Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives


Abstract
1. Introduction
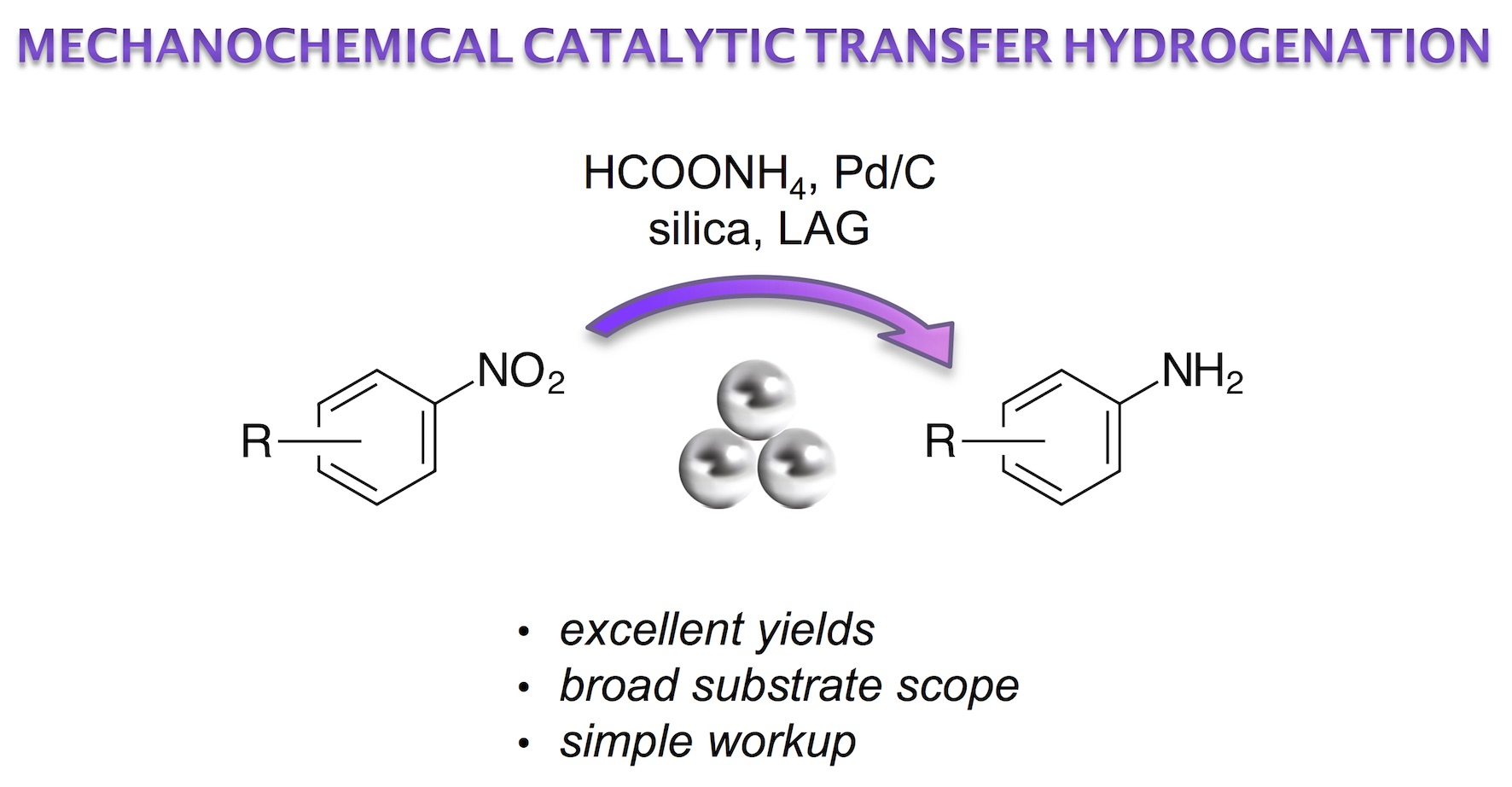
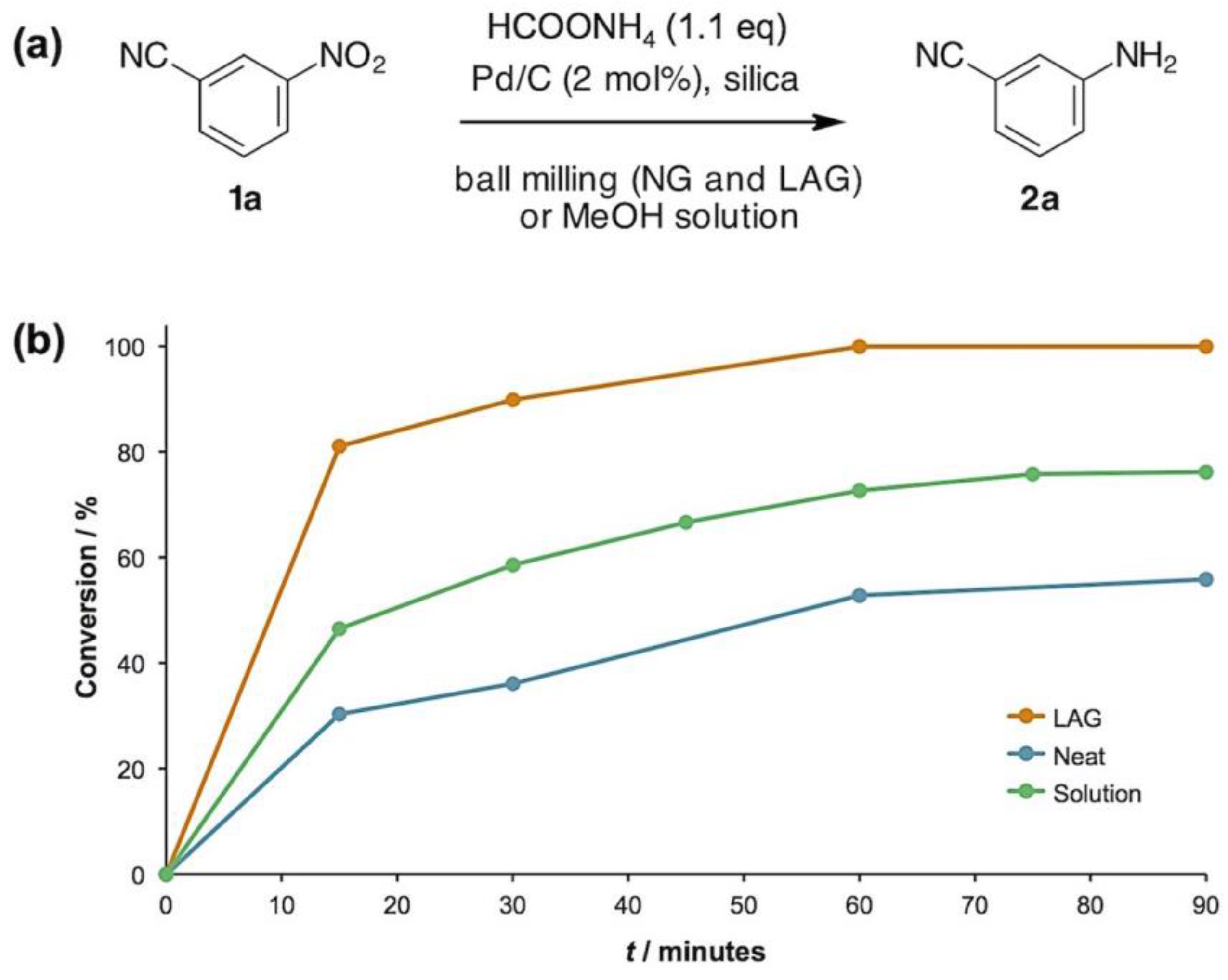
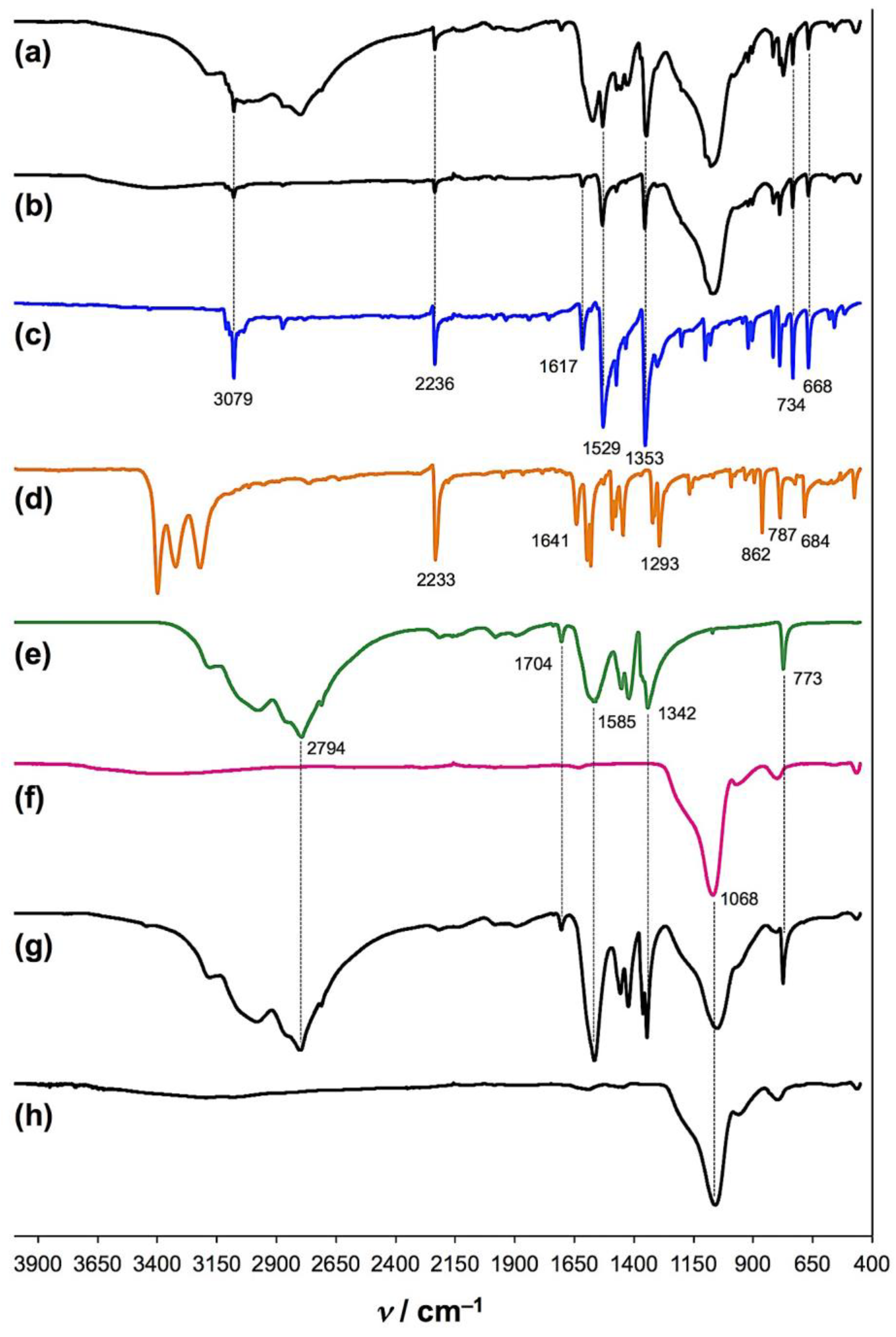
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Brieger, G.; Nestrick, T.J. Catalytic Transfer Hydrogenation. Chem. Rev. 1974, 74, 567–580. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Hagen, J. Heterogeneously Catalyzed Processes in Industry. In Industrial Catalysis: A Practical Approach, 2nd ed.; Hagen, J., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 261–293. ISBN 978-3-527-31144-6. [Google Scholar]
- Ram, S.; Ehrekaufer, R.E. Ammonium Formate in Organic Synthesis: A Versatile Agent in Catalytic Hydrogen Transfer Reductions. Synthesis 1988, 1988, 91–95. [Google Scholar] [CrossRef]
- Zoran, A.; Sasson, Y. Catalytic transfer hydrogenation of unsaturated compounds by solid sodium formate in the presence of palladium on carbon. J. Mol. Catal. 1984, 26, 321–326. [Google Scholar] [CrossRef]
- Paryzek, Z.; Koenig, H.; Tabaczka, B. Ammonium Formate/Palladium on Carbon: A Versatile System for Catalytic Hydrogen Transfer Reductions of Carbon-Carbon Double Bonds. Synthesis 2003, 2023–2026. [Google Scholar] [CrossRef]
- Dobrovolná, Z.; Červeny, L. Ammonium formate decomposition using palladium catalyst. Res. Chem. Intermed. 2000, 26, 489–497. [Google Scholar] [CrossRef]
- Banik, B.K.; Barakat, K.J.; Wagle, D.R.; Manhas, M.S.; Bose, A.K. Microwave-Assisted Rapid and Simplified Hydrogenation. J. Org. Chem. 1999, 64, 5746–5753. [Google Scholar] [CrossRef]
- Vass, A.; Dudás, J.; Tóth, J.; Varma, R.S. Solvent-free reduction of aromatic nitro compounds with alumina-supported hydrazine under microwave irradiation. Tetrahedron Lett. 2001, 42, 5347–5349. [Google Scholar] [CrossRef]
- Berthold, H.; Schotten, T.; Hönig, H. Transfer Hydrogenation in Ionic Liquids under Microwave Irradiation. Synthesis 2002, 1607–1610. [Google Scholar] [CrossRef]
- Do, J.-L.; Friščić, T. Mechanochemistry: A Force of Synthesis. ACS Cent. Sci. 2017, 3, 13–19. [Google Scholar] [CrossRef]
