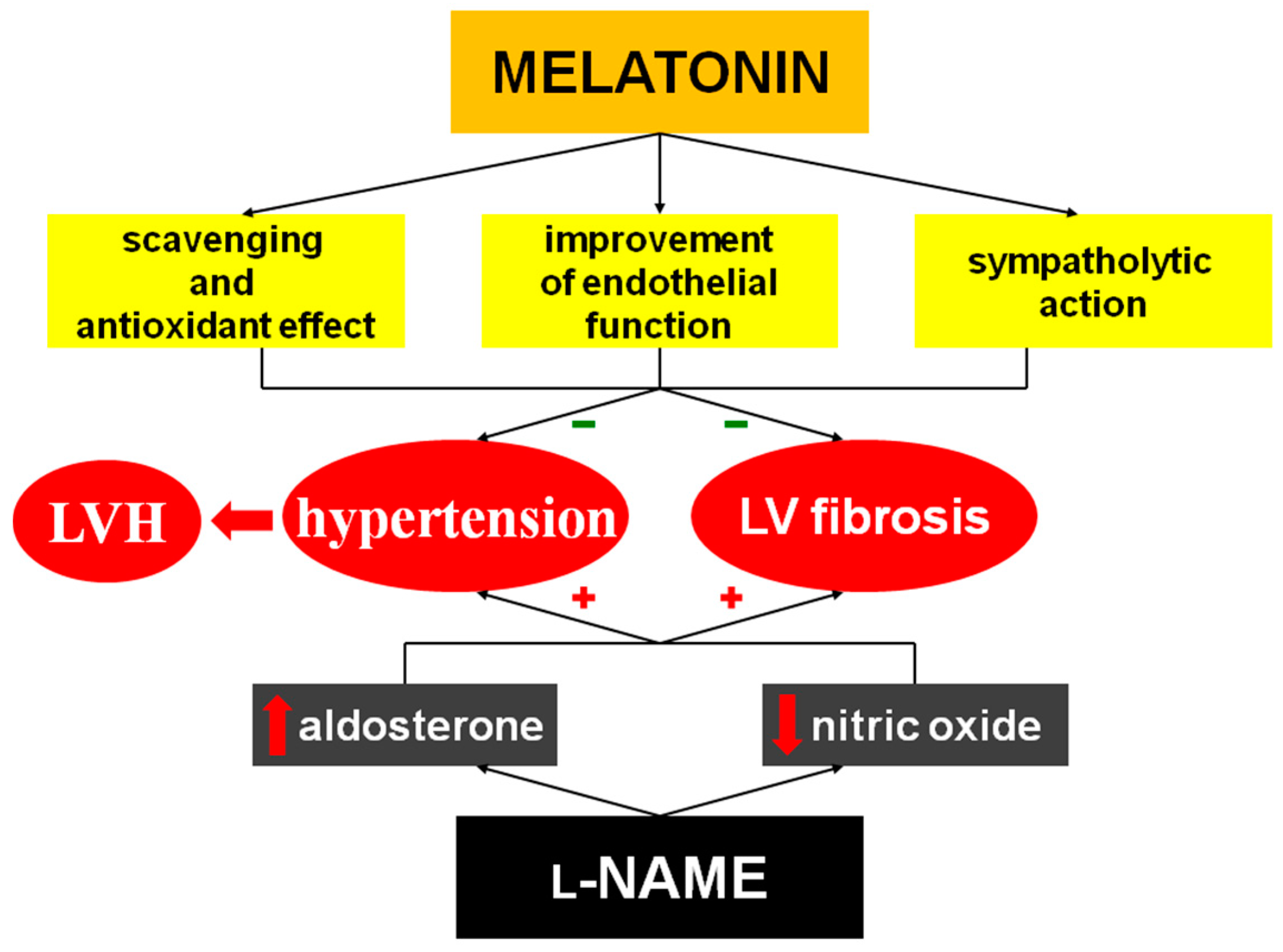
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Determination of Hydroxyproline
4.3. Angiotensins and Aldosterone Analyses
4.4. Statistical Analyses
5. Limitations
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Simko, F.; Simko, J. The potential role of nitric oxide in the hypertrophic growth of the left ventricle. Physiol. Res. 2000, 49, 37–46. [Google Scholar] [PubMed]
- Simko, F. Is NO the king? Pathophysiological benefit with uncertain clinical impact. Physiol. Res. 2007, 56, S1–S6. [Google Scholar] [PubMed]
- Pechanova, O.; Bernatova, I.; Pelouch, V.; Simko, F. Protein remodelling of the heart in NO-deficient hypertension: The effect of captopril. J. Mol. Cell. Cardiol. 1997, 29, 3365–3374. [Google Scholar] [CrossRef] [PubMed]
- Bernatova, I.; Pechanova, O.; Pelouch, V.; Simko, F. Regression of chronic l-NAME-treatment-induced left ventricular hypertrophy: Effect of captopril. J. Mol. Cell. Cardiol. 2000, 32, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bernatova, I.; Pechanova, O.; Simko, F. Captopril prevents NO-deficient hypertension and left ventricular hypertrophy without affecting nitric oxide synthase activity in rats. Physiol. Res. 1996, 45, 311–316. [Google Scholar] [PubMed]
- Holecyova, A.; Torok, J.; Bernatova, I.; Pechanova, O. Restriction of nitric oxide rather than elevated blood pressure is responsible for alterations of vascular responses in nitric oxide-deficient hypertension. Physiol. Res. 1996, 45, 317–321. [Google Scholar] [PubMed]
- Takemoto, M.; Egashira, K.; Usui, M.; Numaguchi, K.; Tomita, H.; Tsutsui, H.; Shimokawa, H.; Sueishi, K.; Takeshita, A. Important role of tissue angiotensin-converting enzyme activity in the pathogenesis of coronary vascular and myocardial structural changes induced by long-term blockade of nitric oxide synthesis in rats. J. Clin. Investig. 1997, 15, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, R.; Node, K.; Akashi, M. Estimation methods for human circadian phase by use of peripheral tissues. Hypertens. Res. 2016, 39, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Simko, F. Chronobiology of blood pressure: Emerging implications of melatonin. Eur. J. Clin. Investig. 2012, 42, 1252–1254. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Paulis, L. Melatonin as a potential antihypertensive treatment. J. Pineal Res. 2007, 42, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Simko, F. Blood pressure modulation and cardiovascular protection by melatonin: Potential mechanisms behind. Physiol. Res. 2007, 56, 671–684. [Google Scholar] [PubMed]
- Reiter, R.J.; Manchester, L.C.; Fuentes-Broto, L.; Tan, D.X. Cardiac hypertrophy and remodelling: Pathophysiological consequences and protective effects of melatonin. J. Hypertens. 2010, 28, S7–S12. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O. Remodelling of the heart and vessels in experimental hypertension: Advances in protection. J. Hypertens. 2010, 28, S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Reiter, R.J.; Pechanova, O.; Paulis, L. Experimental models of melatonin-deficient hypertension. Front. Biosci. 2013, 18, 616–625. [Google Scholar] [CrossRef]
