Recent Studies on Cyclic 1,7-Diarylheptanoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis


Abstract
1. Introduction

2. Isolation, Structural Features, and Biological Properties
2.1. Biphenyl Diarylheptanoids
2.1.1. Asadanin and Related Derivatives




2.1.2. Myricanone, Myricanol and Related Derivatives



2.1.3. Garuganins

2.1.4. Miscellaneous


2.2. Diphenyl Ether Diarylheptanoids
2.2.1. Acerogenins and Acerosides




2.2.2. Garuganins and Garugamblins


2.2.3. Miscellaneous



3. Total Synthesis of Biphenyl Diarylheptanoids
3.1. Biosynthetic Pathway


3.2. Total Synthesis of Biphenyl Heptanoids
3.2.1. Aryl–Aryl Bond Formation via Metal Catalyzed Coupling






3.2.2. Aryl–Aryl Bond Formation via Photochemical Cyclization

3.3. Total Synthesis of Cyclic Diphenyl Ether Heptanoids
3.3.1. Formation of Macrocycles via Oxidative Coupling


3.3.2. Formation of Macrocycles via SNAr Reaction


3.3.3. Formation of Macrocycles via Intramolecular Ullmann Ether Synthesis







3.3.4. Formation of Macrocycle via Formation of Heptane Skeleton
Via Wurtz and/or Wittig Reactions


Via Ring-Closing Metathesis

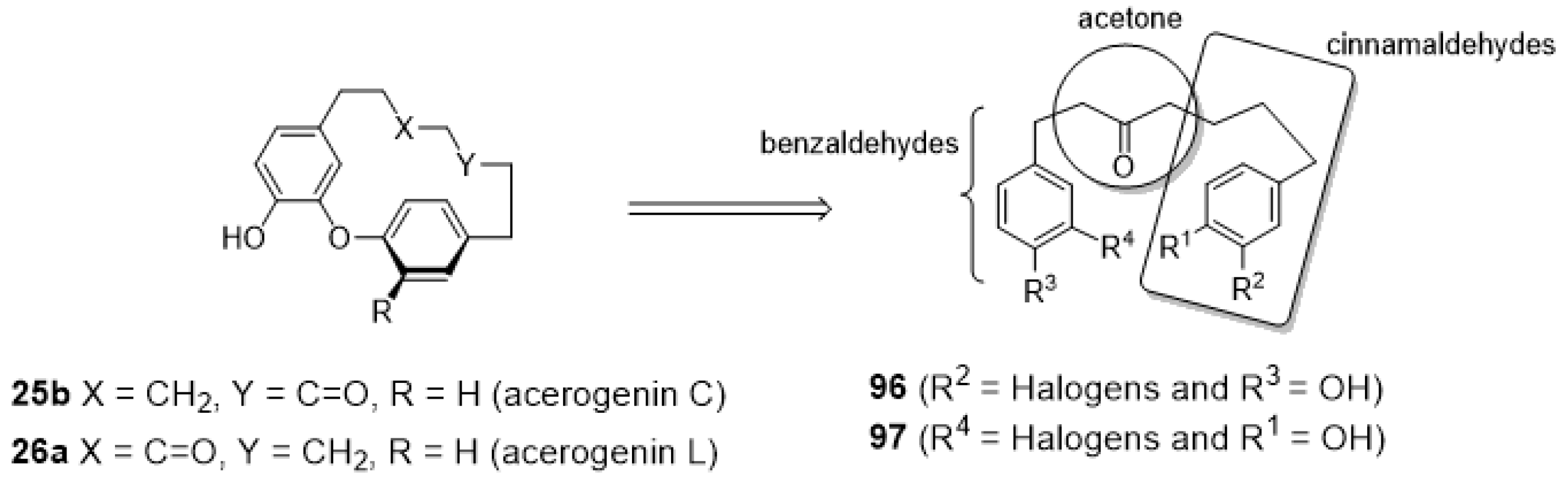
3.3.5. Enantioselective Synthesis of Diaryl Ether Heptanoids




4. Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Although the nomenclature of this system generated from ChemDraw® is 1,2(1,3)-dibenzenacyclononaphane, we used common name system which is more familiar to most of the chemists. Similarly, oxa[7.1]metaparacyclophane was used instead of 2-oxa-1(1,3),3(1,4)-dibenzenacyclodecaphane for type II diaryl ether heptanoids.
- Keserü, G.M.; Nógrádi, M. The chemistry of natural diarylheptanoids. Stud. Nat. Prod. Chem. 1995, 17, 357–394. [Google Scholar]
- Zhu, J.; Islas-Gonzalez, G.; Bois-Choussy, M. Recent progress in isolation, bioactiviy evaluation and total synthesis of diarylheptanoids. Org. Prep. Proced. Int. 2000, 32, 505–546. [Google Scholar] [CrossRef]
- Vogel, H.; Pelletier, J. Examen chimique de la racine de curcuma. J. Pharm. 1815, 1, 289–303. [Google Scholar]
- Milobedzka, J.; van Kostanecki, S.; Lampe, V. Zur Kenntnis des Curcumins. Ber. Dtsch. Chem. Ges. 1910, 43, 2163–2170. [Google Scholar] [CrossRef]
- Maurent, K.; Vanucci-Bacqué, C.; Saffon-Merceron, N.; Baltas, M.; Bedos-Belval, F. Total synthesis of tedarene A. J. Nat. Prod. 2017, 80, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Gao, Y.; Shen, J.; He, C.; Liu, H.; Peng, Y.; Zhang, C.; Xiao, P. Traditional uses, phytochemistry, and pharmacology of the genus Acer (maple): A review. J. Ethnopharm. 2016, 189, 31–60. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, B.-G.; Wu, Z.-J.; Zhang, G.-L. Lupane triterpenoids and a diarylheptanoid from Clematoclethra actinidioides. Planta Med. 2006, 72, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; He, T.; Chang, Y.; Zhao, Y.; Chen, X.; Bai, S.; Wang, L.; She, M.; She, G. The genus Alnus, a comprehensive outline of its chemical constituents and biological activities. Molecules 2017, 22, 1383. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Pandey, M.M.; Rawat, A.K.S. Medicinal plants of the genus Betula—Traditional uses and a phytochemical-pharmacological review. J. Ethnopharm. 2015, 159, 62–83. [Google Scholar] [CrossRef] [PubMed]
- Masullo, M.; Cerulli, A.; Olas, B.; Pizza, C.; Piacente, S. Giffonin A–I, antioxidant cyclized diarylheptanoids from the leaves of hazelnut tree (Corylus avellana), source of the Italian PGI product “Nocciola di Giffoni”. J. Nat. Prod. 2015, 78, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Masullo, M.; Cantone, V.; Cerulli, A.; Lauro, G.; Messano, F.; Russo, G.L.; Pizza, C.; Bifulco, G.; Piacente, S. Giffonin J–P, highly hydroxylated cyclized diarylheptanoids from the leaves of Corylus avellana cultivar “Tonda di Giffoni”. J. Nat. Prod. 2015, 78, 2975–2982. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, H.J.; Park, H.; Lee, Y.S. New diarylheptanoids from the stems of Carpinus cordata. J. Nat. Prod. 2002, 65, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Yasue, M.; Miyazaki, M.; Takahashi, T.; Imamura, H.; Honda, O. Wood extractives. VIII. Constituents of Ostrya japonica wood. J. Japan Wood Res. Soc. 1965, 11, 111–113. [Google Scholar]
- Feng, M.-M.; Zhang, Y.-X.; Xia, B.; He, D.-H.; Ding, L.-S.; Zhou, Y.; Ye, X.-X. Chemical constituents in leaves of Ostryopsis nobilis and their antioxidant activities. Zhongyaocai 2013, 44, 2650–2656. [Google Scholar]
- Zhang, Y.-X.; Xia, B.; Zhou, Y.; Ding, L.-S.; Peng, S.-L. Two new cyclic diarylheptanoids from the stems of Ostryopsis noblis. Chin. Chem. Lett. 2013, 24, 512–514. [Google Scholar] [CrossRef]
- Venkatraman, G.; Mishra, A.K.; Thombare, P.S.; Sabata, B.K. Diarylheptanoids from Garuga pinnata. Phytochemistry 1933, 33, 1221–1225. [Google Scholar] [CrossRef]
- Ara, K.; Rahman, A.H.M.M.; Hasan, C.M.; Iskander, M.N.; Asakawa, Y.; Quang, D.N.; Rashid, M.A. Macrocyclic diarylheptanoids from Garuga pinnata. Phytochemistry 2006, 67, 2659–2662. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.L.N.; Ravinder, K.; Srinivasulu, M.; Goud, T.V.; Reddy, S.M.; Srujankumar, D.; Rao, T.P.; Murty, U.S.; Venkateswarlu, Y. Two new macrocyclic diaryl ether heptanoids from Boswellia ovalifoliolata. Chem. Pharm. Bull. 2003, 51, 1081–1084. [Google Scholar] [CrossRef]
- Kaneda, A.; Kinghorn, A.D.; Farnsworth, N.R.; Tuchinda, P.; Udchachon, J.; Santisuki, T.; Reutrakul, V. Two diarylheptanoids and a lignin from Casuarina junghuhniana. Phytochemistry 1990, 29, 3366–3368. [Google Scholar] [CrossRef]
- Wu, H.-C.; Cheng, M.-J.; Peng, C.-F.; Yang, S.-C.; Chang, H.-S.; Lin, C.-H.; Wang, C.-J.; Chen, I.-S. Secondary metabolites from the stems of Engelhardia roxburghiana and their antitubercular activities. Phytochemistry 2012, 82, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-S.; Li, G.; Kim, S.H.; Lee, C.-S.; Woo, M.-H.; Lee, S.-H.; Jahng, Y.-D.; Son, J.-K. Cytotoxic diarylheptanoids from the roots of Juglans mandshurica. J. Nat. Prod. 2002, 65, 1707–1708. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xu, M.-L.; Choi, H.-G.; Lee, S.-H.; Jahng, Y.-D.; Lee, C.-S.; Moon, D.-C.; Woo, M.-H.; Son, J.-K. Four new diarylheptanoids from the roots of Juglans mandshurica. Chem. Pharm. Bull. 2003, 51, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Jiang, Z.-H.; Isao, K. Distribution of ellagic acid derivatives and a diarylheptanoid in wood of Platycarya strobilacea. Phytochemistry 1998, 47, 851–854. [Google Scholar] [CrossRef]
- Liu, H.B.; Cui, C.B.; Gu, Q.Q.; Zhang, D.Y.; Zhao, Q.C.; Guan, H.S. Pterocarine, a new diarylheptanoid from Pterocarya tonkinesis, its cell cycle inhibition at G0/G1 phase and induction of apoptosis in HCT-15 and K562 cells. Chin. Chem. Lett. 2005, 16, 215–218. [Google Scholar]
- Jiang, Z.-H.; Tanaka, T.; Hirata, H.; Fukuoka, R.; Kouno, I. Three diarylheptanoids from Rhoiptelea chiliantha. Phytochemistry 1996, 43, 1049–1054. [Google Scholar] [CrossRef]
- Whiting, D.; Wood, A. Cyclisation of 1,7-diarylheptanoids through oxidative, reductive, and photochemical radical processes: Total syntheses of the m,m-bridged biaryls myricanone and (±)-myricanol, and a related diaryl ether. Tetrahedron Lett. 1978, 19, 2335–2338. [Google Scholar] [CrossRef]
- Inoue, T.; Arai, Y.; Nagai, M. Diarylheptanoids in the bark of Myrica rubra Sieb et. Zucc. Yakugaku Zasshi 1984, 104, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Makule, E.; Schmidt, T.J.; Heilmann, J.; Kraus, B. Diarylheptanoid glycosides of Morella salicifolia bark. Molecules 2017, 22, 2266. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Gao, G.; Qi, S.; Li, Q.; Zhang, C. Studies on the constituents of Scyphiphora hydrophyllacea (II). Zhongyaocai 2009, 32, 712–714. [Google Scholar] [PubMed]
- Ma, X.-N.; Xie, C.-L.; Miao, Z.; Yang, Q.; Yang, X.-W. An overview of chemical constituents from Alpinia species in the last six decades. RSC Adv. 2017, 7, 14114–14144. [Google Scholar] [CrossRef]
- Claeson, P.; Tuchinda, P.; Reutrakul, V. Naturally occurring 1,7-diarylheptanoids. J. Indian Chem. Soc. 1994, 71, 509–521. [Google Scholar]
- Claeson, P.; Claeson, U.P.; Tuchinda, P.; Reutrakul, V. Occurrence, isolation, and bioactivity of 1,7-diarylheptanoids. Stud. Nat. Prod. Chem. 2002, 26, 881–908. [Google Scholar]
- Lv, H.; She, G. Naturally occurring diarylheptanoids. Nat. Prod. Commun. 2010, 5, 1687–1708. [Google Scholar] [PubMed]
- Lv, H.; She, G. Naturally occurring diarylheptanoids—A supplementary version. Rec. Nat. Prod. 2012, 6, 321–333. [Google Scholar]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Priyadarsini, K.I. The chemistry of curcumin: From extraction to therapeutic agent. Molecules 2014, 19, 20091–20112. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The essential medicinal chemistry of curcumin: Miniperspective. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.F.M.M.; Lu, Y.; Lee, H.-J.; Jo, H.; Yin, W.; Alam, S.M.; Cha, H.; Kadi, A.A.; Kwon, Y.; Jahng, Y. Linear diarylheptanoids as potential anticancer therapeutics: Synthesis, biological evaluation, and structure-activity relationship studies. Arch. Pharm. Res. 2018. [Google Scholar] [CrossRef]
- Liu, J.X.; Di, D.L.; Wei, X.-N.; Han, Y. Cytotoxic diarylheptanoid from the pericarps of walnuts (Juglans regia). Planta Med. 2008, 74, 754–759. [Google Scholar] [CrossRef] [PubMed]
- He, J.-B.; Yan, Y.-M.; Ma, X.-J.; Lu, Q.; Li, X.-S.; Su, J.; Li, Y.; Liu, G.-M.; Cheng, Y.-X. Sesquiterpenoids and diarylheptanoids from Nidus vespae and their inhibitory effects on nitric oxide production. Chem. Biodivers. 2011, 8, 2270–2276. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Fattorusso, E.; Mangoni, A.; Perinu, C.; Teta, R.; Panza, E.; Ianaro, A. Tedarenes A and B: Structural and stereochemical analysis of two new strained cyclic diarylheptanoids from the marine sponge Tedania ignis. J. Org. Chem. 2012, 77, 6377–6383. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-W.; Tanaka, T.; Nonaka, G.-I.; Hahn, D.-R. Phenolic compounds on the leaves of Betula platyphylla var. latifolia. Arch. Pharm. Res. 1992, 15, 211–214. [Google Scholar] [CrossRef]
- Joshi, B.S.; Pelletier, S.W.; Newton, M.G.; Lee, D.; McGaughey, G.B.; Puar, M.S. Extensive 1D, 2D NMR spectra of some[7.0]metacyclophanes and X-ray analysis of (±)-myricanol. J. Nat. Prod. 1996, 59, 759–764. [Google Scholar] [CrossRef]
- Jones, J.R.; Lebar, M.D.; Jinwal, U.K.; Abisambra, J.F.; Koren III, J.; Blair, L.; O’Leary, J.C.; Davey, Z.; Trotter, J.; Johnson, A.G.; et al. The diarylheptanoid (+)-aR,11S-myricanol and two flavones from bayberry (Myrica cerifera) destabilize the microtubule-associated protein tau. J. Nat. Prod. 2011, 74, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Alberti, Á.; Riethmüller, E.; Béni, S. Characterization of diarylheptanoids: An emerging class of bioactive natural products. J. Pharm. Biomed. Anal. 2018, 147, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Yasue, M. Wood extractives. XI. Structure of asadanin. 3. J. Japan Wood Res. Soc. 1965, 11, 202–205. [Google Scholar]
- Yasue, M. Wood extracts of Ostrya japonica. Structures of asadanin and related compounds. Ringyo Shikenjo Kenkyu Hokoku 1968, 209, 77–168. [Google Scholar]
- Singldinger, B.; Dunkel, A.; Hofmann, T. The cyclic diarylheptanoid asadanin as the main contributor to the bitter off-taste in hazelnuts (Corylus avellana L.). J. Agric. Food Chem. 2017, 65, 1677–1683. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Tokoroyama, T. Three new cyclized C9-C1-C9 compounds from Alnus japonica steud. J. Chem. Soc. Chem. Comm. 1974, 65–66. [Google Scholar] [CrossRef]
- Nomura, M.; Tokoroyama, T. Further phenolic components from Alnus japonica. J. Chem. Soc. Chem. Comm. 1975, 316–317. [Google Scholar] [CrossRef]
- Hanawa, F.; Shiro, M.; Hayashi, Y. Heartwood constituents of Betula maximowicziana. Phytochemistry 1997, 45, 589–595. [Google Scholar] [CrossRef]
- Watanabe, N.; Sasaya, T. Extractives of the genus Corylus. II. Novel diarylheptanoids from the wood of Corylus sieboldiana Blume (1). J. Japan Wood Res. Soc. 1994, 40, 199–203. [Google Scholar]
- Watanabe, N.; Sasaya, T.; Sano, Y. Extractives of the genus Corylus. III. Novel diarylheptanoids from the wood of Corylus sieboldiana blume. (2). J. Japan Wood Res. Soc. 1994, 40, 1219–1225. [Google Scholar]
- Sung, S.H.; Lee, M. Anti-adipogenic activity of a new diarylheptanoid isolated from Alnus japonica on 3T3-L1 cells via modulation of PPARγ, C/EBPα and SREBP1c signaling. Bioorg. Med. Chem. Lett. 2015, 25, 4648–4651. [Google Scholar] [CrossRef] [PubMed]
- Fuchino, H.; Satoh, T.; Yokochi, M.; Tanaka, N. Chemical evaluation of Betula species in Japan. III. Constituents of Betula maximowicziana. Chem. Pharm. Bull. 1996, 44, 1748–1753. [Google Scholar] [CrossRef]
- Nagumo, S.; Kaji, N.; Inoue, T.; Nagai, M. Studies on the constituents of Aceraceae plants. XI. Two types of cyclic diarylheptanoid from Acer nikoense. Chem. Pharm. Bull. 1993, 41, 1255–1257. [Google Scholar] [CrossRef]
- Fuchino, H.; Satoh, T.; Tanaka, N. Chemical evaluation of Betula species in Japan. I. Constituents of Betula ermanii. Chem. Pharm. Bull. 1995, 43, 1937–1942. [Google Scholar] [CrossRef]
- Nagumo, S.; Ishizawa, S.; Inoue, T.; Nagai, M. Studies on the constituents of Aceraceae plants. XIII. Diarylheptanoids and other phenolics from Acer nikoense. Chem. Pharm. Bull. 1996, 44, 1086–1089. [Google Scholar] [CrossRef]
- Fuchino, H.; Konishi, S.; Satoh, T.; Yagi, A.; Saitsu, K.; Tatsumi, T.; Tanaka, N. Chemical evaluation of Betula species in Japan. II. Constituents of Betula platyphylla var. japonica. Chem. Pharm. Bull. 1996, 44, 1033–1038. [Google Scholar] [CrossRef]
- Fuchino, H.; Satoh, T.; Hida, J.; Terada, M.; Tanaka, N. Chemical evaluation of Betula species in Japan. IV. Constituents of Betula davurica. Chem. Pharm. Bull. 1998, 46, 166–168. [Google Scholar] [CrossRef]
- Wang, S.; Pei, Y. Diarylheptanoids from leaves of Betula platyphylla. Zhongcaoyao 2001, 32, 99–101. [Google Scholar]
- Takahashi, M.; Fuchino, H.; Sekita, S.; Satake, M. In vitro leishmanicidal activity of some scare natural products. Phytotherapy Res. 2004, 18, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Cerulli, A.; Lauro, G.; Masullo, M.; Cantone, V.; Olas, B.; Kontek, B.; Nazzaro, F.; Bifulco, G.; Piacente, S. Cyclic diarylheptanoids from Corylus avellana green leafy covers: Determination of their absolute configurations and evaluation of their antioxidant and antimicrobial activities. J. Nat. Prod. 2017, 80, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Ichizawa, H.; Kawai, S.; Nishida, T. α-Glucosidase inhibition activity by cyclic diarylheptanoids from Alnus sieboldiana. J. Wood Chem. Tech. 2013, 33, 44–51. [Google Scholar] [CrossRef]
- Inoue, T. Constituents of Acer nikoense and Myrica rubra. On diarylheptanoids. Yakugaku Zhazzhi 1993, 113, 181–197. [Google Scholar] [CrossRef]
- Morikawa, T.; Tao, J.; Toguchida, I.; Matsuda, H.; Yoshikawa, M. Structures of new cyclic diarylheptanoids and inhibitors of nitric oxide production from Japanese folk medicine Acer nikoense. J. Nat. Prod. 2003, 61, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.V.M.; Crombie, L.; Tuck, B.; Whiting, D.A. Isolation and structure of new meta-bridged biphenyls from Myrica nagi. J. Chem. Soc. Chem. Comm. 1970, 1206–1207. [Google Scholar] [CrossRef]
- Begley, M.J.; Whiting, D.A. X-ray study of 16-bromomyricanol; the structure of myricanol, a natural m,m-bridged bent biphenyl. J. Chem. Soc. Chem. Comm. 1970, 1207–1208. [Google Scholar] [CrossRef]
- Begley, M.J.; Campbell, R.V.M.; Crombie, L.; Tuck, B.; Whiting, D.A. Constitution and absolute configuration of meta,meta-bridged stained biphenyls from Myrica nagi; X-ray analysis of 16-bromomyricanol. J. Chem. Soc. 1971, 3634–3642. [Google Scholar]
- Martin, M.D.; Calcul, L.; Smith, C.; Jinwal, U.K.; Fontaine, S.N.; Darling, A.; Seeley, K.; Wojtas, L.; Narayan, M.; Gestwicki, J.E.; et al. Synthesis, stereochemical analysis, and derivatization of myricanol provide new probes that promote autophagic tau clearance. ACS Chem. Biol. 2015, 10, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Smyth, J.E.; Butler, N.M.; Keller, P.A. A twist of nature—The significance of atropisomers in biological systems. Nat. Prod. Rep. 2015, 32, 1562–1583. [Google Scholar] [CrossRef] [PubMed]
- Cahn, R.S.; Ingold, C.; Prelog, V. Specification of molecular chirality. Angew Chem. Int. Ed. Engl. 1966, 5, 385–415. [Google Scholar] [CrossRef]
- Anthonsen, T.; Lorentzen, G.B.; Malterud, K.E. Porson, a new [7,0]-metacyclophane from Myrica gale. Acta Chim. Scand. Ser. B 1975, 29, 529–530. [Google Scholar] [CrossRef]
- Sun, D.W.; Zhao, Z.C.; Wong, H.; Foo, L.Y. Tannins and other phenolics from Myrica esculenta bark. Phytochemistry 1988, 27, 579–583. [Google Scholar]
- Ren, Z.; Tong, Y.; Dai, G.; Chen, X.; Yang, F. Effect of myricanone on tumor in vitro. Zhonghua Zhongyiyao Xuekan 2014, 32, 2423–2425. [Google Scholar]
- Dai, G.; Tong, Y.; Chen, X.; Ren, Z.; Yang, F. In vitro anticancer activity of myricanone in human lung adrenocarcinoma A549 cells. Chemotherapy 2014, 60, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Pual, A.; Das, S.; Das, J.; Samadder, A.; Bishayee, K.; Sadhukhan, R.; Bukhuda-bukhsh, A.R. Diarylheptanoid-myricanone isolated from ethanolic extract of Myrica cerifera showing anticancer effects on HeLa and PC3 cell lines: Signaling pathway and drug-DNA interaction. J. Integrative Med. 2013, 11, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Ting, Y.-C.; Ko, H.-H.; Wang, H.-C.; Peng, C.-F.; Chang, H.-S.; Hsieh, P.-C.; Chen, I.-S. Biological evaluation of secondary metabolites from the roots of Myrica adenophora. Phytochemistry 2014, 103, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ishida, J.; Kozuka, M.; Tokuda, H.; Nishino, H.; Nagumo, S.; Lee, K.-H.; Nagai, M. Chemopreventive potential of cyclic diarylheptanoids. Bioorg. Med. Chem. 2002, 10, 3361–3365. [Google Scholar] [CrossRef]
- Ibrahim, S.R.M.; Mohamed, G.A.; Khedr, A.I.M.; Aljaeid, B.M. Alnuheptanoid B: A new cyclic diarylheptanoid from Alnus japonica stem bark. Rec. Nat. Prod. 2016, 10, 362–368. [Google Scholar]
- Wang, J.; Dong, S.; Wang, Y.; Lu, Q.; Zhong, H.; Du, G.; Zhang, L.; Cheng, Y. Cyclic diarylheptanoids from Myrica nana inhibiting nitric oxide release. Bioorg. Med. Chem. 2008, 16, 8510–8513. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-F.; Lu, Q.; Guo, L.; Mei, R.-Q.; Liang, H.-X.; Luo, D.-Q.; Cheng, Y.-X. Myricananone and myricananadiol: Two new cyclic diarylheptanoids from the roots of Myrica nana. Helv. Chim. Acta 2007, 90, 1691–1696. [Google Scholar] [CrossRef]
- Takeda, Y.; Fujita, T.; Shingu, T.; Ogimi, C. Studies on the bacterial gall of Myrica rubra: Isolation of a new [7.0]-metacyclophan from the gall and dl-β-phenylacetic acid from the culture of gall-forming bacteria. Chem. Pharm. Bull. 1987, 35, 2569–2573. [Google Scholar] [CrossRef]
- Wang, J.-F.; Zhang, C.-L.; Lu, Q.; Yu, Y.-F.; Zhong, H.-M.; Long, C.-L.; Cheng, Y.-X. Three new diarylheptanoids from Myrica nana. Helv. Chim. Acta 2009, 92, 1594–1599. [Google Scholar] [CrossRef]
- Matsuda, H.; Morikawa, T.; Tao, J.; Ueda, K.; Yoshikawa, M. Bioactive constituents of Chinese natural medicines. VII. 1: Inhibition of degranulation in RBL-2H3 cells and absolute stereo structures of three new diarylheptanoid glycosides from the bark of Myrica rubra. Chem. Pharm. Bull. 2002, 50, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, Y.; Sakura, N.; Nagai, M.; Inoue, T. Constituents of Myrica rubra. III. 1: Structures of two glycosides of myricanol. Chem. Pharm. Bull. 1988, 36, 1419–1424. [Google Scholar] [CrossRef]
- Akazawa, H.; Fujita, Y.; Banno, N.; Watanabe, K.; Kimura, Y.; Manosrol, A.; Manosrol, J.; Akihisa, T. Three new cyclic diarylheptanoids and other phenolic compounds from the bark of Myrica rubra and their melanogenesis inhibitory and radical scavenging activities. J. Oleo. Sci. 2010, 59, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Yamakami, S.; Amakura, Y.; Yoshida, T. Diarylheptanoid sulfates and related compounds from Myrica rubra. J. Nat. Prod. 2012, 75, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Sakura, N.; Yaguchi, Y.; Hirakawa, T.; Nagai, M.; Inoue, T. Two myricanol glycosides from Myrica rubra and revision of the structure of isomyricanone. Phytochemistry 1991, 30, 3077–3079. [Google Scholar] [CrossRef]
- Liu, J.X.; Di, D.L.; Huang, X.Y.; Li, C. Two new diarylheptanoids from the pericarps of Juglans regia L. Chin. Chem. Lett. 2007, 18, 943–946. [Google Scholar] [CrossRef]
- Nagai, M.; Dohi, J.; Morihara, M.; Sakurai, N. Diarylheptanoids from Myrica gale var. tomentosa and revised structure of porson. Chem. Pharm. Bull. 1995, 43, 1674–1677. [Google Scholar] [CrossRef]
- Krishnaswamy, S.; Pattabhi, V.; Gabe, E.J. Garuganin-II, an antibiotic. Acta Cryst. 1987, 43, 527–530. [Google Scholar] [CrossRef]
- Navee, C.; Vijay-Kumar, S. Crystal structure of garuganin V, a tricyclononadecane from Garuga pinnata, C21H22O4. Zeit. Krist. 1995, 210, 365–366. [Google Scholar]
- Ara, K.; Kaisar, M.A.; Rahman, M.S.; Chowdhury, S.R.; Islam, F.; Rashid, M.A. Antimicrobial constituents from Garuga pinnata Roxb. Latin Am. J. Pharm. 2012, 31, 1071–1073. [Google Scholar]
- Wang, D.-Y.; Liu, G.-G. A new diarylheptanoid from the bark of Myrica nana. Nat. Prod. Res. 2008, 22, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Chen, L.; Liu, L. A new diarylheptanoid from the bark of Myrica nana. J. Chem. Res. 2009, 337–338. [Google Scholar] [CrossRef]
- Moss, G.P. Basic terminology of stereochemistry. Pure Appl. Chem. 1996, 68, 2193–2222. [Google Scholar] [CrossRef]
- Nagai, M.; Kubo, M.; Fujita, M.; Inoue, T.; Matsuo, M. Acerogenin A, a novel new cyclic diarylheptanoid. J. Chem. Soc. Chem. Comm. 1976, 338–339. [Google Scholar] [CrossRef]
- Nagai, M.; Kubo, M.; Fujita, M.; Inoue, T.; Matsuo, M. Studies on the constituents of Aceraceae plants. II. Structure of acerosides I, a glycoside of novel cyclic diarylheptanoid from Acer nickoense Maxim. Chem. Pharm. Bull. 1978, 26, 2805–2810. [Google Scholar] [CrossRef]
- Kubo, M.; Inoue, T.; Nagai, M. Studies on the constituents Aceraceae plants. III. Structure of acerogenin B from Acer nikoense Maxim. Chem. Pharm. Bull. 1980, 28, 1300–1302. [Google Scholar] [CrossRef]
- Pretsch, E.; Clerc, T.; Seibl, J.; Simon, W. Tables of Spectrral Data for Structure Determination of Organic Compounds, 2nd ed.; Springer: Berlin, Germany, 1989. [Google Scholar]
- Farnum, D.G.; Wilcox, C.F. Use of a model for the ring-current effect in analysis of the nuclear magnetic resonance spectra of di- and triphenylcyclopropenium ions. J. Am. Chem. Soc. 1967, 89, 5379–5383. [Google Scholar] [CrossRef]
- Malterud, K.E.; Anthonsen, T.; Hjorta, J. 14-Oxa-[7.1]-metapara-cyclophane from Myrica gale L. A new class of natural products. Tetrahedron Lett. 1976, 17, 3069–3072. [Google Scholar] [CrossRef]
- Morita, H.; Deguchi, J.; Motegi, Y.; Sato, S.; Aoyama, C.; Takeo, J.; Shiro, M.; Hirasawa, Y. Cyclic diarylheptanoids as Na+-glucose cotransporter (SLGT) inhibitors from Acer nikoense. Bioorg. Med. Chem. Lett. 2010, 20, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Nagai, M.; Inoue, T. Studies on the constituents Aceraceae plants. IV. Carbon-13 nuclear magnetic resonance spectra of acerogenin A, rhododendrol and related compounds, and structure of aceroside IV from Acer nikoense Maxim. Chem. Pharm. Bull. 1983, 31, 1917–1922. [Google Scholar] [CrossRef]
- Akihisa, T.; Taguchi, Y.; Yasukawa, K.; Tokuda, H.; Akazawa, H.; Suzuki, T.; Kimura, Y. Acerogenin M, a cyclic diarylheptanoid and other phenolic compounds from Acer nikoense and their anti-inflammatory and anti-tumor promoting effects. Chem. Pharm. Bull. 2006, 54, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, T.; Lee, J.-W.; Akazawa, H.; Inagaki, M.; Cha, B.-Y.; Nagai, K.; Yagasaki, K.; Akihisa, T.; Woo, J.-T. Osteogenic activity of diphenyl ether cyclic diarylheptanoids derived from Acer nikoense. Bioorg. Med. Chem. Lett. 2011, 21, 3248–3251. [Google Scholar] [CrossRef] [PubMed]
- Akihisa, T.; Takeda, A.; Akazawa, H.; Kikuchi, T.; Yokokawa, S.; Ukiya, M.; Fukatsu, M.; Watanabe, K. Melanogenesis-inhibitory and cytotoxic activities of diaryheptanoids from Acer nikoense bark and their derivatives. Chem. Biodivers. 2012, 9, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-T.; Lee, D.-S.; Chowdhury, A.; Lee, H.; Cha, B.-Y.; Woo, J.-T.; Woo, E.R.; Jang, J.-H. Acergenin C from Acer nikoense exhibits a neuroprotective effect in mouse hippocampal HT22 cell lines through the upregulation of Nrf-2/HO-1 signaling pathways. Mol. Med. Rep. 2017, 16, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, J.; Motegi, Y.; Nakata, A.; Hosoya, T.; Morita, H. Cyclic diarylheptanoids as inhibitors of NO production. J. Nat. Med. 2013, 67, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Morihara, M.; Sakurai, N.; Inoue, T.; Kawai, K.-I.; Nagai, M. Two novel diarylheptanoid glucosides Myrica gale var. tomentosa and absolute structure of plane-chiral galeon. Chem. Pharm. Bull. 1997, 45, 820–823. [Google Scholar]
- Bryant, V.C.; Kishore Kumar, G.D.; Nyong, A.M.; Natarajan, A. Synthesis and evaluation of macrocyclic diarylether heptanoid natural products and their analogs. Bioorg. Med. Chem. Lett. 2012, 22, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Ishidate, Y.; Fuchita, M. Studies on the constituents of Aceraceae plants. I. Constituents in leaves and stem bark of Acer nikoense Maxim. Yakugaku Zasshi 1978, 98, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Kubo, M.; Takahashi, K. Studies on the constituents of Aceraceae plants. V. Two diaylheptanoid glycosides and an arylbutanol apiosylglucosiude from Acer nikoense. Chem. Pharm. Bull. 1983, 31, 1923–1928. [Google Scholar] [CrossRef]
- Liang, J.; Peng, X.; Zhou, J.; Zhou, M.; Ruan, H. Diarylheptanoids from the fresh pericarps of Juglans sigillata. Nat. Prod. Res. 2018, 32, 2457–2463. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Matshda, E.; Inoue, T.; Fujita, M.; Chi, H.J.; Ando, T. Studies on the constituents of Aceraceae plants. VII. Diarylheptanoids from Acer griseum and Acer triflorum. Chem. Pharm. Bull. 1990, 38, 1506–1508. [Google Scholar] [CrossRef]
- Nagai, M.; Kenmochi, N.; Fujita, M.; Furukawa, N.; Inoue, T. Studies on the constituents of Aceraceae plants. VI. Revised stereochemistry of (−)-centrolobol, and new glycosides from Acer nikoense. Chem. Pharm. Bull. 1986, 34, 1056–1060. [Google Scholar] [CrossRef]
- Pattawong, O.; Salih, M.Q.; Rosson, N.T.; Beaudry, C.M.; Cheong, P.H.-Y. The nature of persistent conformational chirality, racemization mechanisms, and predictions in diarylether heptanoid cyclophane natural products. Org. Biomol. Chem. 2014, 12, 3303–3309. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-Y.; Peng, C.-F.; Tsai, I.-L.; Chen, J.-J.; Cheng, M.-J.; Chen, I.-S. Antitubercular constituents from the roots of Engelhardia roxburghiana. Planta Med. 2005, 71, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lee, S.-Y.; Lee, K.-S.; Lee, S.-W.; Kim, S.-H.; Lee, S.-H.; Lee, C.-S.; Woo, M.-H.; Son, J.-K. DNA topoisomerase I and II inhibitory activity of constituents isolated from Juglans mandshuria. Arch. Pharm. Res. 2003, 26, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Kalchhauser, H.; Krishnamurty, H.G.; Talukdar, A.C.; Schmid, W. Isolation and structure determination of two new macrocyclic biaryl ethers from Garuga gamblei. Monatsh. Chem. 1988, 119, 1047–1051. [Google Scholar] [CrossRef]
- Nethaji, M.; Pattabhi, V.; Krishnamurthy, H.G.; Talukdar, A.C. Structures of garugamblin-I and garubamblin-II: Two natural ansa bridged biaryl ethers. Acta Cryst. C Cryst. Struct. Comm. 1990, 46, 307–310. [Google Scholar] [CrossRef]
- Kang, H.-M.; Son, K.-H.; Yang, D.C.; Han, D.C.; Kim, J.H.; Baek, N.-I.; Kwon, B.-M. Inhibitory activity of diarylheptanoids on farnesyl protein transferase. Nat. Prod. Res. 2004, 18, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Pattabhi, V.; Krishnaswami, S.; Gabe, E.J. Garuganin-I, an antibiotic, C22H24O5. Acta Cryst. 1984, 40, 832–834. [Google Scholar] [CrossRef]
- Haribal, M.M.; Mishra, A.K.; Sabata, B.K. Isolation and structure of a new macrocyclic, 15-membered biphenyl ether. Garuganin-I from Garuga pinnata. Tetrahedron 1985, 41, 4949–4951. [Google Scholar] [CrossRef]
- Mishra, A.K.; Haribal, M.M.; Sabata, B.K. Garuganin III, a macrocyclic biphenyl ether from Garuga pinnata. Phytochemistry 1985, 24, 2463–2464. [Google Scholar] [CrossRef]
- Keserü, G.M.; Dienes, Z.; Nógrádi, M.; Kajtár-Peredy, M. Synthesis and revised structure of garuganin III. J. Org. Chem. 1993, 58, 6725–6728. [Google Scholar] [CrossRef]
- Zhu, Z.-Q.; Beaudry, C.M. Structural revision of garuganin IV and 1,9′-didesmethylgaruganin III through total synthesis. J. Org. Chem. 2013, 78, 3336–3341. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-Q.; Salih, M.Q.; Fynn, E.; Bain, A.D.; Beaudry, C.M. The garuganin and garugamblin diarylether heptanoids: Total synthesis and determination of chiral properties using dynamic NMR. J. Org. Chem. 2013, 78, 2881–2896. [Google Scholar] [CrossRef] [PubMed]
- Fuchino, H.; Satoh, T.; Yokochi, M.; Tanaka, N. Chemical evaluation of Betula species in Japan. V. Constituents of Betula ovalifolia. Chem. Pharm. Bull. 1998, 46, 169–170. [Google Scholar] [CrossRef]
- Masullo, M.; Mari, A.; Cerulli, A.; Bottone, A.; Kontek, B.; Olas, B.; Pizza, C.; Piacente, S. Quali-quantitative analysis of the phenolic fraction of the flowers of Corylus avellana, source of the Italian PGI product “Nocciola di Giffoni”: Isolation of antioxidant diarylheptanoids. Phytochemistry 2016, 130, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Singldinger, B.; Dunkel, A.; Bahmann, D.; Bahmann, C.; Kadow, D.; Bisping, B.; Hofmann, T. New taste-active 3-(O-β-d-glucosyl)-2-oxoindole-3-acetic acids and diarylheptanoids in Cimiciato-infected hazelnuts. J. Agric. Food Chem. 2018, 66, 4660–4673. [Google Scholar] [CrossRef] [PubMed]
- Eliel, E.I. Stereochemistry of Carbon Compounds; McGraw-Hill: New York, NY, USA, 1962; p. 156. [Google Scholar]
- Petit, M.; Lapierre, A.J.B.; Curran, D.P. Relaying asymmetry of transition atropisomers of o-iodoanilides by radical cyclization. J. Am. Chem. Soc. 2005, 127, 14994–14995. [Google Scholar] [CrossRef] [PubMed]
- Geissman, T.A.; Crout, D.H.G. Organic Chemistry of Secondary Plant. Products; Freeman Cooper: San Francisco, CA, USA, 1969. [Google Scholar]
- Roughley, P.J.; Whiting, D.A. Diarylheptanoids: The problems in the biosynthesis. Tetrahedron Lett. 1971, 12, 3741–3746. [Google Scholar] [CrossRef]
- Inoue, T.; Kenmochi, N.; Furukawa, N.; Fujita, M. Biosynthesis of acerogenin A, a diarylheptanoid from Acer nikoense. Phytochemistry 1987, 26, 1409–1411. [Google Scholar] [CrossRef]
- Miyaura, N.; Suginome, H.; Suzuki, A. A stereospecific synthesis of conjugated (E,Z)- and (Z,Z)-alkadienes by a palladium-catalyzed cross coupling reaction of 1-alkenoboranes with 1-alkenyl halides. Tetrahedron Lett. 1981, 22, 127–130. [Google Scholar] [CrossRef]
- Sheffy, F.K.; Stille, J.K. Palladium-catalyzed coupling of allyl halides with organotins. J. Am. Chem. Soc. 1983, 105, 7173–7175. [Google Scholar] [CrossRef]
- Negishi, E.; King, A.O.; Okukado, N. Selective carbon-carbon bond formation via transition metal catalysis. 3. A highly selective synthesis of unsymmetrical biaryls and diarylmethanes by the nickel- or palladium-catalyzed reaction of aryl- and benzylzinc derivatives with aryl halides. J. Org. Chem. 1977, 42, 1821–1823. [Google Scholar] [CrossRef]
- Ullmann, F.; Sponagel, P. Über die Phenylirung von Phenolen. Ber. Dtsch. Chem. Ges. 1905, 38, 2211–2212. [Google Scholar] [CrossRef]
- Lindley, J. Copper assisted nucleophilic substitution of aryl halogen. Tetrahedron 1984, 40, 1433–1456. [Google Scholar] [CrossRef]
- Bergley, W.J.; Grimshaw, J. Synthesis of nitidine (8,9-dimethoxy-5-methyl-2,3-methylenedioxybenzo[c]phenanthridinium): A comparison of electrochemical and photochemical methods. J. Chem. Soc. Perkin Trans. 1 1977, 2324–2328. [Google Scholar] [CrossRef]
- Wittig, G.; Geissler, G. Course of reactions of pentaphenyl phosphorous and certain derivatives. Ann. Chem. 1953, 580, 44–57. [Google Scholar] [CrossRef]
- Moore, C.W.; Thorpe, J.F. The formation and reactions of imino-compounds (VI). The formation of derivatives of hydridene from o-phenylenediacetonitrile. J. Chem. Soc. Trans. 1908, 93, 165–187. [Google Scholar] [CrossRef]
- Grubbs, R.H.; Brunck, T.K. Possible intermediate in the tungsten-catalyzed olefin metathesis reaction. J. Am. Chem. Soc. 1972, 94, 2538–2540. [Google Scholar] [CrossRef]
- Dansou, B.; Pichon, C.; Dhal, R.; Brown, E.; Mille, S. Isolation of macrocyclic metacyclophanes from the attempted synthesis of [7.0]metacyclophanes of the myricanone series by Thorpe-Ziegler-intramolecular cyclization of diarys substituted by ω-cyanoalkyl chains. Eur. J. Org. Chem. 2000, 1527–1533. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Ryono, L. Nickel-promoted synthesis of cyclic biphenyls. Total synthesis of alnusone dimethyl ether. J. Am. Chem. Soc. 1975, 97, 3873–3875. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Helquist, P.; Jones, L.D.; Keller, L.; Mendelson, L.; Ryono, L.S.; Smith, J.G.; Stauffer, R.D. Reaction of aryl and vinyl halides with zerovalent nickel—Preparative aspects and the synthesis of alnusone. J. Am. Chem. Soc. 1981, 103, 6460–6471. [Google Scholar] [CrossRef]
- Whiting, D.; Wood, A. Total synthesis of the meta,meta-bridged biphenyls (±)-myricanol and myricanone, and of an isomeric biphenyl ether, a 14-oxa[7.1]metaparacyclophane. J. Chem. Soc. Perkin Trans. 1 1980, 623–628. [Google Scholar] [CrossRef]
- Henley-Smith, P.; Whiting, D.A.; Wood, A.F. Methods for the construction of linear 1,7-diarylheptanoids: Synthesis of di-O-methylcentrolobol and precursors (synthetic and biosynthetic) to the meta,meta-bridged biphenyls myricanol and myricanone. J. Chem. Soc. Perkin 1 1980, 614–622. [Google Scholar] [CrossRef]
- Ogura, T.; Usuki, T. Total synthesis of acerogenin E, G, and K, and centrolobol. Tetrahedron 2013, 69, 2807–2815. [Google Scholar] [CrossRef]
- Carbonnelle, A.-C.; Zhu, J. A novel synthesis of biaryl-containing macrocycles by a domino Miyaura arylboronate formation: Intramolecular Suzuki reaction. Org. Lett. 2000, 2, 3477–3480. [Google Scholar] [CrossRef] [PubMed]
- Darzi, E.R.; White, B.M.; Loventhal, L.K.; Zakharov, L.V.; Jasti, R. An operationally simple and mild oxidative homocoupling of aryl boronic esters to access conformationally constrained macrocycles. J. Am. Chem. Soc. 2017, 139, 3106–3114. [Google Scholar] [CrossRef] [PubMed]
- Riley, A.P.; Prisinzano, T.E. Studies towards the Total Synthesis of (+)-aR,11S-Myricanol. In Proceedings of the 244th ACS National Meeting & Exhibition, Philadelphia, PA, USA, 19–23 August 2012. [Google Scholar]
- Weyer, M.J.; Nye, J.; Ostlund, A.J.; Cook, G.R. Total Synthesis of the Diarylheptanoid (+)-aR,11S-Myricanol. In Proceedings of the 245th ACS National meeting & Exhibition, New Orleans, LA, USA, 7–11 April 2013. [Google Scholar]
- Rybak, T.; Hall, D. Stereoselective and regiodivergent allylic Suzuki-Miyaura cross coupling of 2-ethoxydihydroxypyranyl boronates: Synthesis and confirmation of absolute stereochemistry of diospongin B. Org. Lett. 2015, 17, 4156–4159. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Nishiyama, S. Biomimetic synthesis of isodityrosine natural products, and an approach to chemistry and molecular recognition of secoaglucovancomycin and related oligopeptides. J. Syn. Org. Chem. Jpn. 1997, 55, 1029–1039. [Google Scholar] [CrossRef]
- Zhu, J. SNAr bases macrocyclization via biaryl ether formation: Application in natural product synthesis. Synlett 1997, 2, 133–144. [Google Scholar] [CrossRef]
- Cava, M.P.; Afzali, A. Practical route to bisbenzylisoquinolines by an improved Ullmann diphenyl ether synthesis. J. Org. Chem. 1975, 40, 1553–1556. [Google Scholar] [CrossRef]
- Sambiagio, C.; Marsden, S.P.; Blacker, A.J.; McGowan, P.C. Copper catalyzed Ullmann type chemistry: From mechanistic aspects to modern development. Chem. Soc. Rev. 2014, 43, 3525–3550. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.C. Recent advances in diary ether synthesis. Tetrahedron 2000, 56, 5045–5065. [Google Scholar] [CrossRef]
- Pitsinos, E.N.; Vivaldi, V.P.; Couladouros, E.A. Diaryl ether formation in the synthesis of natural products. Eur. J. Org. Chem. 2011, 1207–1222. [Google Scholar] [CrossRef]
- Salih, M.Q.; Beaudry, C.M. Bio-inspired oxidative phenolic coupling: Total synthesis of the diarylether heptanoids (±)-pterocarine. Tetrahedron Lett. 2017, 58, 2023–2025. [Google Scholar] [CrossRef]
- Omura, K. Rapid conversion of phenols to p-benzoquinones under acidic conditions with lead oxide. Synthesis 1998, 145–1148. [Google Scholar]
- Gonzalez, G.I.; Zhu, J. First total synthesis of acerogenin C and aceroside IV. J. Org. Chem. 1997, 62, 7544–7545. [Google Scholar] [CrossRef]
- Meisenheimer, J. Über Reactionen aromatischer Nitrokörper. Ann. Chem. 1902, 323, 205–246. [Google Scholar] [CrossRef]
- Doyle, M.P.; Dellaria, J.F.; Siegfried Jr., B.; Bishop, S.W. Reductive deamination of arylamines by alkyl nitriles in N,N-dimethylformamide. A direct conversion of arylamines to aromatic hydrovarbons. J. Org. Chem. 1977, 42, 3494–3498. [Google Scholar] [CrossRef]
- Gonzalez, G.I.; Zhu, J. A unified strategy toward the synthesis of acerogenin-type macrocycles: Total syntheses of acerogenins A, B, L, and aceroside IV. J. Org. Chem. 1999, 64, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Keserü, G.M.; Nógrádi, M.; Szöllösy, Á. Synthesis of acerogenin C and (+)-acerogenin A, two macrocyclic diarylheptanoid constituents of Acer nikoense. Eur. J. Org. Chem. 1998, 521–524. [Google Scholar] [CrossRef]
- Jeong, B.-S.; Wang, Q.; Son, J.-K.; Jahng, Y. A versatile synthesis of cyclic diphenyl ether-type diarylheptanoids: Acerogenins, (±)-galeon, and (±)-pterocarine. Eur. J. Org. Chem. 2007, 1338–1344. [Google Scholar] [CrossRef]
- Wang, Q.; Son, J.-K.; Jahng, Y. First total synthesis of cytotoxic diarylheptanoids, galeon and pterocarine. Synth. Comm. 2007, 37, 675–681. [Google Scholar] [CrossRef]
- Shen, L.; Sun, D. Total synthesis and structural revision of engelhardione. Tetrahedron Lett. 2011, 52, 4570–4574. [Google Scholar] [CrossRef] [PubMed]
- Salih, M.Q.; Beaudry, C.M. Chirality in diarylether heptanoids; Synthesis of myricatomentogenin, jugcathanin, and congers. Org. Lett. 2012, 14, 4026–4029. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Tuckemantel, W. The total synthesis of 2,3,3a,4,5,7a-hexahydro-1H-inden-1-ols by intramolecular Diels-Alder reactions of 1,3,8-nonatriene-5-ols. Dependence of product stereochemistry on the substitution pattern. J. Org. Chem. 1991, 56, 2826–2837. [Google Scholar] [CrossRef]
- Vermes, B.; Keserü, G.M.; Mezey-Vándor, G.; Nógrádi, M.; Tóth, G. The total synthesis of garugamblin 1. Tetrahedron 1993, 49, 4893–4900. [Google Scholar] [CrossRef]
- Keserü, G.M.; Nógrádi, M.; Kajtár-Peredy, M. Synthesis garugamblin-2, a macrocyclic diaryheptanoid constituents of Garuga gamblei. Liebigs Ann. Chem. 1994, 361–364. [Google Scholar] [CrossRef]
- Kumar, G.D.K.; Natarajan, A. Total synthesis of ovalifoliolatin B, acerogenins A and C. Tetrahedron Lett. 2008, 49, 2103–2105. [Google Scholar] [CrossRef]
- Salih, M.Q.; Beaudry, C.M. Enantioselective Ullmann ether couplings: Syntheses of (−)-myricantomentogenin, (−)-jugcathanin, (+)-galeon, and pterocarine. Org. Lett. 2013, 15, 4540–4543. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Wang, Q.; He, H.; Cai, Q. Asymmetric synthesis of (−)-pterocarine and (−)-galeon via chiral phase transfer-catalyzed atropselective formation of diarylether cyclophane skeleton. Org. Lett. 2017, 19, 1804–1807. [Google Scholar] [CrossRef] [PubMed]
- Ishida, J.; Ohtsu, A.; Tachibana, Y.; Nakanishi, Y.; Bastow, K.F.; Nagai, M.; Wang, H.-K.; Itokawa, H.; Lee, K.-H. Antitumor agents. Part 214: Synthesis and evaluation of curcumin analogues as cytotoxic agents. Bioorg. Med. Chem. 2002, 10, 3481–3487. [Google Scholar] [CrossRef]
- Shen, L.; Maddox, M.M.; Adhikari, S.; Bruhn, D.F.; Kumar, M.; Lee, R.E.; Hurdle, J.G.; Lee, R.E.; Sun, D. Syntheses and evaluation of macrocyclic engelhardione analogs as antitubercular and antibacterial agents. J. Antiobiot. 2013, 66, 319–325. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 25b, 26a, 90, 96, 97, 98, and 99 are available from the authors. |



