Informing Efforts to Develop Nitroreductase for Amine Production

Abstract
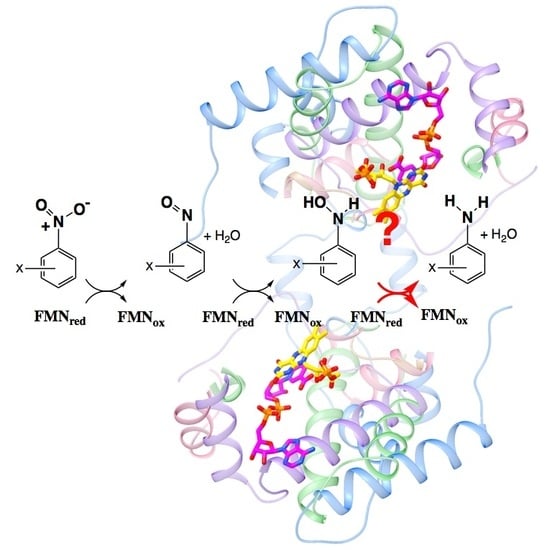
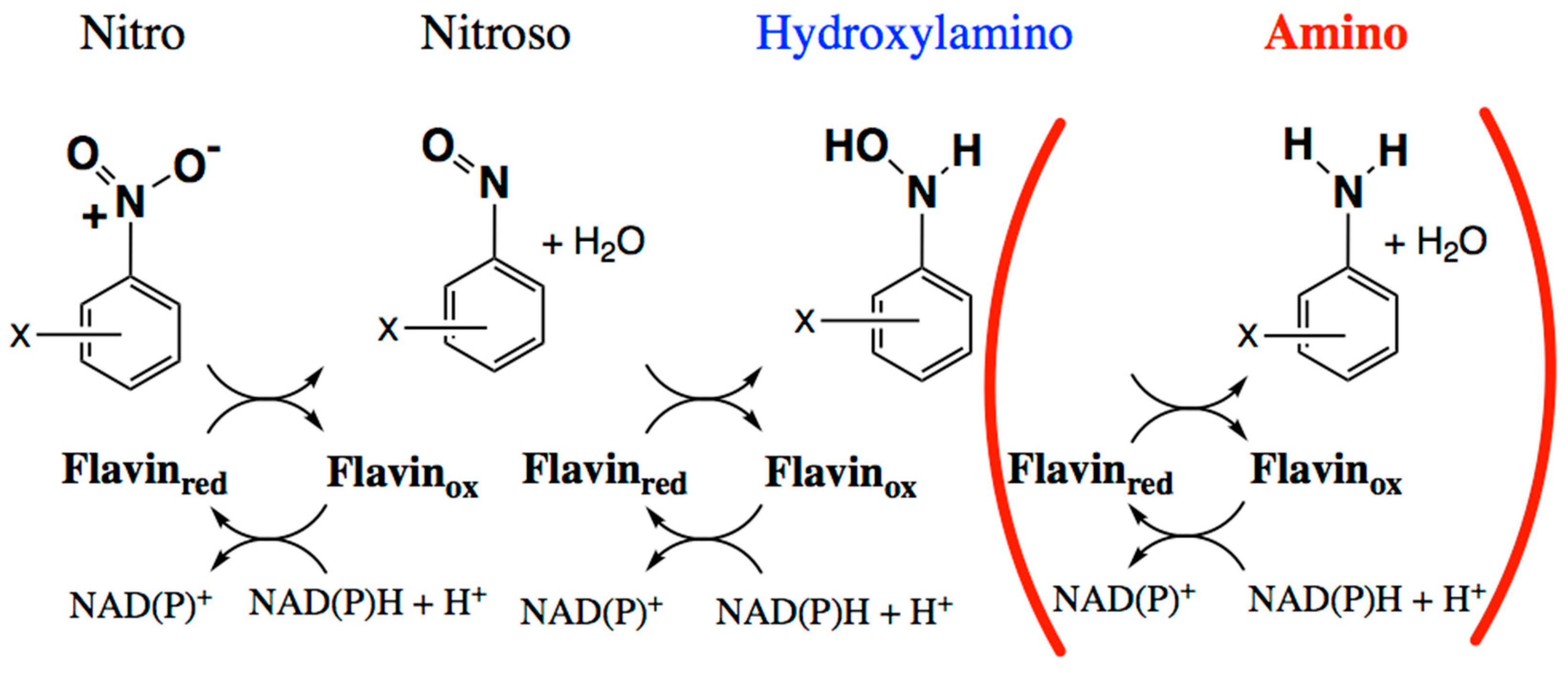
:
1. Introduction
2. Results and Discussion
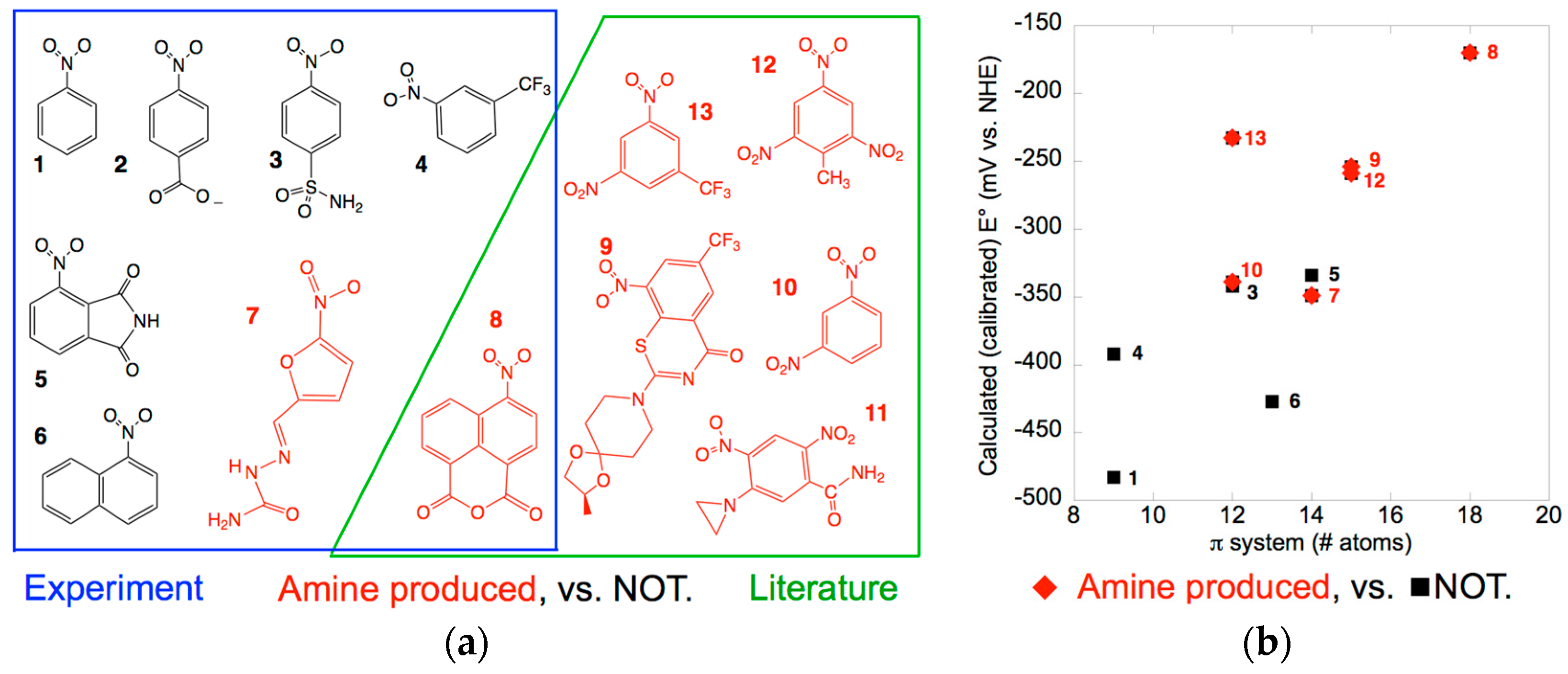
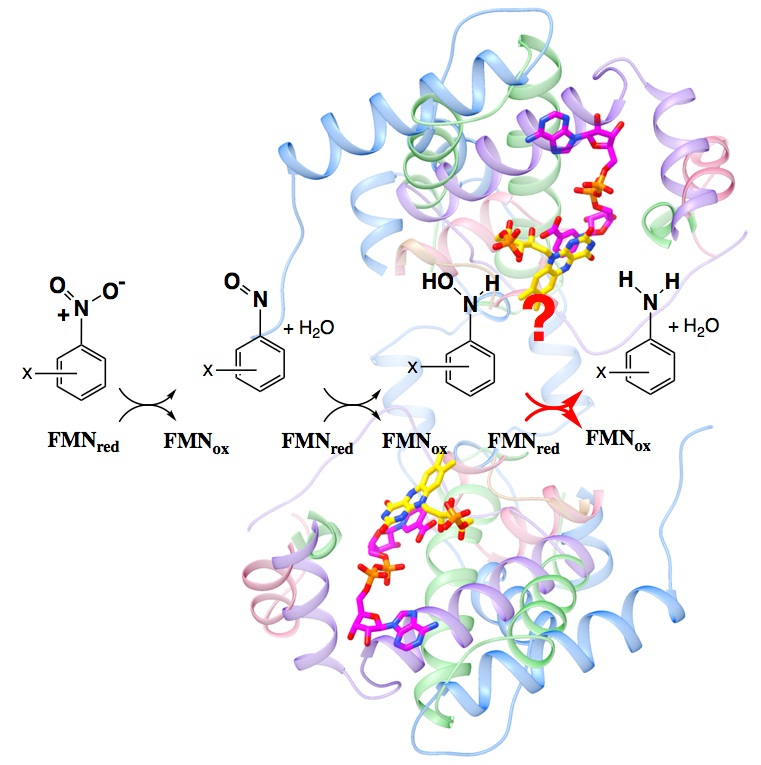
2.1. Amine Product Formation Is Low for Both Enzymes, but Large π Systems Seem Better
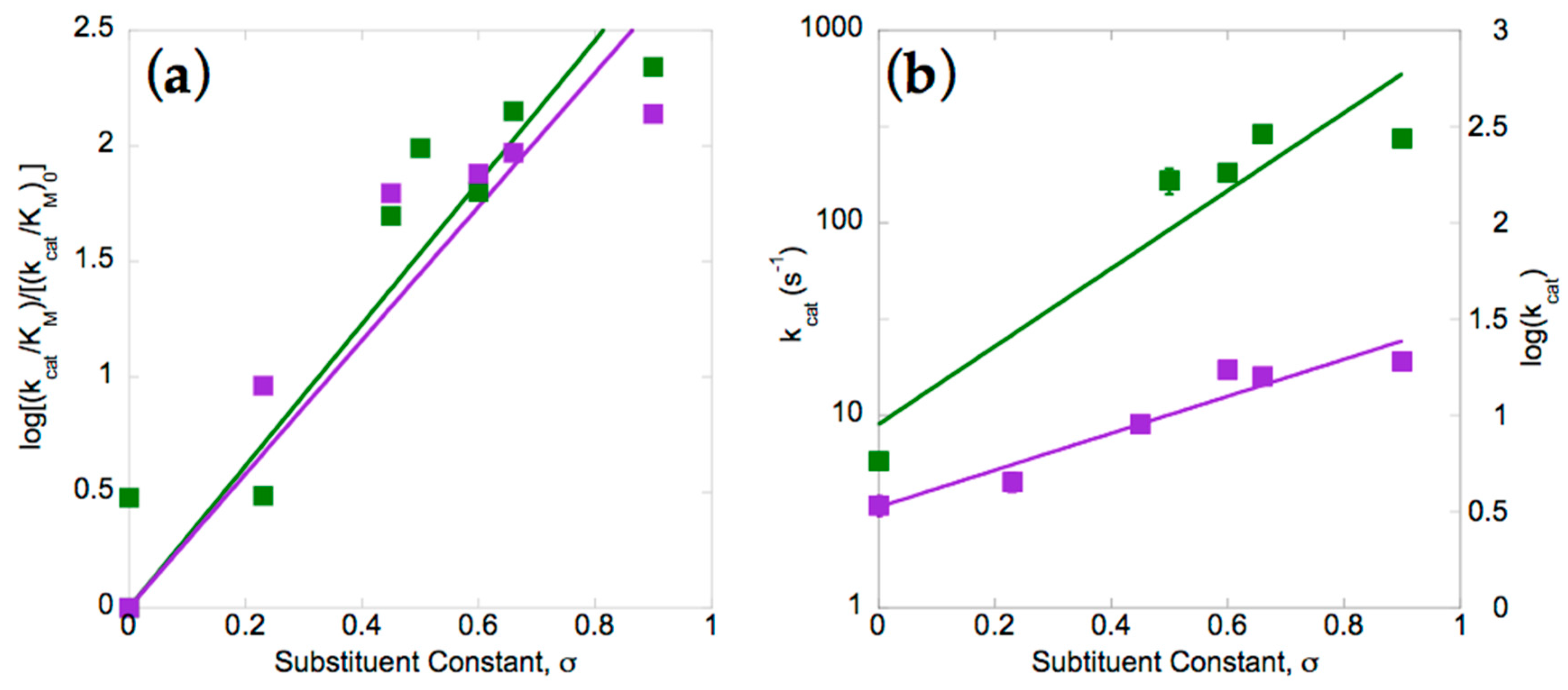
2.2. Electron Withdrawing Groups Favour Reduction of Nitro Substrates
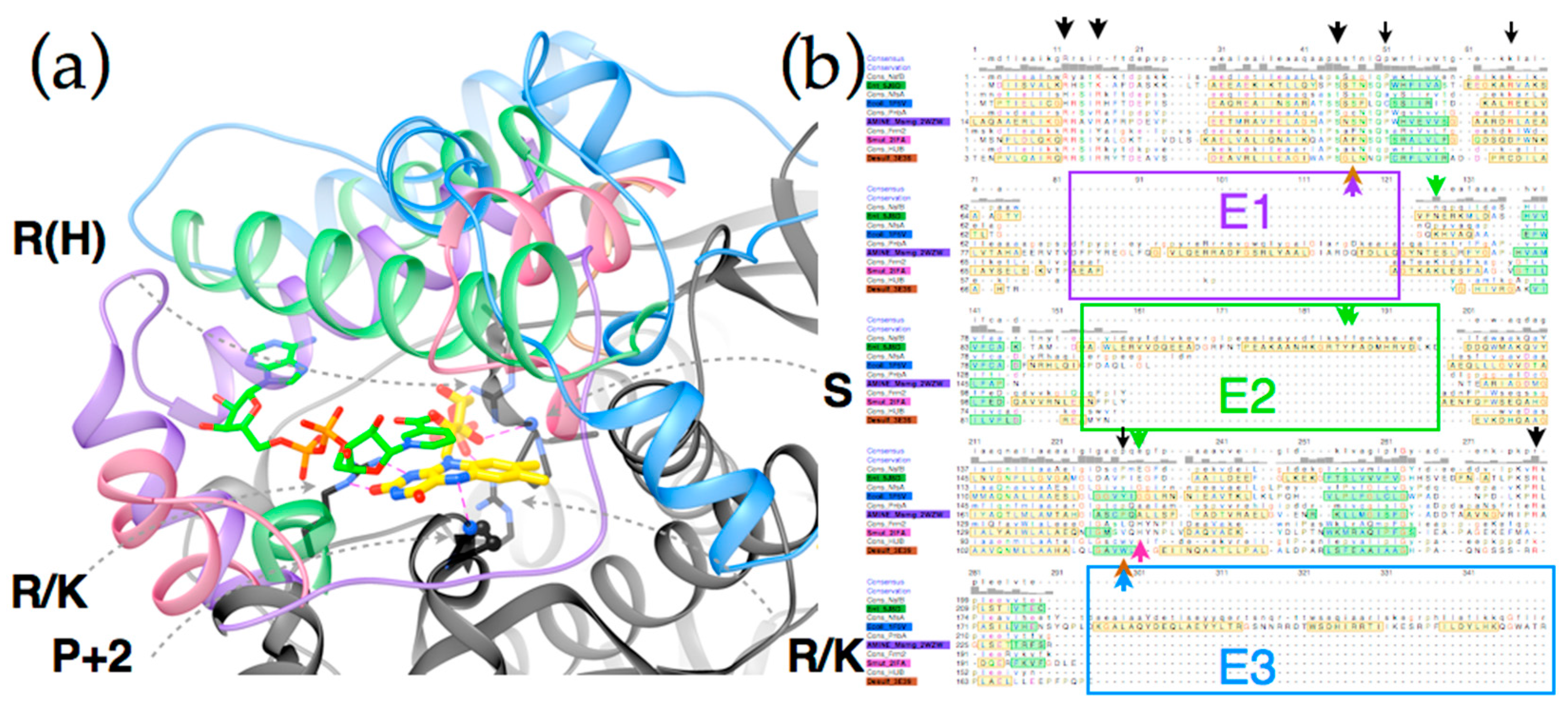
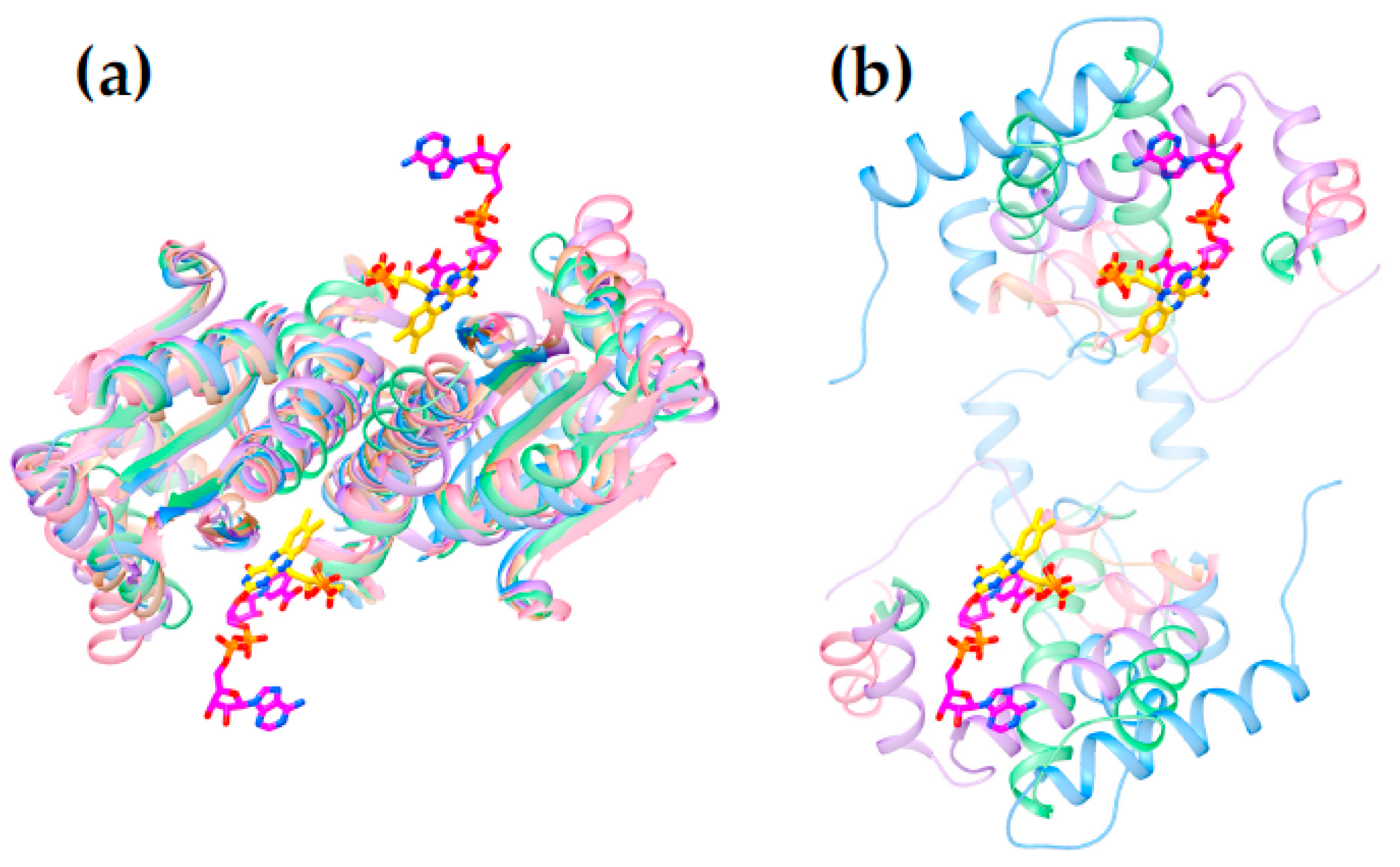
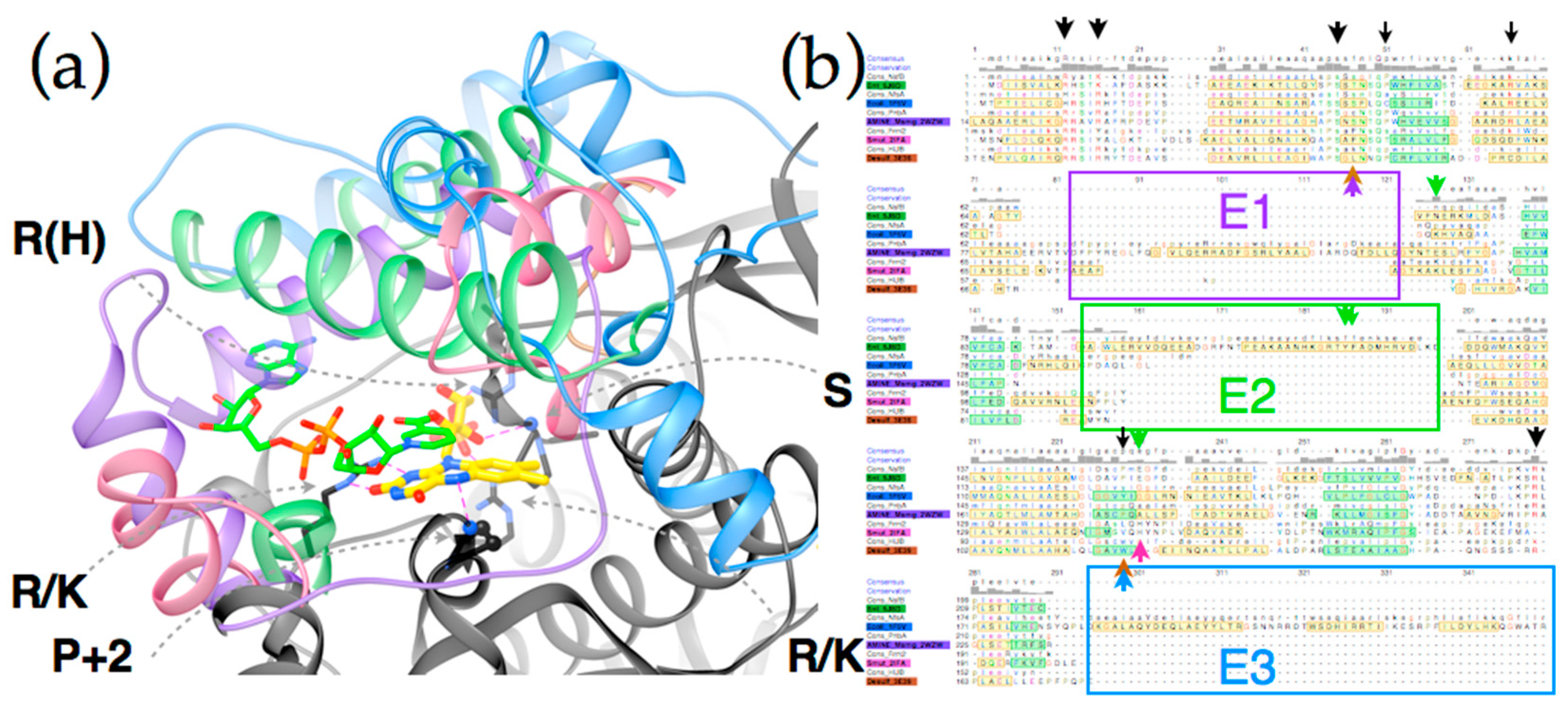
2.3. Differences between Subgroups in the NR Superfamily That Could Affect Nitroreduction Activity
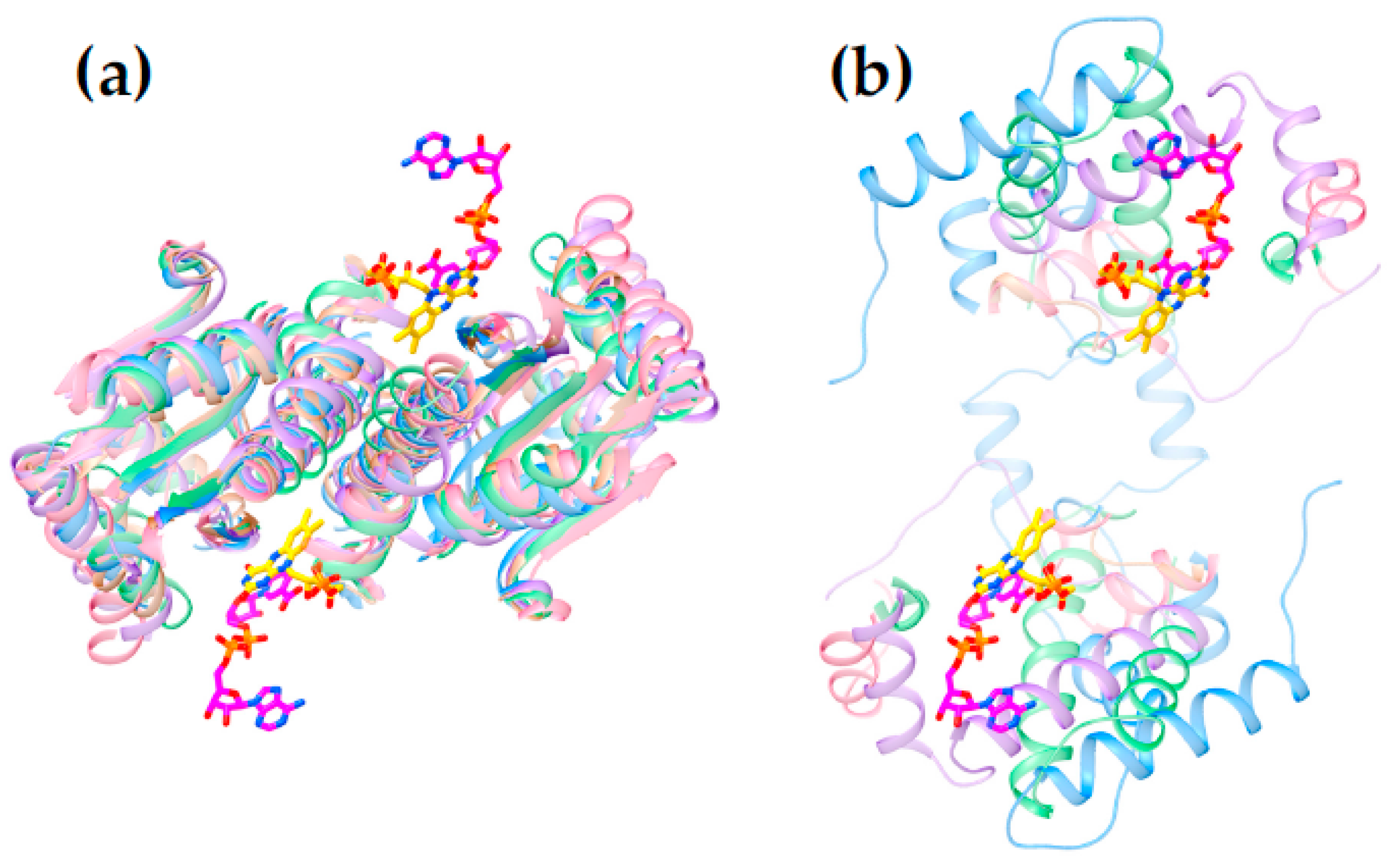
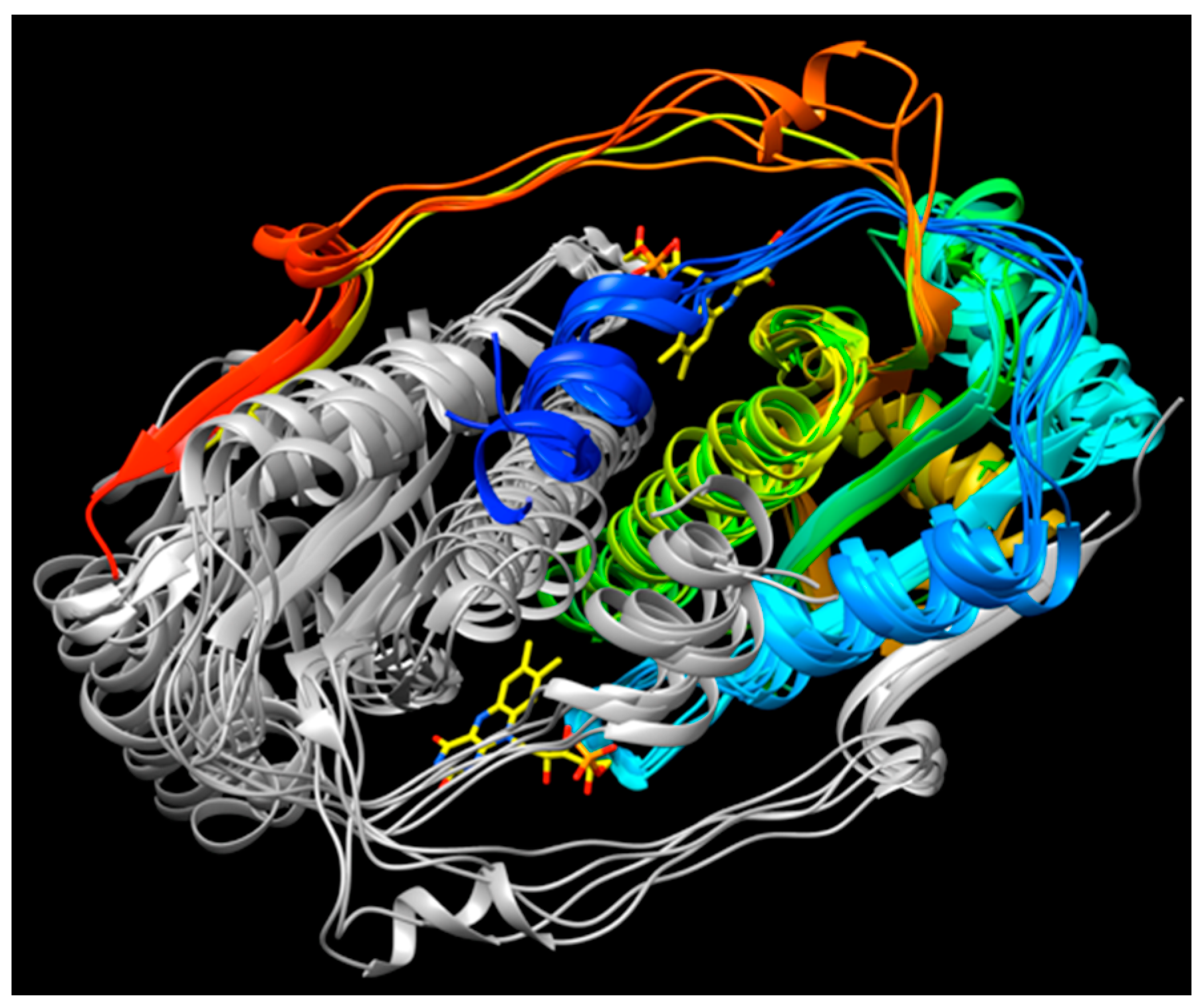
2.4. Active Site Constraints on Substrate Binding Mode and Orientation
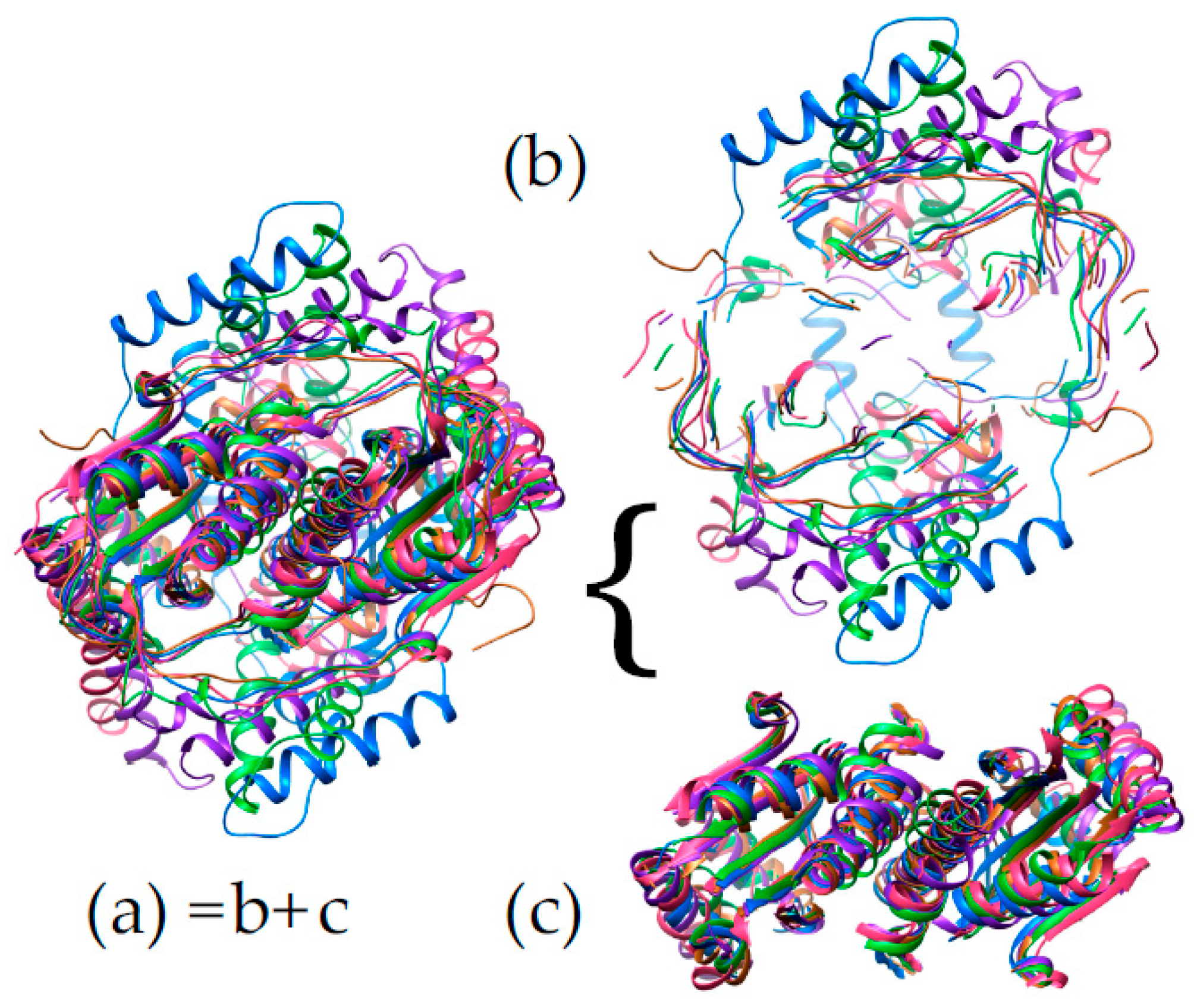
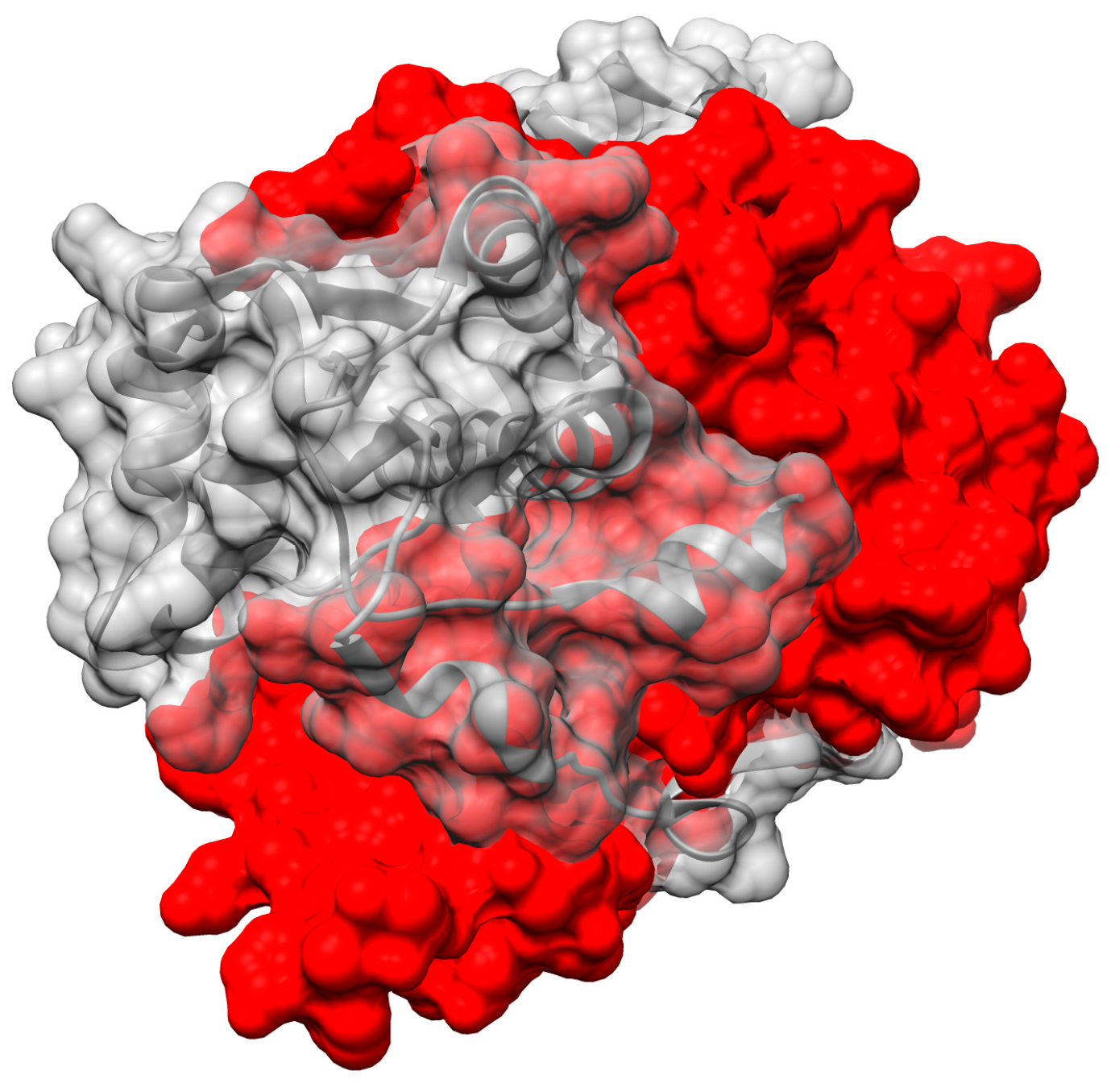
2.5. ‘Intertwining’ of the Two Peptide Chains May Enable the NR Dimer to Tolerate Diverse Substrates and Diverse Interactions in Its Active Site
3. Materials and Methods
3.1. Materials
3.2. Genes
3.3. Protein Expression and Purification
3.4. Measurement of Enzyme Kinetics
3.5. Detection of Amine Products
3.6. Measurement of Enzyme Reduction Potential
3.7. Computations
3.8. Structural Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Williams, E.M.; Little, R.F.; Mowday, A.M.; Rich, M.H.; Chan-Hyams, J.V.E.; Copp, J.N.; Smaill, J.B.; Pattersion, A.V.; Ackerley, D.F. Nitroreductase gene-directed enzyme prodrug therapy: Insights and advances toward clinical utility. Biochem. J. 2015, 471, 131–153. [Google Scholar] [CrossRef] [PubMed]
- Copp, J.N.; Mowday, A.M.; Williams, E.M.; Guise, C.P.; Ashoorzadeh, A.; Sharrock, A.V.; Flanagan, J.U.; Smaill, J.B.; Patterson, A.V.; Ackerley, D.F. Engineering a multifunctional nitroreductase for improved activation of prodrugs and PET probes for cancer gene therapy. Cell Chem. Biol. 2017, 24, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.M.; Lin, B.H.; Chen, S.C.; Wei, S.F.; Chen, C.C.; Yao, C.L.; Chien, C.C. Biodegradation of trinitrotoluene (TNT) by indigenous microorganisms from TNT-containing soil, and their application in TNT bioremediation. Bioremediat. J. 2016, 20, 165–173. [Google Scholar] [CrossRef]
- Spain, J.C. Biodegradation of Nitroaromatics. Annu. Rev. Microbiol. 1995, 49, 523–555. [Google Scholar] [CrossRef] [PubMed]
- Honeycutt, M.E.; Jarvis, A.S.; McFarland, V.A. Cytotoxicity and mutagenicity of 2,4,6-trinitrotoluene and its metabolites. Ecotoxicol. Environ. Saf. 1996, 35, 282–287. [Google Scholar] [CrossRef] [PubMed]
- You, S.H.; Zhu, B.; Han, H.J.; Wang, B.; Peng, R.H.; Yao, Q.H. Phytoremediation of 2,4,6-trinitrotoluene by Arabidopsis plants expressiong a NAD(P)H-flavin nitroreductase from Enterobacter cloacae. Plant Biotechnol. Rep. 2015, 9, 417–430. [Google Scholar] [CrossRef]
- Bai, J.; Yang, J.; Liu, P.; Yang, Q. Transformation pathway of 2,4,6-trinitrotoluene by Escherischia coli nitroreductases and improvement of activity using structure-based mutagenesis. Process Biochem. 2015, 50, 705–711. [Google Scholar] [CrossRef]
