Abstract
DNA double-strand breaks (DSBs) are deleterious lesions that are generated in response to ionizing radiation or replication fork collapse that can lead to genomic instability and cancer. Eukaryotes have evolved two major pathways, namely homologous recombination (HR) and non-homologous end joining (NHEJ) to repair DSBs. Whereas the roles of protein-DNA interactions in HR and NHEJ have been fairly well defined, the functions of small and long non-coding RNAs and RNA-DNA hybrids in the DNA damage response is just beginning to be elucidated. This review summarizes recent discoveries on the identification of non-coding RNAs and RNA-mediated regulation of DSB repair.
1. Introduction
One of the most remarkable advances in molecular biology during the last decade is the annotation of the transcriptome in numerous organisms and the discovery that ~75% of the genome is transcribed into noncoding RNAs, but only ~2% of the transcriptome accounts for proteins [1,2,3]. These noncoding RNAs are involved in a diverse array of biological functions that are only beginning to be understood. A strong connection has been established between several small and long noncoding RNAs (lncRNAs) and human disease, especially cancer. MicroRNA signatures are linked to cancer metastasis and tumor progression. Several microRNAs and lncRNAs are oncogenes or tumor suppressors that regulate the DNA damage response and genomic stability. Understanding the mechanisms by which they act opens up new opportunities for intervention in cancer.
Double-strand DNA breaks (DSBs) are the most lethal form of DNA damage in cells and are predominantly repaired either by Non-Homologous DNA End Joining (NHEJ) [4,5,6] or Homologous Recombination (HR) pathways [7,8]. DSBs can occur endogenously as a result of unrepaired mutations that lead to stalled replication forks, due to inhibition of DNA replication culminating in breaks during mitosis, or because of the harmful effects of ionizing radiation [9,10,11]. Large-scale genome rearrangements such as deletions, insertions, and translocations that are highly genotoxic, are a consequence of defects in DSB repair and are positively correlated with cancer progression [12]. Germ line mutations in proteins involved in HR can cause several diseases such as Fanconi Anemia (FA) [13,14], breast and ovarian cancers [15,16], Bloom syndrome [17,18], Werner syndrome [19], and ataxia telangiectasiam Nijmegen breakage syndrome [20,21], whereas defects in NHEJ can result in chromosomal instability, promoting tumorigenesis, radiosensitive severe combined immunodeficiency (RS-SCID) [22,23], microcephaly, and growth defects in children [24,25,26]. A third mechanism of repair called microhomology-mediated end joining (MMEJ) or Alt-NHEJ is a minor pathway of DSB repair, and is triggered in the event that HR is not capable of repairing breaks [27,28,29,30]. Pathway choice for double strand break repair is complex and is dependent on the type of DSB induced, the cell cycle, and the activity of the repair components [31,32,33]. Understanding the molecular mechanisms involved in repair of DSBs has broad implications for a range of human diseases such as cancer, as well as aging [8,34].
In mammals, NHEJ is the primary DSB repair pathway that is active throughout the cell cycle, although it is more prominent during the G0 and G1 phases of the cell cycle [35,36,37,38]. NHEJ is used to repair double-strand DNA breaks (DSBs) that arise due to V (D) J recombination [39,40], ionizing radiation [41], and reactive oxygen species [42], without the need for a template DNA. The first step in NHEJ involves binding of Ku70–Ku80 heterodimers to both ends of a DNA break [43,44,45], followed by the recruitment and activation of the catalytic subunits of DNA dependent protein kinase (DNA-PKcs) [46,47]. DNA end processing involves phosphorylation of nucleases such as Artemis, and DNA polymerases to replace damaged bases; followed by ligation of blunt DNA ends by the XLF-XRCC4-DNA ligase IV complex. Recently, a triple-strand break repair model has been proposed in which ribonucleotide incorporation at the break termini is important for the ligation step of NHEJ [48]. Due to direct joining of the DNA ends that frequently involves DNA end resection, NHEJ is an error-prone DNA repair mechanism.