{kind=link}
{kind=link}
| Family | Genus | Diarylheptanoids | Reference(s) | ||
|---|---|---|---|---|---|
| Linear (Type I) | Biphenyl (Type II) | Diphenyl Ether (Type III) | |||
| Aceraceae | Acer | + | + | + | [7] |
| Actinidiaceae | Clematoclethra | − | − | + | [8] |
| Betulaceae | Alnus | + | + | + | [9] |
| Betula | + | + | + | [10] | |
| Corylus | − | + | − | [11,12] | |
| Carpinus | − | + | − | [13] | |
| Ostrya | − | + | − | [14] | |
| Ostryopsis | + | + | + | [15,16] | |
| Burseraceae | Garuga | − | + | + | [17,18] |
| Boswellia | − | − | + | [19] | |
| Casuarinaceae | Casuarina | − | + | − | [20] |
| Juglandaceae | Engelhardia | + | − | + | [21] |
| Juglans | + | + | + | [22,23] | |
| Platycarya | − | + | + | [24] | |
| Pterocarya | − | − | + | [25] | |
| Rhoiptelea | + | + | + | [26] | |
| Myricaceae | Myrica | − | + | + | [27,28] |
| Morella | + | + | + | [29] | |
| Rubiaceae | Scyphiphora | − | + | − | [30] |
| Zingiberaceae | Alpinia | + | − | − | [31] |
| Curcuma | + | − | − | [4] | |