- Howard, J.L.; Cao, Q.; Browne, D.L. Mechanochemistry as an emerging tool for molecular synthesis: What can it offer? Chem. Sci. 2018, 9, 3080–3094. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; De, S.; Balu, A.M.; Ojeda, M.; Luque, R. Mechanochemical synthesis of advanced nanomaterials for catalytic applications. Chem. Commun. 2015, 51, 6698–6713. [Google Scholar] [CrossRef]
- Gečiauskaite, A.A.; García, F. Main group mechanochemistry. Beilstein J. Org. Chem. 2017, 13, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- André, V.; Quaresma, S.; da Silva, J.L.F.; Duarte, M.T. Exploring mechanochemistry to turn organic bio-relevant molecules into metal-organic frameworks: A short review. Beilstein J. Org. Chem. 2017, 13, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- Wixtrom, A.; Buhler, J.; Abdel-Fattah, T. Mechanochemical Synthesis of Two Polymorphs of the Tetrathiafulvalene-Chloranil Charge Transfer Salt: An Experiment for Organic Chemistry. J. Chem. Educ. 2014, 91, 1232–1235. [Google Scholar] [CrossRef]
- Crawford, D.E.; Miskimmin, C.K.G.; Albadarin, A.B.; Walker, G.; James, S.L. Organic synthesis by Twin Screw Extrusion (TSE): Continuous, scalable and solvent-free. Green Chem. 2017, 19, 1507–1518. [Google Scholar] [CrossRef]
- Crawford, D.E.; Wright, L.A.; James, S.L.; Abbott, A.P. Efficient continuous synthesis of high purity deep eutectic solvents by twin screw extrusion. Chem. Commun. 2016, 52, 4215–4218. [Google Scholar] [CrossRef]
- Crawford, D.; Casaban, J.; Haydon, R.; Giri, N.; McNally, T.; James, S.L. Synthesis by extrusion: Continuous, large-scale preparation of MOFs using little or no solvent. Chem. Sci. 2015, 6, 1645–1649. [Google Scholar] [CrossRef]
- Michalchuk, A.A.L.; Hope, K.S.; Kennedy, S.R.; Blanco, M.V.; Boldyreva, E.V.; Pulham, C.R. Ball-free mechanochemistry: In situ real-time monitoring of pharmaceutical co-crystal formation by resonant acoustic mixing. Chem. Commun. 2018, 54, 4033–4036. [Google Scholar] [CrossRef]
- Martina, K.; Rotolo, L.; Porcheddu, A.; Delogu, F.; Bysouth, S.R.; Cravotto, G.; Colacino, E. High throughput mechanochemistry: Application to parallel synthesis of benzoxazines. Chem. Commun. 2018, 54, 551–554. [Google Scholar] [CrossRef]
- Štrukil, V.; Sajko, I. Mechanochemically-assisted solid-state photocatalysis (MASSPC). Chem. Commun. 2017, 53, 9101–9104. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. [Google Scholar] [CrossRef]
