
Microwave-Assisted Expeditious Synthesis of 2-Alkyl-2-(N-arylsulfonylindol-3-yl)-3-N-acyl-5-aryl-1,3,4-oxadiazolines Catalyzed by HgCl2 under Solvent-Free Conditions as Potential Anti-HIV-1 Agents


Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Activities
3. Experimental Section
3.1. General Information
3.2. Preparation of 2-Alkyl-2-(N-arylsulfonylindol-3-yl)-3-N-acyl-5-aryl-1,3,4-oxadiazolines (3a–d′)
3.3. Anti-HIV-1 Activity Assay
3.3.1. Virus and Cells
3.3.2. MTT-Based Cytotoxicity Assay
3.3.3. Syncytia Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of interest
References
- Gottlieb, M.S.; Schroff, R.; Schanker, H.M.; Weisman, J.D.; Fan, P.T.; Wolf, R.A.; Saxon, A. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: Evidence of a new acquired cellular immunodeficiency. N. Engl. J. Med. 1981, 305, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, H.; Anné, J.; De Clercq, E. The HIV-1 reverse transcription (RT) process as target for RT inhibitors. Med. Res. Rev. 2000, 20, 129–154. [Google Scholar] [CrossRef]
- Yisma, E.; Dessalegn, B.; Astatkie, A.; Fesseha, N.; Benagiano, G.; Bastianelli, C.; Brusamento, S.; Elmoniry, H.; van Velthoven, M.; Pape, U. Global report: UNAIDS report on the global AIDS epidemic 2013. Reprod. Health 2014, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Boone, L.R. Next-generation HIV-1 non-nucleoside reverse transcriptase inhibitors. Curr. Opin. Investig. Drugs 2006, 7, 128–135. [Google Scholar] [PubMed]
- De Clercq, E. New developments in anti-HIV chemotherapy. Biochim. Biophy. Acta 2002, 1587, 258–275. [Google Scholar] [CrossRef]
- Sluis-Cremer, N.; Wainberg, M.A.; Schinazi, R.F. Resistance to reverse transcriptase inhibitors used in the treatment and prevention of HIV-1 infection. Future Microbiol. 2015, 10, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.Y.; Fan, E.K.; Wu, J.D.; Liu, X.Y. Recent advances in the DABOs family as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Curr. Med. Chem. 2011, 18, 2376–2385. [Google Scholar] [CrossRef] [PubMed]