| R1 | R2 | R3 | X1 | X2 | X3 | X4 | X5 | Chiral Axis | Reference and Biological Activity | |
|---|---|---|---|---|---|---|---|---|---|---|
| alnusonol (5a) | H | H | H | H,H | - | H,H | H,OH (S) | H | α-glucosidase inhibitor [65] | |
| alnusdiol (7a) | H | H | H | H,H | OH (S) | H,H | H,OH (S) | H | aS | [51] |
| 5c | H | H | H | H,H | - | H,H | H,OH | OH | [53] | |
| 5d | H | H | H | H,H | - | H,OH | O | H | anti-adipogenic activity [55] | |
| giffonin L a (7d) | H | H | H | H,OH (S) | OH (S) | H,OH (R) | H,H | OH (S) | [12] | |
| giffonin M (5e) | H | H | H | H,H | - | H,OH (S) | H,H | OH (S) | [12] | |
| giffonin N (5f) | H | H | H | H,H | - | H,OH (S) | H,H | O-β-d-Glc (S) | [12] | |
| giffonin O (7e) | H | H | H | H,OH (R) | H | O | H,OH (R) | OH (S) | [12] | |
| giffonin P a (7f) | H | H | H | H,OH (R) | OH (S) | H,OH | H,OH (S) | OH (S) | [12] | |
| giffonin T (5g) | H | R′ b | H | H,H | - | H,OH (R) | H,OH (S) | OH (R) | aS | [64] |
| giffonin U (7g) | H | H | H | O | OH (R) | H,OH (R) | H,OH (R) | OH (R) | aS | [64] |
| carpinontriol A (5h) | H | H | H | H,OH (R) | - | H,H | H,OH (S) | OH (R) | [13] | |
| carpinontriol B (5i) | H | H | H | H,H | - | H,OH (R) | H,OH (S) | OH (R) | aS | [13], inhibit lipid peroxidation [64] |
| 5j | OH | CH3 | H | H,H | - | H,H | H,H | H | [56], leishmanicidal (IC50 = 17 mg/mL) [63] | |
| aceroside XI c (5k) | H | H | R′ b | H,H | - | H,H | H,H | H | [66] | |
| acerogenin E d (5l) | H | H | H | H,H | - | H,H | H,H | H | NO production inhibitor (IC50 = 24 μM) [67] | |
| acerogenin K (7h) | H | H | H | H,H | OH | H,H | H,H | H | NO production inhibitor (IC50 = 24 μM) [67] | |
| ostryopsitriol (7i) | H | H | H | H,OH | OH | H,H | H,OH | H | [16] | |
| betulatetraol (7j) | H | H | H | H,OH | OH | H,H | H,OH | OH | [43] |