- Hoogenraad, M.; van der Linden, J.B.; Smith, A.A.; Hughes, B.; Derrick, A.M.; Harris, L.J.; Higginson, P.D.; Pettman, A.J. Accelerated process development of pharmaceuticals: Selective catalytic hydrogenations of nitro compounds. Org. Process. Res. Dev. 2004, 8, 469–476. [Google Scholar] [CrossRef]
- Haber, F. Elektrolytische darstellung von phenyl-β-hydroxylamin. Zeitschrift für Elektrochemie 1898, 5, 77–78. [Google Scholar] [CrossRef]
- Haber, F. Über die elektrolytische reduction der Nitrokörper. Angew. Chem. 1900, 13, 433–439. [Google Scholar] [CrossRef]
- Koder, R.L.; Miller, A.F. Steady state kinetic mechanism, stereospecificity, substrate and inhibitor specificity of Enterobacter cloacae nitroreductase. Biochim. Biophys. Acta 1998, 1387, 394–405. [Google Scholar] [CrossRef]
- Race, P.R.; Lovering, A.L.; Green, R.M.; Ossor, A.; White, S.A.; Searle, P.F.; Wrighton, C.J.; Hyde, E.I. Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone: Reversed binding orientations in different redox states of the enzyme. J. Biol. Chem. 2005, 280, 13256–13264. [Google Scholar] [CrossRef] [PubMed]
- Loos, P.; Alex, H.; Hassfeld, J.; Lovis, K.; Platzek, J.; Steinfeldt, N. Selective hydrogenation of halogenated nitroaromatics to haloanilines in batch and flow. Org. Proc. Res. Dev. 2015, 20, 452–464. [Google Scholar] [CrossRef]
- Corma, A.; Concepcion, P.; Serna, P. A different reaction pathway for the reduction of aromatic nitro compounds on gold catalysts. Angew. Chem. Int. Ed. Engl. 2007, 46, 7266–7269. [Google Scholar] [CrossRef] [PubMed]
- Alsante, K.M.; Huynh-Ba, K.C.; Baertschi, S.W.; Reed, R.A.; Landis, M.S.; Furness, S.; Olsen, B.; Mowery, M.; Russo, K.; Iser, R.; et al. Recent trends in product development and regulatory issues on impurities in active pharmaceutical ingredient (API) and drug products. Part 2: Safety considerations of impurities in pharmaceutical products and surveying the impurity landscape. AAPS Pharm. Sci. Tech. 2014, 15, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Bommarius, A.S. Biocatalysis: A status report. Annu. Rev. Chem. Biomol. Eng. 2015, 6, 319–345. [Google Scholar] [CrossRef] [PubMed]
- Yanto, Y.; Winkler, C.K.; Lohr, S.J.; Hall, M.; Faber, K.; Bommarius, A.S. Asymmetric bioreduction of alkenes using ene reductases YersER and KYE1, and effects of organic solvents. Org. Lett. 2011, 13, 2540–2543. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, L.; He, Z.; Spain, J. Production of 2-amino-5-phenoxyphenol from 4-nitrobiphenyl ether using nitrobenzene nitroreductase and hydroxylaminobenzene mutase from Pseudomonas pseudoalcaligenes JS45. J. Ind. Microbiol. Biotechnol. 2000, 24, 301–305. [Google Scholar] [CrossRef]
- Jiang, Y.Y.; Maniar, D.; Woortman, A.J.J.; Loos, K. Enzymatic synthesis of 2,5-furandicarboxylic acid- based semi-aromatic polyamides: Enzymatic polymerization kinetics, effect of diamine chain length and thermal properties. RSC Adv. 2016, 6, 67941–67953. [Google Scholar] [CrossRef]
- Oberleitner, N.; Peters, C.; Muschiol, J.; Kadow, M.; Saß, S.; Bayer, T.; Schaaf, P.; Iqbal, N.; Rudroff, F.; Mihovilovic, M.D.; et al. An enzymatic toolbox for cascade reactions: A showcase for an in vivo redox sequence in asymmetric synthesis. ChemCatChem 2013, 5, 3524–3528. [Google Scholar] [CrossRef]
