
Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma

Abstract
1. Introduction
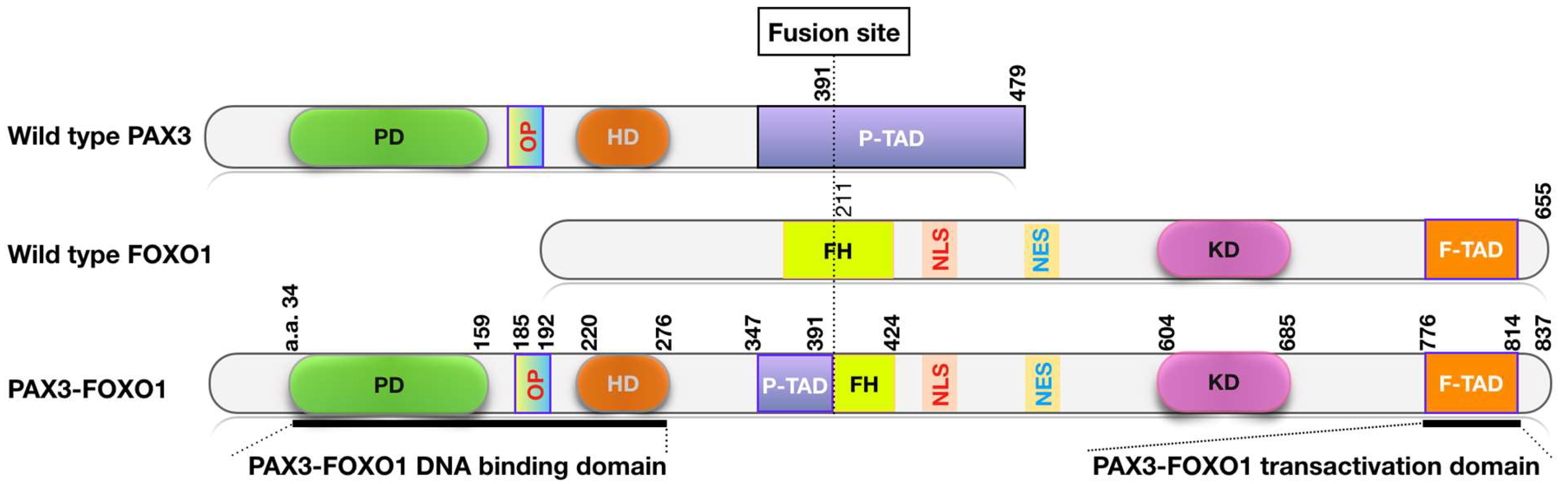
2. PAX-FOXO1 Fusions are Oncogenic Drivers and Therapeutic Targets in FP RMS Tumors
3. Pharmaceutical Targeting of PAX3-FOXO1 Regulatory and Transcriptional Pathways
3.1. Direct Inhibition of PAX3-FOXO1 by Small Molecules
3.2. Inhibition of PAX3-FOXO1 Regulatory Networks
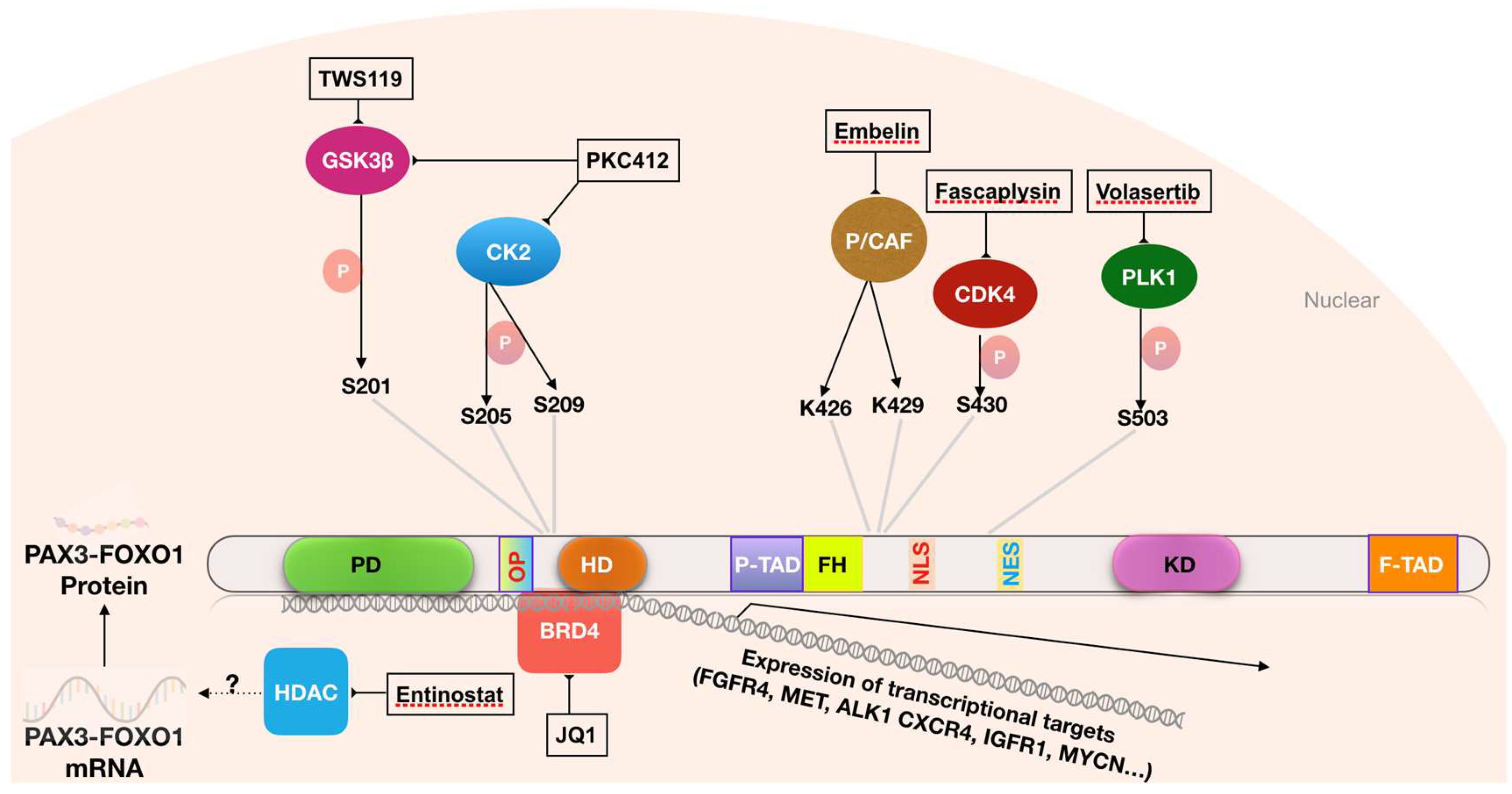
3.2.1. Targeting Phosphorylation of PAX3-FOXO1
3.2.2. Targeting Transcriptional Co-Activators of PAX3-FOXO1
3.2.3. Targeting the Acetylation of PAX3-FOXO1
3.3 Targeting Downstream Effectors of PAX3-FOXO1
4. Immunotherapy Applications to Target PAX3-FOXO1 and Downstream Pathways
4.1. Targeting PAX3-FOXO1 by Immunotherapy
4.2. Targeting Cell Surface Targets of PAX3-FOXO1 by Monoclonal Antibodies
4.3. Targeting Cell Surface Targets of PAX3-FOXO1 by CAR T-Cells
5. Future Approaches in the Treatment of RMS
5.1. Inhibition of PAX3-FOXO1 Expression by Oligonucleotide-Based Technologies
5.2. Identification of New Therapeutic Targets for RMS
6. Ongoing Challenges in the Development of Therapies for RMS
7. Conclusions
Funding
Conflicts of Interest
References
- Linet, M.S.; Ries, L.A.; Smith, M.A.; Tarone, R.E.; Devesa, S.S. Cancer surveillance series: Recent trends in childhood cancer incidence and mortality in the united states. J. Natl. Cancer Inst. 1999, 91, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Ognjanovic, S.; Linabery, A.M.; Charbonneau, B.; Ross, J.A. Trends in childhood rhabdomyosarcoma incidence and survival in the united states, 1975–2005. Cancer 2009, 115, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Weihkopf, T.; Blettner, M.; Dantonello, T.; Jung, I.; Klingebiel, T.; Koscielniak, E.; Lückel, M.; Spix, C.; Kaatsch, P. Incidence and time trends of soft tissue sarcomas in german children 1985–2004–A report from the population-based german childhood cancer registry. Eur. J. Cancer 2008, 44, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA: Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Gatta, G.; Trama, A.; Capocaccia, R.; Hackl, M.; Eycken, E.V.; Henau, K.; Dimitrova, N.; Sekerija, M.; Dušek, L.; Mägi, M.; et al. Epidemiology of rare cancers and inequalities in oncologic outcomes. Eur. J. Surg. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pediatric Bone and Soft Tissue Sarcomas; Pappo, A.S., Ed.; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2006; ISBN 978-3-540-40843-7. [Google Scholar]
- Yang, L.; Takimoto, T.; Fujimoto, J. Prognostic model for predicting overall survival in children and adolescents with rhabdomyosarcoma. BMC Cancer 2014, 14, 395–397. [Google Scholar] [CrossRef] [PubMed]
- O'Neill, J.P.; Bilsky, M.H.; Kraus, D. Head and neck sarcomas. Neurosurg. Clin. N. Am. 2013, 24, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Parham, D.M.; Barr, F.G. Classification of rhabdomyosarcoma and its molecular basis. Adv. Anatomic Pathol. 2013, 20, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G.; Nauta, L.E.; Davis, R.J.; Schafer, B.W.; Nycum, L.M.; Biegel, J.A. In Vivo amplification of the pax3-fkhr and pax7-fkhr fusion genes in alveolar rhabdomyosarcoma. Hum. Mol. Genet. 1996, 5, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J.; Barr, F.G. Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 1997, 94, 8047–8051. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.H.B.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHRand PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J. Clin. Oncol. 2002, 20, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- de Alava, E.; Ladanyi, M.; Rosai, J.; Gerald, W.L. Detection of chimeric transcripts in desmoplastic small round cell tumor and related developmental tumors by reverse transcriptase polymerase chain reaction. A specific diagnostic assay. Am. J. Pathol. 1995, 147, 1584–1591. [Google Scholar] [PubMed]
- Frascella, E.; Toffolatti, L.; Rosolen, A. Normal and rearranged PAX3 expression in human rhabdomyosarcoma. Cancer Genet. Cytogenet. 1998, 102, 104–109. [Google Scholar] [CrossRef]
- Barr, F.G.; Smith, L.M.; Lynch, J.C.; Strzelecki, D.; Parham, D.M.; Qualman, S.J.; Breitfeld, P.P. Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the intergroup rhabdomyosarcoma study-III trial. J. Mol. Diagn. 2006, 8, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Olanich, M.E.; Barr, F.G. A Call to ARMS: Targeting the PAX3-FOXO1gene in Alveolar Rhabdomyosarcoma. Expert Opin. Ther. Targets 2013, 17, 607–623. [Google Scholar] [CrossRef] [PubMed]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G.; Qualman, S.J.; Macris, M.H.; Melnyk, N.; Lawlor, E.R.; Strzelecki, D.M.; Triche, T.J.; Bridge, J.A.; Sorensen, P.H.B. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res. 2002, 62, 4704–4710. [Google Scholar] [PubMed]
- Wachtel, M.; Dettling, M.; Koscielniak, E.; Stegmaier, S.; Treuner, J.; Simon-Klingenstein, K.; Bühlmann, P.; Niggli, F.K.; Schäfer, B.W. Gene expression signatures identify rhabdomyosarcoma subtypes and detect a novel T(2;2)(Q35;P23) translocation fusing PAX3 to NCOA1. Cancer Res. 2004, 64, 5539–5545. [Google Scholar] [CrossRef] [PubMed]
- Chmielecki, J.; Bailey, M.; He, J.; Elvin, J.; Vergilio, J.-A.; Ramkissoon, S.; Suh, J.; Frampton, G.M.; Sun, J.X.; Morley, S.; et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res. 2017, 77, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, J.M.; Knezevich, S.R.; Mathers, J.A.; Sorensen, P.H. Molecular detection of the ETV6-NTRK3 gene fusion differentiates congenital fibrosarcoma from other childhood spindle cell tumors. Am. J. Surg. Pathol. 2000, 24, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, G.; Guanciali Franchi, P.; Stuppia, L.; Rossi, C.; Bianchi, C.; Antonucci, A.; Palka, G. Translocation (8;11)(Q12–13;Q21) in embryonal rhabdomyosarcoma. Cancer Genet. Cytogenet. 1992, 58, 210–211. [Google Scholar] [CrossRef]