| Compound | R1 | R2 | R3 | R4 | X1 | X2 | X3 | Reference(s) and Biological Properties |
|---|---|---|---|---|---|---|---|---|
| 11a (myricanone) | OH | CH3 | H | H | H | H | H | cytotoxicity [76,77], cell apoptosis [78], anti-inflammatory in iNOS assay (IC50 =1.0 μM) [79], radical scavenging (IC50 = 19.6 μM) [79], lowering protein tau level [71], chemo-preventive a (IC50 = 320) [80] |
| 11b | O-β-Glc | CH3 | H | H | H | H | H | DPPH radical scavenging activity (49.09%) [81] |
| 11c (alnuheptanoid B) | O-β-Glc | CH3 | COCH3 | H | H | H | H | DPPH radical scavenging activity (41.16%) [81] |
| 11d (saliciclaireone A) |  | CH3 | H | H | H | H | H | [29] |
| 11e (saliciclaireone C) | OCH3 | H |  | H | H | H | H | [29] |
| 11f |  | CH3 | H | H | H | H | H | [29] |
| myricananin C (11g) | H | H | H | H | H | H | H | NO release inhibition (IC50 = 64.51 μM) [82] |
| myricananin E (11h) | OH | CH3 | H | OH | H | H | OCH3 | [82] |
| myricananone (11i) | OH | CH3 | H | H | OH | H | H | [83] |
| 11j | H | CH3 | H | H | H | H | H | [84], anti-tubercular activity (IC50 = 25.8 μg/mL) [79] |
| 11k | OH | CH3 | H | H | H | H | OH | anti-tubercular activity (IC50 = 35.8 μg/mL) [79] |