- Bucko, M.; Geneiner, P.; Schenkmayerova, A.; Krajcovic, T.; Rudroff, F.; Mihovilovic, M.D. Baeyer-Villiger oxidations: Biotechnological approach. Appl. Microbiol. Biotechnol. 2016, 100, 6585–6599. [Google Scholar] [CrossRef] [PubMed]
- Pitsawong, W.; Hoben, J.P.; Miller, A.F. Understanding the broad substrate repertoire of nitroreductase based on its simple mechanism. J. Biochem. Chem. 2014, 289, 15203–15214. [Google Scholar]
- Kutty, R.; Bennett, G.N. Biochemical characterization of trinitrotoluene transforming oxygen-insensitive nitroreductases from Clostridium acetobutylicum ATCC 824. Arch. Microbiol. 2005, 184, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Song, H.G. Purification and characterization of NAD(P)H-dependent nitroreductase I from Klebsiella sp. C1 and enzymatic transformation of 2,4,6-trinitrotoluene. Appl. Microbiol. Biotechnol. 2005, 68, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Song, H.G. Nitroreductase II involved in 2,4,6-trinitrotoluene degradation: Purification and characterization from Klebsiella sp. C1. J. Microbiol. 2009, 47, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Gwenin, V.V.; Poornima, P.; Halliwell, J.; Ball, P.; Robinson, G.; Gwenin, C.D. Identification of novel nitroreductases from Bacillus cereus and their interaction with th CB1954 prodrug. Biochem. Pharmacol. 2015, 98, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Chaignon, P.; Cortial, S.; Ventura, A.P.; Lopes, P.; Halgand, F.; Laprevote, O.; Ouazzani, J. Purification and identification of a Bacillus nitroreductase: Potential use in 3,5-DNBTF biosensoring system. Enzym. Microb. Technol. 2006, 39, 1499–1506. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, J.; Wei, D. Heterologous overexpression and biochemical characterization of a nitroreductase from Gluconobacter oxydans 621H. Mol. Biotechnol. 2016, 58, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Smets, B.F.; Yin, H.; Esteve-Núñez, A. TNT biotransformation: When chemistry contronts mineralization. Appl. Microbiol. Biotechnol. 2007, 76, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Koder, R.L.; Rodgers, M.E.; Miller, A.F. Flavin binding thermodynamics in Enterobacter cloacae nitroreductase. In Flavins and Flavoproteins; Ghisla, S., Kroneck, P., Macheroux, P., Sund, H., Eds.; Rudolf Weber: Berlin, Germany, 1999; pp. 45–48. [Google Scholar]
- Pearson, J. The reduction of nitrocompounds at the dropping-mercury cathode. Trans. Faraday Soc. 1948, 44, 683–697. [Google Scholar] [CrossRef]
- Bussy, U.; Chung-Davidson, Y.W.; Li, K.; Li, W. Phase I and phase II reductive metabolism simulation of nitro aromatic xenobiotics with electrochemistry coupled with high resolution mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 7253–7260. [Google Scholar] [CrossRef] [PubMed]
- La-Scalea, M.A.; Menezes, C.M.S.; Juliao, M.S.S.; Chung, M.C.; Serrano, S.H.P.; Ferreira, E.I. Voltammetric behavior of nitrofurazone and its hydroxymethyl prodrug with potential anti-chagas activity. J. Braz. Chem. Soc. 2005, 16, 774–782. [Google Scholar] [CrossRef]
- Olson, E.J.; Isley, W.C., III; Brennan, J.E.; Cramer, C.J.; Bühlmann, P. Electrochemical reduction of 2,4-dinitrotoluene in aprotic and pH-buffered media. J. Phys. Chem. 2015, 119, 13088–13097. [Google Scholar] [CrossRef]