- He, B.; Zhao, Y.; Xu, L.; Gao, L.; Su, Y.; Lin, N.; Pu, J. The nuclear melatonin receptor RORα is a novel endogenous defender against myocardial ischemia/reperfusion injury. J. Pineal Res. 2016, 60, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Nduhiraband, I.F.; Lamont, K.; Albertyn, Z.; Opie, L.H.; Lecour, S. Role of toll-like receptor 4 in melatonin-induced cardioprotection. J. Pineal Res. 2016, 60, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O. Recent trends in hypertension treatment: Perspectives from animal studies. J. Hypertens. 2009, 27, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H.; Lecour, S. Melatonin has multiorgan effects. Eur. Heart J. Cardiovasc. Pharmacother. 2016, 2, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Baka, T.; Paulis, L.; Reiter, R.J. Elevated heart rate and nondipping heart rate as potential targets for melatonin: A review. J. Pineal Res. 2016, 61, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Galano, A. Melatonin: Exceeding expectations. Physiology 2014, 29, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Bernatova, I.; Pechanova, O.; Simko, F. Effect of captopril in l-NAME-induced hypertension on the rat myocardium, aorta, brain and kidney. Exp. Physiol. 1999, 84, 1095–1105. [Google Scholar] [PubMed]
- Simko, F.; Matuskova, J.; Luptak, I.; Krajcirovicova, K.; Kucharska, J.; Gvozdjakova, A.; Babal, P.; Pechanova, O. Effect of simvastatin on remodeling of the left ventricle and aorta in l-NAME-induced hypertension. Life Sci. 2004, 23, 1211–1224. [Google Scholar] [CrossRef]
- Simko, F.; Luptak, I.; Matuskova, J.; Krajcirovicova, K.; Sumbalova, Z.; Kucharska, J.; Gvozdjakova, A.; Simko, J.; Babal, P.; Pechanova, O.; et al. l-arginine fails to protect against myocardial remodelling in l-NAME-induced hypertension. Eur. J. Clin. Investig. 2005, 35, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Pechanova, O.; Zicha, J.; Liskova, S.; Celec, P.; Mullerova, M.; Kollar, J.; Behuliak, M.; Kunes, J.; Adamcova, M.; et al. Melatonin improves the restoration of endothelium-derived constricting factor signalling and inner diameter in the rat femoral artery after cessation of l-NAME treatment. J. Hypertens. 2010, 28 (Suppl. 1), S19–S24. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Pechanova, O.; Zicha, J.; Krajcirovicova, K.; Barta, A.; Pelouch, V.; Adamcova, M.; Simko, F. Melatonin prevents fibrosis but not hypertrophy development in the left ventricle of NG-nitro-l-arginine-methyl ester hypertensive rats. J. Hypertens. 2009, 27, S11–S16. [Google Scholar] [CrossRef]
- Simko, F.; Simko, J. Heart failure and angiotensin converting enzyme inhibition: Problems and perspectives. Physiol. Res. 1999, 48, 1–8. [Google Scholar] [PubMed]
- Simko, F.; Simko, J.; Fabryova, M. ACE-inhibition and angiotensin II receptor blockers in chronic heart failure: Pathophysiological consideration of the unresolved battle. Cardiovasc. Drugs Ther. 2003, 17, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Unger, T. Novel therapeutic targets for hypertension. Nat. Rev. Cardiol. 2010, 7, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Nehme, A.; Zibara, K. Efficiency and specificity of RAAS inhibitors in cardiovascular diseases: How to achieve better end-organ protection? Hypertens. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hrenak, J.; Paulis, L.; Simko, F. Angiotensin A/Alamandine/MrgD Axis: Another Clue to Understanding Cardiovascular Pathophysiology. Int. J. Mol. Sci. 2016, 17, 1098. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Poglitsch, M.; Yogasundaram, H.; Thomas, J.; Rowe, B.H.; Oudit, G.Y. Roles of Angiotensin Peptides and Recombinant Human ACE2 in Heart Failure. J. Am. Coll. Cardiol. 