| Compound | R1 | R2 | R3 | R4 | X1 | X2 | X3 | X4 | Reference(s) and Biological Properties |
|---|---|---|---|---|---|---|---|---|---|
| myricanol (12a) | OH | CH3 | H | H | H | H | OH (S) | H | [68], radical scavenging (IC50 = 22.3 μM) [79], anti-Alzheimer’s disease by lowering tau level [71] |
| juglanin B (12ba) | H | H | H | H | H | H | OH (S) | H | cytotoxic (HT-29) [40] |
| salicireneol A (12c) | H | β-Glc | H | H | H | H | OH (S) | H | [29] |
| salicireneol B (12d) | H | H | β-Glc | OH | H | H | OH (S) | H | [29] |
| myricananin A (12e) | H | H | H | H | H | H | OH (S) | OH (S) | NO release inhibitor (IC50 = 45.32 μM) [82] |
| myricananin B (12f) | OH | CH3 | H | H | H | H | OH (S) | OH (S) | [82] |
| myricananin F (12g) | H | H | H | H | H | OH | H | H | [85] |
| myricananin G (12h) | OH | CH3 | CH3 | H | H | H | OH (S) | OH (R) | [85] |
| myricananin H (12i) | OCH3 | CH3 | CH3 | H | H | H | OH (S) | OH (S) | [85] |
| salicimeckol (12j) | O-β-Glc | CH3 | H | H | OH | H | OH (S) | H | [29] |
| 12k | O-β-Glc | CH3 | H | H | H | H | OH (S) | H | anti-allergic activity [86] |
| 12l |  | CH3 | H | H | H | H | OH (S) | H | [87], DPPH radical scavenging (IC50 = 6.8 μM) [88], SOD-like activity (IC50 = 90.5 μg/mL) [89] |
| 12m |  | CH3 | H | H | H | H | OH (S) | [90] | |
| 12n |  | CH3 | H | H | H | H | OH (S) | [87] |