- Tian, D.; Jin, B. FT-IR spectroelectrochemical study of the reduction of 1,4-dinitrobenzene on Au electrode: Hydrogen bonding and protonation in proton donor mixed media. Electrochimica Acta 2011, 56, 9144–9151. [Google Scholar] [CrossRef]
- Baeza, A.; Ortiz, J.L.; González, I. Control of the electrochemical reduction of O-nitrophenol by pH imposition in acetonitrile. J. Electroanal. Chem. 1997, 429, 121–127. [Google Scholar] [CrossRef]
- Schwarzenbach, R.P.; Stierli, R.; Lanz, K.; Zeyer, J. Quinone and iron prophyrin mediated reduction of nitroaromatic compounds in homogeneous aqueous solution. Environ. Sci. Technol. 1990, 24, 1566–1574. [Google Scholar] [CrossRef]
- Bratin, K.; Kissinger, P.T.; Briner, R.C.; Bruntlett, C.S. Determination of nitro aromatic, nitramine, and nitrate ester explosive compounds in explosive mixtures and gunshot residue by liquid chromatography and reductive electrochemical detection. Anal. Chim. Acta 1981, 130, 295–311. [Google Scholar] [CrossRef]
- Luan, F.; Gorski, C.A.; Burgos, W.D. Linear free energy relationships for the biotic and abiotic reduction of nitroaromatic compounds. Environ. Sci. Technol. 2015, 49, 3557–3565. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, T.B.; Heijman, C.G.; Haderlein, S.B.; Holliger, C.; Schwarzenbach, R.P. Complete reduction of TNT and other (polu)nitroaromatic compounds under iron reducing subsurface conditions. Environ. Sci. Technol. 1999, 33, 1479–1487. [Google Scholar] [CrossRef]
- Akiva, E.; Copp, J.N.; Tokuriki, N.; Babbitt, P.C. Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily. Proc. Natl. Acad. Sci. USA 2017, 114, E9549–E9558. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Reinado, E.; Roldán, M.D.; Castillo, F.; Moreno-Vivián, C. The NprA nitroreductase required for 2,4-dinitrophenol reduction in Rhodobacter capsulatus is a dihydropteridine reductase. Environ. Microbiol. 2008, 10, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Taga, M.E.; Larsen, N.A.; Howard-Jones, A.R.; Walsh, C.T.; Walker, G.C. BluB cannibalizes flavin to form the lower ligand of vitamin B12. Nature 2007, 446, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.E.; Watson, J.A.; Lam, D.W.H.; Rokita, S.E. Iodotyrosine deiodinase is the first mammalian member of the NADH oxidase/flavin reductase super-family. J. Biol. Chem. 2006, 281, 2812–2819. [Google Scholar] [CrossRef] [PubMed]
- Gerlt, J.A.; Babbitt, P.C. Divergent evolution of enzymatic function: Mechanisti- cally diverse superfamilies and functionally distinct suprafamilies. Annu. Rev. Biochem. 2001, 70, 209–246. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.Y.; Kim, J.H.; Lee, P.Y.; Bae, K.H.; Lee, J.S.; Kim, P.S.; Lee, D.H.; Myung, P.K.; Park, B.C.; Park, S.G. Confirmation of Frm2 as a novel nitroreductase in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2012, 423, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Mermod, M.; Mourlane, F.; Waltersperger, S.; Oberholzer, A.E.; Baumann, U.; Solioz, M. Structure and function of CinD (YtjD) of Lactococcus lactis, a copper-induced nitroreductase involved in defense against oxidative stress. J. Bacteriol. 2010, 192, 4172–4180. [Google Scholar] [CrossRef] [PubMed]