- Sirvent, N.; Trassard, M.; Ebran, N.; Attias, R.; Pedeutour, F. Fusion of EWSR1 with the DUX4 facioscapulohumeral muscular dystrophy region resulting from T(4;22)(Q35;Q12) in a case of embryonal rhabdomyosarcoma. Cancer Genet. Cytogenet. 2009, 195, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.; Missiaglia, E.; de Reyniès, A.; Pierron, G.; Thuille, B.; Palenzuela, G.; Thway, K.; Orbach, D.; LAÉ, M.; Fréneaux, P.; et al. Fusion gene–negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J. Clin. Oncol. 2010, 28, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Davicioni, E.; Anderson, M.J.; Finckenstein, F.G.; Lynch, J.C.; Qualman, S.J.; Shimada, H.; Schofield, D.E.; Buckley, J.D.; Meyer, W.H.; Sorensen, P.H.B.; et al. Molecular classification of rhabdomyosarcoma-genotypic and phenotypic determinants of diagnosis: A report from the children's oncology group. Am. J. Pathol. 2009, 174, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.-P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Shimose, S.; Fujimori, J.; Furuta, T.; Ochi, M. Prognostic value of PAX3/7–FOXO1 fusion status in alveolar rhabdomyosarcoma: systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2015, 96, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Smith, L.M.; Gustafson, D.M.; Zhang, C.; Dunlevy, M.J.; Gastier-Foster, J.M.; Barr, F.G. Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the children's oncology group. Genes Chromosom. Cancer 2012, 51, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Selfe, J.; Olmos, D.; Al-Saadi, R.; Thway, K.; Chisholm, J.; Kelsey, A.; Shipley, J. Impact of fusion gene status versus histology on risk-stratification for rhabdomyosarcoma: retrospective analyses of patients on uk trials. Pediatr. Blood Cancer 2016, 64, e26386. [Google Scholar] [CrossRef] [PubMed]
- Eguía-Aguilar, P.; López-Martínez, B.; Retana-Contreras, C.; Perezpeña-Diazconti, M. Alveolar Rhabdomyosarcoma: Origin and prognostic implications of molecular findings. Boletín Médico del Hospital Infantil de México 2017, 73, 405–410. [Google Scholar]
- Huh, W.; Egas Bejar, D. Rhabdomyosarcoma in adolescent and young adult patients: Current perspectives. Adolesc. Health Med. Ther. 2014, 5, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, H. Current status of treatment for pediatric rhabdomyosarcoma in the usa and japan. Pediatr. Int. 2016, 58, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaf, W.T.A.; Orbach, D.; Judson, I.R.; Ferrari, A. Soft tissue sarcomas in adolescents and young adults: A comparison with their paediatric and adult counterparts. Lancet Oncol. 2017, 18, e166–e175. [Google Scholar] [CrossRef]
- Ray, A.; Huh, W.W. Current state-of-the-art systemic therapy for pediatric soft tissue sarcomas. Curr. Oncol. Rep. 2012, 14, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Crist, W.M.; Garnsey, L.; Beltangady, M.S.; Gehan, E.; Ruymann, F.; Webber, B.; Hays, D.M.; Wharam, M.; Maurer, H.M. Prognosis in children with rhabdomyosarcoma: a report of the intergroup rhabdomyosarcoma studies I and II. Intergroup rhabdomyosarcoma committee. J. Clin. Oncol. 1990, 8, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Hibbitts, E.; Hawkins, D.S.; Arndt, C.; Chi, Y.Y. Risk stratification including FOXO1 fusion status (FOXO1) in patients with rhabdomyosarcoma (RMS) treated on six recent frontline trials: A report from the children's oncology group. J. Clin. Oncol. 2017, 35 (Suppl. S15), 10528. [Google Scholar] [CrossRef]
- Breneman, J.C.; Lyden, E.; Pappo, A.S.; Link, M.P.; Anderson, J.R.; Parham, D.M.; Qualman, S.J.; Wharam, M.D.; Donaldson, S.S.; Maurer, H.M.; et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma—A report from the intergroup rhabdomyosarcoma study IV. J. Clin. Oncol. 2003, 21, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.A.S.; Stoner, J.A.; Hawkins, D.S.; Rodeberg, D.A.; Hayes-Jordan, A.A.; Paidas, C.N.; Parham, D.M.; Teot, L.A.; Wharam, M.D.; Breneman, J.C.; et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children’s oncology group study D9803. J. Clin. Oncol. 2009, 27, 5182–5188. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.J.; Schwartz, C. Survivorship. J. Surg. Oncol. 2015, 111, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 2001, 20, 5736–5746. [Google Scholar] [CrossRef] [PubMed]
- Linardic, C.M. PAX3–FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett. 2008, 270, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G.; Duan, F.; Smith, L.M.; Gustafson, D.; Pitts, M.; Hammond, S.; Gastier-Foster, J.M. Genomic and clinical analyses of 2p24 and 12q13-Q14 amplification in alveolar rhabdomyosarcoma: A report from the children’s oncology group. Genes Chromosom. Cancer 2009, 48, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Olanich, M.E.; Sun, W.; Hewitt, S.M.; Abdullaev, Z.; Pack, S.D.; Barr, F.G. CDK4 amplification reduces sensitivity to cdk4/6 inhibition in fusion-positive rhabdomyosarcoma. Clin. Cancer Res. 2015, 21, 4947–4959. [Google Scholar] [CrossRef] [PubMed]