- Štrukil, V. Mechanochemical Organic Synthesis: The Art of Making Chemistry Green. Synlett 2018, 29, 1281–1288. [Google Scholar] [CrossRef]
- Tan, D.; Friščić, T. Mechanochemistry for Organic Chemists: An Update. Eur. J. Org. Chem. 2018, 18–33. [Google Scholar] [CrossRef]
- Colacino, E.; Porcheddu, A.; Halasz, I.; Charnay, C.; Delogu, F.; Guerra, R.; Fullenwarth, J. Mechanochemistry for “no solvent, no base” preparation of hydantoin-based active pharmaceutical ingredients: Nitrofurantoin and dantrolene. Green Chem. 2018, 20, 2973–2977. [Google Scholar] [CrossRef]
- Andersen, J.; Mack, J. Mechanochemistry and organic synthesis: From mystical to practical. Green Chem. 2018, 20, 1435–1443. [Google Scholar] [CrossRef]
- Chauhan, P.; Chimni, S.S. Mechanochemistry assisted asymmetric organocatalysis: A sustainable approach. Beilstein J. Org. Chem. 2012, 8, 2132–2141. [Google Scholar] [CrossRef]
- Rodríguez, B.; Rantanen, T.; Bolm, C. Solvent-Free Asymmetric Organocatalysis in a Ball Mill. Angew. Chem. Int. Ed. 2006, 45, 6924–6926. [Google Scholar] [CrossRef]
- Rantanen, T.; Schiffers, I.; Bolm, C. Solvent-Free Asymmetric Anhydride Opening in a Ball Mill. Org. Process Res. Dev. 2007, 11, 592–597. [Google Scholar] [CrossRef]
- Hernández, J.G.; Friščić, T. Metal-catalyzed organic reactions using mechanochemistry. Tetrahedron Lett. 2015, 56, 4253–4265. [Google Scholar] [CrossRef]
- Hernández, J.G. C−H Bond Functionalization by Mechanochemistry. Chem. Eur. J. 2017, 23, 17157–17165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, Y.; Liu, C.; Zhao, Y. Recent advances in mechanochemical C–H functionalization reactions. Tetrahedron Lett. 2018, 59, 317–324. [Google Scholar] [CrossRef]
- Leonardi, M.; Villacampa, M.; Menéndez, J.C. Multicomponent mechanochemical synthesis. Chem. Sci. 2018, 9, 2042–2064. [Google Scholar] [CrossRef] [PubMed]
- Delogu, F.; Mulas, G.; Garroni, S. Hydrogenation of carbon monoxide under mechanical activation conditions. Appl. Catal. A Gen. 2009, 366, 201–205. [Google Scholar] [CrossRef]
- Mack, J.; Fulmer, D.; Stofel, S.; Santos, N. The first solvent-free method for the reduction of esters. Green Chem. 2007, 9, 1041–1043. [Google Scholar] [CrossRef]
- Kaupp, G. Organic Solid-State Reactions with 100% Yield. Top. Curr. Chem. 2005, 254, 95–183, (pp. 117, unpublished results). [Google Scholar] [CrossRef]
