
Next Generation Sequencing-Based Molecular Marker Development: A Case Study in Betula Alnoides

 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Information of Sequencing Data
2.2. Distribution of SSR in the Genome
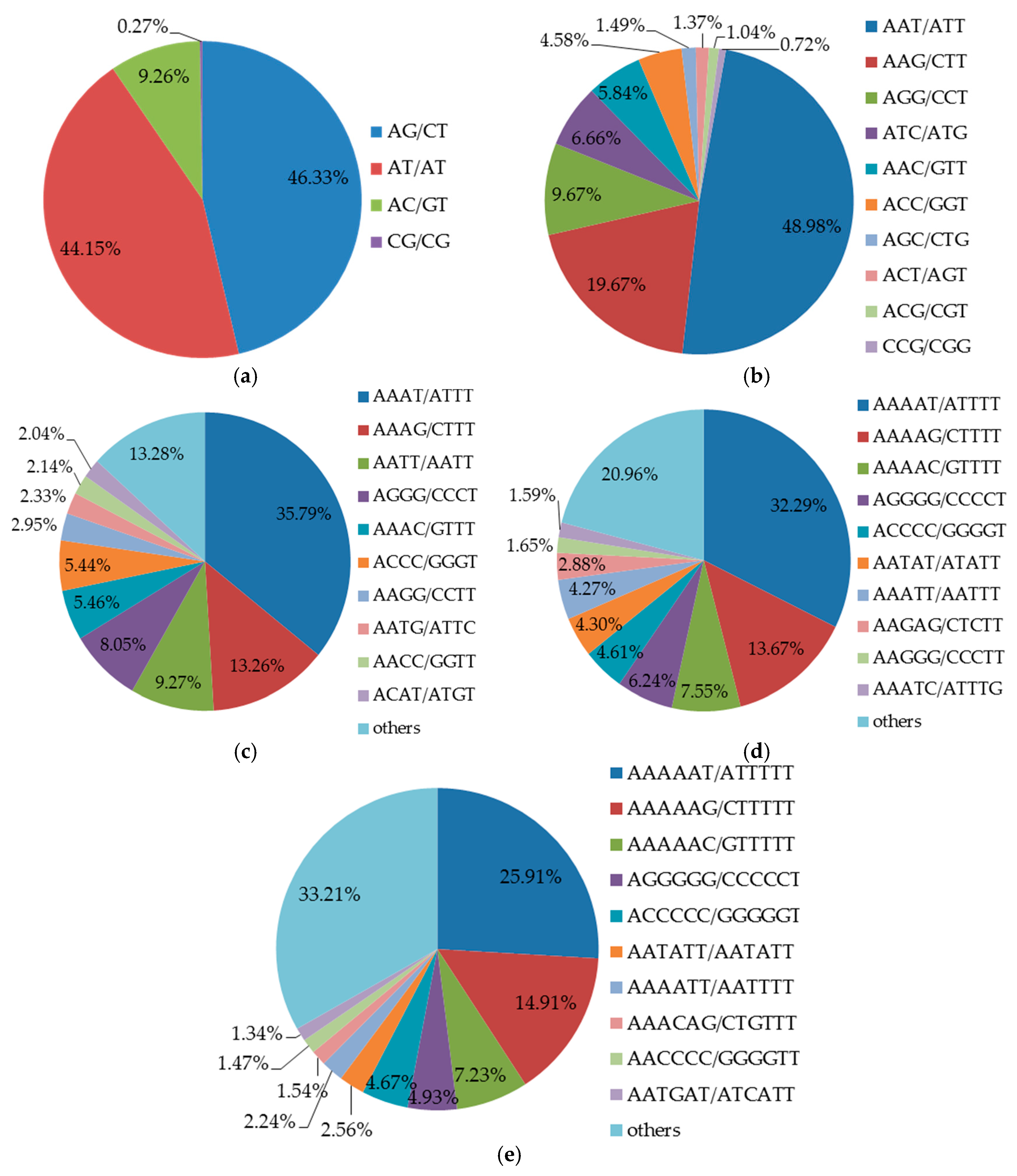
2.3. Characteristics of SSR Sequences
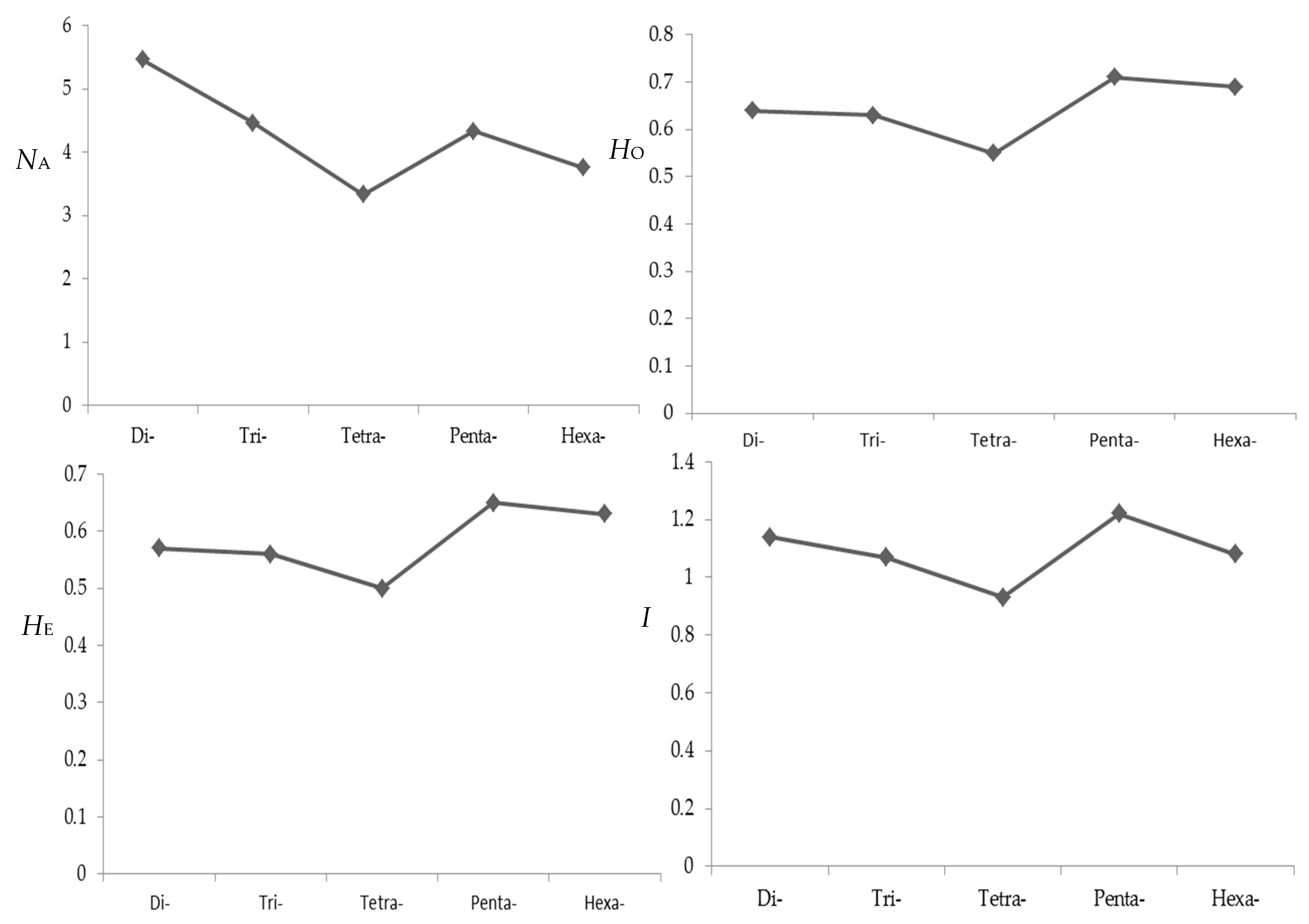
2.4. Primer Screening and SSR Markers Polymorphism Detecting
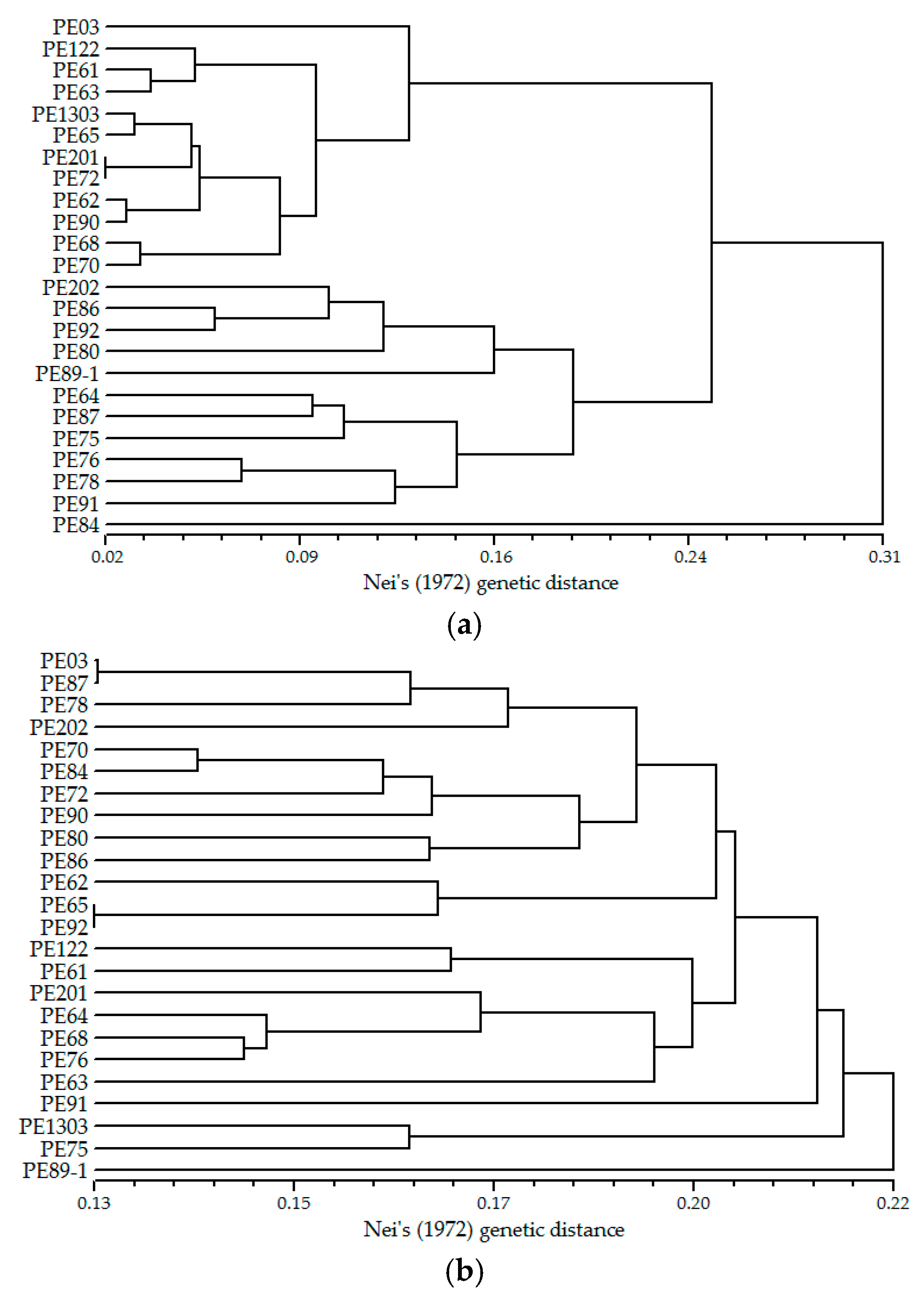
2.5. Cross-Species Transferability
3. Materials and Methods
3.1. Plant Material and DNA Isolation
3.2. Genome Sequencing, SSR Finding and Survey
3.3. SSR Primer Design and Screening
3.4. SSR Markers Polymorphism Detection
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zeng, J.; Zhen, H.S.; Weng, Q.J. Geographic distributions and ecological conditions of Betula alnoides in China. For. Res. 1999, 12, 479–484, (In Chinese with English Abstract). [Google Scholar]
- Wang, C.S.; Zhao, Z.G.; Hein, S.; Zeng, J.; Schuler, J.; Guo, J.J. Effect of planting density on knot attributes and branch occlusion of Betula alnoides under natural pruning in southern China. Forests 2015, 6, 1343–1361. [Google Scholar] [CrossRef]
- Sur, T.K.; Pandit, S.; Battacharyya, D.; Kumar, C.K.A.; Lakshmi, S.M.; Chatttopadhyay, D.; Mandal, S.C. Studies on the anti-inflammatory activity of Betula alnoides bark. Phytother. Res. 2002, 16, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.D.A.; Malarvili, T.; Velavan, S. Restorative effect of Betula alnoides bark on hepatic metabolism in high fat diet fed Wistar rats. Int. J. Pharma BioSci. 2015, 6, 1281–1288. [Google Scholar]
- Velavan, S. Antioxidant activity of Betula alnoides bark extract in high fat diet fed Wistar rats. Int. J. Chemtech. Res. 2015, 7, 2391–2398. [Google Scholar]