Unlike NHEJ, HR occurs only in rapidly dividing cells during the S and G2/M phases of the cell cycle and requires a sister chromatid as a template for repair [35,49,50]. The early events in HR can be characterized as (1) signaling to the DSB (2) relaxing chromatin to open the site of the break for repair, and (3) resection of the 5’ end of the DNA end [51,52,53]. The kinetics of the reaction measured in vivo, based on fluorescence recovery after photobleaching (FRAP) experiments after UV-based laser irradiation experiments, is rapid [54]. Within ~1 sec of the damage, the DSB is recognized by poly (ADP-ribose) polymerase 1 (PARP1), followed by recruitment of the chromatin remodeler Alc1 to the PAR chains, relaxation of chromatin allowing access of the helicase-nuclease “sensor” Mre11-Rad50-Nbs1 (MRN) complex to the site of damage [55,56,57]. Recruitment of MRN to the DSB site has been reported to occur within ~13 s and appears to be via direct association with PAR chains. Single-molecule localization experiments show that the temporal dynamics and spatial distribution of MRN foci are cell type-specific, and the MRN complex is recruited after γH2AX foci have already formed [58,59]. In addition to MRN, the phosphatidylinositol 3-kinase-like protein kinase (PIKK) ATM is activated by autophosphorylation [60]. ATM phosphorylates the DNA repair factors BRCA1, CtIP, EXO1 that act downstream in the reaction mechanism to promote resection, as well as the histone variant H2AX, which is phosphorylated at Ser139 to generate γH2AX [50]. Deposition of γH2AX occurs at the site of the DSB and is amplified, spreading for hundreds of kilobases from the damage site. These initial factors recruited to the sites of DNA damage form distinct DNA repair foci, the nature of which remains ambiguous. The amplification of this damage is dependent on formation of the γH2AX-MDC1 complex that recruits the ubiquitin E3 ligase RNF8 [61]. RNF8-mediated ubiquitylation of histones H2A and H2AX results in decondensation of chromatin and recruitment of RAP80, ABRA1, and BRCA1 [61]. RNF8 also interacts with other chromatin remodeling complexes such as CHD4, a component of the nucleosome remodeling and deacetylase complex, NuRD [62]. After repair has occurred, chromatin is remodeled to its initial state. Resection of the 5’ ends of the DSB is promoted by MRN, BRCA1, CtIP, and Exo1 nuclease and helicase activities, leaving ssDNA, which is rapidly bound by replication protein A (RPA) [50,63]. RPA is then exchanged for Rad51 in the presence of BRCA2 [50]. The later steps in HR involve strand invasion to the homologous template followed by DNA synthesis. This can occur by either the double-strand break repair pathway (DSBR) that results in chromosomal crossover, or the synthesis-dependent strand-annealing (SDSA) pathway in which there is strand displacement after DNA synthesis. Precise assembly of a multitude of protein factors and macromolecular machines is required for repair to occur. Regulators such as kinases, ubiquitin ligases, and acetyl-transferases that remodel chromatin are also essential to temporally and spatially control DSB repair.
HR and NHEJ DNA repair mechanisms are conserved in evolution and occur in both eukaryotes and prokaryotes. There are several excellent reviews on the detailed mechanisms and protein factors involved in DSB repair [64,65,66,67,68,69]. Recent studies indicate that non-coding RNAs (ncRNAs), that include long ncRNAs (lncRNAs) as well as small ~21 nt RNAs such as microRNAs (miRNAs), DNA damage induced RNAs (diRNAs), and Drosha- and Dicer-dependent small RNAs (DDRNAs) play important roles in the DNA damage response [70,71,72,73]. This review focuses on emerging themes in the roles of ncRNAs in DSB repair that are observed in humans and have direct cancer relevance.