| Compound | R1 | R2 | R3 | X1 | X2 | X3 | X4 | Reference and Biological Properties |
|---|---|---|---|---|---|---|---|---|
| acerogenin A (23b) | H | H | H | H,H | H,H | H,OH (R) | H,H | anti-inflammatory (IC50 = 0.32 mg/ear) [107], osteogenic activity [108] |
| acerogenin B (24a) | H | H | H | H,H | H,OH | H,H | H,H | cytotoxicity (IC50 = 25.1 μM) of (R)-isomer against HL60 [109], Na+-glucose cotransporter inhibitor [105] |
| acerogenin C (25b) | H | H | H | H,H | H,H | O | H,H | antibacterial and neuroprotective [110], NO production inhibitor (IC50 = 61.4 μM) [111] |
| acerogenin D (25c) | H | H | H | H,H | H,OH | O | H,H | radical scavenging activity (IC50 = 40 μM) [62] |
| acerogenin F (23e) a | H | H | H | H,H | H,H | H,OH (R) | H,OH (R) | [60] |
| acerogenin H (23f) | H | H | H | O | H,H | H,OH | H,H | [64] |
| acerogenin I (24b) | H | H | H | H,H | H,OH | H,H | H,OH | [64] |
| acerogenin J (23g) | H | H | H | H,H | H,H | H,OH (R) | H,OH (S) | [64] |
| acerogenin L (26a) | H | H | H | H,H | O | H,H | H,H | [64] |
| acerogenin M (24c) | H | H | H | O | H,OH | H,H | H,H | antitumor promoting activity [107] |
| pterocarine (26b) | H | OH | H | H,H | O | H,H | H,H | cytotoxic against K562 (51% @100 μg/mL) [25] |
| galeon (26c) | H | OCH3 | H | H,H | O | H,H | H,H | [104] |
| 10-hydroxygaleon (26d) | H | OCH3 | H | H,OH | O | H,H | H,H | [104] |
| 24d | H | OCH3 | H | H,H | H,OH (R) | H,H | H,H | [23] |
| myricatomentogenin (26e) | H | OCH3 | OH | H,H | O | H,H | H,H | [112] |
| jugcathanin (juglanin A, 26f) | CH3 | OCH3 | OH | H,H | O | H,H | H,H | cytotoxicity [40,91] |
| platycarynol (24e) | CH3 | OCH3 | H | H,H | H,OH (R) | H,H | H,H | [24], cytotoxic against A549 (IC50 = 11.5 μg/mL) [22], NF-kB inhibitor [113] |
| aceroside B1 (24fa) | β-d-Glc | H | H | H,H | H,OH (R) | H,H | H,H | [61], osteogenic activity [108] |
| aceroside B2 (24fb) | β-d-Glc | H | H | H,H | H,OH (S) | H,H | H,H | [61] |
| aceroside I (23h) | β-d-Glc | H | H | H,H | H,H | H,OH (R) | H,H | [114], osteogenic activity [108] |
| aceroside II (24g) | H | H | H | H,H | H, O-β-d-Glc | H,H | H,H | [60] |
| aceroside III (23i) | H | H | H | H,H | H,H | H,Y c | H,H | radical scavenging activity (IC50 = 40 μM), osteogenic activity [108] |
| aceroside IV (25a) | β-d-Glc | H | H | H,H | H,H | O | H,H | [106] |
| aceroside V (25d) | β-d-Glc | H | H | H,H | H,OH | O | H,H | [62] |
| aceroside VI b (23j) | H | H | H | H,H | H,H | H,O-β-d-Glc | H,H | [115] |
| 9-oxoacerogenin A (23k) | H | H | H | H,H | O | H,OH | H,H | anti-melanogenesis (17.6% @100 μM) [109] |
| maximowicziol A (24h) | H | H | H | H,H | H,OH (S) | H,H | H,OH (S) | [52] |
| 24i | CH3 | OCH3 | OH | H,H | H,OH | H,H | H,H | cytotoxic against A549 (IC50 = 11.5 μg/mL) [22] |
| jugsigin A (24j) | H | H | OH | H,H | H,OH (S) | H,H | H,H | cytotoxic against HT-29 [116] |
| 2-methylacerogenin A (23l) | CH3 | H | H | H,H | H,H | H,OH (S) | H,H | [100] |

