
Quantitative Assessment of rPM6 for Fluorine- and Chlorine-Containing Metal Complexes: Comparison with Experimental, First-Principles, and Other Semiempirical Results


Abstract
1. Introduction
2. Computational Details
2.1. rPM6 Parameters for F and Cl
- 1.
- Geometric parameters of four open-shell and 11 closed-shell species [9,57,58] with 72 reference data points. Nine subsets for general chemical problems taken from the GMTKN30 database with 52 reference data points, which covered ionization potential (G21IP), electron affinity (G21EA), atomization energies (W4-08), decomposition energies (MB08-165), proton affinities (PA), barrier heights and reaction energies (BH76, BH76RC, and G2RC), and radical stabilization energies (RSE43) [59,60,61]. The total number of reference data points (N) is 124.
- 2.
- Geometric parameters of three open-shell and 15 closed-shell species [56,62,63,64] with 86 reference data points. Nine subsets for general chemical problems taken from the GMTKN30 database with 50 reference data, which covered ionization potential (G21IP), electron affinity (G21EA), atomization energies (W4-08), decomposition energies (MB08-165), barrier heights and reaction energies (BH76, BH76RC, and G2RC), self-interaction error related problems (SIE11), and isomerization energies (ISOL22) [59,60,61]. The number of reference data points (N) totaled 136.
2.2. Evaluation of rPM6 on Calculating Magnetic Interactions, Reaction Energies, and Barrier Heights
3. Results and Discussion
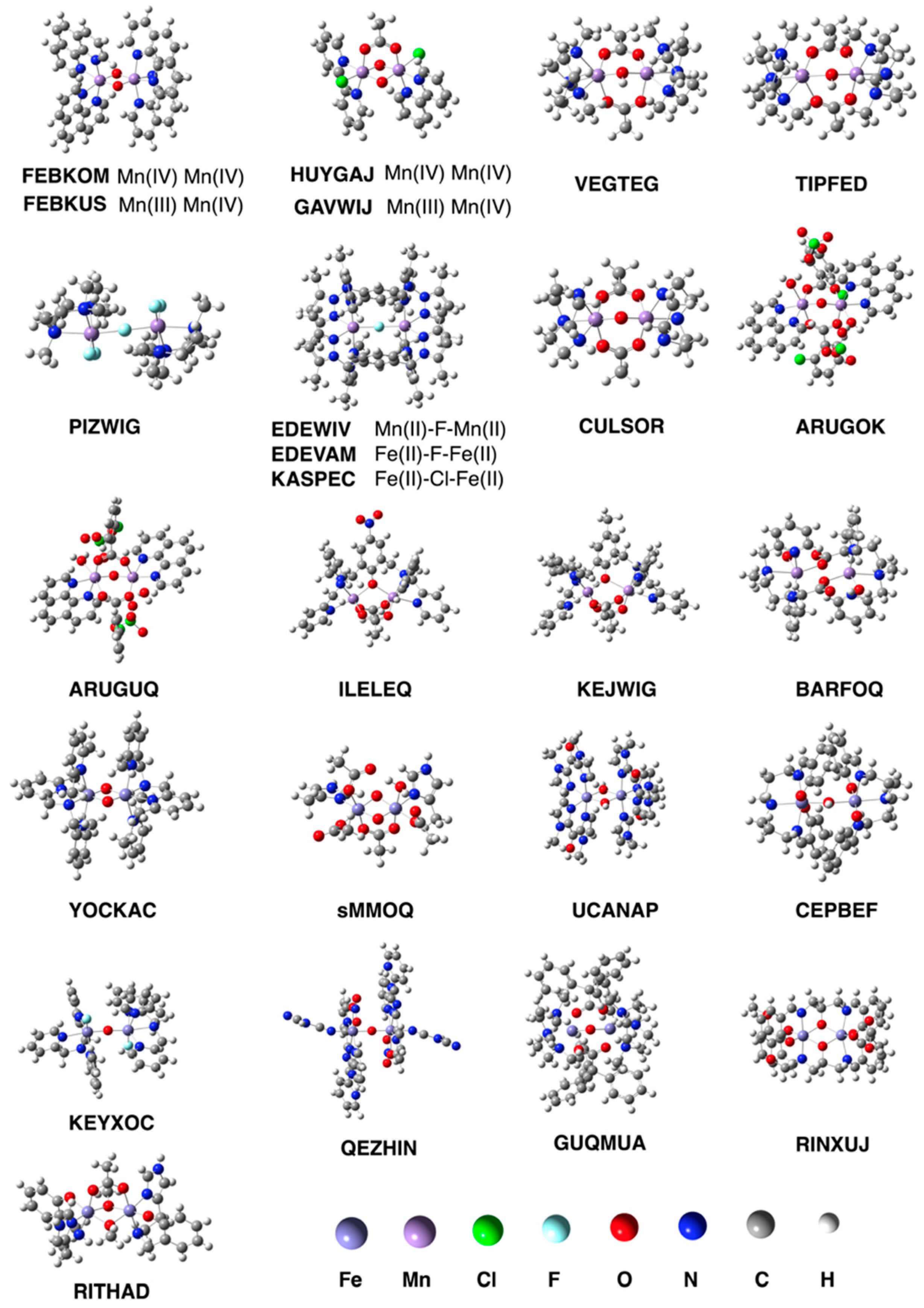
3.1. Test Set (i): Magnetic Interactions
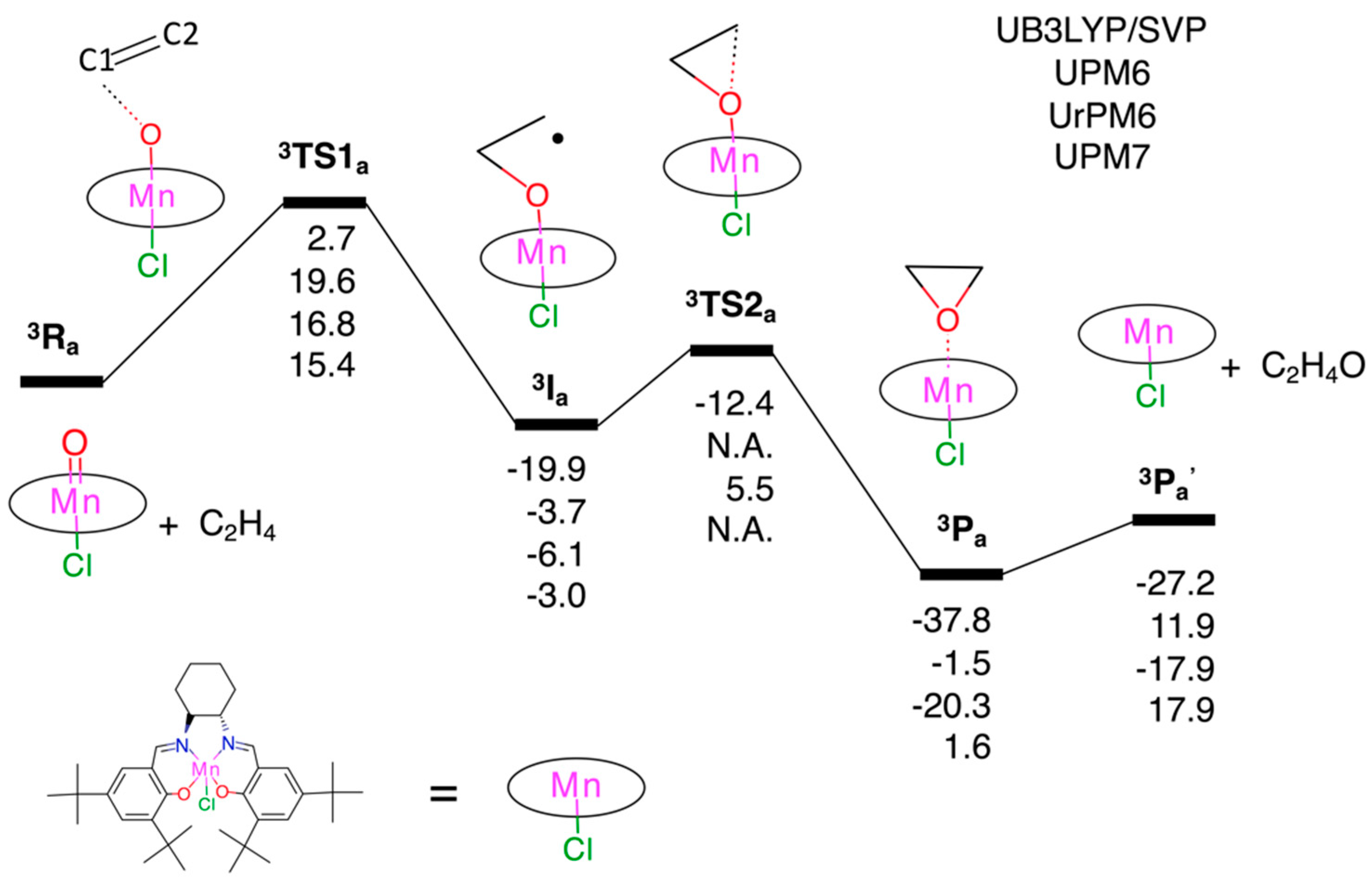
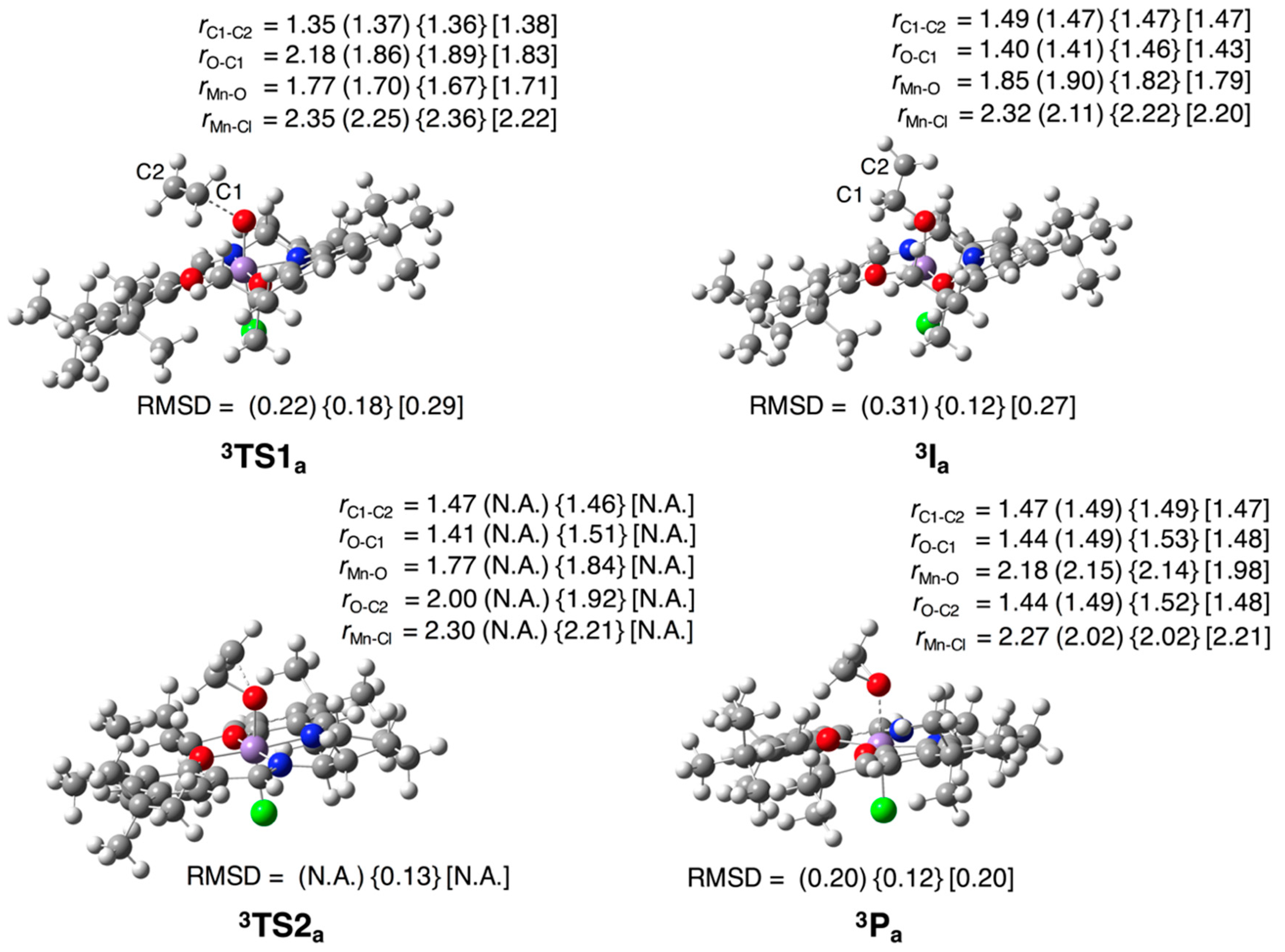
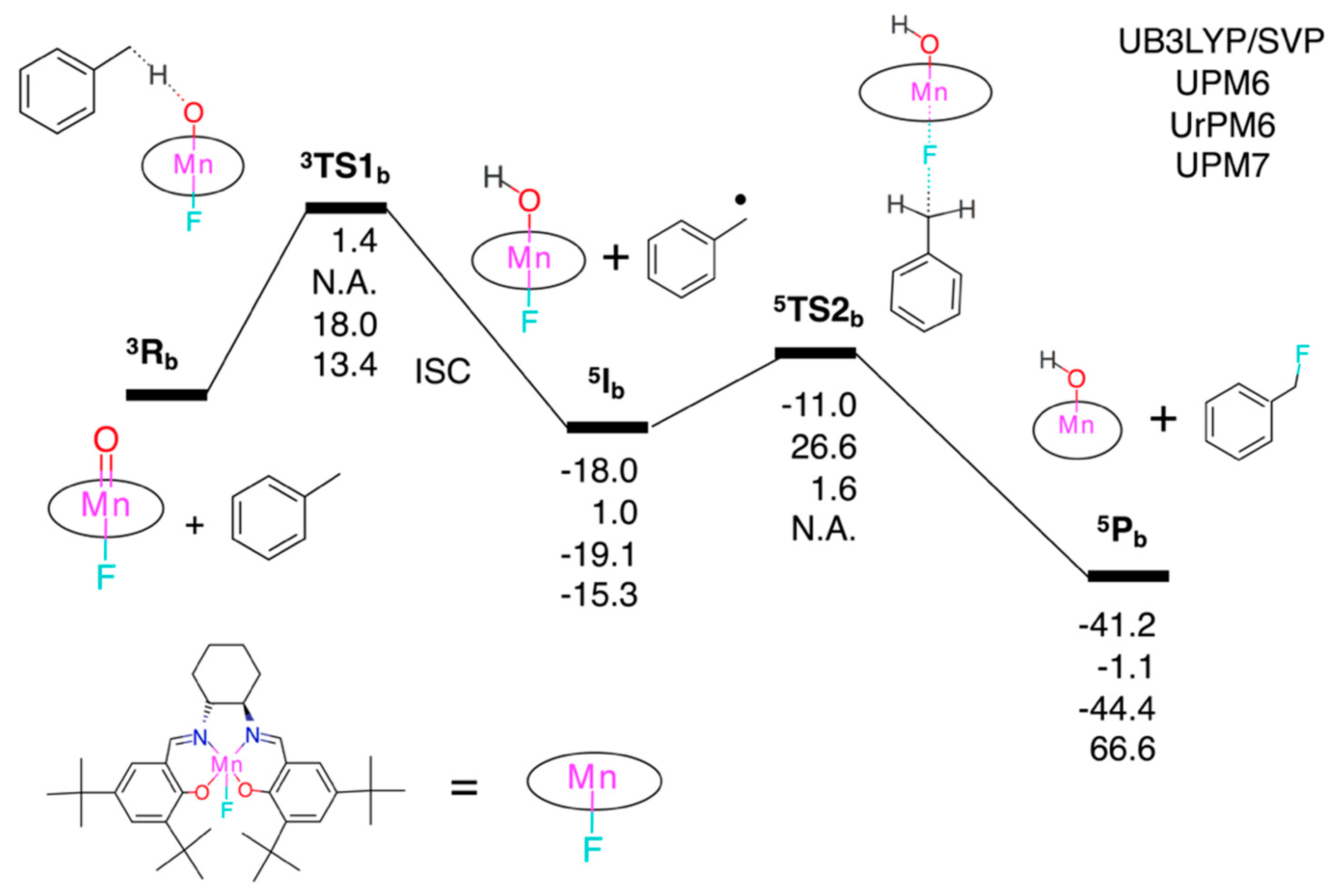
3.2. Test Set (ii): Oxidation Reactions
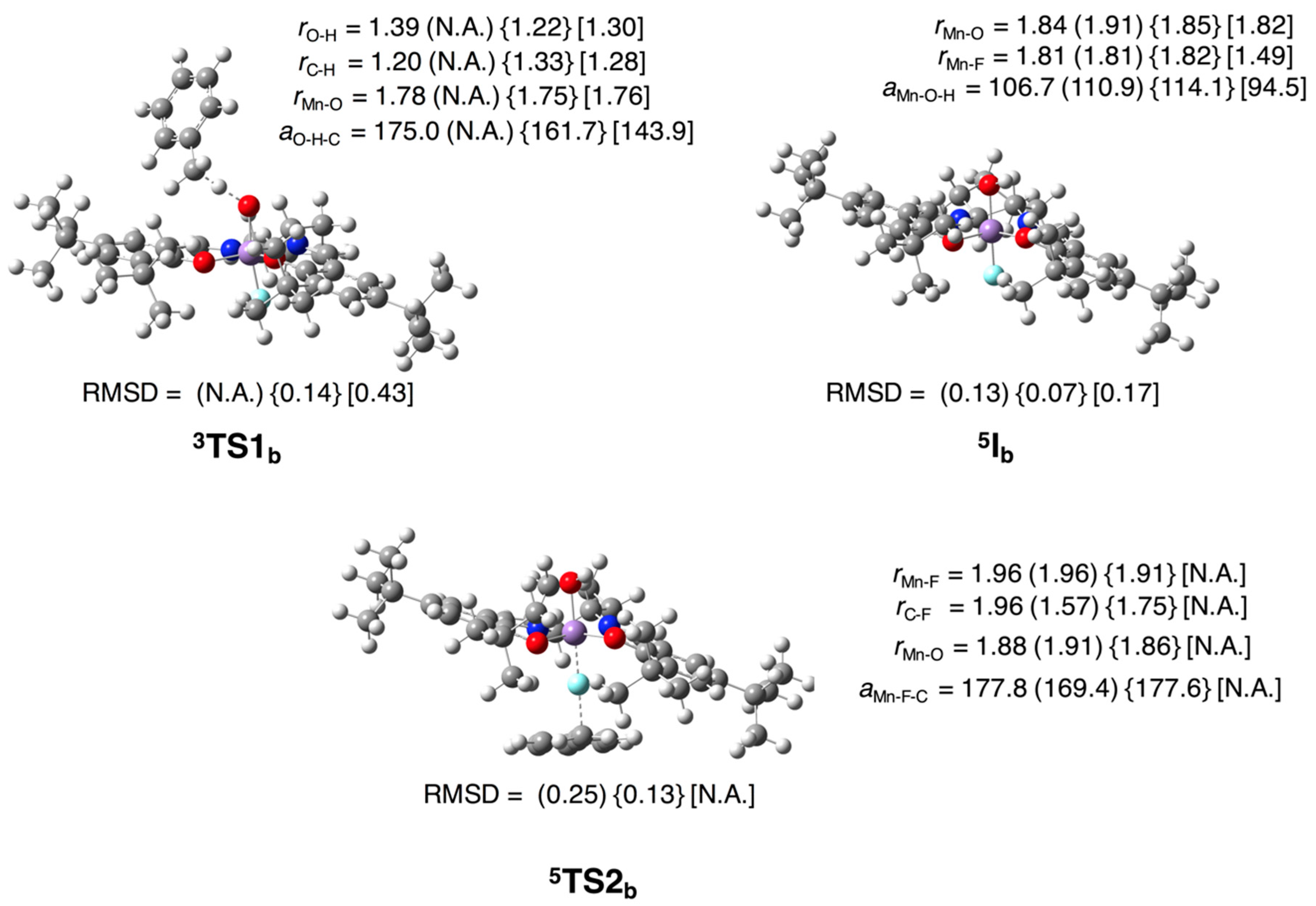
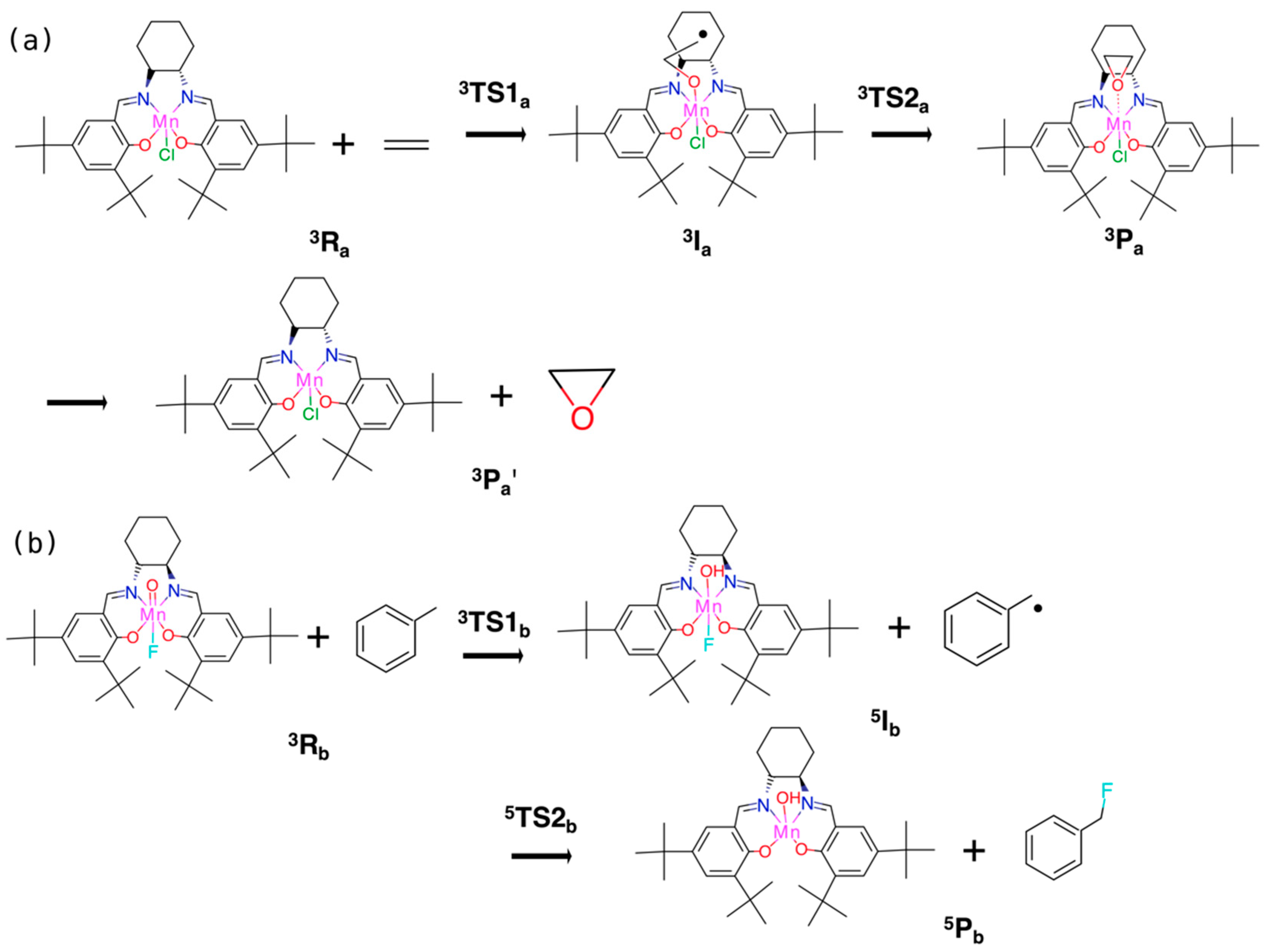
3.2.1. Epoxidation of Ethylene
3.2.2. Fluorination of Toluene
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Que, L., Jr.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen managanese complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Irie, R.; Noda, K.; Ito, Y.; Katsuki, T. Enantioselective epoxidation of unfunctionalized olefins using chiral (salen)manganese(III) complexes. Tetrahedron Lett. 1991, 32, 1055–1058. [Google Scholar] [CrossRef]
- Samuelsen, S.V.; Santilli, C.; Ahlquist, M.S.G.; Madsen, R. Development and mechanistic investigation of the manganese(III) salen-catalyzed dehydrogenation of alcohols. Chem. Sci. 2018, in press. [Google Scholar] [CrossRef]
- De Visser, S.P. The axial ligand effect of oxo-iron porphyrin catalysts. How does chloride compare to thiolate. J. Biol. Inorg. Chem. 2006, 11, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Furuya, T.; Kamlet, A.S.; Ritter, T. Catalysis for fluorination and trifluoromethylation. Nature 2011, 473, 470–477. [Google Scholar] [CrossRef]