- Birke, V.; Mattik, J.; Runne, D. Mechanochemical reductive dehalogenation of hazardous polyhalogenated contaminants. J. Mat. Sci. 2004, 39, 5111–5116. [Google Scholar] [CrossRef]
- Lu, S.; Huang, J.; Peng, Z.; Li, X.; Yan, J. Ball milling 2,4,6-trichlorophenol with calcium oxide: Dechlorination experiment and mechanism considerations. Chem. Eng. J. 2012, 195, 62–68. [Google Scholar] [CrossRef]
- Loiselle, S.; Branca, M.; Mulas, G.; Cocco, G. Selective Mechanochemical Dehalogenation of Chlorobenzenes over Calcium Hydride. Environ. Sci. Technol. 1996, 31, 261–265. [Google Scholar] [CrossRef]
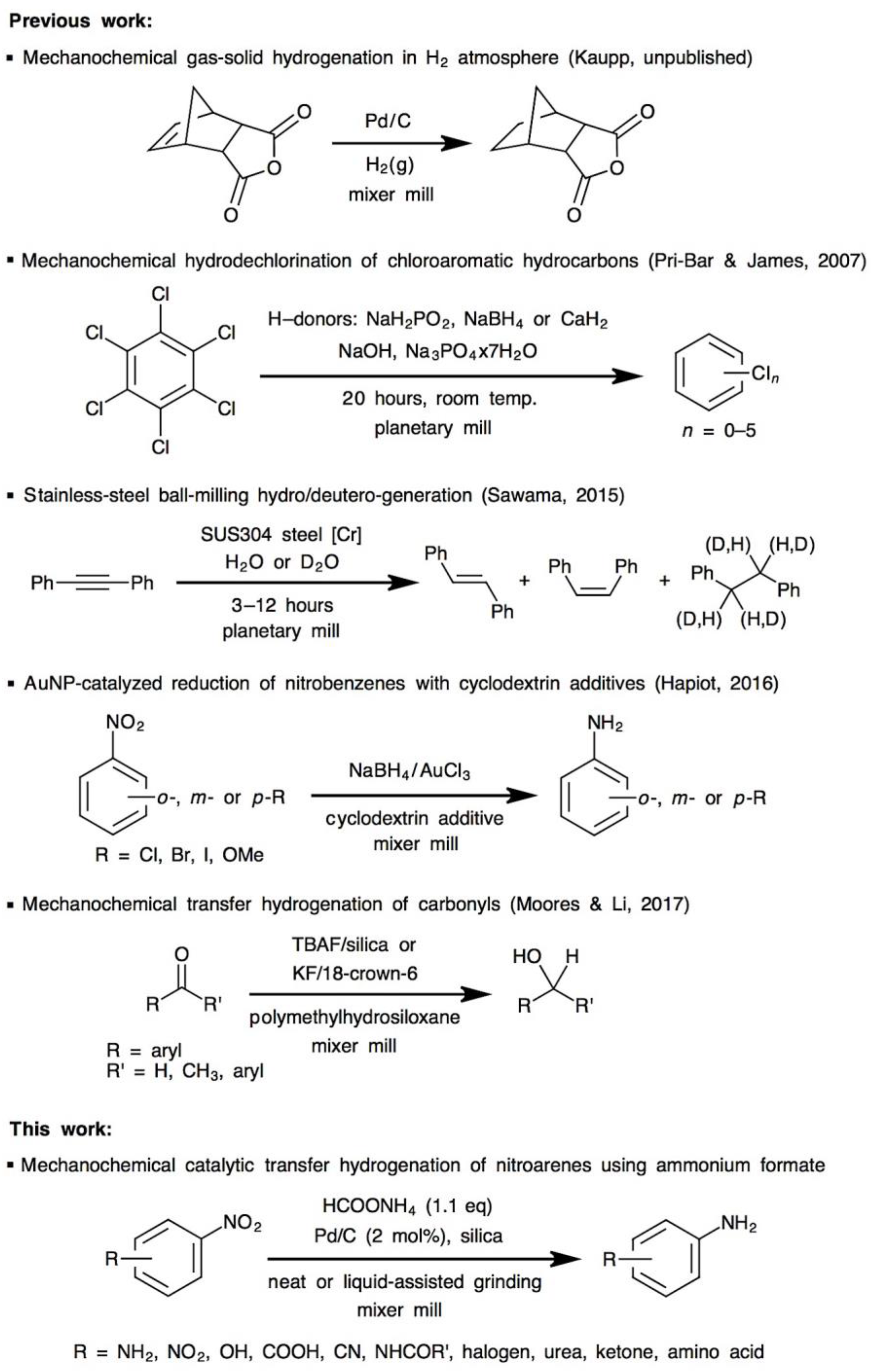
- Pri-Bar, I.; James, B.R. Mechanochemical, solvent free, palladium-catalyzed hydrodechlorination of chloroaromatic hydrocarbons. J. Mol. Catal. A Chem. 2007, 264, 135–139. [Google Scholar] [CrossRef]
- Sawama, Y.; Kawajiri, T.; Niikawa, M.; Goto, R.; Yabe, Y.; Takahashi, T.; Marumoto, T.; Itoh, M.; Kimura, Y.; Monguchi, Y.; et al. Stainless-Steel Ball-Milling Method for Hydro-/Deutero-genation using H2O/D2O as a Hydrogen/Deuterium Source. ChemSusChem 2015, 8, 3773–3776. [Google Scholar] [CrossRef] [PubMed]
- Sawama, Y.; Niikawa, M.; Yabe, Y.; Goto, R.; Kawajiri, T.; Marumoto, T.; Takahashi, T.; Itoh, M.; Kimura, Y.; Sasai, Y.; et al. Stainless-Steel-Mediated Quantitative Hydrogen Generation from Water under Ball Milling Conditions. ACS Sustain. Chem. Eng. 2015, 3, 683–689. [Google Scholar] [CrossRef]
- Sawama, Y.; Yasukawa, N.; Ban, K.; Goto, R.; Niikawa, M.; Monguchi, Y.; Itoh, M.; Sajiki, H. Stainless Steel-Mediated Hydrogen Generation from Alkanes and Diethyl Ether and Its Application for Arene Reduction. Org. Lett. 2018, 20, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- Menuel, S.; Léger, B.; Addad, A.; Monflier, E.; Hapiot, F. Cyclodextrins as effective additives in AuNP-catalyzed reduction of nitrobenzene derivatives in a ball-mill. Green Chem. 2016, 18, 5500–5509. [Google Scholar] [CrossRef]
- Li, A.Y.; Segalla, A.; Li, C.-J.; Moores, A. Mechanochemical Metal-Free Transfer Hydrogenation of Carbonyls Using Polymethylhydrosiloxane as the Hydrogen Source. ACS Sustain. Chem. Eng. 2017, 5, 11752–11760. [Google Scholar] [CrossRef]