- Zeng, J.; Guo, W.F.; Zhao, Z.G.; Weng, Q.J.; Yin, G.T.; Zhen, H.S. Domestication of Betula alnoides in China: Current Status and Perspectives. For. Res. 2006, 19, 379–384, (In Chinese with English Abstract). [Google Scholar]
- Wang, C.S.; Hein, S.; Zhao, Z.G.; Guo, J.J.; Zeng, J. Branch occlusion and discoloration of Betula alnoides, under artificial and natural pruning. For. Ecol. Manag. 2016, 375, 200–210. [Google Scholar] [CrossRef]
- Sakiyama, N.S.; Ramos, H.C.C.; Caixeta, E.T.; Pereira, M.G. Plant breeding with marker-assisted selection in Brazil. Crop Breed. Appl. Biotechnol. 2014, 14, 54–60. [Google Scholar] [CrossRef]
- Zargar, S.M.; Raatz, B.; Sonah, H.; Bhat, J.A.; Dar, Z.A.; Agrawal, G.K.; Rakwal, R. Recent advances in molecular marker techniques: Insight into QTL mapping, GWAS and genomic selection in plants. J. Crop Sci. Biotechnol. 2015, 18, 293–308. [Google Scholar] [CrossRef]
- Zhao, D.W.; Yang, J.B.; Yang, S.X.; Kato, K.; Luo, J.P. Genetic diversity and domestication origin of tea plant Camellia taliensis (Theaceae) as revealed by microsatellite markers. BMC Plant Biol. 2014, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, P.; Kang, D.; Mo, X.; Tian, E.; Hu, Y.; Huang, R. Development and characterization of High-throughput Est-based SSR markers for Pogostemon cablin using transcriptome sequencing. Molecules 2018, 23, 2014. [Google Scholar] [CrossRef] [PubMed]
- Payn, K.G.; Dvorak, W.S.; Janse, B.J.H.; Myburg, A.A. Microsatellite diversity and genetic structure of the commercially important tropical tree species Eucalyptus urophylla, endemic to seven islands in eastern Indonesia. Tree Genet. Genomes 2008, 4, 519–530. [Google Scholar] [CrossRef]