- Chander, S.; Wang, P.; Ashok, P.; Yang, L.M.; Zheng, Y.T.; Murugesan, S. Rational design, synthesis, anti-HIV-1 RT and antimicrobial activity of novel 3-(6-methoxy-3,4-dihydroquinolin-1(2H)-yl)-1-(piperazin-1-yl) propan-1-one derivatives. Bioorg. Chem. 2016, 67, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yu, D.L.; Ho, P.; Lee, K.H.; Chen, C.H. Synthesis and anti-HIV activity of bi-functional triterpene derivatives. Lett. Drug Des. Discov. 2007, 4, 471–478. [Google Scholar] [CrossRef]
- Polanski, J.; Niedbala, H.; Musiol, R.; Podeszwa, B.; Tabak, D.; Palka, A.; Mencel, A.; Finster, J.; Mouscadet, J.F.; Le Bret, M. 5-Hydroxy-6-quinaldic acid as a novel molecular scaffold for HIV-1 integrase inhibitors. Lett. Drug Des. Discov. 2006, 3, 175–178. [Google Scholar] [CrossRef]
- Safakish, M.; Hajimahdi, Z.; Zabihollahi, R.; Aghasadeghi, M.R.; Vahabpour, R.; Zarghi, A. Design, synthesis, and docking studies of new 2-benzoxazolinone derivatives as anti-HIV-1 agents. Med. Chem. Res. 2017, 26, 2718–2726. [Google Scholar] [CrossRef]
- Ke, S.Y.; Liu, F.Y.; Wang, N.; Yang, Q.; Qian, X.H. 1,3,4-Oxadiazoline derivatives as novel potential inhibitors targeting chitin biosynthesis: Design, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2009, 19, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Coley, H.M.; Sarju, J.; Wagner, G. Synthesis and characterization of platinum(II) oxadiazoline complexes and their in vitro antitumor activity in platinum-sensitive and -resistant cancer cell lines. J. Med. Chem. 2008, 51, 135–141. [Google Scholar] [CrossRef] [PubMed]
- El-Emam, A.A.; Al-Deeb, O.A.; Al-Omar, M.; Lehmann, J. Synthesis, antimicrobial, and anti-HIV-1 activity of certain 5-(1-adamantyl)-2-substituted thio-1,3,4-oxadiazoles and 5-(1-adamantyl)-3-substituted aminomethyl-1,3,4-oxadiazoline-2-thiones. Bioorg. Med. Chem. 2004, 12, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- Che, Z.P.; Huang, N.; Yu, X.; Yang, L.M.; Ran, J.Q.; Zhi, X.Y.; Xu, H.; Zheng, Y.T. Microwave-assisted combinatorial synthesis of 2-alkyl-2-(N-arylsulfonylindol-3-yl)-3-N-acyl-5-aryl-1,3,4-oxadiazolines as anti-HIV-1 agents. Comb. Chem. High Throughput Scr. 2013, 16, 400–407. [Google Scholar] [CrossRef]
