
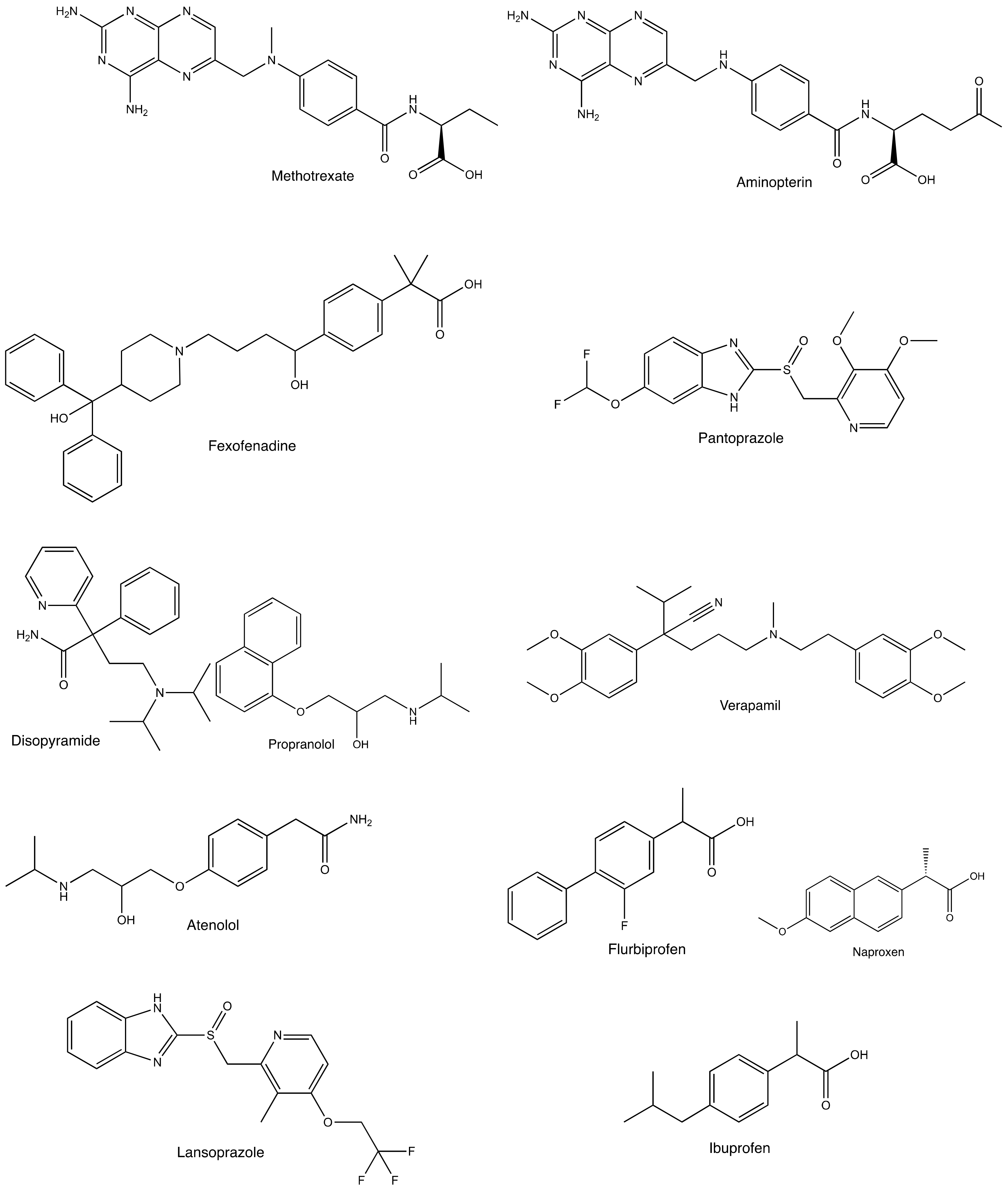
Enantioselective Drug Recognition by Drug Transporters

Abstract
1. Introduction
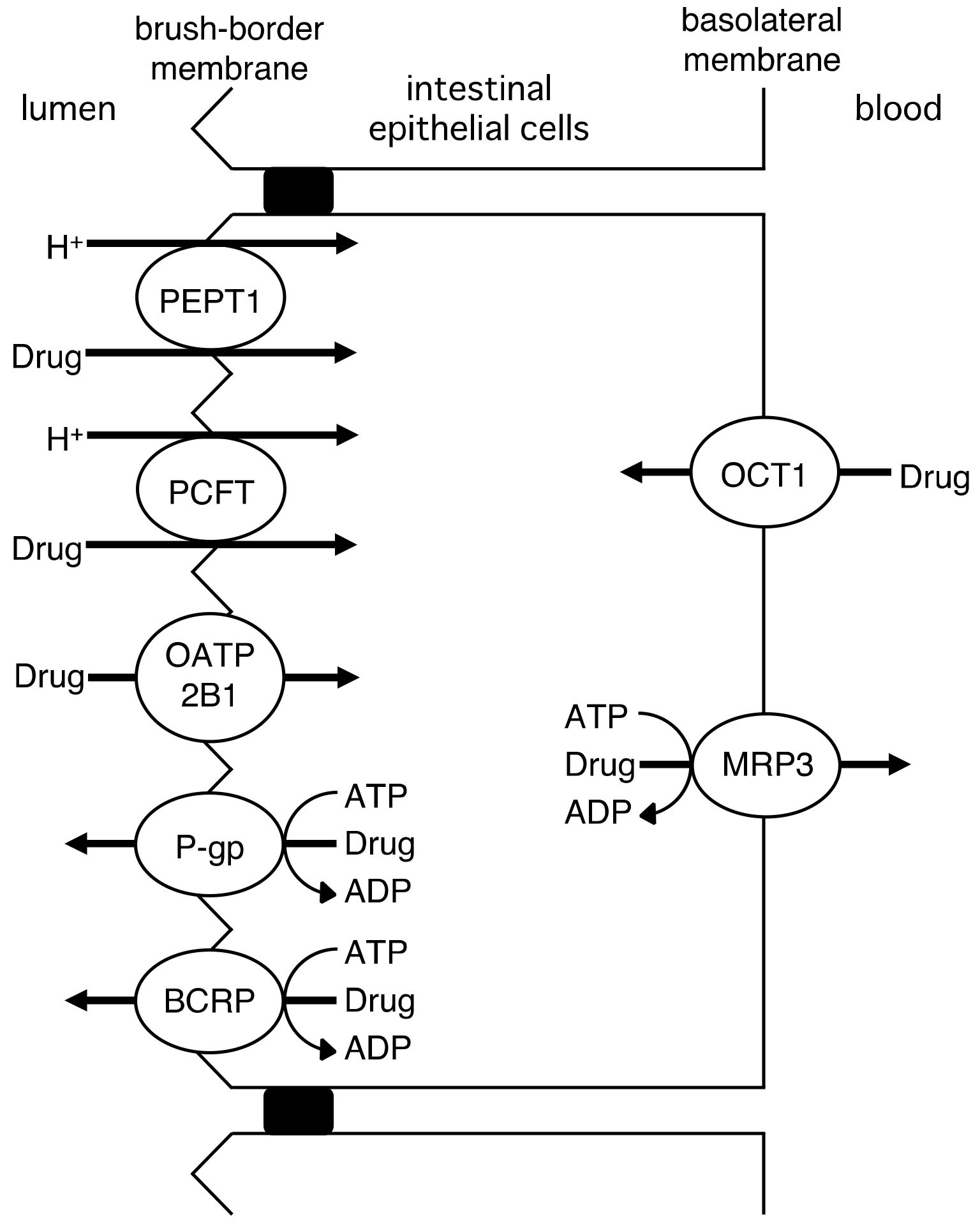
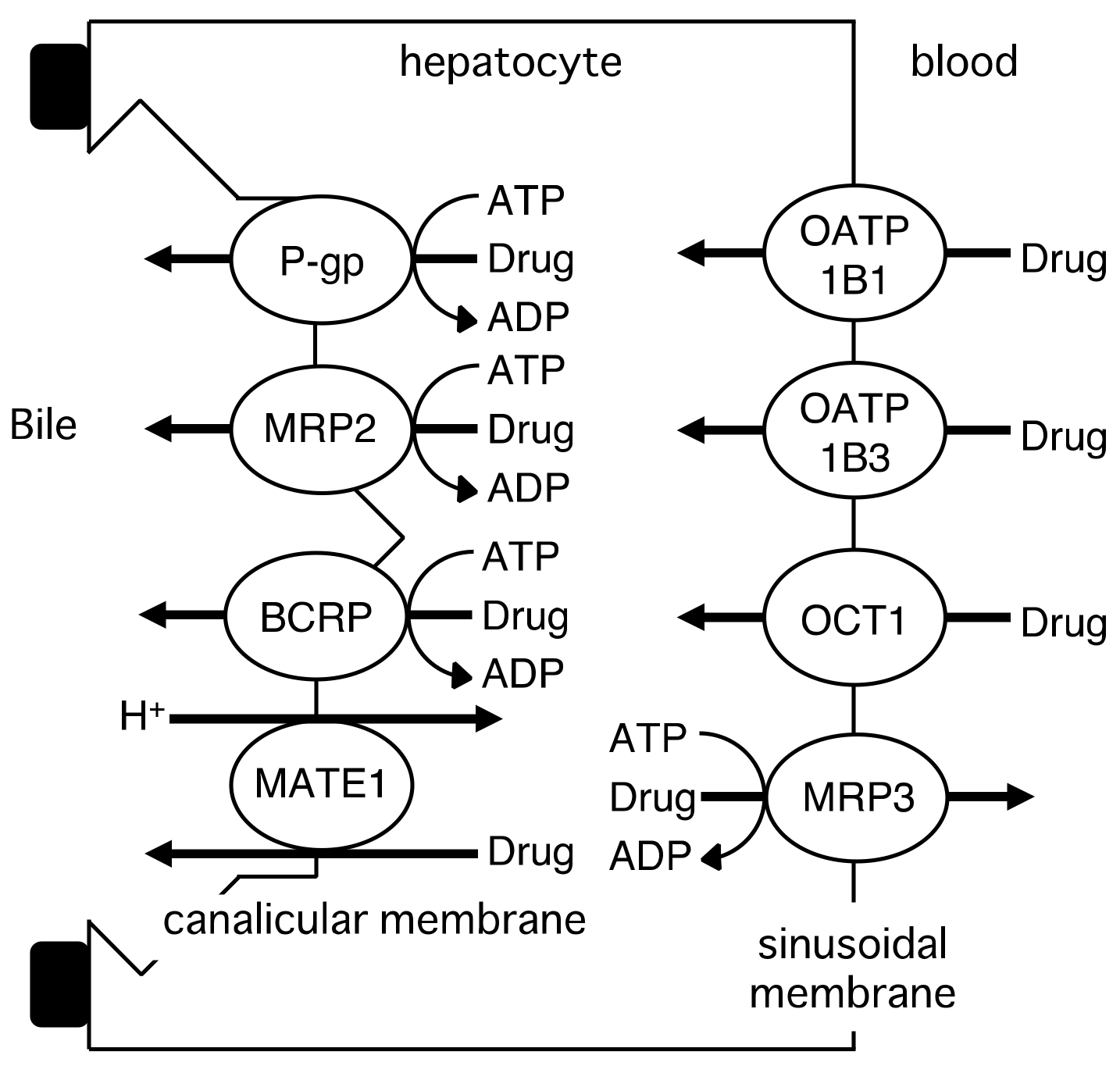
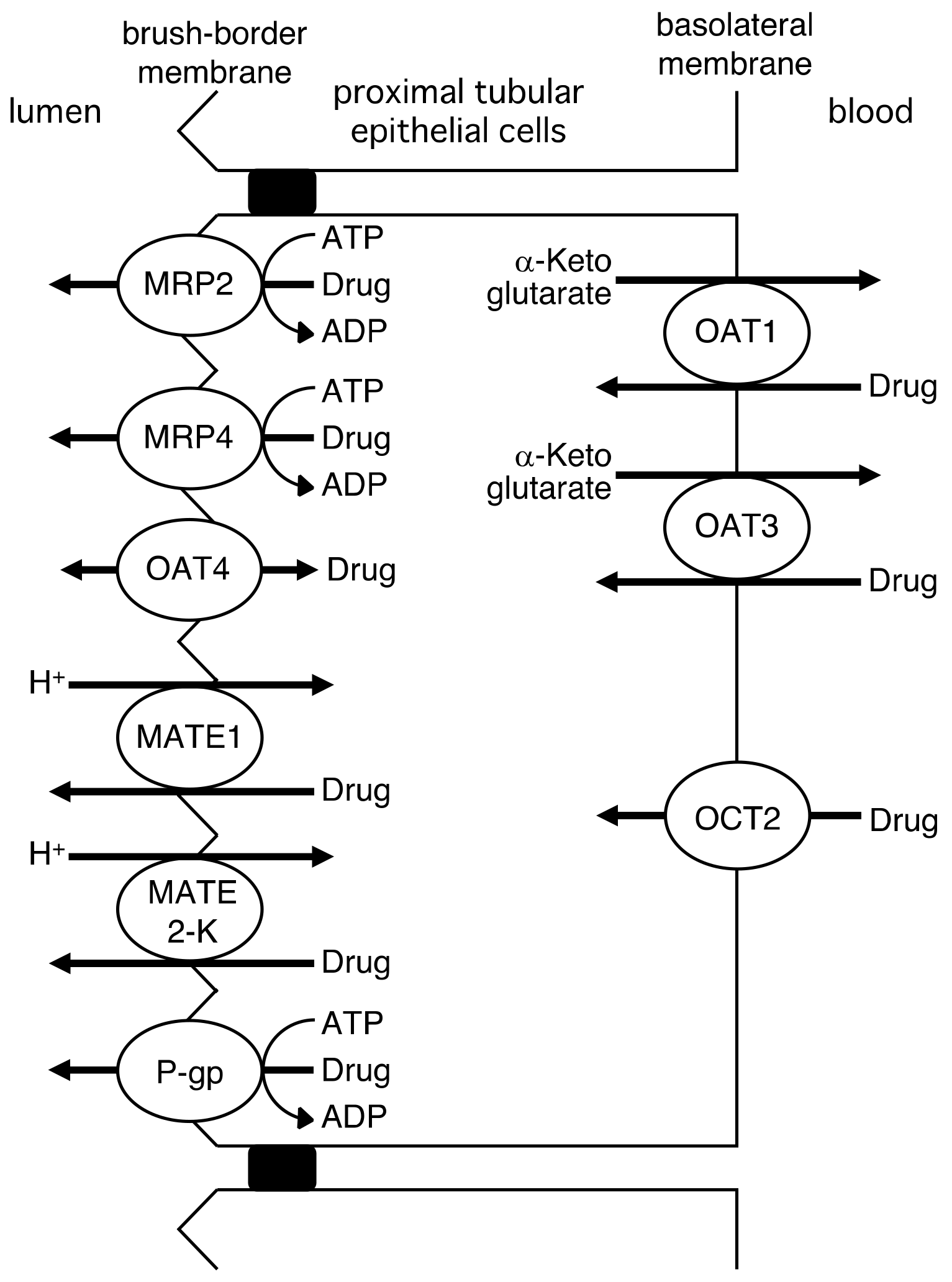
2. Role of Drug Transporters in the Intestinal Absorption, Biliary Excretion, and Renal Tubular Secretion of Drugs
2.1. Intestinal Drug Absorption by Drug Transporters
2.2. Hepatic Transport of Drugs by Drug Transporters
2.3. Renal Tubular Secretion of Drugs
3. Enantioselective Drug Transport by Drug Transporters
3.1. Enantioselective Transport of Antifolates by PCFT
3.2. Enantioselectivity in the Pharmacokinetics of Fexofenadine and Its Transport by OATP2B1
3.3. Enantioselective Secretion of Pantoprazole into Milk by BCRP
4. Enantioselective Inhibitory Effects of Drugs on Drug Transporters
4.1. Enantioselectivity in Inhibitory Effects of Drugs on OCT1, and Binding Affinities
4.2. Enantioselective Inhibitory Effects of NSAIDs and Lansoprazole on OAT1 and OAT3
4.3. Enantioselective Inhibitory Effects of NSAIDs on MRP2 and MRP4
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Carabaza, A.; Cabré, F.; Rotllan, E.; Gómez, M.; Gutiérrez, M.; García, M.L.; Mauleón, D. Stereoselective inhibition of inducible cyclooxygenase by chiral nonsteroidal antiinflammatory drugs. J. Clin. Pharmacol. 1996, 36, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Uwai, Y.; Matsumoto, M.; Kawasaki, T.; Nabekura, T. Enantioselective effect of flurbiprofen on lithium disposition in rats. Pharmacology 2017, 99, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K. Warfarin: Metabolism and mode of action. Biochem. Pharmacol. 1988, 37, 19–27. [Google Scholar] [CrossRef]
- Kaminsky, L.S.; Zhang, Z.Y. Human P450 metabolism of warfarin. Pharmacol. Ther. 1997, 73, 67–74. [Google Scholar] [CrossRef]
- Kendall, M.J. Review article: Esomeprazole—The first proton pump inhibitor to be developed as an isomer. Aliment. Pharmacol. Ther. 2003, 17, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Andersson, T.; Hassan-Alin, M.; Hasselgren, G.; Röhss, K.; Weidolf, L. Pharmacokinetic studies with esomeprazole, the (S)-isomer of omeprazole. Clin. Pharmacokinet. 2001, 40, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Vilbois, F.; Hardwick, J.P.; McBride, O.W.; Nebert, D.W.; Gelboin, H.V.; Meyer, U.A. Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22. Genomics 1988, 2, 174–179. [Google Scholar] [CrossRef]
- Terada, T.; Inui, K. Peptide transporters: Structure, function, regulation and application for drug delivery. Curr. Drug Metab. 2004, 5, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, M.; Huang, W.; Fei, Y.J.; Leibach, F.H.; Ganapathy, V.; Ganapathy, M.E. Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J. Pharm. Sci. 2000, 89, 781–789. [Google Scholar] [CrossRef]