- Du, Q.; Wang, B.; Wei, Z.; Zhang, D.; Li, B. Genetic diversity and population structure of Chinese White poplar (Populus tomentosa) revealed by SSR markers. J. Hered. 2012, 103, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.W.; Zhao, T.T.; Ma, Q.H.; Liang, L.S.; Wang, G.X. Assessment of genetic diversity and population genetic structure of Corylus mandshurica in China using SSR markers. PLoS ONE 2015, 10, e0137528. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Zhang, M.Y.; Liu, Q.Z.; Li, L.T.; Song, Y.; Wang, L.F.; Zhang, S.L.; Wu, J. Transferability of newly developed pear SSR markers to other Rosaceae species. Plant Mol. Biol. Rep. 2013, 31, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, N.; Zhao, K.G.; Chen, Y.X.; Tang, R.J.; Chen, L.Q. Development and primer selection of EST-SSR molecular markers based on transcriptome sequencing of Chimonanthus praecox. J. Beijing For. Univ. 2013, 35, 25–32, (In Chinese with English Abstract). [Google Scholar]
- Wang, Z.; Fang, B.; Chen, J.; Zhang, X.; Luo, Z.; Huang, L.; Chen, X.; Li, Y. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas). BMC Genom. 2010, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Singh, A.K.; Singh, S.; Singh, B.D. Next-generation sequencing (NGS) tools and impact in plant breeding. In Advances in Plant Breeding Strategies: Breeding, Biotechnology and Molecular Tools; Springer: Cham, Switzerland, 2015; pp. 563–612. [Google Scholar]
- Taheri, S.; Abdullah, T.L.; Jain, S.M.; Sahebi, M.; Azizi, P. TILLING, high-resolution melting (HRM), and next-generation sequencing (NGS) techniques in plant mutation breeding. Mol. Breed. 2017, 37, 40. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, D.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Mcallister, H.A.; Bartlett, P.R.; Buggs, R.J.A. Molecular phylogeny and genome size evolution of the genus Betula (Betulaceae). Ann. Bot. 2016, 117, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zou, Y.; Bai, J.; Zheng, H. RAPD analysis of genetic variation in natural populations of Betula alnoides from Guangxi, China. Euphytica 2003, 134, 33–41. [Google Scholar] [CrossRef]