2. LncRNAs
Long non-coding RNAs are > 200 nt RNA Pol-II transcripts that, in general, are not translated into proteins [74,75]. It is projected that >30,000 lncRNAs are expressed in humans, greater than the number of protein-coding genes (~20,000) in the genome [76,77,78]. These transcripts are polyadenylated, capped at the 5’ end, and spliced into mature RNAs, exported into the cytoplasm and are regulated like protein-coding mRNAs, although the mechanisms of RNA processing might differ from mRNAs [79]. Based on their proximity to protein coding genes, they have been classified as sense, antisense, pseudogenes, intronic, and intergenic (also called lincRNAs) [80,81]. Although the functions of most lncRNAs are poorly understood, in several cases such as the Xist RNA [82,83] that is required for mammalian dosage compensation and X-chromosome inactivation, they are not artifacts of pervasive transcription of “junk DNA”, and play important biological roles in control of the cell cycle, in development, and have been linked to cancer progression [84,85].
The mechanisms by which lncRNAs act are varied. Some lncRNAs exert epigenetic functions by acting as scaffolds for chromatin-modifying complexes, e.g., HOTAIR is associated with chromatin and recruits the polycomb repressive complex 2 (PRC2) and the LSD1/CoREST/REST demethylase complexes to specific loci in the genome for gene silencing [86,87]. Chromatin-associated lncRNAs such as promotor RNAs (pRNAs) are tethered to specific sites in the genome by forming RNA-DNA hybrids thereby acting in cis, where they recruit PARP1, the ATP-remodeling complex NoRC, and the methyltransferase Dnmt3b to silence rDNA [88,89]. LncRNAs can also directly regulate transcription by acting as transcription coregulators as in the case of the steroid receptor RNA activator SRA that activates transcription with nuclear receptors [90,91]. An example of a corepressor is the ncRNAs transcribed from the CCND1 gene (cyclin D1) locus that can bind the TLS protein that in turn turns-off the histone acetyl transferase activity of p300/CBP [92]. Another role of lncRNAs is in formation of nuclear bodies or foci. Paraspeckles are nuclear bodies that trap adenosine-to-inosine edited RNAs and paraskeckle formation relies on the lncRNA NEAT1 [93,94]. Similarly, MALAT1 (or NEAT2) is a component of nuclear speckles where it retains serine/arginine (SR) splicing factors and affects splicing [95,96]. Besides their roles in the nucleus, several antisense lncRNAs can hybridize with the 3’ untranslated regions (3’ UTRs) of mRNAs to regulate their stability in the cytoplasm and/or interact with RNA decay factors and miRNAs. Examples of antisense transcripts are lncRNAs produced from the 3’ UTR of iNOS [97] and half-STAU1-binding site RNAs (1/2sbsRNAs) [98]. The lncRNAs PTENP1 and KRASP1 [99] sequester miRNAs and act as miRNA sponges thereby affecting RNA levels of a range of mRNAs in the cell.
3. LncRNAs and the DNA Damage Response
Given the large number of lncRNAs and their diverse functions, it is not surprising that several lncRNAs are induced upon DNA damage and have been proposed to play a role in double-strand break repair (Table 1). Oncogenes as well as tumor suppressor lncRNAs have been documented. The first lncRNAs (ncRNACCND1) identified as being transcribed in response to DNA damage signals (ionizing radiation), were from the CCND1 gene (cyclin D1) locus [92,100]. The ncRNACCND1 (between 200–330 nts and greater) exist on chromatin as RNA: DNA hybrids as well as ssRNA.

Table 1.
LncRNAs involved in homologous recombination (HR) or non-homologous end joining (NHEJ).
RNA immunoprecipitation and ChIP experiments established TLS (translocated in liposarcoma) as the protein that was recruited to ncRNACCND1. RNA bound TLS results in an allosteric change that allows TLS to bind and inhibit the histone acetyltransferase complex CBP/p300, resulting in transcription repression. Since then, several damage-induced lncRNAs have been identified.