- Kadam, H.K.; Tilve, S.G. Advancement in methodologies for reduction of nitroarenes. RSC Adv. 2015, 5, 83391–83407. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, H.; Li, Y.; Guo, X. Cobalt-modified molybdenum carbide as an efficient catalyst for chemoselective reduction of aromatic nitro compounds. Green Chem. 2014, 16, 1274–1281. [Google Scholar] [CrossRef]
- Yang, X.-J.; Chen, B.; Zheng, L.-Q.; Wu, L.-Z.; Tung, C.-H. Highly efficient and selective photocatalytic hydrogenation of functionalized nitrobenzenes. Green Chem. 2014, 16, 1082–1086. [Google Scholar] [CrossRef]
- Wang, L.; Li, P.; Wu, Z.; Yan, J.; Wang, M.; Ding, Y. Reduction of Nitroarenes to Aromatic Amines with Nanosized Activated Metallic Iron Powder in Water. Synthesis 2003, 2001–2004. [Google Scholar] [CrossRef]
- Wienhöfer, G.; Baseda-Krüger, M.; Ziebart, C.; Westerhaus, F.A.; Baumann, W.; Jackstell, R.; Junge, K.; Beller, M. Hydrogenation of nitroarenes using defined iron–phosphine catalysts. Chem. Commun. 2013, 49, 9089–9091. [Google Scholar] [CrossRef]
- Vaidya, M.J.; Kulkarni, S.M.; Chaudhari, R.V. Synthesis of p-Aminophenol by Catalytic Hydrogenation of p-Nitrophenol. Org. Process Res. Dev. 2003, 7, 202–208. [Google Scholar] [CrossRef]
- Baron, M.; Métay, E.; Lemaire, M.; Popowycz, F. Reduction of aromatic and aliphatic nitro groups to anilines and amines with hypophosphites associated with Pd/C. Green Chem. 2013, 15, 1006–1015. [Google Scholar] [CrossRef]
- Orlandi, M.; Brenna, D.; Harms, R.; Jost, S.; Benaglia, M. Recent Developments in the Reduction of Aromatic and Aliphatic Nitro Compounds to Amines. Org. Process Res. Dev. 2018, 22, 430–445. [Google Scholar] [CrossRef]
- Ram, S.; Ehrekaufer, R.E. A general procedure for mild and rapid reduction of aliphatic and aromatic nitro compounds using ammonium formate as a catalytic hydrogen transfer agent. Tetrahedron Lett. 1984, 25, 3415–3418. [Google Scholar] [CrossRef]
- Ram, S.; Ehrekaufer, R.E. A Facile Synthesis of α-Amino Esters via Reduction of α-Nitro Esters Using Ammonium Formate as a Catalytic Hydrogen Transfer Agent. Synthesis 1986, 133–135. [Google Scholar] [CrossRef]
- HPLC analysis showed that the CTH of 3-nitrobenzonitrile (1a) (1.0 mmol) in methanol solution (10 mL) with 3 eq of ammonium formate (9.9 mmol) reached quantitative yield in 30 minutes.