- Zeng, J.Z.R.; Wang, S.L.; Zhou, J.Y.; Bai, J.; Zheng, H.S. Allozyme variation and population genetic structure of Betula alnoides from Guangxi, China. Biochem. Genet. 2003, 41, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Huang, S.X.; Chen, R.; Gui, R.Y. Study on genetic diversity of Betula alnoides. J. Zhejiang For. Sci. Technol. 2010, 30, 50–52, (In Chinese with English Abstract). [Google Scholar]
- Guo, J.J.; Zeng, J.; Zhou, S.L.; Zhan, Z.G. Isolation and characterization of 19 microsatellite markers in a tropical and warm-subtropical birch, Betula alnoides Buch.-Ham. Ex D. Don. Mol. Ecol. Resour. 2008, 8, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.Z.; Zhen, Y.; Qiu, Y.P.; Nie, Y.L. Genetic diversity analysis of four strains of Betula alnoides. J. Southwest For. Univ. 2017, 37, 21–25, (In Chinese with English Abstract). [Google Scholar]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.H.; Huang, C.; You, C.Y.; Li, W.; Zhao, W.X.; Shen, C.; Zhang, B.B.; Wang, H.T.; Yan, Z.H.; Dai, B.S.; et al. Genome-wide SSR-based association mapping for fiber quality in nation-wide upland cotton inbreed cultivars in China. BMC Genom. 2016, 17, 35. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yan, J.; Zhao, J.; Song, W.; Zhang, X.; Xiao, Y.; Zheng, Y. Genome-wide association study (GWAS) of resistance to head smut in maize. Plant Sci. 2012, 196, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Csencsics, D.; Brodbeck, S.; Holderegger, R. Cost-effective, species-specific microsatellite development for the endangered dwarf bulrush (Typha minima) using next-generation sequencing technology. J. Hered. 2010, 101, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Kkm, K.; Pekkinen, M.; Varvio, S. Twenty-three microsatellite primer pairs for Betula pendula (Betulaceae). Mol. Ecol. Resour. 2004, 4, 471–473. [Google Scholar]