- Bridge, J.A.; Liu, J.; Qualman, S.J.; Suijkerbuijk, R.; Wenger, G.; Zhang, J.; Wan, X.; Baker, K.S.; Sorensen, P.; Barr, F.G. Genomic gains and losses are similar in genetic and histologic subsets of rhabdomyosarcoma, whereas amplification predominates in embryonal with anaplasia and alveolar subtypes. Genes Chromosom. Cancer 2002, 33, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Nishimura, R.; Yoshida, K.; Shimamura, T.; Shiraishi, Y.; Sato, Y.; Kato, M.; Chiba, K.; Tanaka, H.; Hoshino, N.; et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat. Commun. 2015, 6, 3395–3398. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.G.; Galili, N.; Holick, J.; Biegel, J.A.; Rovera, G.; Emanuel, B.S. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 3, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to pax3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Relaix, F. PAX3 and PAX7 as upstream regulators of myogenesis. Semin. Cell Dev. Biol. 2015, 44, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Fredericks, W.J.; Galili, N.; Mukhopadhyay, S.; Rovera, G.; Bennicelli, J.; Barr, F.G.; Rauscher, F.J., III. The PAX3-FKHR fusion protein created by the T(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol. Cell. Biol. 1995, 15, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Begum, S.; Emani, N.; Cheung, A.; Wilkins, O.; Der, S.; Hamel, P.A. Cell-Type-Specific regulation of distinct sets of gene targets by Pax3 and Pax3/FKHR. Oncogene 2005, 24, 1860–1872. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.; Arenkiel, B.R.; Coffin, C.M.; El-Bardeesy, N.; DePinho, R.A.; Capecchi, M.R. Alveolar rhabdomyosarcomas in conditional pax3:fkhr mice: Cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004, 18, 2614–2626. [Google Scholar] [CrossRef] [PubMed]
- Nishijo, K.; Chen, Q.-R.; Zhang, L.; McCleish, A.T.; Rodriguez, A.; Cho, M.J.; Prajapati, S.I.; Gelfond, J.A.L.; Chisholm, G.B.; Michalek, J.E.; et al. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer Res. 2009, 69, 2902–2911. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Tsuchiya, K.; Otabe, O.; Gotoh, T.; Tamura, S.; Katsumi, Y.; Yagyu, S.; Tsubai-Shimizu, S.; Miyachi, M.; Iehara, T.; et al. Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem. Biophys. Res. Commun. 2008, 365, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.R.; Chatterjee, B.; Olanich, M.E.; Khan, J.; Miettinen, M.M.; Hewitt, S.M.; Barr, F.G. PAX3-FOXO1 is essential for tumour initiation and maintenance but not recurrence in a human myoblast model of rhabdomyosarcoma. J. Pathol. 2017, 241, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.H.; Zhao, H.; Womer, R.B.; Barr, F.G. Proliferative and apoptotic differences between alveolar rhabdomyosarcoma subtypes: A comparative study of tumors containing PAX3-FKHR or PAX7-FKHR gene fusions. Med. Pediatr. Oncol. 2001, 37, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Ramsay, A.; Gould, S.; Pritchard-Jones, K. PAX3-FKHR induces morphological change and enhances cellular proliferation and invasion in rhabdomyosarcoma. Am. J. Pathol. 2001, 159, 1089–1096. [Google Scholar] [CrossRef]
- Bernasconi, M.; Remppis, A.; Fredericks, W.J.; Rauscher, F.J.; Schafer, B.W. Induction of Apoptosis in Rhabdomyosarcoma Cells Through Down-Regulation of PAX Proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 13164–13169. [Google Scholar] [CrossRef] [PubMed]
- Ayyanathan, K.; Fredericks, W.J.; Berking, C.; Herlyn, M.; Balakrishnan, C.; Gunther, E.; Rauscher, F.J. Hormone-dependent tumor regression in vivo by an inducible transcriptional repressor directed at the PAX3-FKHR oncogene. Cancer Res. 2000, 60, 5803–5814. [Google Scholar] [PubMed]
- Keller, C.; Capecchi, M.R. New genetic tactics to model alveolar rhabdomyosarcoma in the mouse. Cancer Res. 2005, 65, 7530–7532. [Google Scholar] [CrossRef] [PubMed]
- Scheidler, S.; Fredericks, W.J.; Rauscher, F.J.; Barr, F.G.; Vogt, P.K. The hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts in culture. Proc. Natl. Acad. Sci. USA 1996, 93, 9805–9809. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.-X.; Finckenstein, F.G.; Abdueva, D.A.; Shahbazian, V.; Chung, B.; Weinberg, K.I.; Triche, T.J.; Shimada, H.; Anderson, M.J. Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res. 2008, 68, 6587–6597. [Google Scholar] [CrossRef] [PubMed]
- Naini, S.; Etheridge, K.T.; Adam, S.J.; Qualman, S.J.; Bentley, R.C.; Counter, C.M.; Linardic, C.M. Defining the cooperative genetic changes that temporally drive alveolar rhabdomyosarcoma. Cancer Res. 2008, 68, 9583–9588. [Google Scholar] [CrossRef] [PubMed]
- Diller, L.; Sexsmith, E.; Gottlieb, A.; Li, F.P.; Malkin, D. Germline P53 mutations are frequently detected in young children with rhabdomyosarcoma. J. Clin. Investig. 1995, 95, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Obana, K.; Yang, H.-W.; Piao, H.-Y.; Taki, T.; Hashizume, K.; Hanada, R.; Yamamoto, K.; Tanaka, Y.; Toyoda, Y.; Takita, J.; et al. Aberrations of p16INK4A, p14ARF and p15INK4B genes in pediatric solid tumors. Int. J. Oncol. 2003, 23, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Faienza, M.F.; Coppola, B.; Rosolen, A.; Basso, G.; Della Ragione, F.; Schettini, F. Analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B, and CDKN2C) in childhood rhabdomyosarcoma. Genes Chromosom. Cancer 1996, 15, 217–222. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Clayton, S.J.; Girdler, F.; Brotherick, I.; Shenton, B.; Browell, D.; Stuart, M.; Fox, J.C.; Ceuppens, P.; Foy, C.A.; et al. ARMS allele-specific amplification-based detection of mutant P53 DNA and mRNA in tumors of the breast. Clin. Chem. 2001, 47, 774–778. [Google Scholar] [PubMed]