The mechanisms by which lncRNAs modulate DSB repair include: (i) modulating the activity of p53 either at the level of transcription, translation or posttranslational modifications; (ii) by acting as tethers for recruitment of chromatin remodeling complexes to the site of damage; (iii) by sequestering negative regulators of DNA repair away from the damage site as decoys; (iv) by acting as scaffolds that directly interact with several DNA repair proteins such as Ku70/Ku80, BRCA1, 53BP1, Mre11, PARP1; (v) by sequestering miRNAs that regulate the stability of DNA repair proteins, thereby modulating mRNA expression levels. As noted below and in Table 1, a subset of lncRNAs are part of a p53 network i.e., either under direct p53 control or they modulate the activity or expression of p53. In this section, I highlight some of the unique lncRNAs that have clear cancer relevance. A summary of the roles of DNA damage induced lncRNAs in specific disease states and development is given in Table 2 (at the end of Section 3).
Table 2.
Relevance of lncRNAs to development, disease, and inborn errors of metabolism.
3.1. p53 Linked LncRNAs
The transcription factor p53 plays key roles in normal cell proliferation and tumor suppression and is mutated in over 50% of human cancers. In response to DNA damage, it is involved in transactivation or repression of target genes to induce either cell cycle arrest or apoptosis. Not surprisingly, several lncRNAs have been discovered that are either transcription targets of p53 or regulate the p53-dependent gene expression signature [129,130]. Notably, the intergenic lncRNA that resides ~15 Kb upstream of the p53 target CDKN1A (or p21), called lincRNA-p21 [101,104,131] was identified as a repressor of the p53 transcriptional response, decreasing gene expression of hundreds of p53 target genes and triggering apoptosis. lincRNA-p21 is a large ~3.1 Kb transcript that is a distinct gene with its own promoter and is transcribed in the opposite orientation to CDKN1A in response to DNA damage. LincRNA-p21 likely functions through its interaction with heterogeneous nuclear ribonucleoprotein-K (hnRNP-K), which is a component of repressor complexes, and is recruited to promoters of p53 target genes. In addition to its roles in the nucleus, lincRNA-p21 is also reported to regulate translation in the cytoplasm [132]. A similar mechanism is employed by the DNA damage induced lncRNA called PANDA (for p21 associated ncRNA DNA damage activated) which is also induced by p53 and is transcribed antisense ~5 Kb upstream of CDKN1A [104]. PANDA acts as a decoy for the nuclear transcription factor Y subunit alpha (NF-YA) to repress the pro-apoptotic genes FAS and BIK. The APELA lncRNA is a positive regulator of p53 [104,133]. It binds to heterogeneous nuclear ribonucleoprotein-L (hnRNP-L) to inhibit the hnRNP-L-p53 interaction, thereby promoting p53-induced apoptosis. A more recent study [105] identified the lncRNA DINO (Damage Induced Noncoding), a ~951 bp RNA that is induced by p53 and is also located upstream of CDKN1A. Intriguingly, DINO interacts directly with p53 and stabilizes it, promoting the transactivation functions of p53. DINO knockout mice are also deficient in p53-dependent gene expression in response to DNA damage. Several lncRNAs have been shown to bind the repressor PRC2 that has H3K27 trimethylase activity. PINT is a lincRNA that is transcriptionally activated by p53 and is a positive regulator of cell proliferation and cell survival in mouse cells and a negative regulator in human cells. In both mice and humans, PINT targets PRC2 to specific gene loci for repression, thereby affecting cell proliferation, although the outcomes appear to be different in mice vs. humans [102,103]. TUG1 is a lincRNA transcript that is upregulated by p53 and may act on chromatin to downregulate p53 mediated transcriptional pathways [112,113]. TUG1 interacts with PRC2 and additional corepressor complexes that include histone methyltransferases, demethylases, and chromatin modifiers [112,113].