- Shi, J.; Dai, X.; Chen, Y.; Chen, J.; Shi, J.; Yin, T. Discovery and experimental analysis of microsatellites in an oil woody plant Camellia chekiangoleosa. Plant Syst. Evol. 2013, 299, 1387–1393. [Google Scholar] [CrossRef]
- Gupta, P.K.; Balyan, H.S.; Sharma, P.C.; Ramesh, B. Microsatellites in plants: A new class of molecular markers. Curr. Sci. India 1996, 70, 45–54. [Google Scholar]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Schorderet, D.F.; Gartler, S.M. Analysis of CpG suppression in methylated and nonmethylated species. Proc. Natl. Acad. Sci. USA 1992, 89, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Kantety, R.V.; La, R.M.; Matthews, D.E.; Sorrells, M.E. Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat. Plant Mol. Biol. 2002, 48, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, S.J.; Liu, H.J.; Zhou, B.; Wang, X.; Jiang, T. Development of SSR markers and genetic diversity in white birch (Betula platyphylla). PLoS ONE 2015, 10, e0129758. [Google Scholar]
- Wang, H.X.; Walla, J.A.; Zhong, S.B.; Huang, D.Q.; Dai, W.H. Development and cross-species genera transferability of microsatellite markers discovered using 454 genome sequencing in chokecherry (Prunus virginiana L.). Plant Cell Rep. 2012, 31, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Baird, R.E.; Wadl, P.A.; Allen, T.; McNeill, D.; Wang, X.; Moulton, J.K.; Rinehart, T.A.; Abbas, H.K.; Shier, T.; Trigiano, R.N. Variability of united states isolates of Macrophomina phaseolina based on simple sequence repeats and cross genus transferability to related genera within Botryosphaeriaceae. Mycopathologia 2010, 170, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Zou, Y.P.; Bai, J.Y.; Zheng, H.S. Preparation of Total DNA from “Recalcitrant Plant Taxa”. Acta Bot. Sin. 2002, 44, 694–697. [Google Scholar]