- Qiu, A.; Jansen, M.; Sakaris, A.; Min, S.H.; Chattopadhyay, S.; Tsai, E.; Sandoval, C.; Zhao, R.; Akabas, M.H.; Goldman, I.D. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 2006, 127, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Nakai, Y.; Ueda, S.; Kamigaso, S.; Ohta, K.Y.; Hatakeyama, M.; Hayashi, Y.; Otagiri, M.; Yuasa, H. Functional characterization of PCFT/HCP1 as the molecular entity of the carrier-mediated intestinal folate transport system in the rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G660–G668. [Google Scholar] [CrossRef] [PubMed]
- Menter, A.; Thrash, B.; Cherian, C.; Matherly, L.H.; Wang, L.; Gangjee, A.; Morgan, J.R.; Maeda, D.Y.; Schuler, A.D.; Kahn, S.J.; et al. Intestinal transport of aminopterin enantiomers in dogs and humans with psoriasis is stereoselective: Evidence for a mechanism involving the proton-coupled folate transporter. J. Pharmacol. Exp. Ther. 2012, 342, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Yuasa, H. Molecular basis for pharmacokinetics and pharmacodynamics of methotrexate in rheumatoid arthritis therapy. Drug Metab. Pharmacokinet. 2014, 29, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Maeda, K.; Ikejiri, K.; Yoshida, K.; Horie, T.; Sugiyama, Y. Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: Their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 2013, 34, 45–78. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I.; Nakanishi, T. OATP transporter-mediated drug absorption and interaction. Curr. Opin. Pharmacol. 2013, 13, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J. 2015, 17, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; van Tol, H.; Boersma, A.W.; Brok, M.; Wiemer, E.A.; Stoter, G.; Nooter, K. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Blood 2004, 104, 2940–2942. [Google Scholar] [CrossRef] [PubMed]
- Elkind, N.B.; Szentpétery, Z.; Apáti, A.; Ozvegy-Laczka, C.; Várady, G.; Ujhelly, O.; Szabó, K.; Homolya, L.; Váradi, A.; Buday, L.; et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib). Cancer Res. 2005, 65, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Scharenberg, C.; Dohse, M.; Robey, R.W.; Bates, S.E.; Shukla, S.; Ambudkar, S.V.; Wang, Y.; Wennemuth, G.; Burchert, A.; et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia 2007, 21, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [PubMed]
- Jonker, J.W.; Wagenaar, E.; Mol, C.A.; Buitelaar, M.; Koepsell, H.; Smit, J.W.; Schinkel, A.H. Reduced hepatic uptake and intestinal excretion of organic cations in mice with a targeted disruption of the organic cation transporter 1 (Oct1 [Slc22a1]) gene. Mol. Cell. Biol. 2001, 21, 5471–5477. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Kusuhara, H.; Sugiyama, Y. Functional characterization of multidrug resistance-associated protein 3 (Mrp3/Abcc3) in the basolateral efflux of glucuronide conjugates in the mouse small intestine. J. Pharmacol. Exp. Ther. 2010, 332, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Kusuhara, H.; Sugiyama, Y. In vitro-in vivo extrapolation of transporter-mediated clearance in the liver and kidney. Drug Metab. Pharmacokinet. 