2017, 69, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1–7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Papinska, A.M.; Mordwinkin, N.M.; Meeks, C.J.; Jadhav, S.S.; Rodgers, K.E. Angiotensin-(1–7) administration benefits cardiac, renal and progenitor cell function in db/db mice. Br. J. Pharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Joviano-Santos, J.V.; Santos-Miranda, A.; Joca, H.C.; Cruz, J.S.; Ferreira, A.J. New insights into the elucidation of angiotensin-(1–7) in vivo antiarrhythmic effects and its related cellular mechanisms. Exp. Physiol. 2016, 101, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, H.J.; Zhou, P.; Kitzman, D.W.; Ferrario, C.M.; Li, W.M.; Cheng, C.P. Cellular basis of angiotensin-(1–7)-induced augmentation of left ventricular functional performance in heart failure. Int. J. Cardiol. 2017, 236, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yuan, K.; Phuong, H.T.; Park, B.M.; Kim, S.H. Angiotensin-(1-5), an active mediator of renin-angiotensin system, stimulates ANP secretion via Mas receptor. Peptides 2017, 86, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Benter, I.F.; Yousif, M.H.; Anim, J.T.; Cojocel, C.; Diz, D.I. Angiotensin-(1–7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with l-NAME. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H684–H691. [Google Scholar] [CrossRef] [PubMed]
- Brilla, C.G.; Weber, K.T. Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. J. Lab. Clin. Med. 1992, 120, 893–901. [Google Scholar] [PubMed]
- Young, M.J.; Rickard, A.J. Mineralocorticoid receptors in the heart: Lessons from cell-selective transgenic animals. J. Endocrinol. 2015, 224, R1–R13. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Fuller, P.J.; Young, M.J. Mechanisms of mineralocorticoid action. Hypertension 2005, 46, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Young, M.J. Mechanisms of mineralocorticoid receptor-mediated cardiac fibrosis and vascular inflammation. Curr. Opin. Nephrol. Hypertens. 2008, 17, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.F.; Warin, L.; Michel, J.B. Determinants of aortic cyclic guanosine monophosphate in hypertension induced by chronic inhibition of nitric oxide synthase. J. Clin. Investig. 1992, 90, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Ichiki, T.; Katoh, M.; Egashira, K.; Takeshita, A. Regulation of angiotensin II receptor expression by nitric oxide in rat adrenal gland. Hypertension 1998, 32, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Matuskova, J.; Luptak, I.; Pincikova, T.; Krajcirovicova, K.; Stvrtina, S.; Pomsar, J.; Pelouch, V.; Paulis, L.; Pechanova, O. Spironolactone differently influences remodeling of the left ventricle and aorta in l-NAME-induced hypertension. Physiol. Res. 2007, 56, S25–S32. [Google Scholar] [PubMed]
- Mulrow, P.J. Angiotensin II and aldosterone regulation. Regul. Pept. 1999, 17, 27–32. [Google Scholar] [CrossRef]
- Faulkner, J.L.; Bruder-Nascimento, T.; Belin de Chantemèle, E.J. The regulation of aldosterone secretion by leptin: Implications in obesity-related cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Rebuffat, P.; Malendowicz, L.K.; Nussdorfer, G.G.; Mazzocchi, G. Stimulation of endogenous nitric oxide production is involved in the inhibitory effect of adrenomedullin on aldosterone secretion in the rat. Peptides 2001, 22, 923–936. [Google Scholar] [CrossRef]