- Linardic, C.M.; Naini, S.; Herndon, J.E.; Kesserwan, C.; Qualman, S.J.; Counter, C.M. The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res. 2007, 67, 6691–6699. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Lagutina, I.; Grosveld, G.C. PAX3-FOXO1 induces cannabinoid receptor 1 to enhance cell invasion and metastasis. Cancer Res. 2011, 71, 7471–7480. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Grosveld, G.C. Alveolar rhabdomyosarcoma–the molecular drivers of PAX3/7-FOXO1-induced tumorigenesis. Skelet Muscle 2012, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Ebauer, M.; Wachtel, M.; Niggli, F.K.; Schafer, B.W. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene 2007, 26, 7267–7281. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A Chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 2013, 3, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Babiceanu, M.; Kumar, S.; Jia, Y.; Qin, F.; Barr, F.G.; Li, H. Fusion transcriptome profiling provides insights into alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 13126–13131. [Google Scholar] [CrossRef] [PubMed]
- Redell, M.S.; Tweardy, D.J. Targeting transcription factors in cancer: challenges and evolving strategies. Drug Discov Today Technol. 2006, 3, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the“Undruggable”Cancer Targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.J.; Hollenbach, A.D. The oncogenic fusion protein Pax3–FKHR has a greater post-translational stability relative to pax3 during early myogenesis. Biochim. Biophys. Acta 2007, 1770, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Amstutz, R.; Wachtel, M.; Troxler, H.; Kleinert, P.; Ebauer, M.; Haneke, T.; Oehler-Janne, C.; Fabbro, D.; Niggli, F.K.; Schafer, B.W. Phosphorylation regulates transcriptional activity of PAX3/FKHR and reveals novel therapeutic possibilities. Cancer Res. 2008, 68, 3767–3776. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.-Y.; Dong, H.; Cui, J.; Liu, L.; Chen, T. Glycogen synthase kinase 3 regulates PAX3–FKHR-mediated cell proliferation in human alveolar rhabdomyosarcoma cells. Biochem. Biophys. Res. Commun. 2010, 391, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wu, J.; Ong, S.S.; Chen, T. Cyclin-dependent kinase 4 phosphorylates and positively regulates PAX3-FOXO1 in human alveolar rhabdomyosarcoma cells. PLoS ONE 2013, 8, e58193. [Google Scholar] [CrossRef] [PubMed]
- Thalhammer, V.; Lopez-Garcia, L.A.; Herrero-Martin, D.; Hecker, R.; Laubscher, D.; Gierisch, M.E.; Wachtel, M.; Bode, P.; Nanni, P.; Blank, B.; et al. PLK1 phosphorylates PAX3-FOXO1, the inhibition of which triggers regression of alveolar rhabdomyosarcoma. Cancer Res. 2015, 75, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Loupe, J.M.; Miller, P.J.; Ruffin, D.R.; Stark, M.W.; Hollenbach, A.D. Inhibiting phosphorylation of the oncogenic PAX3-FOXO1 reduces alveolar rhabdomyosarcoma phenotypes identifying novel therapy options. Oncogenesis 2015, 4, e145. [Google Scholar] [CrossRef] [PubMed]
- Dietz, K.N.; Miller, P.J.; Iyengar, A.S.; Loupe, J.M.; Hollenbach, A.D. Identification of serines 201 and 209 as sites of Pax3 phosphorylation and the altered phosphorylation status of Pax3-FOXO1 during early myogenic differentiation. Int. J. Biochem. Cell Biol. 2011, 43, 936–945. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, A.S.; Loupe, J.M.; Miller, P.J.; Hollenbach, A.D. Identification of CK2 as the kinase that phosphorylates Pax3 at Ser209 in early myogenic differentiation. Biochem. Biophys. Res. Commun. 2012, 428, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Jothi, M.; Mal, M.; Keller, C.; Mal, A.K. Small molecule inhibition of PAX3-FOXO1 through AKT activation suppresses malignant phenotypes of alveolar rhabdomyosarcoma. Mol. Cancer Ther. 2013, 12, 2663–2674. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Yohe, M.E.; Chou, H.-C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3–FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Bharathy, N.; Suriyamurthy, S.; Rao, V.K.; Ow, J.R.; Lim, H.J.; Chakraborty, P.; Vasudevan, M.; Dhamne, C.A.; Chang, K.T.E.; Min, V.L.K.; et al. P/CAF mediates PAX3-FOXO1-dependent oncogenesis in alveolar rhabdomyosarcoma. J. Pathol. 2016, 240, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Nuñez-Álvarez, Y.; Hettmer, S.; Carrió, E.; Chen, H.-I.H.; Nishijo, K.; Huang, E.T.; Prajapati, S.I.; Walker, R.L.; Davis, S.; et al. Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Dev. 2014, 28, 1578–1591. [Google Scholar] [CrossRef] [PubMed]
- Herrero Martín, D.; Boro, A.; Schäfer, B.W. Cell-based small-molecule compound screen identifies fenretinide as potential therapeutic for translocation-positive rhabdomyosarcoma. PLoS ONE 2013, 8, e55072. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lee, B.H.; Williams, I.R.; Kutok, J.L.; Mitsiades, C.S.; Duclos, N.; Cohen, S.; Adelsperger, J.; Okabe, R.; Coburn, A.; et al. FGFR3 as a therapeutic target of the small molecule inhibitor PKC412 in hematopoietic malignancies. Oncogene 2005, 24, 8259–8267. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, K.V.; Mayerhofer, M.; Aichberger, K.J.; Derdak, S.; Sonneck, K.; Böhm, A.; Gruze, A.; Samorapoompichit, P.; Manley, P.W.; Fabbro, D.; et al. PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: Comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 2006, 107, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Bahlis, N.J.; Miao, Y.; Koc, O.N.; Lee, K.; Boise, L.H.; Gerson, S.L. N-Benzoylstaurosporine (PKC412) inhibits akt kinase inducing apoptosis in multiple myeloma cells. Leuk. Lymphoma 2009, 46, 899–908. [Google Scholar] [CrossRef] [PubMed]