- Guo, J.J.; Shang, S.B.; Wang, C.S.; Zhao, Z.G.; Zeng, J. Twenty microsatellite markers for the endangered Vatica mangachapoi (Dipterocarpaceae). Appl. Plant Sci. 2017, 5, 1600134. [Google Scholar] [CrossRef] [PubMed]
- Boutinganache, I.; Raposo, M.; Raymond, M.; Deschepper, C.F. M13-tailed primers improve the readability and usability of microsatellite analyses performed with two different allele-sizing methods. Biotechniques 2001, 31, 24–28. [Google Scholar]
- Hulce, D.; Li, X.; Snyderleiby, T.; Johathan-Liu, C.S. Genemarker® genotyping software: Tools to increase the statistical power of DNA fragment analysis. J. Biomol. Tech. 2011, 22, S35. [Google Scholar]
- Puyvelde, K.V.; Geert, AV.; Triest, L. Atetra, a new software program to analyse tetraploid microsatellite data: Comparison with TETRA and TETRASAT. Mol. Ecol. Resour. 2010, 10, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Rohlf, F.J. NTSYS-pc numerical taxonomy and multivariate analysis system. Am. Stat. 2000, 41, 330. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |





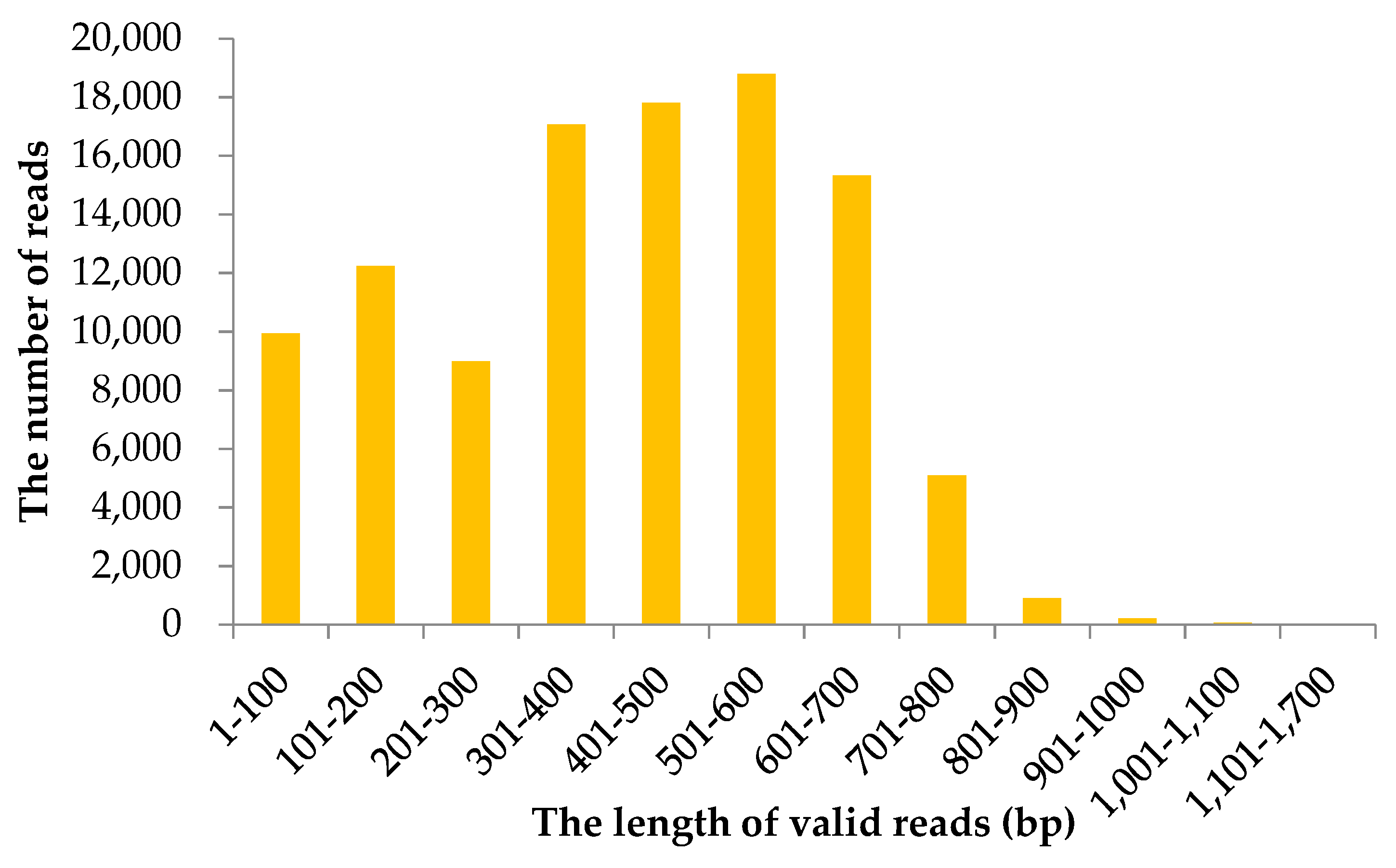{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repeat Type | Repeat Number | Total | Frequency (%) | |||||
|---|---|---|---|---|---|---|---|---|
| 3–8 | 9–14 | 15–20 | 21–26 | 27–32 | ≥33 | |||
| Mono-nucleotide | - | 106,526 | 16,966 | 2328 | 821 | 4 | 126,645 | 77.05 |
| Di-nucleotide | 13,777 | 2601 | 296 | 37 | 15 | - | 16,726 | 10.18 |
| Tri-nucleotide | 5675 | 163 | 15 | 3 | - | - | 5856 | 3.56 |
| Tetra-nucleotide | 10,053 | 2 | - | - | - | - | 10,055 | 6.12 |
| Penta-nucleotide | 3509 | 1 | 2 | - | - | - | 3512 | 2.14 |