2009, 24, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.S.; Jonker, J.W.; Kato, Y.; Kusuhara, H.; Schinkel, A.H.; Sugiyama, Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J. Pharmacol. Exp. Ther. 2002, 302, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, M.V.; Saadatmand, A.R.; Bokelmann, K.; Meineke, I.; Kaiser, R.; Brockmöller, J. Effects of OCT1 polymorphisms on the cellular uptake, plasma concentrations and efficacy of the 5-HT3 antagonists tropisetron and ondansetron. Pharmacogenomics J. 2012, 12, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, J.; Kuron, D.; Faltraco, F.; Knoch, T.; Dos Santos Pereira, J.N.; Abu Abed, M.; Prukop, T.; Brockmöller, J.; Tzvetkov, M.V. OCT1 mediates hepatic uptake of sumatriptan and loss-of-function OCT1 polymorphisms affect sumatriptan pharmacokinetics. Clin. Pharmacol. Ther. 2016, 99, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Tzvetkov, M.V.; Matthaei, J.; Pojar, S.; Faltraco, F.; Vogler, S.; Prukop, T.; Seitz, T.; Brockmöller, J. Increased systemic exposure and stronger cardiovascular and metabolic adverse reactions to fenoterol in individuals with heritable OCT1 deficiency. Clin. Pharmacol. Ther. 2018, 103, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Toh, S.; Wada, M.; Uchiumi, T.; Inokuchi, A.; Makino, Y.; Horie, Y.; Adachi, Y.; Sakisaka, S.; Kuwano, M. Genomic structure of the canalicular multispecific organic anion-transporter gene (MRP2/cMOAT) and mutations in the ATP-binding-cassette region in Dubin-Johnson syndrome. Am. J. Hum. Genet. 1999, 64, 739–746. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Rost, D.; Cui, Y.; Keppler, D. Characterization of the human multidrug resistance protein isoform MRP3 localized to the basolateral hepatocyte membrane. Hepatology 1999, 29, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Hirouchi, M.; Kusuhara, H.; Schuetz, J.D.; Sugiyama, Y. Increasing systemic exposure of methotrexate by active efflux mediated by multidrug resistance-associated protein 3 (mrp3/abcc3). J. Pharmacol. Exp. Ther. 2008, 327, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Ivanyuk, A.; Livio, F.; Biollaz, J.; Buclin, T. Renal drug transporters and drug interactions. Clin. Pharmacokinet. 2017, 56, 825–892. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.; Manganel, M.; Montrose-Rafizadeh, C.; Werner, D.; Roch-Ramel, F. Transport of urate and p-aminohippurate in rabbit renal brush-border membranes. Am. J. Physiol. 1990, 58, F1145–F1153. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, K.; Takano, M.; Okano, T.; Maeda, S.; Inui, K.; Hori, R. p-Aminohippurate transport in rat renal brush-border membranes: A potential-sensitive transport system and an anion exchanger. Biol. Pharm. Bull. 1993, 16, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Inui, K.I.; Masuda, S.; Saito, H. Cellular and molecular aspects of drug transport in the kidney. Kidney Int. 2000, 58, 944–958. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, T.; Kusuhara, H.; Adachi, M.; Schuetz, J.D.; Takeuchi, K.; Sugiyama, Y. Functional involvement of multidrug resistance-associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol. Pharmacol. 2007, 71, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kusuhara, H.; Adachi, M.; Schuetz, J.D.; Takeuchi, K.; Sugiyama, Y. Multidrug resistance-associated protein 4 is involved in the urinary excretion of hydrochlorothiazide and furosemide. J. Am. Soc. Nephrol. 2007, 18, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Ci, L.; Kusuhara, H.; Adachi, M.; Schuetz, J.D.; Takeuchi, K.; Sugiyama, Y. Involvement of MRP4 (ABCC4) in the luminal efflux of ceftizoxime and cefazolin in the kidney. Mol. Pharmacol. 2007, 71, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Smeets, P.H.; van Aubel, R.A.; Wouterse, A.C.; van den Heuvel, J.J.; Russel, F.G. Contribution of multidrug resistance protein 2 (MRP2/ABCC2) to the renal excretion of p-aminohippurate (PAH) and identification of MRP4 (ABCC4) as a novel PAH transporter. J. Am. Soc. Nephrol. 2004, 15, 2828–2835. [Google Scholar] [CrossRef] [PubMed]
- Ekaratanawong, S.; Anzai, N.; Jutabha, P.; Miyazaki, H.; Noshiro, R.; Takeda, M.; Kanai, Y.; Sophasan, S.; Endou, H. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J. Pharmacol. Sci. 2004, 94, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Inui, K. Organic cation transporter OCTs (SLC22) and MATEs (SLC47) in the human kidney. AAPS J. 2013, 15, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Hori, R.; Okamura, N.; Aiba, T.; Tanigawara, Y. Role of P-glycoprotein in renal tubular secretion of digoxin in the isolated perfused rat kidney. J. Pharmacol. Exp. Ther. 1993, 266, 1620–1625. [Google Scholar] [PubMed]
- Narawa, T.; Itoh, T. Stereoselective transport of amethopterin enantiomers by the proton-coupled folate transporter. Drug Metab. Pharmacokinet. 2010, 25, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Kusuhara, H.; Fuse, E.; Sugiyama, Y. P-glycoprotein plays a major role in the efflux of fexofenadine in the small intestine and blood-brain barrier, but only a limited role in its biliary excretion. Drug Metab. Dispos. 2005, 33, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Uno, T.; Tateishi, T.; Suzuki, T. Pharmacokinetics of fexofenadine enantiomers in healthy subjects. Chirality 2007, 19, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Sakugawa, T.; Miura, M.; Hokama, N.; Suzuki, T.; Tateishi, T.; Uno, T. Enantioselective disposition of fexofenadine with the P-glycoprotein inhibitor verapamil. Br. J. Clin. Pharmacol. 2009, 67, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, M.; Leake, B.; Fromm, M.F.; Wilkinson, G.R.; Kim, R.B. OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab. Dispos. 1999, 27, 866–871. [Google Scholar] [PubMed]