- Akiva, E.; Brown, S.; Almonacid, D.E.; Barber, A.E.; Custer, A.F.; Hicks, M.A.; Huang, C.C.; Lauck, F.; Mashiyama, S.T.; Meng, E.C.; et al. The Structure-function linkage database. Nucleic Acids Res. 2014, 42, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Rokita, S.E. Single amino acid switch between a flavin-dependent dehalogenase and nitroreductase. J. Am. Chem. Soc. 2015, 137, 15342–15345. [Google Scholar] [CrossRef] [PubMed]
- Pitsawong, W.; Haynes, C.A.; Koder, R.L.; Rodgers, C.T.; Miller, A.F. Mechanism-informed refinement reveals altered substrate-binding mode for catalytically competent nitroreductase. Structure 2017, 29, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Koder, R.L.; Haynes, C.A.; Rodgers, M.E.; Rodgers, D.W.; Miller, A.F. Flavin thermodynamics explain the oxygen insensitivity of enteric nitroreductases. Biochemistry 2002, 41, 14197–14205. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.I.; Lovering, A.L.; Guise, C.P.; Race, P.R.; Wrighton, C.J.; White, S.A.; Hyde, E.I.; Searle, P.F. Generation of Escherichia coli nitroreductase mutants conferring improved cell sensitization to the prodrug CB1954. Cancer Res. 2003, 63, 5532–5537. [Google Scholar] [PubMed]
- Race, P.R.; Lovering, A.L.; White, S.A.; Grove, J.I.; Searle, P.F.; Wrighton, C.W.; Hyde, E.I. Kinetic and structural characterisation of Escherichia coli nitroreductase mutants showing improved efficacy for the prodrug substrate CB1954. J. Mol. Biol. 2007, 368, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Jarrom, D.; Jaberipour, M.; Guise, C.P.; Daff, S.N.; White, S.A.; Searle, P.F.; Hyde, E.I. Steady-state and stopped-flow kinetic studies of three Escherichia coli NfsB mutants with enhanced activity for the prodrug CB1954. Biochemistry 2009, 48, 7665–7672. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Su, Q.; Rokita, S.E. The distribution and mechanism of iodotyrosine deiodinase defied expectations. Arch. Biochem. Biophys. 2017, 632, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Manina, G.; Bellinzoni, M.; Pasca, M.R.; Neres, J.; Milano, A.; Ribeiro, A.L.J.L.; Buroni, S.; Skovierova, H.; Dianiskova, P.; Mikusova, K.; et al. Biological and structural characterization of the Mycobacterium smegmatis nitroreductase NfnB, and its role in benzothiozinone resistance. Mol. Microbiol. 2010, 77, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Yanto, Y.; Hall, M.; Bommarius, A.S. Nitroreductase from Salmonella typhimurium: Characterization and catalytic activity. Org. Biomol. Chem. 2010, 8, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Wood, T.K.; Smets, B.F. Reductive transformation of TNT by Escherichia coli: Pathway description. Appl. Microbiol. Biotechnol. 2005, 67, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Yang, J.; Yang, Q. Isolation and characterization of an efficient nitro- reducing bacterium, Streptomyces mirabilis DUT001, from soil. World J. Microb. Biotechnol. 2010, 26, 855–862. [Google Scholar] [CrossRef]