- Joshi, S.D.; Vagdevi, H.M.; Vaidya, V.P.; Gadaginamath, G.S. Synthesis of new 4-pyrrol-1-yl benzoic acid hydrazide analogs and some derived oxadiazole, triazole and pyrrole ring systems: A novel class of potential antibacterial and antitubercular agents. Eur. J. Med. Chem. 2008, 43, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Cottineau, B.; Renaux, S.; Chenault, J.; Guillaumet, G. New synthesis of pyrazolyl-1,3,4-oxadiazole and 1,3,4-oxadiazoline derivatives. Lett. Org. Chem. 2005, 2, 599–601. [Google Scholar] [CrossRef]
- Li, C.K.; Ma, Y.J.; Cao, L.H. Synthesis of novel 3-acetyl-2-aryl-5-(3-aryl-1-phenyl-pyrazol-4-yl)-2,3- dihydro-1,3,4-oxadiazoles. J. Chin. Chem. Soc. 2009, 56, 182–185. [Google Scholar] [CrossRef]
- Xu, H.; Che, Z.P.; Wang, Q. An efficiently sonochemical synthesis of 2-(N-aryl-sulfonylindol-3-yl)-3-N-acyl-5-phenyl-1,3,4-oxadiazolines. Heterocycles 2011, 42, 825–832. [Google Scholar] [CrossRef]
- Gedye, R.; Smith, F.; Westaway, K.; Humera, A.; Baldisera, L.; Laberge, L.; Rousell, L. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Giguere, R.J.; Bray, T.L.; Duncan, S.M.; Majetich, G. Application of commercial microwave ovens to organic synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Koziej, D.; Floryan, C.; Sperling, R.A.; Ehrlicher, A.J.; Issadore, D.; Westervelt, R.; Weitz, D.A. Microwave dielectric heating of non-aqueous droplets in a microfluidic device for nanoparticle synthesis. Nanoscale 2013, 5, 5468–5475. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.L.; Tofteng, A.P.; Malik, L.; Jensen, J. Microwave heating in solid-phase synthesis. Chem. Soc. Rev. 2012, 41, 1826–1844. [Google Scholar] [CrossRef] [PubMed]
- Westman, J. An efficient combination of microwave dielectric heating and the use of solid-supported triphenylphosphine for Wittig reactions. Org. Lett. 2001, 3, 3745–3747. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, A.; Rajaguru, K.; Muthusubramanian, S.; Bhuvanesh, N. Microwave-assisted catalyst-free synthesis of tetrasubstituted pyrroles using dialkyl acetylenedicarboxylates and monophenacylanilines. Syn. Commun. 2016, 47, 805–812. [Google Scholar] [CrossRef]
- Wang, S.L.; Cheng, C.; Wu, F.Y.; Jiang, B.; Shi, F.; Tu, S.J.; Rajale, T.; Li, G. Microwave-assisted multi-component reaction in water leading to highly regioselective formation of benzo[f]azulen-1-ones. Tetrahedron 2011, 67, 4485–4493. [Google Scholar] [CrossRef] [PubMed]