- Liu, W.; Huang, X.Y.; Cheng, M.J.; Nielsen, R.J.; Goddard, W.A.; Groves, J.T. Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science 2012, 337, 1322–1325. [Google Scholar] [CrossRef]
- Huang, X.; Liu, W.; Ren, H.; Neelamegam, R.; Hooker, J.M.; Groves, J.T. Late Stage Benzylic C-H Fluorination with [18F] Fluoride for PET Imaging. J. Am. Chem. Soc. 2014, 136, 6842–6845. [Google Scholar] [CrossRef]
- Acheson, J.F.; Bailey, L.J.; Brunold, T.C.; Fox, B.G. In-crystal reaction cycle of a toluene–bound diiron hydroxylase. Nature 2017, 544, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen–evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Kondo, M.; Kuga, R.; Kurashige, Y.; Tanai, T.; Hayami, S.; Praneeth, V.K.K.; Yoshida, M.; Yoneda, K.; Kawata, S.; et al. A pentanuclear iron catalyst designed for water oxidation. Nature 2015, 530, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Castillo, R.G.; Banerjee, R.; Allpress, C.J.; Rohde, G.T.; Bill, E.; Que, L.; Lipscomb, J.D.; DeBeer, S. High-Energy-Resolution Fluorescence-Detected X-ray Absorption of the Q Intermediate of Soluble Methane Monooxygenase. J. Am. Chem. Soc. 2017, 139, 18024–18033. [Google Scholar] [CrossRef] [PubMed]
- Cutsall, G.E., III; Banerjee, R.; Zhou, A.; Que, L., Jr.; Lipscomb, J.D.; DeBeer, S. High-Resolution Extended X-ray Absorption Fine Structure Analysis Provides Evidence for a Longer Fe···Fe Distance in the Q Intermediate of Methane Monooxygenase. J. Am. Chem. Soc. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Leuenberger, M.N.; Loss, D. Quantum computing in molecular magnets. Nature 2001, 410, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Vitali, L.; Fabris, S.; Conte, A.M.; Brink, S.; Ruben, M.; Baroni, S.; Kern, K. Electronic Structure of Surface-supported Bis(phthalocyaninato) terbium(III) Single Molecular Magnets. Nano Lett. 2008, 8, 3364–3368. [Google Scholar] [CrossRef]
- Stebler, M.; Ludi, A.; Buergi, H.B. Tetrakis(phenanthroline)di-.mu.-oxodimanganese(III,IV) tris(hexafluorophosphate).cntdot.acetonitrile and tetrakis(phenanthroline)di-.mu.-oxodimanganese(IV) tetraperchlorate.cntdot.acetonitrile: Crystal structure analyses at 100 K, interpretation of disorder, and optical, magnetic, and electrochemical results. Inorg. Chem. 1986, 25, 4743–4750. [Google Scholar] [CrossRef]
- Bhaduri, S.; Tasiopoulos, A.J.; Bolcar, M.A.; Abboud, K.A.; Streib, W.E.; Christou, G. Symmetric and Asymmetric Dinuclear Manganese(IV) Complexes Possessing a [MnIV2(μ-O)2(μ-O2CMe)]3+ Core and Terminal Cl– Ligands. Inorg. Chem. 2003, 42, 1483–1492. [Google Scholar] [CrossRef]
- Bashkin, J.S.; Schake, A.R.; Vincent, J.B.; Chang, H.-R.; Li, Q.; Huffman, J.C.; Christou, G.; Hendrickson, D.N. Mixed valence manganese-(II, III) and -(III, IV) dinuclear complexes: Preparation, structure, magnetochemistry, and e.s.r. spectra of Mn2(biphen)2(biphenH)(bpy)2 and Mn2O2Cl2 (OAc)(bpy)2(biphenH2 = 2,2′-biphenol, bpy = 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1988, 700–702. [Google Scholar] [CrossRef]