3.1.1. WRAP53 (WD Repeat Containing Antisense to p53)
A fascinating example of a lncRNA that is partially antisense to p53 and induces p53 levels upon DNA damage is WRAP53 [134,135] (Figure 1).
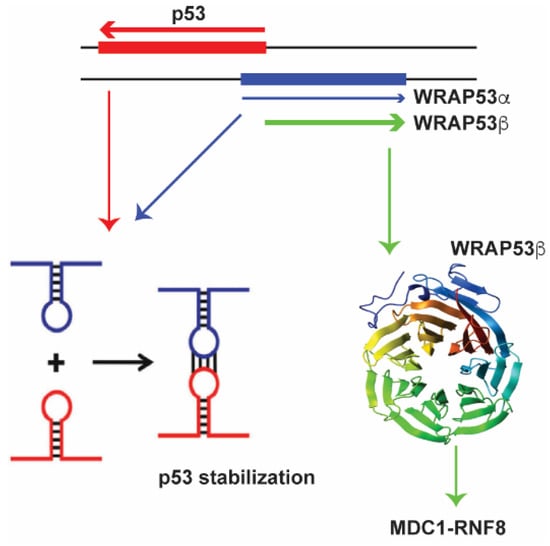
Figure 1.
The antisense lncRNA WRAP53α (shown in blue) directly interacts with the p53 mRNA (shown in red) to stabilize it, affecting p53 protein levels. The WRAP53β (in green) recruits and stabilizes RNF8 at double strand breaks.
Antisense lncRNA transcripts of p53 that overlap with the first p53 exon are called WRAP53α [135] and those that overlap with the first intron are called WRAP53γ. The WRAP53α RNA directly interacts with the p53 mRNA via RNA-RNA interactions to affect p53 protein levels [135]. WRAP53 an interesting example of gene that encodes both a lncRNA and is also transcribed into the protein WRAP53β (also known as WDR79 or TCAB1) [136,137,138] WRAP53β has multiple functions in the cell one of which is to target RNF8 to DNA double-strand breaks. Therefore both the protein, as well as the lncRNA is involved in the DNA damage response, albeit through different mechanisms.
3.1.2. LINP1
The long non-coding RNA “lncRNA in non-homologous end joining pathway” LINP1 [106,139,140] forms an RNA scaffold that, at least in cell extracts, interacts with both Ku70–Ku80 and DNA-PKcs on chromatin to promote end joining (Figure 2). LINP1 is overexpressed in triple negative breast cancer (TNBC) tumors, in the TNBC cell lines MDA-MB-231 and MDA-MB-468, and in triple-negative immortalized mammary cells MCF10A. LINP1 is not detected in ER-positive MCF7 cells. NHEJ reporter assays show that knockdown of LINP1 in TNBC cells decreases NHEJ and overexpression of LINP1 in MCF7 cells increases NHEJ activity [106]. Knockdown of LINP1 also sensitizes mice to ionizing radiation due to defects in DNA repair via the NHEJ pathway. Activation of EGFR signaling via MEK and JNK kinases up regulates LINP1 whereas tumor suppressor p53 represses LINP1 via the microRNA miR-29. Recent studies show that LINP1 levels are also increased in cervical cancer [141] as well as advanced prostate cancer [139] and are correlated with poor prognosis.
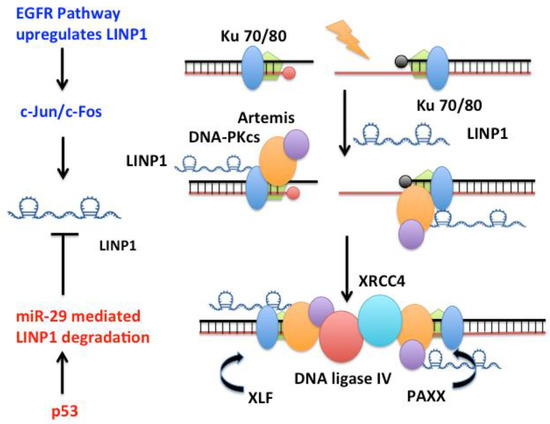
Figure 2.