- Watrous, M.M.; Clark, S.; Kutty, R.; Huang, S.; Rudolph, F.B.; Hughes, J.B.; Bennett, G.N. 2,4,6-trinitrotoluene reduction by an Fe-only hydrogenase in Clostridium acetobutylicum. Appl. Environ. Microbiol. 2003, 69, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Lindahl, P.A.; Wang, C.; Bennett, G.N.; Rudolph, F.B.; Hughes, J.B. 2,4,6-trinitrotoluene reduction by carbon monoxide dehydrogenase from Clostridium thermoaceticum. Appl. Environ. Microbiol. 2000, 66, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Fiorella, P.D.; Spain, J.C. Transformation of 2,4,6-trinitrotoluene by Pseudomonas pseudoalcaligenes JS52. Appl. Environ. Microbiol. 1997, 63, 2007–2015. [Google Scholar] [PubMed]
- Oh, B.-T.; Sarath, G.; Shea, P.J. TNT nitroreductase from a Pseudomonas aeruginosa strain isolated from TNT-contaminated soil. Soil Biol. Biochem. 2001, 33, 875–881. [Google Scholar] [CrossRef]
- Krumholz, L.R.; Li, J.; Clarkson, W.W.; Wilber, G.G.; Sulfita, J.M. Transformations of TNT and related aminotoluenes in goundwater aquifer slurries under different electron accepting conditions. J. Ind. Microbiol. Biotechnol. 1997, 18, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Phillips, K.L.; Sandler, S.I.; Chiu, P.C. A method to calculate the one-electron reduction potentials for nitroaromatic compounds based on gas-phase quantum mechanics. J. Comput. Chem. 2011, 32, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Salter-Blanc, A.J.; Bylaska, E.J.; Johnston, H.J.; Tratnyek, P.G. Predicting reduction rates of energetic nitroaromatic compounds using calculated one-electron reduction potentials. Environ. Sci. Technol. 2015, 49, 3778–3786. [Google Scholar] [CrossRef] [PubMed]
- Zubatyuk, R.I.; Gorb, L.; Shishkin, O.V.; Qasim, M.; Leszczynski, J. Exploration of density functional methods for one-electron reduction potential of nitrobenzenes. J. Comput. Chem. 2010, 31, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Nandigama, R.K.; Edmondson, D.E. Structure-activity relations in the oxidation of phenethylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry 2000, 39, 15258–15265. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Edmondson, D. Structure-activity relationships in the oxidation of para-substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry 1999, 38, 13670–13683. [Google Scholar] [CrossRef] [PubMed]
- Bommarius, A.S.; Wang, D.I.C.; Hatton, T.A. Xanthine oxidase reactivity in reversed micellar systems: A contribution to the prediction of enzymatic activity in organized media. J. Am. Chem. Soc. 1995, 117, 4515–4523. [Google Scholar] [CrossRef]
- Fujita, T.; Iwasa, J.; Hansch, C. A new substituent constant, π, derived from partition coefficients. J. Am. Chem. Soc. 1964, 86, 5175–5180. [Google Scholar] [CrossRef]
- Nivinskas, H.; Koder, R.L.; Anusevicius, Z.; Sarlauskas, J.; Miller, A.F.; Cenas, N. Quantitative structure-activity relationships in two-electron reduction of nitroaromatic compounds by Enterobacter cloacae NAD(P)H:Nitroreductase. Arch. Biochem. Biophys. 2001, 385, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A. Enzyme Structure and Mechanism, 2nd ed.; Freeman & Company: London, UK, 1985. [Google Scholar]
- Jencks, W.P. Structure-reactivity correlations and general acid-base catalysis in enzymic transacylation reactions. Cold Spring Harbor Symp. Quant. Biol. 1971, 36, 1–11. [Google Scholar] [CrossRef]