- Bossek, U.; Hummel, H.; Weyhermüller, T.; Wieghardt, K.; Russell, S.; van derWolf, L.; Kolb, U. The [Mn(μ -O)(μ-PhBO2)2]2+ Unit: A New Structural Model for Manganese-Containing Metalloproteins. Angew. Chem. Int. Ed. 1996, 35, 1552–1554. [Google Scholar] [CrossRef]
- Bossek, U.; Wieghardt, K.; Nuber, B.; Weiss, J. Bioinorganic model complexes for the active site in manganese containing catalases. The crystal structures of [L2MnII2(μ-OH)(μ-O2CCH3)2] (PF6)•CH3OH and [L′2MnIII2(μ-O)(μ-O2CCH3)2] (I3)I•H2O. Inorg. Chim. Acta 1989, 165, 123–129. [Google Scholar] [CrossRef]
- Gultneh, Y.; Tesema, Y.T.; Yisgedu, T.B.; Butcher, R.J.; Wang, G.; Yee, G.T. Studies of a Dinuclear Manganese Complex with Phenoxo and Bis-acetato Bridging in the Mn2(II,II) and Mn2(II,III) States: Coordination Structural Shifts and Oxidation State Control in Bridged Dinuclear Complexes. Inorg. Chem. 2006, 45, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, S.; Blain, G.; Rivière, E.; Nierlich, M.; Blondin, G. Temperature Dependence of X- and Q-Band EPR Spectra of the Dinuclear Manganese(II) Complex [(NO2Bpmp)Mn2(μ-OAc)2]+: Determination of the Exchange Constant and of the Spin Parameters for the S = 1, 2, and 3 Spin States. Chem. Eur. J. 2003, 9, 4260–4268. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.S.; Sigrist, M.; Weihe, H.; Bond, A.D.; Thuesen, C.A.; Simonsen, K.P.; Birk, T.; Mutka, H.; Barra, A.-L.; Bendix, J. Magnetic Interactions through Fluoride: Magnetic and Spectroscopic Characterization of Discrete, Linearly Bridged [MnIII2(μ-F)F4(Me3tacn)2](PF6). Inorg. Chem. 2014, 53, 5013–5019. [Google Scholar] [CrossRef] [PubMed]
- Reger, D.L.; Pascui, A.E.; Smith, M.D.; Jezierska, J.; Ozarowski, A. Dinuclear Complexes Containing Linear M-F-M [M = Mn(II), Fe(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II)] Bridges: Trends in Structures, Antiferromagnetic Superexchange Interactions, and Spectroscopic Properties. Inorg. Chem. 2012, 51, 11820–11836. [Google Scholar] [CrossRef] [PubMed]
- Wieghardt, K.; Bossek, U.; Ventur, D.; Weiss, J. Assembly and structural characterization of binuclear μ-oxo-di-μ-acetato bridged complexes of manganese(III). Analogues of the di-iron(III) centre in hemerythrin. J. Chem. Soc. Chem. Commun. 1985, 347–349. [Google Scholar] [CrossRef]
- Gómez, V.; Corbella, M.; Aullón, G. Two Temperature-Independent Spinomers of the Dinuclear Mn(III) Compound [Mn(H2O)(phen)2(μ-2-ClC6H4COO)2(μ-O)](ClO4)2. Inorg. Chem. 2010, 49, 1471–1480. [Google Scholar] [CrossRef]
- Baffert, C.; Collomb, M.-N.; Deronzier, A.; Kjaergaard-Knudsen, S.; Latour, J.-M.; Lund, K.H.; McKenzie, C.J.; Mortensen, M.; Nielsen, L.P.; Thorup, N. Biologically relevant mono- and di-nuclear manganese ii/iii/iv complexes of mononegative pentadentate ligands. Dalton Trans. 2003, 1765–1772. [Google Scholar] [CrossRef]