- Đud, M.; Magdysyuk, O.V.; Margetić, D.; Štrukil, V. Synthesis of monosubstituted thioureas by vapour digestion and mechanochemical amination of thiocarbamoyl benzotriazoles. Green Chem. 2016, 18, 2666–2674. [Google Scholar] [CrossRef]
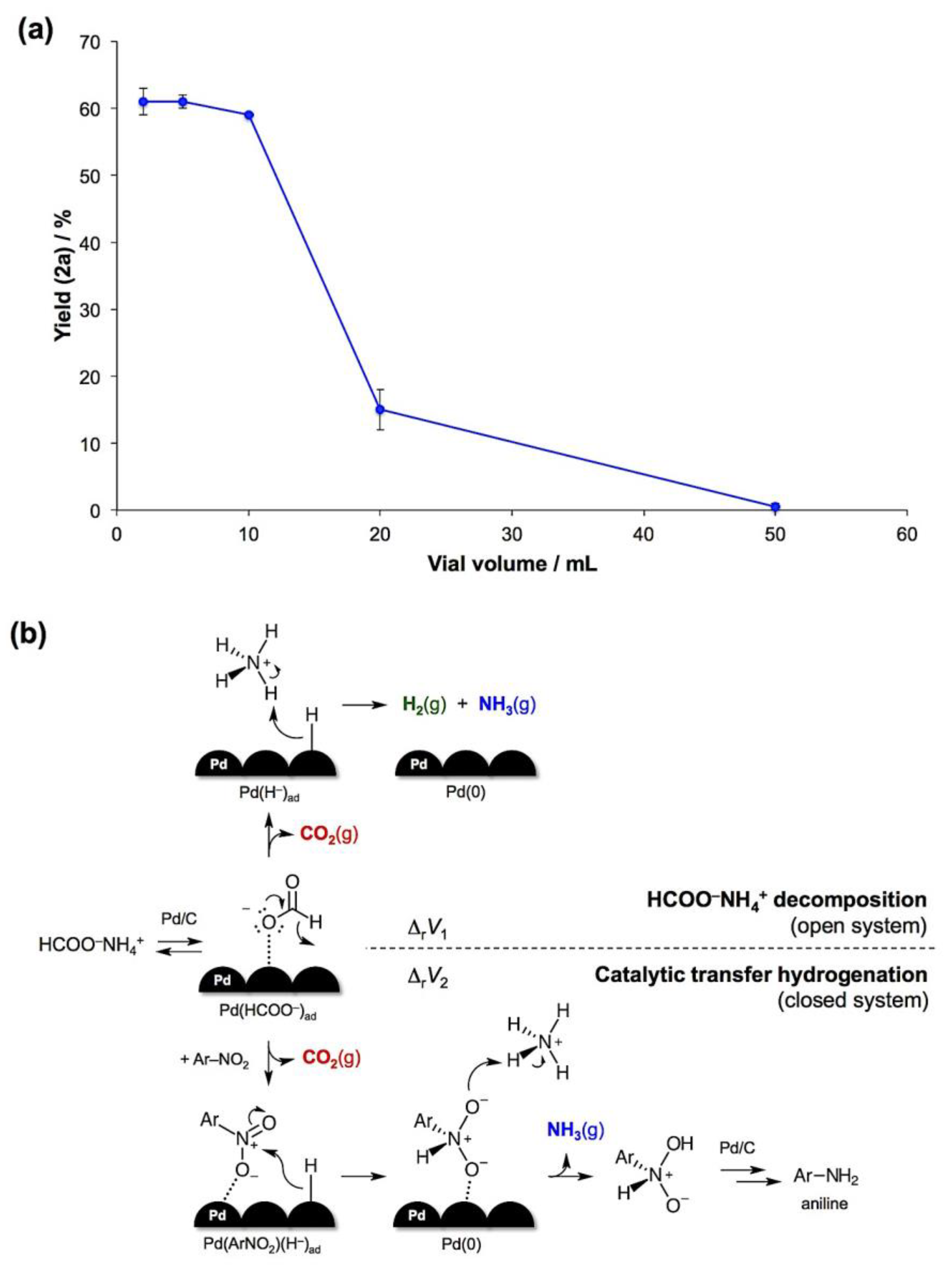
- The reaction mass loss upon opening the jars indicated ca. 60% conversion after 1 hour and quantitative decomposition after 2 hours in air.
- Rajagopal, S.; Anwer, M.K.; Spatola, A.F. Catalytic transfer hydrogenation and hydrogenolysis by formic acid and its salts. In Peptides Design, Synthesis, and Biological Activity, 1st ed.; Basava, C., Anantharamaiah, G.M., Eds.; Birkhäuser: Boston, MA, USA, 1994; Chapter 2; pp. 11–26. ISBN 978-1-4615-8178-9. [Google Scholar]
- Byun, E.; Hong, B.; De Castro, K.A.; Lim, M.; Rhee, H. One-Pot Reductive Mono-N-alkylation of Aniline and Nitroarene Derivatives Using Aldehydes. J. Org. Chem. 2007, 72, 9815–9817. [Google Scholar] [CrossRef]
- Tuteja, J.; Nishimura, S.; Ebitani, K. Base-free chemoselective transfer hydrogenation of nitroarenes to anilines with formic acid as hydrogen source by a reusable heterogeneous Pd/ ZrP catalyst. RSC Adv. 2014, 4, 38241–38249. [Google Scholar] [CrossRef]
- The reaction volume change is defined as the difference between the sums of molar volumes of products and reactants, ΔrV = ∑Vm(products) − ∑Vm(reactants). For solids, the difference in molar volumes is typically small and can be neglected. With this assumption, the CTH will generate 2 moles of gaseous products per 1 mole of the formate salt, while the complete decomposition of HCOONH4 will result in 3 moles of gases per 1 mole of the salt. Therefore, ΔrV1 (decomposition) > ΔrV2 (CTH).