- Vaddula, B.R.; Varma, R.S.; Leazer, J. Mixing with microwaves: Solvent-free and catalyst-free synthesis of pyrazoles and diazepines. Tetrahedron Lett. 2013, 54, 1538–1541. [Google Scholar] [CrossRef]
- De-la-Torre, P.; Osorio, E.; Alzate-Morales, J.H.; Caballero, J.; Trilleras, J.; Astudillo-Saavedra, L.; Brito, L.; Cárdenas, A.; Quiroga, J.; Gutiérrez, M. Ultrasound-assisted phase-transfer catalysis method in an aqueous medium to promote the Knoevenagel reaction: Advantages over the conventional and microwave-assisted solvent-free/catalyst-free method. Ultrason. Sonochem. 2014, 21, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Driowya, M.; Saber, A.; Marzag, H.; Demange, L.; Bougrin, K.; Benhida, R. Microwave-assisted syntheses of bioactive seven-membered, macro-sized heterocycles and their fused derivatives. Molecules 2016, 21, 1032. [Google Scholar] [CrossRef] [PubMed]
- Vera, G.; Diethelm, B.; Terraza, C.A.; Recabarren-Gajardo, G. Suzuki-type cross-coupling reaction of unprotected 3-iodoindazoles with pinacol vinyl boronate: An expeditive C-3 vinylation of indazoles under microwave irradiation. Molecules 2018, 23, 2051. [Google Scholar] [CrossRef] [PubMed]
- Kantar, G.K.; Baltas, N.; Mentese, E.; Sasmaz, S. Microwave-assisted synthesis and investigation of xanthine oxidase inhibition of new phthalonitrile and phthalocyanines containing morpholino substituted 1,2,4-triazole-3-one. J. Organomet. Chem. 2015, 787, 8–13. [Google Scholar] [CrossRef]
- Saber, A.; Marzag, H.; Benhida, R.; Bougrin, K. Microwave-assisted cycloaddition reactions in carbo- and heterocyclic chemistry. Curr. Org. Chem. 2014, 18, 2139–2180. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3a–r′ are available from the authors. |


{kind=link}

| Entry | Amount of (mmol) | Catalyst (5 mol%) | ta (min) | Isolated Yield b (%) | ||
|---|---|---|---|---|---|---|
| 1a | 2a | 3a | 1a | |||
| 1 | 0.5 | 1.5 | MgCl2.6H2O | 10 × 3 | 38 | 37 |
| 2 | 0.5 | 1.5 | ZnCl2 | 10 × 3 | 48 | 23 |
| 3 | 0.5 | 1.5 | AlCl3 | 10 × 3 | 53 | 9 |
| 4 | 0.5 | 1.5 | SnCl2.2H2O | 10 × 3 | 63 | 15 |
| 5 | 0.5 | 1.5 | FeCl3 | 10 × 3 | 68 | 18 |
| 6 | 0.5 | 1.5 | HgCl2 | 10 × 3 | 91 | 0 |