- Reger, D.L.; Pascui, A.E.; Foley, E.A.; Smith, M.D.; Jezierska, J.; Wojciechowska, A.; Stoian, S.A.; Ozarowski, A. Dinuclear Metallacycles with Single M-X-M Bridges (X = Cl–, Br–; M = Fe(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II)): Strong Antiferromagnetic Superexchange Interactions. Inorg. Chem. 2017, 56, 2884–2901. [Google Scholar] [CrossRef]
- Bruijnincx, P.C.A.; Buurmans, I.L.C.; Huang, Y.; Juhász, G.; Viciano-Chumillas, M.; Quesada, M.; Reedijk, J.; Lutz, M.; Spek, A.L.; Münck, E.; et al. Mono- and Dinuclear Iron Complexes of Bis(1-methylimidazol-2-yl)ketone (bik): Structure, Magnetic Properties, and Catalytic Oxidation Studies. Inorg. Chem. 2011, 50, 9243–9255. [Google Scholar] [CrossRef] [PubMed]
- Jullien, J.; Juhász, G.; Mialane, P.; Dumas, E.; Mayer, C.R.; Marrot, J.; Rivière, E.; Bominaar, E.L.; Münck, E.; Sécheresse, F. Structure and Magnetic Properties of a Non-Heme Diiron Complex Singly Bridged by a Hydroxo Group. Inorg. Chem. 2006, 45, 6922–6927. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Dong, Y.; Que, L.; Kauffmann, K.; Muenck, E. The First Bis(.mu.-oxo)diiron(III) Complex. Structure and Magnetic Properties of [Fe2(.mu.-O)2(6TLA)2](ClO4)2. J. Am. Chem. Soc. 1995, 117, 1169–1170. [Google Scholar] [CrossRef]
- Mitra, M.; Maji, A.K.; Ghosh, B.K.; Raghavaiah, P.; Ribas, J.; Ghosh, R. Catecholase activity of a structurally characterized dinuclear iron(III) complex [FeIII2(L)2] [H3L=N,N′-bis(3-methoxysalicylaldimine)-1,3-diaminopropan-2-ol]. Polyhedron 2014, 67, 19–26. [Google Scholar] [CrossRef]
- Min, K.S.; Arif, A.M.; Miller, J.S. Synthesis, structure and magnetic properties of an oxo-bridged dinuclear iron(III) complex [(TPyA)FFeIIIOFeIIIF(TPyA)](BF4)2•0.5MeOH (TPyA=tris(2-pyridylmethyl)amine) containing the FFeIIIOFeIIIF linkage. Inorg. Chim. Acta 2007, 360, 1854–1858. [Google Scholar] [CrossRef]
- Majumder, A.; Pilet, G.; El Fallah, M.S.; Ribas, J.; Mitra, S. Isolation of a singly oxo-bridged Fe(III) dinuclear complex: Synthesis, crystal structure, spectroscopic and magnetic studies. Inorg. Chim. Acta 2007, 360, 2307–2312. [Google Scholar] [CrossRef]
- Scarpellini, M.; Neves, A.; Bortoluzzi, A.J.; Vencato, I.; Drago, V.; Ortiz, W.A.; Zucco, C. A new FeIII(μ-OCH3)2(μ-OAc)FeIII complex containing phenolate and imidazole ligands as a structural model for the active site of non-heme diiron enzymes. J. Chem. Soc. Dalton Trans. 2001, 2616–2623. [Google Scholar] [CrossRef]
- Payne, S.C.; Hagen, K.S. Steric Control of Reactivity of Non-Heme μ-Hydroxo Diiron(II) Complexes with Oxygen: Isolation of a Strongly Coupled μ-Oxo Fe(II)Fe(III) Dimer. J. Am. Chem. Soc. 2000, 122, 6399–6410. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 46, 10757–10816. [Google Scholar] [CrossRef]
- Ruiz, E.; Rodriguez-Fortea, A.; Tercero, J.; Cauchy, T. Exchange coupling in transition-metal complexes via density-functional theory: Comparison and reliability of different basis set approaches. J. Chem. Phys. 2005, 123, 074102. [Google Scholar] [CrossRef]