- Maegawa, T.; Fujiwara, Y.; Ikawa, T.; Hisashi, H.; Monguchi, Y.; Sajiki, H. Novel deprotection method of Fmoc group under neutral hydrogenation conditions. Amino Acids 2009, 36, 493–499. [Google Scholar] [CrossRef]
- Albers, P.; Pietsch, J.; Parker, S.F. Poisoning and deactivation of palladium catalysts. J. Mol. Catal. A Chem. 2001, 173, 275–286. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Sane, M.G. Poisoning of Pd–carbon catalysts by sulphur, chloro and heavy metal compounds in liquid phase hydrogenation of o-nitrophenol to O-aminophenol. J. Chem. Technol. Biotechnol. 1998, 73, 336–340. [Google Scholar] [CrossRef]
- Kulla, H.; Fischer, F.; Benemann, S.; Rademann, K.; Emmerling, F. The effect of the ball to reactant ratio on mechanochemical reaction times studied by in situ PXRD. Cryst. Eng. Comm. 2017, 19, 3902–3907. [Google Scholar] [CrossRef]
- As suggested by preliminary DFT calculations (B97D/6-31G(d) method), the reason behind the observed poor reactivity of these substrates could be a strong - interaction of polyaromatic molecules with graphite-like structure of activated carbon, where the formation of 1:1 or 1:2 sandwich complexes might interfere with the reduction of the nitro group. The stability of such 1:1 and 1:2 complexes increases with the number of condensed aromatic rings in nitroarene substrates. For instance, the relative energies of 1:1 complexes of nitrobenzene, 1-amino and 2-aminonaphthalene, 1-amino and 2-aminoanthracene and 1-aminopyrene, with coronene as a graphite-like model structure are −13.6, −18.3, −16.1, −21.0, −21.7 and −24.1 kcal mol–1, respectively. By employing the M062X/6-31G(d) method, these values were found to follow the same trend, but with less pronounced relative energy difference: −12.1, −15.7, −16.5, −17.9, −18.9 and −19.3 kcal mol–1.
- Shannon, S.K.; Peacock, M.J.; Kates, S.A.; Barany, G. Solid-Phase Synthesis of Lidocaine and Procainamide Analogues Using Backbone Amide Linker (BAL) Anchoring. J. Comb. Chem. 2003, 5, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Loots, L.; Friščić, T. Towards medicinal mechanochemistry: Evolution of milling from pharmaceutical solid form screening to the synthesis of active pharmaceutical ingredients (APIs). Chem. Commun. 2016, 52, 7760–7781. [Google Scholar] [CrossRef]
- Métro, T.-X.; Bonnamour, J.; Reidon, T.; Sarpoulet, J.; Martinez, J.; Lamaty, F. Mechanosynthesis of amides in the total absence of organic solvent from reaction to product recovery. Chem. Commun. 2012, 48, 11781–11783. [Google Scholar] [CrossRef]
- Bonnamour, J.; Métro, T.-X.; Martinez, J.; Lamaty, F. Environmentally benign peptide synthesis using liquid-assisted ball-milling: Application to the synthesis of Leu-enkephalin. Green Chem. 2013, 15, 1116–1120. [Google Scholar] [CrossRef]
- Estévez, V.; Villacampa, M.; Menéndez, J.C. Concise synthesis of atorvastatin lactone under high-speed vibration milling conditions. Org. Chem. Front. 2014, 1, 458–463. [Google Scholar] [CrossRef]
- Tan, D.; Štrukil, V.; Mottillo, C.; Friščić, T. Mechanosynthesis of pharmaceutically relevant sulfonyl-(thio)ureas. Chem. Commun. 2014, 50, 5248–5250. [Google Scholar] [CrossRef]
- Rajput, L.; Banerjee, R. Mechanochemical Synthesis of Amide Functionalized Porous Organic Polymers. Cryst. Growth Des. 2014, 14, 2729–2732. [Google Scholar] [CrossRef]
- Yang, Y.; Bu, F.; Liu, J.; Shakir, I.; Xu, Y. Mechanochemical synthesis of two-dimensional aromatic polyamides. Chem. Commun. 2017, 53, 7481–7484. [Google Scholar] [CrossRef]