| Hexa-nucleotide | 1562 | 1 | - | - | - | - | 1563 | 0.95 |
| Total | 34,576 | 109,294 | 17,279 | 2368 | 836 | 4 | 164,357 | 100 |
| Frequency (%) | 21.04 | 66.50 | 10.51 | 1.44 | 0.51 | 0.24 × 10−4 | ||
| Related Species | Collection Locality | Longitude (°E) | Latitude (°N) |
|---|---|---|---|
| B. platyphylla | Baihuashan Mountain, Beijing | 115.57 | 39.85 |
| B. austro-sinensis | Yangdongshan Shierdushui Provincial Nature Reserve, Lechang, Guangdong | 113.39 | 25.28 |
| B. cylindrostachya | Hill behind Cizhong Church, Weixi County, Yunnan | 98.92 | 28.48 |
| B. fujianensis | Luoboyan Natural Reserve, Fujian | 117.57 | 27.43 |
| B. hainanensis | Jianfengling Natural Reserve, Hainan | 109.17 | 18.73 |
| B. luminifera | Qinwang Mountain, Tianlin County, Guangxi | 106.62 | 23.91 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, J.; Guo, J.-J.; Yin, M.-Y.; Wang, H.; Dong, W.-P.; Zeng, J.; Zhou, S.-L. Next Generation Sequencing-Based Molecular Marker Development: A Case Study in Betula Alnoides. Molecules 2018, 23, 2963. https://doi.org/10.3390/molecules23112963
Tan J, Guo J-J, Yin M-Y, Wang H, Dong W-P, Zeng J, Zhou S-L. Next Generation Sequencing-Based Molecular Marker Development: A Case Study in Betula Alnoides. Molecules. 2018; 23(11):2963. https://doi.org/10.3390/molecules23112963
Chicago/Turabian StyleTan, Jing, Jun-Jie Guo, Ming-Yu Yin, Huan Wang, Wen-Pan Dong, Jie Zeng, and Shi-Liang Zhou. 2018. "Next Generation Sequencing-Based Molecular Marker Development: A Case Study in Betula Alnoides" Molecules 23, no. 11: 2963. https://doi.org/10.3390/molecules23112963
APA StyleTan, J., Guo, J.-J., Yin, M.-Y., Wang, H., Dong, W.-P., Zeng, J., & Zhou, S.-L. (2018). Next Generation Sequencing-Based Molecular Marker Development: A Case Study in Betula Alnoides. Molecules, 23(11), 2963. https://doi.org/10.3390/molecules23112963