- Valero, R.; Illas, F.; Truhlar, D.G. Magnetic Coupling in Transition-Metal Binuclear Complexes by Spin-Flip Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2011, 7, 3523–3531. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Yasuda, N.; Nishihara, S.; Yamanaka, S.; Kitagawa, Y.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Broken-symmetry natural orbital (BS-NO)-Mk-MRCC study on the exchange coupling in the binuclear copper(II) compounds. Chem. Phys. Lett. 2011, 505, 11–15. [Google Scholar] [CrossRef]
- Yamanaka, S.; Kanda, K.; Saito, T.; Kitagawa, Y.; Kawakami, T.; Ehara, M.; Oku- mura, M.; Nakamura, H.; Yamaguchi, K. Does B3LYP correctly describe magnetism of manganese complexes with various oxidation numbers and various structural motifs? Chem. Phys. Lett. 2012, 519–520, 134–140. [Google Scholar] [CrossRef]
- Thiel, W. Semiempirical quantum–chemical methods. WIREs Comput. Mol. Sci. 2014, 4, 145–157. [Google Scholar] [CrossRef]
- Gaus, M.; Cui, Q.; Elstner, M. Density Functional Tight Binding: Application to Organic and Biological Molecules. WIREs Comput. Mol. Sci. 2014, 4, 49–61. [Google Scholar] [CrossRef]
- Dral, P.O.; Wu, X.; Spörkel, L.; Koslowski, A.; Thiel, W. Semiempirical Quantum chemical Orthogonalization-corrected Methods: Benchmarks for Ground-state Properties. J. Chem. Theory Comput. 2016, 12, 1097–1120. [Google Scholar] [CrossRef] [PubMed]
- Gruden, M.; Andjeklović, L.; Jissy, A.K.; Stepanović, S.; Zlatar, M.; Cui, Q.; Elstner, M. Benchmarking density functional tight binding models for barrier heights and reaction energetics of organic molecules. J. Comput. Chem. 2017, 38, 2171–2185. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kitagawa, Y.; Takano, Y. Reparameterization of PM6 Applied to Organic Diradical Molecules. J. Phys. Chem. A 2016, 120, 8750–8760. [Google Scholar] [CrossRef]
- Saito, T.; Takano, Y. rPM6 Parameters for Manganese and Application to Transition State Search for Oxidation Reactions of Cyclohexene by Manganese (IV)-Oxo Species. Chem. Lett. 2017, 46, 1567–1569. [Google Scholar] [CrossRef]
- Saito, T.; Kitagawa, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M.; Takano, Y. Assessment of semi-empirical molecular orbital calculations for describing magnetic interactions. Polyhedron 2017, 136, 52–57. [Google Scholar] [CrossRef]
- Saito, T.; Takano, Y. rPM6 Parameters for Phosphorous and Sulphur-containing Open-shell Molecules. Mol. Phys. 2018, 116, 602–610. [Google Scholar] [CrossRef]
- Saito, T.; Takano, Y. Transition State Search Using rPM6: Iron- and Manganese-catalyzed Oxidation Reactions as a Test Case. Bull. Chem. Soc. Jpn. 2018, 91, 1377–1389. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods V: Modification of NDDO Approximations and Application to 70 Elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.; Voityuk, A.A. Extension of the MNDO formalism to d or bitals: Integral approximations and preliminary numerical results. Theor. Chim. Acta 1992, 81, 391–404. [Google Scholar] [CrossRef]