- Pitsawong, W.; Jeerus, S.; Methinee, P.; Tan, T.C.; Spadiut, O.; Haltrich, D.; Divne, C.; Chaiyen, P. A conserved active-site threonine is important for both sugar and flavin oxidations of pyranose 2-oxidase. J. Biol. Chem. 2010, 285, 9697–9705. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, J.F. Linear Free Energy Relationships in Enzymology. In Advances in Linear Free Energy Relationships; Chapman, N.B., Shorter, J., Eds.; Plenum Press: London, UK, 1972. [Google Scholar]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Park, J.T.; Ferguson, K.L.; Pitsawong, W.; Miller, A.F.; Bommarius, A.S. Insights from Variation of Substrates and Protein Platforms in Nitroreductase. Proceedings of 19th International Symposium on Flavins and Flavoproteins, Groningen, The Netherlands, 2–6 July 2017. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.A.; Koder, R.L.; Miller, A.F.; Rodgers, D.W. Structures of nitroreductase in three States: Effects of Inhibitor binding and reduction. J. Biol. Chem. 2002, 277, 11513–11520. [Google Scholar] [CrossRef] [PubMed]
- Prosser, G.A.; Copp, J.N.; Mowday, A.M.; Guise, C.P.; Syddall, S.P.; Williams, E.M.; Horvat, C.N.; Swe, P.M.; Ashoorzadeh, A.; Denny, W.A.; et al. Creation and screening of a multi-family bacterial oxidoreductase library to discover novel nitroreductases that efficiently activate the bioreductive prodrugs CB1954 and PR-104-A. Biochem. Pharmacol. 2013, 85, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, S.S.; Wodak, S.J. Landscape of intertwined associations in multi-domain homo-oligomeric proteins. J. Mol. Biol. 2015, 427, 350–370. [Google Scholar] [CrossRef] [PubMed]
- Wodak, S.J.; Malevanets, A.; MacKinnon, S.S. The landscape of intertwined associations in homooligomeric proteins. Biophys. J. 2015, 109, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Ponstingl, H.; Henrick, K.; Thornton, J.M. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins Struct. Funct. Genet. 2000, 41, 47–57. [Google Scholar] [CrossRef]
- Vallone, B.; Miele, A.E.; Vecchini, P.; Chiancone, E.; Brunori, M. Free energy of burying hydrophobic residues in the interface between protein subunits. Proc. Natl. Acad. Sci. USA 1998, 95, 6103–6107. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, F.B.; Honig, B. On the role of electrostatic interactions in the design of protein-protein interfaces. J. Mol. Biol. 2002, 318, 161–177. [Google Scholar] [CrossRef]
- Watanabe, M.; Ishidate, M.; Nohmi, T. Nucleotide sequence of Salmonella typhimurium nitroreductase gene. Nucleic Acids. Res. 1990, 18, 1059. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T. Enzymatic Reduction of Nitro Compounds to Amines with Nitroreductases. Ph.D. Thesis, Georgia Institute of Technology, Atlanta, GA, USA, 2014. [Google Scholar]
- Koder, R.L.; Miller, A.F. Overexpression, isotopic labeling and spectral characterization of Enterobacter cloacae nitroreductase. Protein Expr. Purif. 1998, 13, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, L.; Menten, M.L. Die kinetik der invertinwirkung. Biochim. Z. 1913, 49, 333–369. [Google Scholar]
- Massey, V. A simple method for determination of redox potentials. In Flavins and Flavoproteins; Curti, B., Ronchi, S., Zanetti, G., Eds.; Walter de Gruyer: Berlin, Germany, 1991; pp. 59–66. [Google Scholar]