LINP1 is a 917 nt long RNA that promotes the assembly of NHEJ factors to enhance DNA repair in Triple Negative Breast Cancers (TNBC). LINP1 directly associates with Ku70/Ku80 as well as DNA-PKcs to increase NHEJ activity in TNBC tumors. Activation of EGFR signaling via MEK and JNK kinases up regulates LINP1 whereas the tumor suppressor TP53 represses LINP1 via the microRNA miR-29.
3.1.3. MALAT1 or NEAT2
Metastasis-associated lung adenocarcinoma transcript 1 or MALAT1 [110,111] is a ~7 Kb lincRNA that is evolutionarily conserved and is associated with many cancers [142]. It was originally identified in a screen for lncRNAs in early-stage non-small cell lung cancers that were metastatic. MALAT1 performs a wide array of functions. It co-localizes with nuclear speckles [143] and is important for pre-mRNA processing and splicing [96,144,145]. The longer MALAT1 transcript can be further processed into a short 61 nt tRNA-like fragment called MALAT1-associated small cytoplasmic RNA (mascRNA) that regulates translation in the cytoplasm [146]. MALAT1 is one of the few lncRNAs for which structural information is available for at least a portion of the RNA. The 3’ end of MALAT1 forms an expression and nuclear retention element (ENE)+A, a triple helix element for which a 3.1 Å crystal structure is available (Figure 3A). The structure shows how the triple helix sequesters the 3’ end of MALAT1 in a U-A-U base triple, and protects the 3’ end of the lncRNA from decay [147].
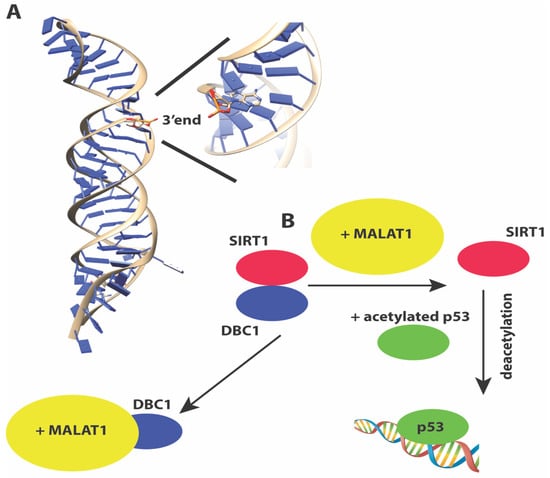
Figure 3.
(A) Structure of the 3’ end of MALAT1 reveals a triple helix with the 3’ end sequestered in a U-A-U base triple, protecting the 3’ end from nucleolytic degradation (B) MALAT1 sequesters the negative regulator DBC1, releasing the deacetylase SIRT1 to activate p53 for transactivation.
Like TUG1, MALAT1 is also upregulated by p53 upon DNA damage. A proteomics screen has identified numerous proteins involved in transcription, RNA processing, translation, protein degradation, and metabolism as MALAT1 interacting proteins [148]. MALAT1 indirectly regulates p53 protein transactivation function by sequestering DBC1, a partner for the deacetylase SIRT1, which targets p53 for deacetylation (Figure 3B). Therefore similar to several lncRNAs discussed here, p53 creates a feedback loop to regulate the expression of several lncRNAs, that in turn control p53 function. Besides regulating p53 activity, MALAT1 has also been reported to exist in a complex with PARP1 and Lig3 in vivo, and may regulate the Alt-NHEJ pathway of double-strand break repair [149].