- del Peso, L.; González, V.M.; Hernández, R.; Barr, F.G.; Núñez, G. Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene 1999, 18, 7328–7333. [Google Scholar] [CrossRef] [PubMed]
- Zabidi, M.A.; Stark, A. Regulatory enhancer–core-promoter communication via transcription factors and cofactors. Trends Genet. 2016, 32, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Böhm, M.; Wachtel, M.; Marques, J.G.; Streiff, N.; Laubscher, D.; Nanni, P.; Mamchaoui, K.; Santoro, R.; Schäfer, B.W. Helicase CHD4 is an epigenetic coregulator of PAX3-FOXO1 in alveolar rhabdomyosarcoma. J. Clin. Investig. 2016, 126, 4237–4249. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wong, J.; Moreno, G.T.; Young, M.K.; Côté, J.; Wang, W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol. Cell 1998, 2, 851–861. [Google Scholar] [CrossRef]
- Zhou, M.-M.; Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 39, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M.; Heinrich, R. Biological control through regulated transcriptional coactivators. Cell 2004, 119, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Heinicke, U.; Kupka, J.; Fichter, I.; Fulda, S. Critical role of mitochondria-mediated apoptosis for JNJ-26481585-induced antitumor activity in rhabdomyosarcoma. Oncogene 2015, 35, 3729–3741. [Google Scholar] [CrossRef] [PubMed]
- Heinicke, U.; Kupka, J.; Fulda, S. JNJ-26481585 primes rhabdomyosarcoma cells for chemotherapeutics by engaging the mitochondrial pathway of apoptosis. Oncotarget 2015, 6, 37836–37851. [Google Scholar] [CrossRef] [PubMed]
- Haydn, T.; Metzger, E.; Schuele, R.; Fulda, S. Concomitant epigenetic targeting of LSD1 and HDAC synergistically induces mitochondrial apoptosis in rhabdomyosarcoma cells. Cell Death Dis. 2017, 8, e2879. [Google Scholar] [CrossRef] [PubMed]
- Enßle, J.C.; Boedicker, C.; Wanior, M.; Vogler, M.; Knapp, S.; Fulda, S. Co-targeting of BET proteins and hdacs as a novel approach to trigger apoptosis in rhabdomyosarcoma cells. Cancer Lett. 2018, 428, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Modak, R.; Basha, J.; Bharathy, N.; Maity, K.; Mizar, P.; Bhat, A.V.; Vasudevan, M.; Rao, V.K.; Kok, W.K.; Natesh, N.; et al. Probing P300/CBP associated factor (PCAF)-dependent pathways with a small molecule inhibitor. ACS Chem. Biol. 2013, 8, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Aoyama, H.; Yoshimochi, K.; Fukamizu, A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. USA 2005, 102, 11278–11283. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Dilworth, F.J.; Seaver, K.J.; Fishburn, A.L.; Htet, S.L.; Tapscott, S.J. In Vitro Transcription system delineates the distinct roles of the coactivators pCAF and P300 during MyoD/E47-dependent transactivation. Proc. Natl. Acad. Sci. USA 2004, 101, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Sartorelli, V.; Puri, P.L.; Hamamori, Y.; Ogryzko, V.; Chung, G.; Nakatani, Y.; Wang, J.Y.; Kedes, L. Acetylation of myod directed by PCAF is necessary for the execution of the muscle program. Mol. Cell 1999, 4, 725–734. [Google Scholar] [CrossRef]
- Davicioni, E.; Finckenstein, F.G.; Shahbazian, V.; Buckley, J.D.; Triche, T.J.; Anderson, M.J. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006, 66, 6936–6946. [Google Scholar] [CrossRef] [PubMed]
- Laé, M.; Ahn, E.H.; Mercado, G.E.; Chuai, S.; Edgar, M.; Pawel, B.R.; Olshen, A.; Barr, F.G.; Ladanyi, M. Global gene expression profiling of PAX-FKHR fusion-positive alveolar and PAX-FKHR fusion-negative embryonal rhabdomyosarcomas. J. Pathol. 2007, 212, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Crose, L.E.S.; Etheridge, K.T.; Chen, C.; Belyea, B.; Talbot, L.J.; Bentley, R.C.; Linardic, C.M. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin. Cancer Res. 2012, 18, 3780–3790. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Dong, J.; Sun, L.; Geng, L.; Wang, J.; Zheng, J.; Li, Y.; Bridge, J.; Hinrichs, S.H.; Ding, S.-J. Inhibition of phosphorylated C-Met in rhabdomyosarcoma cell lines by a small molecule inhibitor SU11274. J. Transl. Med. 2011, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Prajapati, S.I.; Nishijo, K.; Schaffer, B.S.; Taniguchi, E.; Kilcoyne, A.; McCleish, A.T.; Nelon, L.D.; Giles, F.G.; Efstratiadis, A.; et al. Evasion mechanisms to Igf1r inhibition in rhabdomyosarcoma. Mol. Cancer Ther. 2011, 10, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Manara, M.C.; Nicoletti, G.; Lollini, P.-L.; Lukas, S.; Benini, S.; Croci, S.; Perdichizzi, S.; Zambelli, D.; Serra, M.; et al. Antitumor activity of the insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 in musculoskeletal tumors. Cancer Res. 2005, 65, 3868–3876. [Google Scholar] [CrossRef] [PubMed]
- García-Echeverría, C.; Pearson, M.A.; Marti, A.; Meyer, T.; Mestan, J.; Zimmermann, J.; Gao, J.; Brueggen, J.; Capraro, H.-G.; Cozens, R.; et al. In vivo antitumor activity of NVP-AEW541—A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell 2004, 5, 231–239. [Google Scholar] [CrossRef]