- Dresser, G.K.; Bailey, D.G.; Leake, B.F.; Schwarz, U.I.; Dawson, P.A.; Freeman, D.J.; Kim, R.B. Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin. Pharmacol. Ther. 2002, 71, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Imai, K.; Nezu, J.; Tsuji, A.; Tamai, I. Functional characterization of pH-sensitive organic anion transporting polypeptide OATP-B in humans. J. Pharmacol. Exp. Ther. 2004, 308, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Fuse, K.; Okudaira, K.; Nishigaki, R.; Maeda, K.; Kusuhara, H.; Sugiyama, Y. Contribution of OATP (organic anion-transporting polypeptide) family transporters to the hepatic uptake of fexofenadine in humans. Drug Metab. Dispos. 2005, 33, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.G.; Dresser, G.K.; Leake, B.F.; Kim, R.B. Naringin is a major and selective clinical inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juice. Clin. Pharmacol. Ther. 2007, 81, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Maeda, K.; Ishiguro, N.; Igarashi, T.; Sugiyama, Y. Investigation of the inhibitory effects of various drugs on the hepatic uptake of fexofenadine in humans. Drug Metab. Dispos. 2008, 36, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Maeda, K.; Hayashi, H.; Debori, Y.; Schinkel, A.H.; Schuetz, J.D.; Kusuhara, H.; Sugiyama, Y. Involvement of multiple efflux transporters in hepatic disposition of fexofenadine. Mol. Pharmacol. 2008, 73, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Tahara, H.; Kusuhara, H.; Maeda, K.; Koepsell, H.; Fuse, E.; Sugiyama, Y. Inhibition of oat3-mediated renal uptake as a mechanism for drug-drug interaction between fexofenadine and probenecid. Drug Metab. Dispos. 2006, 34, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Maeda, K.; Inoue, K.; Ohta, K.Y.; Yuasa, H.; Kondo, T.; Nakayama, H.; Horita, S.; Kusuhara, H.; Sugiyama, Y. The inhibition of human multidrug and toxin extrusion 1 is involved in the drug-drug interaction caused by cimetidine. Drug Metab. Dispos. 2009, 37, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Akamine, Y.; Miura, M.; Komori, H.; Saito, S.; Kusuhara, H.; Tamai, I.; Ieiri, I.; Uno, T.; Yasui-Furukori, N. Effects of one-time apple juice ingestion on the pharmacokinetics of fexofenadine enantiomers. Eur. J. Clin. Pharmacol. 2014, 70, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Kusuhara, H.; Miura, M.; Yasui-Furukori, N.; Yoshida, K.; Akamine, Y.; Yokochi, M.; Fukizawa, S.; Ikejiri, K.; Kanamitsu, K.; Uno, T.; et al. Effect of coadministration of single and multiple doses of rifampicin on the pharmacokinetics of fexofenadine enantiomers in healthy subjects. Drug Metab. Dispos. 2013, 41, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Merino, G.; Musters, S.; van Herwaarden, A.E.; Bolscher, E.; Wagenaar, E.; Mesman, E.; Dale, T.C.; Schinkel, A.H. The breast cancer resistance protein BCRP (ABCG2) concentrates drugs and carcinogenic xenotoxins into milk. Nat. Med. 2005, 11, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Van Herwaarden, A.E.; Wagenaar, E.; Karnekamp, B.; Merino, G.; Jonker, J.W.; Schinkel, A.H. Breast cancer resistance protein (Bcrp1/Abcg2) reduces systemic exposure of the dietary carcinogens aflatoxin B1, IQ and Trp-P-1 but also mediates their secretion into breast milk. Carcinogenesis 2006, 27, 123–130. [Google Scholar] [CrossRef] [PubMed]