- Sainz, J.M.; Reche, C.; Rabano, M.A.; Mondillo, C.; Patrignani, Z.J.; Macarulla, J.M.; Pignataro, O.P.; Trueba, M. Effects of nitric oxide on aldosterone synthesis and nitric oxide synthase activity in glomerulosa cells from bovine adrenal gland. Endocrine 2004, 24, 61–71. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Holmes, B.B.; McCoy, M.J.; Hillard, C.J.; Campbell, W.B. Chronic administration of nitric oxide reduces angiotensin II receptor type 1 expression and aldosterone synthesis in Zona glomerulosa cells. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E820–E827. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Tsuruya, K.; Toyonaga, J.; Masutani, K.; Hayashida, H.; Hirakata, H.; Iida, M. Spironolactone suppresses inflammation and prevents l-NAME-induced renal injury in rats. Kidney Int. 2009, 75, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Muldowney, J.A.; Davis, S.N.; Vaughan, D.E.; Brown, N.J. NO synthase inhibition increases aldosterone in humans. Hypertension 2004, 44, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Suehiro, T.; Tsuruya, K.; Ikeda, H.; Toyonaga, J.; Yamada, S.; Noguchi, H.; Tokumoto, M.; Kitazono, T. Systemic aldosterone, but not angiotensin II, plays a pivotal role in the pathogenesis of renal injury in chronic nitric oxide-deficient male rats. Endocrinology 2015, 156, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Chaswal, M.; Das, S.; Prasad, J.; Katyal, A.; Fahim, M. Cardiac autonomic function in acutely nitric oxide deficient hypertensive rats: Role of the sympathetic nervous system and oxidative stress. Can. J. Physiol. Pharmacol. 2011, 89, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Escames, G.; Venegas, C.; Diaz-Casado, M.E.; LimaCabello, E.; Lopez, L.C.; Rosales-Corall, S.; Tan, D.X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Reiter, R.J. CSF generation by pineal gland results in a robust melatonin circadian rhythm in the third ventricle as an unique light/dark signal. Med. Hypotheses 2016, 86, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.X.; Manchester, L.C.; Esteban-Zubero, E.; Zhou, Z.; Reiter, R.J. Melatonin as a Potent and Inducible Endogenous Antioxidant: Synthesis and Metabolism. Molecules 2015, 20, 18886–18906. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Ghosh, A.K.; Dutta, M.; Mitra, E.; Mallick, S.; Saha, B.; Reiter, R.J.; Bandyopadhyay, D. Mechanisms of isoproterenol-induced cardiac mitochondrial damage: Protective actions of melatonin. J. Pineal Res. 2015, 58, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Agabiti-Rosei, C.; Favero, G.; De Ciuceis, C.; Rossini, C.; Porteri, E.; Rodella, L.F.; Franceschetti, L.; Maria Sarkar, A.; Agabiti-Rosei, E.; Rizzoni, D.; et al. Effect of long-term treatment with melatonin on vascular markers of oxidative stress/inflammation and on the anticontractile activity of perivascular fat in aging mice. Hypertens. Res. 2017, 40, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O. Potential roles of melatonin and chronotherapy among the new trends in hypertension treatment. J. Pineal Res. 2009, 47, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Paulis, L. Antifibrotic effect of melatonin—Perspective protection in hypertensive heart disease. Int. J. Cardiol. 2013, 168, 2876–2877. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; Piccolo, R.; Galasso, G.; Reiter, R.J. Melatonin is associated with reverse remodeling after cardiac resynchronization therapy in patients with heart failure and ventricular dyssynchrony. Int. J. Cardiol. 2016, 221, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; de la Torre-Hernandez, J.M.; Gonzalez-Gonzalez, J.; Garcia-Camarero, T.; Consuegra-Sanchez, L.; Garcia-Saiz, M.D.; Aldea-Perona, A.; Virgos-Aller, T.; Azpeitia, A.; et al. MARIA Investigators. Effect of intravenous and intracoronary melatonin as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: Results of the Melatonin Adjunct in the acute myocaRdial Infarction treated with Angioplasty trial. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef]
- Hu, W.; Ma, Z.; Jiang, S.; Fan, C.; Deng, C.; Yan, X.; Di, S.; Lv, J.; Reiter, R.J.; Yang, Y. Melatonin: The dawning of a treatment for fibrosis? J. Pineal Res. 2016, 60, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, L.; Yang, Y.; Guo, Y.; Fan, Y.; Zhang, M.; Man, W.; Gao, E.; Hu, W.; Reiter, R.J.; et al. Melatonin alleviates postinfarction cardiac remodeling and dysfunction by inhibiting Mst1. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef]
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Mullerova, M.; Bednarova, K.; Adamcova, M.; Paulis, L. Effect of melatonin, captopril, spironolactone and simvastatin on blood pressure and left ventricular remodelling in spontaneously hypertensive rats. J. Hypertens. 2009, 27, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Paulis, L.; Pechanova, O.; Zicha, J.; Barta, A.; Gardlik, R.; Celec, P.; Kunes, J.; Simko, F. Melatonin interactions with blood pressure and vascular function during l-NAME-induced hypertension. J. Pineal Res. 2010, 48, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Repova-Bednarova, K.; Krajcirovicova, K.; Celec, P.; Kamodyova, N.; Zorad, S.; Kucharska, J.; Gvozdjakova, A.; Adamcova, M.; et al. Hypertension and cardiovascular remodelling in rats exposed to continuous light: Protection by ACE-inhibition and melatonin. Mediat. Inflamm. 2014, 2014, 703175. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Celec, P.; Palffy, R.; Bednarova, K.; Vrankova, S.; Adamcova, M.; Paulis, L. Continuous light and l-NAME-induced left ventricular remodelling: Different protection with melatonin and captopril. J. Hypertens. 2010, 28, S13–S18. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Bednarova-Repova, K.; Krajcirovicova, K.; Hrenak, J.; Celec, P.; Kamodyova, N.; Gajdosechova, L.; Zorad, S.; Adamcova, M. Melatonin reduces cardiac remodeling and improves survival in rats with isoproterenol-induced heart failure. J. Pineal Res. 2014, 57, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Simko, F.; Pechanova, O.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Celec, P.; Tothova, L.; Vrankova, S.; Balazova, L.; Zorad, S.; et al. Lactacystin-Induced Model of Hypertension in Rats: Effects of Melatonin and Captopril. Int. J. Mol. Sci. 2017, 25, 1612. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.Y. Neural control of the pineal gland. Behav. Brain Res. 1996, 73, 125–130. [Google Scholar] [CrossRef]
- Reiter, R.J. Pineal melatonin: Cell biology of its synthesis and of its physiological interactions. Endocr. Rev. 1991, 12, 151–180. [Google Scholar] [CrossRef] [PubMed]
- Pechanova, O.; Paulis, L.; Simko, F. Peripheral and central effects of melatonin on blood pressure regulation. Int. J. Mol. Sci. 2014, 15, 17920–17937. [Google Scholar] [CrossRef] [PubMed]