- Faqar-Uz-Zaman, S.F.; Heinicke, U.; Meister, M.T.; Vogler, M.; Fulda, S. BCL-xl–Selective BH3 mimetic sensitizes rhabdomyosarcoma cells to chemotherapeutics by activation of the mitochondrial pathway of apoptosis. Cancer Lett. 2018, 412, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Peron, M.; Lovisa, F.; Poli, E.; Basso, G.; Bonvini, P. Understanding the interplay between expression, mutation and activity of ALK receptor in rhabdomyosarcoma cells for clinical application of small-molecule inhibitors. PLoS ONE 2015, 10, e0132330. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; McDowell, H.P.; Camero, S.; Mannarino, O.; Ceccarelli, S.; Paiano, M.; Losty, P.D.; Pizer, B.; Shukla, R.; Pizzuti, A.; et al. Crizotinib-induced antitumour activity in human alveolar rhabdomyosarcoma cells is not solely dependent on ALK and MET inhibition. J. Exp. Clin. Cancer Res. 2015, 34, 112. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010, 70, 6497–6508. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.A.; Song, B.; Lakkis, M.; Wang, C. Tumor-specific PAX3-FKHR transcription factor, but not PAX3, activates the platelet-derived growth factor alpha receptor. Mol. Cell. Biol. 1998, 18, 4118–4130. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, E.; Nishijo, K.; McCleish, A.T.; Michalek, J.E.; Grayson, M.H.; Infante, A.J.; Abboud, H.E.; LeGallo, R.D.; Qualman, S.J.; Rubin, B.P.; et al. PDGFR-a is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene 2008, 27, 6550–6560. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.E.; Xia, S.J.; Zhang, C.; Ahn, E.H.; Gustafson, D.M.; Laé, M.; Ladanyi, M.; Barr, F.G. Identification of PAX3-FKHR-regulated genes differentially expressed between alveolar and embryonal rhabdomyosarcoma: focus on MYCN as a biologically relevant target. Genes Chromosom. Cancer 2008, 47, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Walters, Z.S.; Villarejo-Balcells, B.; Olmos, D.; Buist, T.W.S.; Missiaglia, E.; Allen, R.; Al-Lazikani, B.; Garrett, M.D.; Blagg, J.; Shipley, J. JARID2 is a direct target of the PAX3-FOXO1 fusion protein and inhibits myogenic differentiation of rhabdomyosarcoma cells. Oncogene 2014, 33, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Oesch, S.; Walter, D.; Wachtel, M.; Pretre, K.; Salazar, M.; Guzmán, M.; Velasco, G.; Schäfer, B.W. Cannabinoid receptor 1 is a potential drug target for treatment of translocation-positive rhabdomyosarcoma. Mol. Cancer Ther. 2009, 8, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.-D.; Wu, J.; Cui, J.; Chen, T. Carnitine palmitoyltransferase 1A (CPT1A): A transcriptional target of PAX3-FKHR and mediates PAX3-FKHR-dependent motility in alveolar rhabdomyosarcoma cells. BMC Cancer 2012, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Q.; Cheuk, A.T.; Shern, J.F.; Song, Y.K.; Hurd, L.; Liao, H.; Wei, J.S.; Khan, J. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor Ponatinib (AP24534). PLoS ONE 2013, 8, e76551–e76559. [Google Scholar] [CrossRef] [PubMed]
- VI, J.G.T.; Cheuk, A.T.; Tsang, P.S.; Chung, J.-Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.-R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Baskar, S. Targeting FGFR4 with monoclonal antibodies as therapeutic agents for the treatment of rhabdomyosarcoma. Cancer Res. 2016, 76 (Suppl. S14), 4996. [Google Scholar] [CrossRef]
- Shivaprasad, N.; Xiong, Y.; Yohe, M.; Schneider, D.; Shern, J.; Baskar, S.; Dimitrov, D.; Sorenson, P.; Orentas, R.; Khan, J. Developing FGFR4 chimeric antigen receptor CAR T cell therapy against rhabdomyosarcoma. Mol. Ther. 2016, 24, S257–S258. [Google Scholar] [CrossRef]
- Dolgikh, N.; Fulda, S. Rhabdomyosarcoma cells are susceptible to cell death by LDK378 alone or in combination with sorafenib independently of anaplastic lymphoma kinase status. Anti-Cancer Drugs 2017, 28, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- van Erp, A.E.M.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Fleuren, E.D.G.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Targeting Anaplastic Lymphoma Kinase (ALK) in Rhabdomyosarcoma (RMS) with the Second-Generation ALK Inhibitor Ceritinib. Target. Oncol. 2017, 12, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Beltran, P.J.; Chung, Y.A.; Moody, G.; Mitchell, P.; Cajulis, E.; Vonderfecht, S.; Kendall, R.; Radinsky, R.; Calzone, F.J. Efficacy of Ganitumab (AMG 479), alone and in combination with rapamycin, in Ewing's and osteogenic sarcoma models. J. Pharmacol. Exp. Ther. 2011, 337, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Atzori, F.; Tabernero, J.; Cervantes, A.; Prudkin, L.; Andreu, J.; Rodríguez-Braun, E.; Domingo, A.; Guijarro, J.; Gamez, C.; Rodon, J.; et al. A phase I pharmacokinetic and pharmacodynamic study of Dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2011, 17, 6304–6312. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Vassal, G.; Crowley, J.J.; Bolejack, V.; Hogendoorn, P.C.W.; Chugh, R.; Ladanyi, M.; Grippo, J.F.; Dall, G.; Staddon, A.P.; et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a sarcoma alliance. Cancer 2014, 120, 2448–2456. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, M.; Tkacz, M.; Zgutka, K.; Bujak, J.; Kopytko, P.; Pawlik, A. Picropodophyllin (PPP) is a potent rhabdomyosarcoma growth inhibitor both in vitro and in vivo. BMC Cancer 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McDermott, U.; Ames, R.Y.; Iafrate, A.J.; Maheswaran, S.; Stubbs, H.; Greninger, P.; McCutcheon, K.; Milano, R.; Tam, A.; Lee, D.Y.; et al. Ligand-dependent platelet-derived growth factor receptor (PDGFR)- activation sensitizes rare lung cancer and sarcoma cells to PDGFR kinase inhibitors. Cancer Res. 2009, 69, 3937–3946. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Chua, Y.X.; Glover, J.M.; Tyner, J.W.; Loriaux, M.M.; Kilcoyne, A.; Giles, F.J.; Nelon, L.D.; Carew, J.S.; Ouyang, Y.; et al. An adaptive Src–PDGFRA–Raf axis in rhabdomyosarcoma. Biochem. Biophys. Res. Commun. 