- Repasky, M.P.; Chandrasekhar, J.; Jorgensen, W.L. PDDG/PM3 and PDDG/MNDO: Improved Semiempirical Methods. J. Comput. Chem. 2002, 23, 1601–1622. [Google Scholar] [CrossRef] [PubMed]
- Voityuk, A.A.; Rösch, N. AM1/d Parameters for Molybdenum. J. Phys. Chem. A 2000, 104, 4089–4094. [Google Scholar] [CrossRef]
- Berry, J.F.; Bill, E.; García-Serres, R.; Neese, F.; Weyhermüller, T.; Wieghardt, K. Effect of N-Methylation of Macrocyclic Amine Ligands on the Spin State of Iron(III): A Tale of Two Fluoro Complexes. Inorg. Chem. 2006, 45, 2027–2037. [Google Scholar] [CrossRef]
- Kubař, T.; Bodrog, Z.; Gaus, M.; Kühler, C.; Aradi, B.; Frauenheim, T.; Elstner, M. Parametrization of the SCC-DFTB Method for Halogens. J. Chem. Theory Comput. 2013, 9, 2939–2949. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. A General Database for Main Group Thermochemistry, Kinetics, and Noncovalent Interactions-Assessment of Common and Reparameterized (meta-)GGA Density Functionals. J. Chem. Theory Comput. 2010, 6, 107–126. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals―Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2011, 7, 291–309. [Google Scholar] [CrossRef]
- Mulliken Center for Theoretical Chemistry. Available online: https://www.chemie.uni-bonn.de/pctc/mulliken-center/software/GMTKN/gmtkn30 (accessed on 26 November 2018).
- Duboc, C.; Phoeung, T.; Zein, S.; Pécaut, J.; Collomb, M.-N.; Neese, F. Origin of the Zero-Field Splitting in Mononuclear Octahedral Dihalide MnII Complexes: An Investigation by Multifrequency High-Field Electron Paramagnetic Resonance and Density Functional Theory. Inorg. Chem. 2007, 46, 4905–4916. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Latifi, R.; Tahsini, L.; Nam, W. The Axial Ligand Effect on Aliphatic and Aromatic Hydroxylation by Non-heme Iron(IV)-oxo Biomimetic Complexes. Chem. Asian J. 2011, 6, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Harriman, K.L.M.; Kühne, I.A.; Leitch, A.A.; Korobkov, I.; Clérac, R.; Murugesu, M.; Brusso, J.L. Halide Influence on Molecular and Supramolecular Arrangements of Iron Complexes with a 3,5-Bis(2-Pyridyl)-1,2,4,6-Thiatriazine Ligand. Inorg. Chem. 2016, 55, 5375–5383. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional Exchange-energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-energy Formula into a Functional of the Electron Density. Phys. Rev. A. 1988, 37, 785. [Google Scholar] [CrossRef]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Cavallo, L.; Jacobsen, H. Manganese-Salen Complexes as Oxygen-Transfer Agents in Catalytic Epoxidations – A Density Functional Study of Mechanistic Aspects. Eur. J. Inorg. Chem. 2003, 892–902. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Jensen, F.; Dorigo, A.; Houk, K.N. A spin correction procedure for unrestricted Hartree-Fock and Møller-Plesset wavefuncitons for singlet diradicals and polyradicals. Chem. Phys. Lett. 1988, 149, 537–542. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Saito, T.; Ito, M.; Shoji, M.; Koizumi, K.; Yamanaka, S.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Approximately spin-projected geometry optimization method and its application to di-chromium systems. Chem. Phys. Lett. 2007, 442, 445–450. [Google Scholar] [CrossRef]