- van Herwaarden, A.E.; Wagenaar, E.; Merino, G.; Jonker, J.W.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Multidrug transporter ABCG2/breast cancer resistance protein secretes riboflavin (vitamin B2) into milk. Mol. Cell. Biol. 2007, 27, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; McNamara, P.J. Stereoselective interaction of pantoprazole with ABCG2. I. Drug accumulation in rat milk. Drug Metab. Dispos. 2012, 40, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leggas, M.; Empey, P.E.; McNamara, P.J. Stereoselective interaction of pantoprazole with ABCG2. II. In vitro flux analysis. Drug Metab. Dispos. 2012, 40, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Schaner, M.E.; Giacomini, K.M. Functional characterization of an organic cation transporter (hOCT1) in a transiently transfected human cell line (HeLa). J. Pharmacol. Exp. Ther. 1998, 286, 354–361. [Google Scholar] [PubMed]
- Moaddel, R.; Patel, S.; Jozwiak, K.; Yamaguchi, R.; Ho, P.C.; Wainer, I.W. Enantioselective binding to the human organic cation transporter-1 (hOCT1) determined using an immobilized hOCT1 liquid chromatographic stationary phase. Chirality 2005, 17, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Moaddel, R.; Yamaguchi, R.; Ho, P.C.; Patel, S.; Hsu, C.P.; Subrahmanyam, V.; Wainer, I.W. Development and characterization of an immobilized human organic cation transporter based liquid chromatographic stationary phase. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 818, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Thyss, A.; Milano, G.; Kubar, J.; Namer, M.; Schneider, M. Clinical and pharmacokinetic evidence of a life-threatening interaction between methotrexate and ketoprofen. Lancet 1986, 327, 256–258. [Google Scholar] [CrossRef]
- Maiche, A.G. Acute renal failure due to concomitant action of methotrexate and indomethacin. Lancet 1986, 327, 1390. [Google Scholar] [CrossRef]
- Uwai, Y.; Saito, H.; Inui, K. Interaction between methotrexate and nonsteroidal anti-inflammatory drugs in organic anion transporter. Eur. J. Pharmacol. 2000, 409, 31–36. [Google Scholar] [CrossRef]
- Takeda, M.; Khamdang, S.; Narikawa, S.; Kimura, H.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J. Pharmacol. Exp. Ther. 2002, 302, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Uwai, Y.; Taniguchi, R.; Motohashi, H.; Saito, H.; Okuda, M.; Inui, K. Methotrexate-loxoprofen interaction: Involvement of human organic anion transporters hOAT1 and hOAT3. Drug Metab. Pharmacokinet. 