2012, 426, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The role of small molecule platelet-derived growth factor receptor (PDGFR) inhibitors in the treatment of neoplastic disorders. Pharmacol. Res. 2018, 129, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.P.; Todd, J.R.; Finetti, M.A.; McCarthy, F.; Broncel, M.; Vyse, S.; Luczynski, M.T.; Crosier, S.; Ryall, K.A.; Holmes, K.; et al. Dual targeting of PDGFRα and FGFR1 displays synergistic efficacy in malignant rhabdoid tumors. Cell Rep. 2016, 17, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Leruste, A.; Tauziede-Espariat, A.; Andrianteranagna, M.; Surdez, D.; Lescure, A.; Han, Z.-Y.; Anthony, E.; Richer, W.; Baulande, S.; et al. High-throughput drug screening identifies Pazopanib and Clofilium Tosylate as promising treatments for malignant rhabdoid tumors. Cell Rep. 2017, 21, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Lowery, C.D.; Blosser, W.; Dowless, M.; Knoche, S.; Stephens, J.; Li, H.; Surguladze, D.; Loizos, N.; Luffer-Atlas, D.; Oakley, G.J.; et al. Olaratumab exerts antitumor activity in preclinical models of pediatric bone and soft tissue tumors through inhibition of platelet-derived growth factor receptor Α. Clin. Cancer Res. 2018, 24, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Miekus, K.; Lukasiewicz, E.; Jarocha, D.; Sekula, M.; Drabik, G.; Majka, M. The decreased metastatic potential of rhabdomyosarcoma cells obtained through MET receptor downregulation and the induction of differentiation. Cell Death Dis. 2013, 4, e459. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Watanabe, M.; Sato, Y.; Hata, J.; Ishii, N.; Aoki, Y. Inhibition of metastasis of rhabdomyosarcoma by a novel neutralizing antibody to CXC chemokine receptor-4. Cancer Sci. 2014, 105, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, R.; McIntyre, A.; Camerin, C.; Walters, Z.S.; Di Leo, K.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor activity of sustained N-Myc reduction in rhabdomyosarcomas and transcriptional block by antigene therapy. Clin. Cancer Res. 2012, 18, 796–807. [Google Scholar] [CrossRef] [PubMed]
- AHN, E.H.; Lee, M.B.; Seo, D.J.; Lee, J.; Kim, Y.; Gupta, K. Sphingosine induces apoptosis and down-regulation of MYCN in PAX3-FOXO1-positive alveolar rhabdomyosarcoma cells irrespective of TP53 mutation. Anticancer Res. 2018, 38, 71–76. [Google Scholar] [PubMed]
- Luo, X.; Liu, Y.; Kubicek, S.; Myllyharju, J.; Tumber, A.; Ng, S.; Che, K.H.; Podoll, J.; Heightman, T.D.; Oppermann, U.; et al. A selective inhibitor and probe of the cellular functions of Jumonji C domain-containing histone demethylases. J. Am. Chem. Soc. 2011, 133, 9451–9456. [Google Scholar] [CrossRef] [PubMed]
- Hernlund, E.; Ihrlund, L.S.; Khan, O.; Ates, Y.O.; Linder, S.; Panaretakis, T.; Shoshan, M.C. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-Deoxyglucose and etomoxir. Int. J. Cancer 2008, 123, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Dagher, R.; Long, L.M.; Read, E.J.; Leitman, S.F.; Carter, C.S.; Tsokos, M.; Goletz, T.J.; Avila, N.; Berzofsky, J.A.; Helman, L.J.; et al. Pilot trial of tumor-specific peptide vaccination and continuous infusion Interleukin-2 in patients with recurrent ewing sarcoma and alveolar rhabdomyosarcoma: An inter-institute NIH study. Med. Pediatr. Oncol. 2002, 38, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Rodeberg, D.A.; Nuss, R.A.; Heppelmann, C.J.; Celis, E. Lack of effective T-Lymphocyte response to the PAX3/FKHR translocation area in alveolar rhabdomyosarcoma. Cancer Immunol. Immunother. 2005, 54, 526–534. [Google Scholar] [CrossRef] [PubMed]
- van den Broeke, L.T.; Pendleton, C.D.; Mackall, C.; Helman, L.J.; Berzofsky, J.A. Identification and epitope enhancement of a PAX-FKHR fusion protein breakpoint epitope in alveolar rhabdomyosarcoma cells created by a tumorigenic chromosomal translocation inducing CTL capable of lysing human tumors. Cancer Res. 2006, 66, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Mackall, C.L.; Rhee, E.H.; Read, E.J.; Khuu, H.M.; Leitman, S.F.; Bernstein, D.; Tesso, M.; Long, L.M.; Grindler, D.; Merino, M.; et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin. Cancer Res. 2008, 14, 4850–4858. [Google Scholar] [CrossRef] [PubMed]
- Mayeenuddin, L.H.; Yu, Y.; Kang, Z.; Helman, L.J.; Cao, L. Insulin-like growth factor 1 receptor antibody induces rhabdomyosarcoma cell death via a process involving AKT and Bcl-xL. Oncogene 2010, 29, 6367–6377. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.-P.; Chawla, S.P.; Toner, G.C.; Maki, R.G.; Meyers, P.A.; Chugh, R.; et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory ewing sarcoma family of tumors: results of a phase II sarcoma alliance for research through collaboration study. J. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef] [PubMed]
- Vela, M. Anti CXCR4 antibody combined with activated and expanded natural killer cells for sarcoma immunotherapy. J. Clin. Oncol. 2018, 36 (Suppl. S15), 11541. [Google Scholar] [CrossRef]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T Cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Wolchok, J.D.; Chan, T.A.; Mellman, I.; Palucka, K.; Banchereau, J.; Rosenberg, S.A.; Dane Wittrup, K. Immunotherapy: The path to win the war on cancer? Cell 2015, 161, 185–186. [Google Scholar] [PubMed]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using sleeping beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Frey, N.V.; Grupp, S.A.; Maude, S.L. CAR T cell therapy in acute Lymphoblastic Leukemia and potential for chronic lymphocytic leukemia. Curr. Treat. Options Oncol. 2016, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Riddell, S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to bench-to-bedside efficacy. J. Immunol. 