- Perlovich, G.L.; Volkova, T.V.; Bauer-Brandl, A. Polymorphism of paracetamol. J. Therm. Anal. Calorim. 2007, 89, 767–774. [Google Scholar] [CrossRef]
- Di Martino, P.; Conflant, P.; Drache, M.; Huvenne, J.-P.; Guyot-Hermann, A.-M. Preparation and physical characterization of forms II and III of paracetamol. J. Therm. Anal. 1997, 48, 447–458. [Google Scholar] [CrossRef]
- Telford, R.; Seaton, C.C.; Clout, A.; Buanz, A.; Gaisford, S.; Williams, G.R.; Prior, T.J.; Okoye, C.H.; Munshi, T.; Scowen, I.J. Stabilisation of metastable polymorphs: The case of paracetamol form III. Chem. Commun. 2016, 52, 12028–12031. [Google Scholar] [CrossRef] [PubMed]
- Štrukil, V.; Igrc, M.D.; Eckert-Maksić, M.; Friščić, T. Click Mechanochemistry: Quantitative Synthesis of “Ready to Use” Chiral Organocatalysts by Efficient Two-Fold Thiourea Coupling to Vicinal Diamines. Chem. Eur. J. 2012, 18, 8464–8473. [Google Scholar] [CrossRef] [PubMed]
- Štrukil, V.; Igrc, M.D.; Fábián, L.; Eckert-Maksić, M.; Childs, S.L.; Reid, D.G.; Duer, M.J.; Halasz, I.; Mottillo, C.; Friščić, T. A model for a solvent-free synthetic organic research laboratory: Click-mechanosynthesis and structural characterization of thioureas without bulk solvents. Green Chem. 2012, 14, 2462–2473. [Google Scholar] [CrossRef]
- Štrukil, V.; Margetić, D.; Igrc, M.D.; Eckert-Maksić, M.; Friščić, T. Desymmetrisation of aromatic diamines and synthesis of non-symmetrical thiourea derivatives by click-mechanochemistry. Chem. Commun. 2012, 48, 9705–9707. [Google Scholar] [CrossRef] [PubMed]
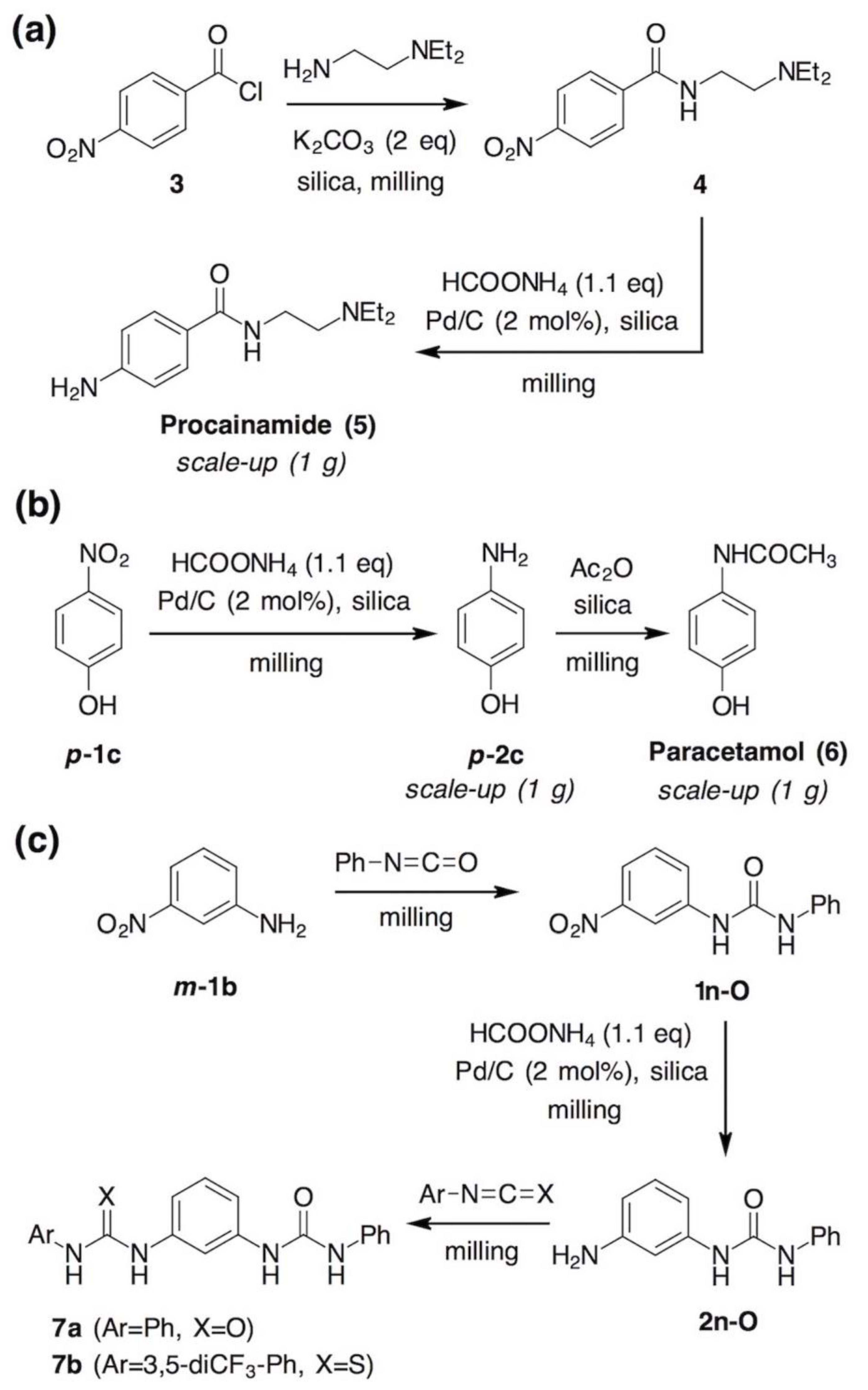
- Neat grinding of equimolar amounts of m-phenylenediamine (m-2b) and phenyl isocyanate for 30 minutes using one 12 mm stainless steel ball led to a mixture of unreacted diamine (21%), isocyanate (4%), amino-urea 2n-O (21%) and bis(urea) 7a (54%), based on 1H NMR analysis. For comparison, sterically more hindered o-phenylenediamine (o-2b) afforded the respective amino-urea in 78% under the same conditions.
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Product | Yield 2 | Product | Yield |
 | o-2b, 99% (99%) m-2b, 98% (96%) p-2b, 95% (96%) |  | 2n-S, n.r. (X=S) 2n-O, 96% (X=O) |
 | o-2c, 99% m-2c, 94% p-2c, 99% |  | 2o, 95% 3 |
 | o-2d, 98% m-2d, 99% p-2d, 97% |  | p-2p, 17% |
 | 2e, 99% |  | o-2p, 42% |
 | 2f, 99% |  | 2q, 37% |
 | 2g, 94% |  | m-2r, 99% (R=Ph) p-2r, 99% (R=Ph) |
 | 2h, 93% |  | 2s, 95% |
 | 2i, 99% |  | 2t, 95% (R=H) 2u, 84% (R=Me) |
 | 2j, 99% |  | 2v, 90% |
 | 2k, 99% |  | 2w, 40% (97%) 3 |
 | 2l-Phe, 93% 3 |  | 2x, 18% (37%) |
 | 2l-Val, 90% 3 |  | 2y, traces |
 | 2m, 97% |  | 2z, n.r. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portada, T.; Margetić, D.; Štrukil, V. Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives. Molecules 2018, 23, 3163. https://doi.org/10.3390/molecules23123163
Portada T, Margetić D, Štrukil V. Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives. Molecules. 2018; 23(12):3163. https://doi.org/10.3390/molecules23123163
Chicago/Turabian StylePortada, Tomislav, Davor Margetić, and Vjekoslav Štrukil. 2018. "Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives" Molecules 23, no. 12: 3163. https://doi.org/10.3390/molecules23123163
APA StylePortada, T., Margetić, D., & Štrukil, V. (2018). Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives. Molecules, 23(12), 3163. https://doi.org/10.3390/molecules23123163