3.2. p53-independent lncRNAs
Several lncRNAs (Table 1) are transcribed independent of p53, and act indirectly in DDR by altering signaling pathways or via epigenetic mechanisms that repress transcription of DNA repair genes. The lncRNAs ANRIL (antisense noncoding RNA in the INK4 locus), MDC1-AS and lncRNA-JADE are induced by ATM activation. ANRIL is encoded from the INK4B-ARF-INK4A locus on chromosome 9p21 and this locus is the most frequent copy number alteration across tumors. ANRIL recruits the PRC complex to repress transcription of tumor suppressors INK4B, ARF, INK4A at this locus and indirectly affects HR by altering cell cycle checkpoints.
The CUPID1 and CUPID2 lncRNAs are transcribed in hormone-positive breast cancers and regulate pathway choice in vivo, switching repair from the error-prone NHEJ to HR. However the exact mechanism is unclear. The TODRA RNA (transcribed in opposite direction of RAD51) increases the expression of RAD51 in an E2F1-dependent manner and hence facilitates HR.
3.2.1. DNA Damage-Sensitive RNA1 (DDSR1)
The lincRNA DDSR1 was identified in microarray screens of RNA isolated from hTert-immortalized human skin fibroblasts treated with the DNA damaging agents neocarzinostatin, campothecin, or etoposide [115]. DDSR1 was identified as a 1616 nt inter-genic transcript that was upregulated by ~2.5 fold in all samples treated with the DNA damaging drugs. DDSR1 expression is induced by ATM and under control of NF-κB, however p53 is not required for expression. Although DDSR1 expression is not under p53 control, DDSR1 was found to regulate the expression of p53 target genes and DDSR1 knockdown significantly upregulated p53 targeted mRNAs, particularly those involved in cell proliferation and cell survival. Intriguingly, DDSR1 knockdown impaired HR by ~50% and DDSR1 knockdown cells were also more sensitive to the PARP1 inhibitor Olaparib. RNA pull-down experiments identified heterogeneous nuclear ribonucleoprotein U-like 1 (hnRNPUL1) as an RNA binding protein that specifically associated with DDSR1, is recruited to sites of double-strand breaks and is important for end resection during HR [150]. In vivo imaging experiments show that depletion of DDSR1 or hnRNPUL1 leads to an increase in BRCA1-RAP80 at DSBs. This leads to a model in which the DDSR1-hnRNPUL1 complex regulates recruitment of BRCA1-RAP80 to sites of DNA breaks.
3.2.2. Prostate Cancer Associated Transcript 1 (PCAT-1)
PCAT-1 is the first lincRNA identified that participates in DSB repair in prostate cancer [118], and extends the “BRCA-ness” paradigm to include lncRNAs, in addition to mutations in DNA repair genes. Increased PCAT-1 levels downregulate BRCA2 and impairs HR, resulting in an increase in γ-H2AX foci formation [116]. PCAT-1 is being used as a biomarker for a predictive response to treatment with PARP1 inhibitors due to synthetic lethality. PCAT-1 levels are inversely correlated with RAD51 foci formation when prostate cancer cells are treated with the PARP1 inhibitors olaparib or ABT-888. The mechanism by which PCAT-1 down regulates BRCA2 is unique compared to other lincRNAs that act predominantly via epigenetic pathways. PCAT-1 is a predominantly cytoplasmic lincRNA and the first 250 nt from the 5’ end of the PCAT-1 RNA were sufficient to downregulate the 3’ untranslated region of BRCA2 mRNA via posttranscriptional mechanisms, likely at the level of RNA stability, although the exact mechanism is unknown. PCAT-1 can also regulate c-Myc levels via a microRNA sequestering mechanism [117]. It will be interesting to see whether a similar mechanism of regulation applies to BRCA2 mRNA.