| Entry | Amount of (mmol) | HgCl2 (mol%) | ta (min) | Isolated Yield b (%) | ||
|---|---|---|---|---|---|---|
| 1a | 2a | 3a | 1a | |||
| 1 | 0.5 | 2.5 | 5 | 10 × 2 | 97 | 0 |
| 2 | 0.5 | 2.0 | 5 | 10 × 2 | 90 | 0 |
| 3 | 0.5 | 1.5 | 5 | 10 × 3 | 91 | 0 |
| 4 | 0.5 | 1.0 | 5 | 10 × 4 | 41 | 55 |
| 5 | 0.5 | 2.5 | 0 | 10 × 12 | 0 | 100 |
| 6 | 0.5 | 2.5 | 2.5 | 10 × 5 | 69 | 26 |
| 7 | 0.5 | 2.5 | 3 | 10 × 3 | 78 | 17 |
| 8 | 0.5 | 2.5 | 4 | 10 × 3 | 91 | 0 |
| 9 | 0.5 | 1.5 | 4 | 10 × 3 | 90 | 0 |

| 3a–d′ | 1 | 2 | tb (min) | Yield c (%) | |||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | |||
| 3a | H | H | Me | H | Me | 10 × 3 | 90 |
| 3b | H | p-Me | Me | H | Me | 10 × 3 | 90 |
| 3c | 6-Me | p-Me | Me | H | Me | 10 × 3 | 95 |
| 3d | H | p-Cl | Me | H | Me | 10 × 4 | 87 |
| 3e | 6-Me | p-Cl | Me | H | Me | 10 × 4 | 90 |
| 3f | H | p-Cl, m-NO2 | Me | H | Me | 10 × 4 | 90 |
| 3g | 6-Me | p-Cl, m-NO2 | Me | H | Me | 10 × 4 | 89 |
| 3h | H | m-NO2 | Me | H | Me | 10 × 4 | 85 |
| 3i | 6-Me | m-NO2 | Me | H | Me | 10 × 4 | 84 |
| 3j | 5-CN | m-NO2 | Me | H | Me | 10 × 4 | 81 |
| 3k | 5-CN | p-Me | Me | H | Me | 10 × 4 | 84 |
| 3l | H | p-OMe | Me | H | Me | 10 × 3 | 90 |
| 3m | 6-Me | p-OMe | Me | H | Me | 10 × 3 | 93 |
| 3n | H | p-Me | Et | H | Me | 10 × 3 | 87 |
| 3o | H | H | n-Pr | H | Me | 10 × 3 | 80 |
| 3p | H | p-Me | n-Pr | H | Me | 10 × 3 | 86 |
| 3c | H | p-Cl | Et | H | Me | 10 × 3 | 88 |
| 3r | H | H | Me | m-Me | Me | 10 × 3 | 84 |
| 3s | H | p-Me | Me | m-Me | Me | 10 × 3 | 87 |
| 3t | 6-Me | m-NO2 | Me | H | Et | 10 × 3 | 81 |
| 3u | H | p-Me | Et | H | Et | 10 × 3 | 88 |
| 3v | H | p-Me | n-Pr | H | Et | 10 × 3 | 87 |
| 3w | H | H | Me | m-Me | Et | 10 × 3 | 85 |
| 3s | 5-NO2 | H | Me | H | Me | 10 × 4 | 90 |
| 3y | 6-Me | H | Me | H | Me | 10 × 3 | 81 |
| 3z | 5-CN | H | Me | H | Me | 10 × 4 | 83 |
| 3a′ | 5-CN | p-Cl | Me | H | Me | 10 × 4 | 80 |
| 3b′ | 5-NO2 | p-Me | Me | H | Me | 10 × 4 | 87 |
| 3c′ | 5-NO2 | p-Cl | Me | H | Me | 10 × 4 | 88 |
| 3d′ | H | H | Et | H | Me | 10 × 3 | 82 |

| Entry | Amount of (mmol) | HgCl2 (mol%) | t (min) | Isolated Yield a (%) | |
|---|---|---|---|---|---|
| 1a | 2a | 3a | |||
| 1 | 3 | 9 | 4 | 5 | 98 |
| 2 | 3 | 9 | 4 | 6 | 98 |
| 3 | 3 | 9 | 4 | 8 | 97 |