2004, 19, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Kusuhara, H.; Kondo, T.; Iwaki, M.; Shiroyanagi, Y.; Nakayama, H.; Horita, S.; Nakazawa, H.; Okano, T.; Sugiyama, Y. Species difference in the inhibitory effect of nonsteroidal anti-inflammatory drugs on the uptake of methotrexate by human kidney slices. J. Pharmacol. Exp. Ther. 2007, 322, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Uwai, Y.; Honjo, H.; Iwamoto, K. Inhibitory effect of selective cyclooxygenase-2 inhibitor lumiracoxib on human organic anion transporters hOAT1 and hOAT3. Drug Metab. Pharmacokinet. 2010, 25, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Honjo, H.; Uwai, Y.; Aoki, Y.; Iwamoto, K. Stereoselective inhibitory effect of flurbiprofen, ibuprofen and naproxen on human organic anion transporters hOAT1 and hOAT3. Biopharm. Drug Dispos. 2011, 32, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Beorlegui, B.; Aldaz, A.; Ortega, A.; Aquerreta, I.; Sierrasesúmega, L.; Giráldez, J. Potential interaction between methotrexate and omeprazole. Ann. Pharmacother. 2000, 34, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Doki, K.; Homma, M.; Tamaki, H.; Hori, S.; Ohtani, H.; Sawada, Y.; Kohda, Y. Co-administration of proton pump inhibitors delays elimination of plasma methotrexate in high-dose methotrexate therapy. Br. J. Clin. Pharmacol. 2009, 67, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Chioukh, R.; Noel-Hudson, M.S.; Ribes, S.; Fournier, N.; Becquemont, L.; Verstuyft, C. Proton pump inhibitors inhibit methotrexate transport by renal basolateral organic anion transporter hOAT3. Drug Metab. Dispos. 2014, 42, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, K.; Hamada, Y.; Kaya, C.; Enokiya, T.; Muraki, Y.; Nakahara, H.; Fujimoto, H.; Kobayashi, T.; Iwamoto, T.; Okuda, M. Lansoprazole exacerbates pemetrexed-mediated hematologic toxicity by competitive inhibition of renal basolateral human organic anion transporter 3. Drug Metab. Dispos. 2016, 44, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Hamada, Y.; Ikemura, K.; Iwamoto, T.; Okuda, M. Stereoselective inhibition of renal basolateral human organic anion transporter 3 by lansoprazole enantiomers. Pharmacology 2018, 101, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Miura, M.; Tada, H.; Yasui-Furukori, N.; Uno, T.; Sugawara, K.; Tateishi, T.; Suzuki, T. Pharmacokinetic differences between the enantiomers of lansoprazole and its metabolite, 5-hydroxylansoprazole, in relation to CYP2C19 genotypes. Eur. J. Clin. Pharmacol. 2004, 60, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Kim, M.J.; Park, J.Y.; Shon, J.H.; Yoon, Y.R.; Lee, S.S.; Liu, K.H.; Chun, J.H.; Hyun, M.H.; Shin, J.G. Stereoselective metabolism of lansoprazole by human liver cytochrome P450 enzymes. Drug Metab. Dispos. 2003, 31, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Kawase, A.; Yamamoto, T.; Egashira, S.; Iwaki, M. Stereoselective inhibition of methotrexate excretion by glucuronides of nonsteroidal anti-inflammatory drugs via multidrug resistance proteins 2 and 4. J. Pharmacol. Exp. Ther. 2016, 356, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Murayama, N.; Yamazaki, H. Stereoselectivity of human cytochrome p450 in metabolic and inhibitory activities. Curr. Drug Metab. 