- Saito, T.; Nishihara, S.; Kataoka, Y.; Nakanishi, Y.; Matsui, T.; Kitagawa, Y.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Transition state optimization based on approximate spin-projection (AP) method. Chem. Phys. Lett. 2009, 483, 168–171. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Saito, T.; Yamaguchi, K. Approximate Spin Projection for Broken-Symmetry Method and Its Application. In Symmetry (Group Theory) and Mathematical Treatment in Chemistry; Takashiro, A., Ed.; IntechOpen: London, UK, 2018; pp. 121–139. ISBN 978-1-78923-315-5. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Gherman, B.F.; Baik, M.-H.; Lippard, S.J.; Friesner, R.A. Dioxygen Activation in Methane Monooxygenase: A Theoretical Study. J. Am. Chem. Soc. 2004, 126, 2978–2990. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Lynch, B.J.; Truhlar, D.G. How Well Can Hybrid Density Functional Methods Predict Transition State Geometries and Barrier Heights? J. Phys. Chem. A 2001, 105, 2936–2941. [Google Scholar] [CrossRef]
- Zhao, Y.; González-García, N.; Truhlar, D.G. Benchmark Database of Barrier Heights for Heavy Atom Transfer, Nucleophilic Substitution, Association, and Unimolecular Reactions and Its Use to Test Theoretical Methods. J. Phys. Chem. A 2005, 109, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nishihara, S.; Kataoka, Y.; Nakanishi, Y.; Kitagawa, Y.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Reinvestigation of the Reaction of Ethylene and Singlet Oxygen by the Approximate Spin Projection Method. Comparison with Multireference Coupled-Cluster Calculations. J. Phys. Chem. A 2010, 114, 7967–7974. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| rPM6 | PM6 | |||
|---|---|---|---|---|
| αF-H | 3.293972 | 3.136740 | ||
| xF-H | 0.851422 | 0.815802 | ||
| αF-C | 2.984681 | 3.027600 | ||
| xF-C | 0.736305 | 0.732968 | ||
| αF-N | 2.899491 | 2.856646 | ||
| xF-N | 0.567813 | 0.635854 | ||
| αF-O | 3.051565 | 3.015444 | ||
| xF-O | 0.694847 | 0.674251 | ||
| αF-Mn | 2.920150 | 2.799150 | ||
| xF-Mn | 1.077996 | 1.113070 | ||
| αF-Fe | 4.261457 | 4.294707 | ||
| xF-Fe | 3.657350 | 3.657350 | ||
| αCl-H | 2.575944 | 2.402886 | ||
| xCl-H | 0.946833 | 0.754831 | ||
| αCl-C | 2.208931 | 2.162197 | ||
| xCl-C | 0.550541 | 0.515787 | ||
| αCl-N | 2.198862 | 2.172134 | ||
| xCl-N | 0.586460 | 0.520745 | ||
| αCl-O | 2.379828 | 2.323236 | ||
| xCl-O | 0.642703 | 0.585510 | ||
| αCl-Mn | 1.951202 | 1.657010 | ||
| xCl-Mn | 0.162889 | 0.201850 | ||
| αCl-Fe | 1.574455 | 1.229793 | ||
| xCl-Fe | 0.004971 | 0.019473 | ||
| F | Cl | F | Cl | |
| Uss | −137.738801 | −63.941830 | −140.225636 | −61.389930 |