| 4 | 3 | 9 | 4 | 9 | 98 |
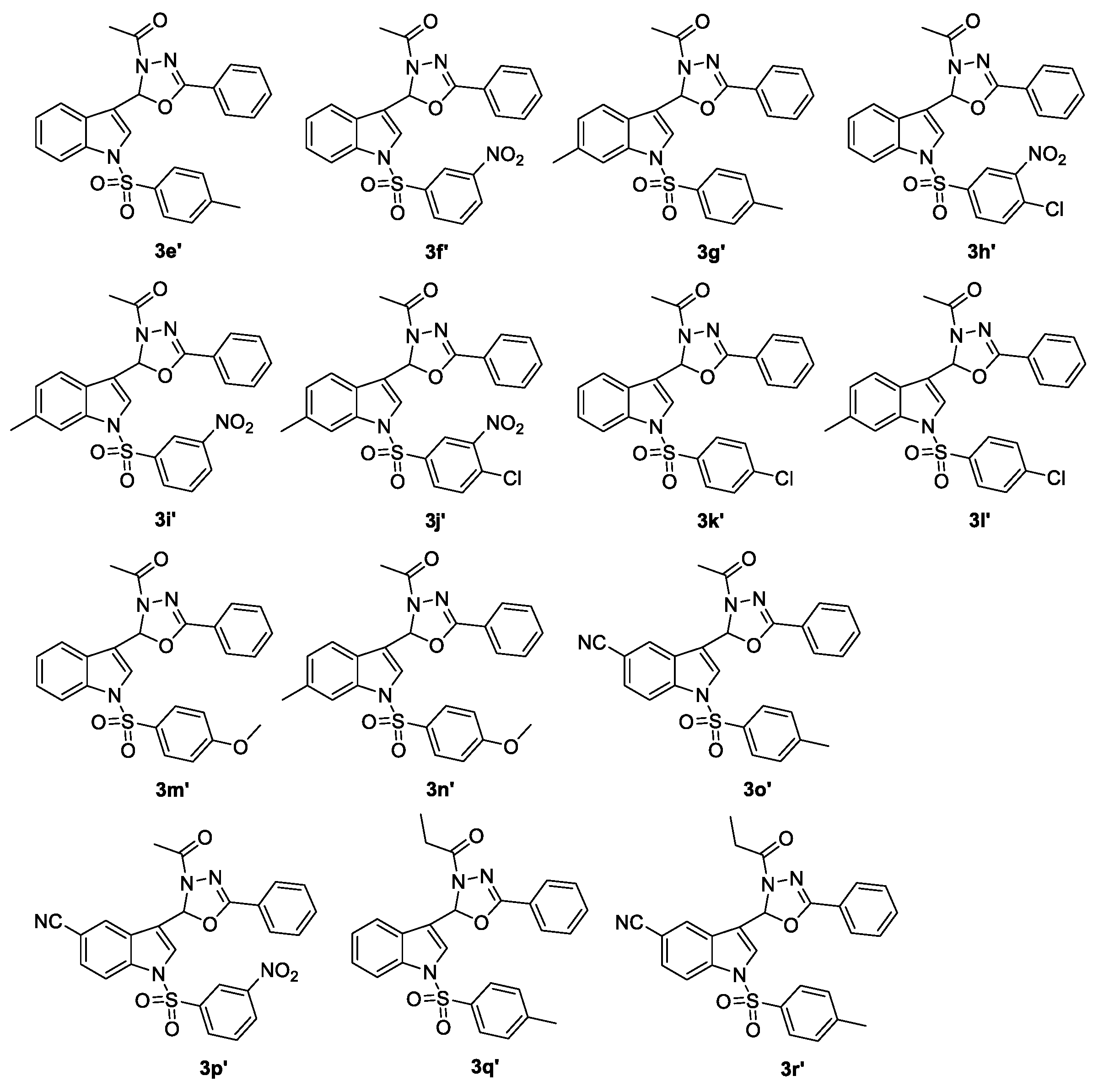
| 3s–r′ | CC50 b (μg/mL) | EC50 c (μg/mL) | TI d |
|---|---|---|---|
| 3s | 109.42 | 3.35 | 32.66 |
| 3y | >200 | 6.12 | >32.68 |
| 3z | 53.03 | 17.57 | 3.01 |
| 3a′ | 113 | 3.63 | 31.22 |
| 3b′ | 132.88 | 9.54 | 13.94 |
| 3c′ | 53.4 | 6.01 | 8.89 |
| 3d′ | 105.21 | 20.33 | 5.18 |
| 3e′ | 15.09 | 15.52 | 0.97 |
| 3f′ | 43.45 | 1.79 | 24.27 |
| 3g′ | 80.79 | 22.48 | 3.59 |
| 3h′ | 4.8 | 3.53 | 1.36 |
| 3i′ | 20.19 | 0.51 | 39.59 |
| 3j′ | 2.26 | 12.42 | 0.18 |
| 3k′ | 16.57 | 19.37 | 0.86 |
| 3l′ | 181.84 | 57.49 | 3.16 |
| 3m′ | 12.33 | 22.48 | 0.55 |
| 3n′ | 11.19 | 16.56 | 0.68 |
| 3o′ | 151.65 | 18.14 | 8.36 |
| 3p′ | 134.98 | 63.8 | 2.12 |
| 3q′ | 78.04 | 3.00 | 26.01 |
| 3r′ | 98.31 | 4.01 | 24.51 |
| AZT e | 1373.17 | 0.00199 | 690,035 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che, Z.; Tian, Y.; Liu, S.; Jiang, J.; Hu, M.; Chen, G. Microwave-Assisted Expeditious Synthesis of 2-Alkyl-2-(N-arylsulfonylindol-3-yl)-3-N-acyl-5-aryl-1,3,4-oxadiazolines Catalyzed by HgCl2 under Solvent-Free Conditions as Potential Anti-HIV-1 Agents. Molecules 2018, 23, 2936. https://doi.org/10.3390/molecules23112936
Che Z, Tian Y, Liu S, Jiang J, Hu M, Chen G. Microwave-Assisted Expeditious Synthesis of 2-Alkyl-2-(N-arylsulfonylindol-3-yl)-3-N-acyl-5-aryl-1,3,4-oxadiazolines Catalyzed by HgCl2 under Solvent-Free Conditions as Potential Anti-HIV-1 Agents. Molecules. 2018; 23(11):2936. https://doi.org/10.3390/molecules23112936
Chicago/Turabian StyleChe, Zhiping, Yuee Tian, Shengming Liu, Jia Jiang, Mei Hu, and Genqiang Chen. 2018. "Microwave-Assisted Expeditious Synthesis of 2-Alkyl-2-(N-arylsulfonylindol-3-yl)-3-N-acyl-5-aryl-1,3,4-oxadiazolines Catalyzed by HgCl2 under Solvent-Free Conditions as Potential Anti-HIV-1 Agents" Molecules 23, no. 11: 2936. https://doi.org/10.3390/molecules23112936
APA StyleChe, Z., Tian, Y., Liu, S., Jiang, J., Hu, M., & Chen, G. (2018). Microwave-Assisted Expeditious Synthesis of 2-Alkyl-2-(N-arylsulfonylindol-3-yl)-3-N-acyl-5-aryl-1,3,4-oxadiazolines Catalyzed by HgCl2 under Solvent-Free Conditions as Potential Anti-HIV-1 Agents. Molecules, 23(11), 2936. https://doi.org/10.3390/molecules23112936