2011, 12, 549–569. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Ecker, G.F.; Chiba, P.; Swaan, P.W. Future directions for drug transporter modelling. Xenobiotica 2007, 37, 1152–1170. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, P.; Kolinski, M.; Moaddel, R.; Jozwiak, K.; Wainer, I.W. Determination and modelling of stereoselective interactions of ligands with drug transporters: A key dimension in the understanding of drug disposition. Xenobiotica 2008, 38, 656–675. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Km or Kt (R)-enantiomer | Km or Kt (S)-enantiomer | Km or Kt (R)/(S) | Vmax (R)-enantiomer | Vmax (S)-enantiomer | Vmax (R)/(S) | Ref. |
|---|---|---|---|---|---|---|---|
| Methotrexate | 211 µM | 4.98 µM | 42.4 | 909 pmol/mg/min | 891 pmol/mg/min | 1.02 | [43] |
| Aminopterin | 15.0 µM | 0.69 µM | 21.7 | 42.9 pmol/mg/2 min | 68.8 pmol/mg/2 min | 0.624 | [12] |
| Drug | IC50 Value (µM) (R)-enantiomer | IC50 Value (µM) (S)-enantiomer | (R)/(S) | Ref. |
|---|---|---|---|---|
| Disopyramide | 15.4 | 29.9 | 0.515 | [63] |
| Propranolol | 41.7 | 15.1 | 2.76 | [64] |
| Drug | Kd Value (µM) (R)-enantiomer | Kd Value (µM) (S)-enantiomer | (R)/(S) | Ref. |
|---|---|---|---|---|
| Verapamil | 0.05 | 3.46 | 0.0145 | [65] |
| Atenolol | 0.98 | 0.46 | 2.13 | [64] |
| Propranolol | 2.85 | 0.95 | 3.00 | [64] |
| Drug | Transporter | Substrate | IC50 Value (µM) (R)-Enantiomer | IC50 Value (µM) (S)-Enantiomer | (R)/(S) | Ref. |
|---|---|---|---|---|---|---|
| Flurbiprofen | OAT1 | p-aminohippurate | 2.35 | 0.615 | 3.82 | [73] |
| OAT3 | estrone sulfate | 2.13 | 1.80 | 1.18 | [73] | |
| MRP2 | methotrexate | 133 | 58.4 | 2.28 | [81] | |
| MRP4 | methotrexate | 10.6 | 37.2 | 0.285 | [81] | |
| Ibuprofen | OAT1 | p-aminohippurate | 6.14 | 2.84 | 2.16 | [73] |
| OAT3 | estrone sulfate | 2.04 | 1.20 | 1.70 | [73] | |
| MRP2 | methotrexate | 303 | 139 | 2.18 | [81] | |
| MRP4 | methotrexate | 129 | 267 | 0.483 | [81] | |
| Naproxen | OAT1 | p-aminohippurate | 5.26 | 1.93 | 2.73 | [73] |
| OAT3 | estrone sulfate | 8.09 | 6.79 | 1.19 | [73] | |
| MRP2 | methotrexate | 510 | 7.11 | 71.7 | [81] | |
| MRP4 | methotrexate | 8.06 | 49.8 | 0.162 | [81] | |
| Lansoprazole | OAT1 | p-aminohippurate | 43.8 | 33.6 | 1.30 | [78] |
| OAT3 | estrone sulfate | 1.75 | 0.61 | 2.87 | [78] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uwai, Y. Enantioselective Drug Recognition by Drug Transporters. Molecules 2018, 23, 3062. https://doi.org/10.3390/molecules23123062
Uwai Y. Enantioselective Drug Recognition by Drug Transporters. Molecules. 2018; 23(12):3062. https://doi.org/10.3390/molecules23123062
Chicago/Turabian StyleUwai, Yuichi. 2018. "Enantioselective Drug Recognition by Drug Transporters" Molecules 23, no. 12: 3062. https://doi.org/10.3390/molecules23123062
APA StyleUwai, Y. (2018). Enantioselective Drug Recognition by Drug Transporters. Molecules, 23(12), 3062. https://doi.org/10.3390/molecules23123062