- Loach, P.A. Oxidation-reduction potentials: Absorbance bands and molar absorbance of compounds used in biochemical studies. In Handbook of Biochemistry Selected Data for Molecular Biology; Sorber, H.A., Ed.; The Chemical Rubber Co.: Cleveland, OH, USA, 1973; pp. 33–40. [Google Scholar]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Meisel, D.; Neta, P.J. One-electron redox potentials of nitro-compounds and radiosensitizers - correlation with spin-densities of their radical-anions. J. Am. Chem. Soc. 1975, 97, 5198–5203. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acid Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Babbitt, P.C. Topological variation in the evolution of new reactions in functionally diverse enzyme superfamilies. Curr. Opin. Struct. Biol. 2011, 21, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Huang, C.C.; Ferrin, T.E. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7, 339. [Google Scholar]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Richards, F.M. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 379–400. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Compound (ChemSpider ID) 1 | Product(s) | Analysis 2 | Detection |
|---|---|---|---|---|
| 1 | nitrobenzene (7138) | ~NHOH | HPLC (40%) | 235 & 265 nm |
| 2 | 4-nitrobenzoic acid (5882) | ~NHOH | HPLC (30%) | 280 nm |
| 3 | 4-nitrobenzenesulfonamide (21360) | ~NHOH | HPLC (30%) LC-MS (50%) ~NHOH M−1 = 185 | 260 nm (−)- mode |
| 4 | 3-trifluoromethyl nitrobenzene (7108) | ~NHOH | HPLC (50%) | 245 nm |
| 5 | 3-nitrophthalimide (11286) | ~NHOH | HPLC (30%) LC-MS (50%) ~NHOH M(+1) = 179 Da | 230 nm (+)-mode |
| 6 | 1-nitronaphthalene (6588) | ~NHOH | HPLC (40%) | 215 nm |
| 7 | 3-nitrofurazone (4566720) | ~NHOH, ~NH2 | HPLC (15%) LC-MS (20%) ~NHOH M(+1) = 185 Da, ~NH2 M(+1) = 169 Da | 260 & 300 nm (+)-mode |
| 8 | 4-nitro-1,8-naphthalic anhydride (73216) | ~NHOH, ~NH2 | HPLC (50%) LC-MS (50%) ~NHOH M(+1) = 230, ~NH2 M(+1) = 214 Da, and [59]. | 270 & 345 nm (+)-mode |
| 9 | BTZ043 (24747357) | ~NH2 ≈ 30% yield | [56] | |
| 10 | 1,3-dinitrobenzene (7172) | 3-nitroaniline | [28] | |
| 11 | CB1954 = 5-(1-aziridinyl)-2,4-dinitrobenzamide (CAS 21919-05-1) | amine products detected | [28] | |
| 12 | 2,4,6-trinitrotoluene (8073) | 2- and 4-amino dinitrotoluene | [23,24,25] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, A.-F.; Park, J.T.; Ferguson, K.L.; Pitsawong, W.; Bommarius, A.S. Informing Efforts to Develop Nitroreductase for Amine Production. Molecules 2018, 23, 211. https://doi.org/10.3390/molecules23020211
Miller A-F, Park JT, Ferguson KL, Pitsawong W, Bommarius AS. Informing Efforts to Develop Nitroreductase for Amine Production. Molecules. 2018; 23(2):211. https://doi.org/10.3390/molecules23020211
Chicago/Turabian StyleMiller, Anne-Frances, Jonathan T. Park, Kyle L. Ferguson, Warintra Pitsawong, and Andreas S. Bommarius. 2018. "Informing Efforts to Develop Nitroreductase for Amine Production" Molecules 23, no. 2: 211. https://doi.org/10.3390/molecules23020211
APA StyleMiller, A.-F., Park, J. T., Ferguson, K. L., Pitsawong, W., & Bommarius, A. S. (2018). Informing Efforts to Develop Nitroreductase for Amine Production. Molecules, 23(2), 211. https://doi.org/10.3390/molecules23020211