2018, 200, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Skarzynski, M.; Shivaprasad, N.; Subramanian, B.; Azorsa, D.; Zhu, Z.; Dimitrov, D.; Khan, J. Abstract 693: Antibody-based targeting of the cell surface receptor tyrosine kinase FGFR4 in rhabdomyosarcoma and other cancers. Cancer Res. 2017, 77 (Suppl. S13), 693. [Google Scholar] [CrossRef]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-specific CAR T cells as a potential therapy for high risk sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, C. Identification of a new class of PAX3-FKHR target promoters: A role of the Pax3 paired box DNA binding domain. Oncogene 2006, 26, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.; Yang, H.; Chen, E. Targeted ablation of essential oncogenes in rhabdomyosarcoma with CRISPR/Cas9 gene therapy. Cancer Res. 2017, 77 (Suppl. S13), 5096. [Google Scholar] [CrossRef]
- Baquir, B.; Hancock, R.E.W. Exosomes, your body’s answer to immune health. Ann. Transl. Med. 2017, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.M.; Nguyen, Q.H.; Singh, M.; Razorenova, O.V. Approaches to identifying synthetic lethal interactions in cancer. Yale J. Biol. Med. 2015, 88, 145–155. [Google Scholar] [PubMed]
- Scholl, C.; Fröhling, S.; Dunn, I.F.; Schinzel, A.C.; Barbie, D.A.; Kim, S.Y.; Silver, S.J.; Tamayo, P.; Wadlow, R.C.; Ramaswamy, S.; et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009, 137, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; McMillan, E.; Kim, H.S.; Venkateswaran, N.; Makkar, G.; Rodriguez-Canales, J.; Villalobos, P.; Neggers, J.E.; Mendiratta, S.; Wei, S.; et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016, 538, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.A.; Li, X.; Xie, W.; Huang, S. MYC-mediated synthetic lethality for treating tumors. Curr. Cancer Drug Targets 2015, 15, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.P.; Varma, S.; Gottesman, M.M. The clinical relevance of cancer cell lines. J. Natl. Cancer Inst. 2013, 105, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wei, X.; Lin, S.; Qin, L.; Cheng, L.; Li, P. Current status and perspectives of patient-derived Xenograft models in cancer research. J. Hematol. Oncol. 2017, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. Patient-derived orthotopic Xenografts: Better mimic of metastasis than subcutaneous Xenografts. Nat. Rev. Cancer 2015, 15, 451–452. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Histologic Pattern | Fusion Status | Frequency | Associated Genetic Changes | Outcome |
|---|---|---|---|---|
| ARMS | FP | ~20% | ● PAX3/7-FOXO1 fusions [16,41,42] ● Amplification [43,44]: 2p24 (MYCN) 12q13-14 (CDK4) 13q31-32 (MIR17HG) | Poor |
| FN | ~5% | ● Point mutations [17]: RAS genes TP53 PIK3CA FGFR4 NF1 BCOR FBXW7 ● Loss of heterozygosity [17]: 11p15.5 9p21.3 (CDKN2A) ● Amplification [17]: 12q15 (MDM2) ● Chromosome copy number gains [45,46]: 2, 8 and 12 | Good | |
| ERMS | ~75% |
| Small Molecules | Relevant Molecular Targets | References |
|---|---|---|
| 1. Kinase inhibitors | ||
| TWS119 | GSK3 | [78] |
| SB212763 | GSK3 | [78] |
| TDZD-8 | GSK3 | [78] |
| Fascaplysin | CDK4 | [79] |
| PKC412 | Multiple kinases including CK2 and GSK3 | [19] |
| BI-2536 | PLK1 | [80] |
| BI-6727 (volasertib) | PLK1 | [80] |
| Thapsigargin | Ca2+ATPases | [84] |
| 2. Inhibitors of chromatin modifying complexes | ||
| JQ1 | BRD4 and BET proteins | [85] |
| OTX015 | BRD4 and BET proteins | [85] |
| iBET151 | BRD4 and BET proteins | [85] |
| iBET726 | BRD4 and BET proteins | [85] |
| iBET762 | BRD4 and BET proteins | [85] |
| Embelin | P/CAF | [86] |
| Entinostat | HDAC | [87] |
| SAHA | HDAC | [87] |
| 3. Unknown inhibitors | ||
| Fenretinide | Unknown | [88] |
| Genes | Targeting Agents | References | Clinical Trials #1 |
|---|---|---|---|
| 1. FGFR4 | Ponatinib | [124] | |
| PD173074 | [109,125] | ||
| FGF401 | NCT02325739 (Phase II) | ||
| FGFR4 MAB #2 | [126] | ||
| FGFR4 CAR T | [127] | ||
| 2. ALK1 | Crizotinib | [116] | NCT02034981 (II) |
| LDK378 | [128,129] | NCT01742286 (I) | |
| 3. IGF1R | Ganitumab #2 | [130] | NCT03041701(II) |
| Dalotuzumab #2 | [131] | NCT00694356 (I) | |
| R1507 | [132] | NCT00642941 (II) | |
| Picropodophyllin | [133] | ||
| 4. PDGFRα | Sorafenib | [134,135] | NCT01502410 (II) |
| Dasatinib | [119] | NCT00464620 (II) | |
| Sunitinib | [134] | NCT00474994 (II) | |
| Axitinib | [136] | NCT01140737 (II) | |
| Pazopanib | [137,138] | NCT01956669 (II) | |
| Olaratumab #2 | [139] | NCT01185964 (II) | |
| 5. MET | Crizotinib | [116] | NCT02034981 (II) |
| Tivantinib | [140] | ||
| 6. CXCR4 | MAB CF172 #2 | [141] | |
| 7. MYCN | PNA-MYCN | [142] | |
| Sphingosine | [143] | ||
| 8. JARID2 | JHDM inhibitor | [144] | |
| 9. CB1 | AM251 | [69] | |
| HU210 | [122] | ||
| THC | [122] | ||
| 10. CPT1A | Etomoxir | [145] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.H.; Barr, F.G. Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma. Molecules 2018, 23, 2798. https://doi.org/10.3390/molecules23112798
Nguyen TH, Barr FG. Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma. Molecules. 2018; 23(11):2798. https://doi.org/10.3390/molecules23112798
Chicago/Turabian StyleNguyen, Thanh Hung, and Frederic G. Barr. 2018. "Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma" Molecules 23, no. 11: 2798. https://doi.org/10.3390/molecules23112798
APA StyleNguyen, T. H., & Barr, F. G. (2018). Therapeutic Approaches Targeting PAX3-FOXO1 and Its Regulatory and Transcriptional Pathways in Rhabdomyosarcoma. Molecules, 23(11), 2798. https://doi.org/10.3390/molecules23112798