| R1 | R2 | R3 | X1 | X2 | X3 | Reference | |
|---|---|---|---|---|---|---|---|
| giffonin A (36a) | OCH3 | OH | OCH3 | - | - | H | [11] |
| giffonin B (36b) | OCH3 | OH | OCH3 | - | - | OH (S) | [11] |
| giffonin C (37a) | OCH3 | OH | OCH3 | H | H,OH (R) | H | [11] |
| giffonin D (37b) | OCH3 | OH | OCH3 | H | O | H | [11] |
| giffonin E (37c) | OCH3 | OH | OCH3 | OH (R) | H,H | H | [11] |
| giffonin F (37d) | OCH3 | OH | OCH3 | OH (R) | O | H | [11] |
| giffonin G (38a) | OCH3 | OH | OCH3 | - | H,OH (S) | H | [11] |
| giffonin H (38b) | H | OH | OCH3 | - | H,OH (S) | H | [11] |
| giffonin J a (37e) | OCH3 | OH | OCH3 | H | H,OH (R) | OH (S) | [12] |
| giffonin K (38c) | OCH3 | OH | OCH3 | - | H,OH (S) | OH (S) | [12] |
| giffonin Q (39) | H | H | H | - | O | H | [132] |
| giffonin R (38d) | H | H | OH | - | O | H | [132] |
| giffonin S (38e) | H | OCH3 | OH | - | O | H | [132] |
| Ligand | Base | Yield (%) a | Ratio (pR : pS) b |
|---|---|---|---|
 | Cs2CO3 | 18 | 38: 62 |
 | Cs2CO3 | 22 | 44: 56 |
 | Cs2CO3 | 41 | 68: 32 |
 | Cs2CO3 | 34 | 57: 43 |
 | K3PO4 | 39 | 72: 28 |

| Solvent | Base (aq. Solution) | Yield (%) b) | Er c) |
|---|---|---|---|
| toluene (5 mL) | 20% CsOH | 47 | 87: 13 |
| DMF (5 mL) | 20% CsOH | 90 | 50: 50 |
| xylene (5 mL) | 20% CsOH | 47 | 82: 18 |
| toluene (5 mL) | 20% CsF | <10 | 69: 31 |
| toluene (5 mL) | 20% KOH | 17 | 64: 36 |
| toluene (5 mL) | 20% CsOH | 46 | 89: 11 |
| toluene (5 mL) d) | 20% CsOH | 80 | 91: 9 |
| toluene (5 mL) d) | 20% CsOH | 47 | 91.5: 8.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahng, Y.; Park, J.G. Recent Studies on Cyclic 1,7-Diarylheptanoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis. Molecules 2018, 23, 3107. https://doi.org/10.3390/molecules23123107
Jahng Y, Park JG. Recent Studies on Cyclic 1,7-Diarylheptanoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis. Molecules. 2018; 23(12):3107. https://doi.org/10.3390/molecules23123107
Chicago/Turabian StyleJahng, Yurngdong, and Jae Gyu Park. 2018. "Recent Studies on Cyclic 1,7-Diarylheptanoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis" Molecules 23, no. 12: 3107. https://doi.org/10.3390/molecules23123107
APA StyleJahng, Y., & Park, J. G. (2018). Recent Studies on Cyclic 1,7-Diarylheptanoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis. Molecules, 23(12), 3107. https://doi.org/10.3390/molecules23123107