- Girouard, H.; Denault, C.; Chulak, C.; de Champlain, J. Treatment by N-acetylcysteine and melatonin increases cardiac baroreflex and improves antioxidant reserve. Am. J. Hypertens. 2004, 17, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Arangino, S.; Cagnacci, A.; Angiolucci, M.; Vacca, A.M.; Longu, G.; Volpe, A.; Melis, G.B. Effects of melatonin on vascular reactivity, catecholamine levels, and blood pressure in healthy men. Am. J. Cardiol. 1999, 83, 1417–1419. [Google Scholar] [CrossRef]
- Pelouch, V.; Milerova, M.; Ostadal, B.; Samanek, M.; Hucan, B. Protein profiling of human atrial and ventricular musculature: The effect of normoxaemia and hypoxaemia in congenital heart diseases. Physiol. Res. 1993, 42, 235–242. [Google Scholar] [PubMed]
- Reddy, K.; Enwemeka, C.S. A simplified method for the analysis of hydroxyproline in biological tissues. Clin. Biochem. 1996, 29, 225–229. [Google Scholar] [CrossRef]
- Sharp, S.; Poglitsch, M.; Zilla, P.; Davies, N.H.; Sturrock, E.D. Pharmacodynamic effects of C-domain-specific ACE inhibitors on the renin-angiotensin system in myocardial infarcted rats. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Pavo, N.; Goliasch, G.; Wurm, R.; Novak, J.; Strunk, G.; Gyöngyösi, M.; Poglitsch, M.; Säemann, M.D.; Hülsmann, M. Low- and High-renin Heart Failure Phenotypes with Clinical Implications. Clin Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pechanova, O.; Zicha, J.; Paulis, L.; Zenebe, W.; Dobesova, Z.; Kojsova, S.; Jendekova, L.; Sladkova, M.; Dovinova, I.; Simko, F.; et al. The effect of N-acetylcysteine and melatonin in adult spontaneously hypertensive rats with established hypertension. Eur. J. Pharmacol. 2007, 30, 129–136. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BW (g) | LVW (mg) | RVW (mg) | RVW/BW (mg/g) | |
|---|---|---|---|---|
| c | 346.50 ± 6.19 | 425.30 ± 14.05 | 164.50 ± 6.08 | 0.48 ± 0.03 |
| Mel | 318.00 ± 7.50 | 427.00 ± 17.08 | 160.00 ± 8.40 | 0.50 ± 0.03 |
| LN | 318.50 ± 7.64 | 461.90 ± 17.98 | 155.00 ± 7.04 | 0.49 ± 0.03 |
| LN + Mel | 304.00 ± 11.57 * | 457.50 ± 22.53 | 158.60 ± 9.50 | 0.52 ± 0.03 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simko, F.; Baka, T.; Krajcirovicova, K.; Repova, K.; Aziriova, S.; Zorad, S.; Poglitsch, M.; Adamcova, M.; Reiter, R.J.; Paulis, L. Effect of Melatonin on the Renin-Angiotensin-Aldosterone System in l-NAME-Induced Hypertension. Molecules 2018, 23, 265. https://doi.org/10.3390/molecules23020265
Simko F, Baka T, Krajcirovicova K, Repova K, Aziriova S, Zorad S, Poglitsch M, Adamcova M, Reiter RJ, Paulis L. Effect of Melatonin on the Renin-Angiotensin-Aldosterone System in l-NAME-Induced Hypertension. Molecules. 2018; 23(2):265. https://doi.org/10.3390/molecules23020265
Chicago/Turabian StyleSimko, Fedor, Tomas Baka, Kristina Krajcirovicova, Kristina Repova, Silvia Aziriova, Stefan Zorad, Marko Poglitsch, Michaela Adamcova, Russel J. Reiter, and Ludovit Paulis. 2018. "Effect of Melatonin on the Renin-Angiotensin-Aldosterone System in l-NAME-Induced Hypertension" Molecules 23, no. 2: 265. https://doi.org/10.3390/molecules23020265
APA StyleSimko, F., Baka, T., Krajcirovicova, K., Repova, K., Aziriova, S., Zorad, S., Poglitsch, M., Adamcova, M., Reiter, R. J., & Paulis, L. (2018). Effect of Melatonin on the Renin-Angiotensin-Aldosterone System in l-NAME-Induced Hypertension. Molecules, 23(2), 265. https://doi.org/10.3390/molecules23020265