| Upp | −98.295972 | −54.599897 | −98.778044 | −54.482801 |
| Udd | −33.686857 | −38.258155 | ||
| βs | −71.770655 | −4.283409 | −69.922593 | −2.367988 |
| βp | −29.432906 | −12.99524 | −30.448165 | −13.802139 |
| βd | −1.426944 | −4.037751 | ||
| ζs | 5.975874 | 2.645837 | 6.043849 | 2.637050 |
| ζp | 2.932020 | 2.084940 | 2.906722 | 2.118146 |
| ζd | 1.291613 | 1.324033 | ||
| System | Expt. | UPM6 | UrPM6 | UPM7 | |
|---|---|---|---|---|---|
| FEBKOM | MnIV2(μ–O)2 | −144 | −9 | −130 | −39 |
| FEBKUS | MnIIIMnIV(μ–O)2 | −148 | −49 | −132 | −32 |
| HUYGAJ | MnIV2(μ–O)2 | −45 | 14 | −70 | −30 |
| GAVWIJ | MnIIIMnIV(μ–O)2 | −114 | −10 | −110 | −21 |
| VEGTEG | MnII–OH–MnII | −9 | −6 | −6 | −14 |
| TIPFED | MnII–OH–MnIII | −9 | −0.4 | −5 | −17 |
| PIZWIG | MnIII–F–MnIII | −17 | −16 | −12 | −470 |
| EDEWIV | MnII–F–MnII | −3 | −354 | −5 | −121 |
| CULSOR | MnIII–O–MnIII | 9 | −5 | 3 | −18 |
| ARUGOK | MnIII–O–MnIII | 3 | −5 | 7 | −25 |
| ARUGUQ | MnIII–O–MnIII | −13 | −8 | −35 | N.A. 1 |
| ILELEQ | MnIIMnII | −4 | 7 | −6 | −5 |
| KEJWIG | MnIIMnIII | −4 | 6 | 6 | 2 |
| BARFOQ | MnIIMnIII | −1 | N.A. 1 | −27 | N.A. 1 |
| YOCKAC | FeIII2(μ–O)2 | −27 | −955 | −95 | −804 |
| sMMOQ 2 | FeIV2(μ–O)2 | −224 3 | −1311 | −201 | −27 |
| UCANAP | FeIII2(μ–OH)2 | −18 | −1217 | −33 | −104 |
| CEPBEF | FeIII–OH–FeIII | −42 | −1252 | −27 | −91 |
| EDEVAM | FeII–F–FeII | −8 | −194 | −0.2 | −191 |
| KASPEC | FeII–Cl–FeII | −17 | −38 | −16 | −187 |
| KEYXOC | FeIII–O–FeIII | −106 | −1149 | −93 | −72 |
| QEZHIN | FeIII–O–FeIII | −212 | −360 | −118 | −98 |
| GUQMUA | FeII–O–FeIII | −119 | −459 | −114 | −70 |
| RINXUJ | FeIIIFeIII | −29 | −648 | −51 | −74 |
| RITHAD | FeIIIFeIII | −10 | −1010 | −98 | −78 |
| MAE 4 | 373 | 19 | 119 | ||
| MARE 5 | 18.5 | 1.0 | 7.0 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, T.; Fujiwara, M.; Takano, Y. Quantitative Assessment of rPM6 for Fluorine- and Chlorine-Containing Metal Complexes: Comparison with Experimental, First-Principles, and Other Semiempirical Results. Molecules 2018, 23, 3332. https://doi.org/10.3390/molecules23123332
Saito T, Fujiwara M, Takano Y. Quantitative Assessment of rPM6 for Fluorine- and Chlorine-Containing Metal Complexes: Comparison with Experimental, First-Principles, and Other Semiempirical Results. Molecules. 2018; 23(12):3332. https://doi.org/10.3390/molecules23123332
Chicago/Turabian StyleSaito, Toru, Manami Fujiwara, and Yu Takano. 2018. "Quantitative Assessment of rPM6 for Fluorine- and Chlorine-Containing Metal Complexes: Comparison with Experimental, First-Principles, and Other Semiempirical Results" Molecules 23, no. 12: 3332. https://doi.org/10.3390/molecules23123332
APA StyleSaito, T., Fujiwara, M., & Takano, Y. (2018). Quantitative Assessment of rPM6 for Fluorine- and Chlorine-Containing Metal Complexes: Comparison with Experimental, First-Principles, and Other Semiempirical Results. Molecules, 23(12), 3332. https://doi.org/10.3390/molecules23123332