3.2.3. Telomeric Repeat-Containing RNAs (TERRA)
Telomeres are structures assembled at the ends of chromosomes that protect the chromosome ends from being recognized as double-strand breaks and hence inhibit activation of the DNA damage response [174]. Telomeres have a unique “end problem” in that activation of DDR could either result in end degradation or trigger incorrect recombination events. Telomere ends also need to be replicated, and progressive shortening of telomeres is correlated with aging. TERRA RNAs are unique class of lncRNAs that are transcribed from the ends of chromosomes and consist of the G-rich telomeric repeats (TTAGGG)n in mammalian cells. TERRA RNAs form stacked G-quadruplex structures [175] that can interact with a number of proteins that are part of the shelterin complex for telomere capping, as well as DNA repair factors [123,176,177]. TERRA lncRNAs regulate the activity of telomerase, form heterochromatin at chromosome ends, and are also involved in capping of chromosome ends to protect telomere genomic integrity. During telomere replication, the ssDNA is bound by RPA, which triggers the ATR checkpoint for repair. The shelterin component POT1 is believed to compete with RPA and inhibit the ATR mediated checkpoint [178]. TERRA RNAs play a role in switching RPA for POT1. In addition, upon depletion of the shelterin protein, telomeric repeat factor 2, TERRA RNA levels increase and TERRA interacts with the hisone demethylase LSD1 [122]. This interaction stimulates the exonuclease activity of Mre11 to trim the 3’ G overhangs of uncapped telomeres and form heterochromatin at telomeric ends [122,177]. Although TERRA RNAs are a very specialized lncRNA family, structure/function studies of TERRA interactions with chromatin modifiers and DNA repair proteins provides important insights into lncRNA function in DSB repair.
4. Small ncRNAs
In mammals, two classes of small non-coding RNAs have been identified as major players in the DNA damage response. The first class consists of microRNAs (miRNAs) which are small, phylogenetically conserved ~22 nt RNAs that negatively regulate the translation and stability of mRNAs via their association with Argonaute (Ago) proteins. miRNAs associate with 3’ untranslated regions (3’UTRs) of mRNAs via RNA-RNA interactions to trigger translational repression or mRNA decay. The second class consisting of ~21 nt small RNAs has recently been identified in response to DNA damage, and are produced from sequences around the DSB. These ncRNAs are called DSB-induced small RNAs or diRNAs in plants and Drosha- and Dicer-dependent small RNAs (DDRNAs) in mammals. Although the same enzymes that are involved in the miRNA pathway generate DDRNAs and diRNAs, their biogenesis and mechanism is distinct from miRNAs, and is discussed in greater detail below.
4.1. miRNAs Involved in the DNA Damage Response
miRNAs are generated from precursor RNA Pol II transcripts that can either be intergenic or intragenic in origin [179,180]. Intergenic miRNAs have their own promoters and are transcribed into pri-miRNA precursors that are capped, and polyadenylated. More than 50% of miRNAs can be transcribed from intergenic regions as multicistronic transcripts [181]. Intragenic miRNAs are predominantly transcribed from introns of a host gene and released during splicing of the host gene. The pri-miRNAs have stem-loop structures that harbor the mature miRNA. The mature miRNA is generated by endonucleolytic cleavage by the nuclear Drosha/DGCR8 heterodimer to release a ~70 nt pre-miRNA hairpin with a 2 nt 3’ overhang. Exportin-5 (XPO5) and Ran-GTP facilitate nuclear export of the pre-miRNA hairpin by recognizing the 3’ overhang. In the cytoplasm, the pre-miRNA hairpin is cleaved by the RNAse III enzyme Dicer to generate the mature ~22 nt miRNA duplex. These mature miRNA duplexes associate with Ago proteins to form the RNA-induced silencing complex (RISC). Assembly of RISC requires degradation of one of the miRNA strands (passenger strand) while retaining the guide strand that ultimately base pairs with the mRNA 3’ UTR. The loading of the Ago-miRNA complex onto the mRNA recruits the mRNA degradation machinery that then targets the mRNA for either translational repression or decay (Figure 4A).
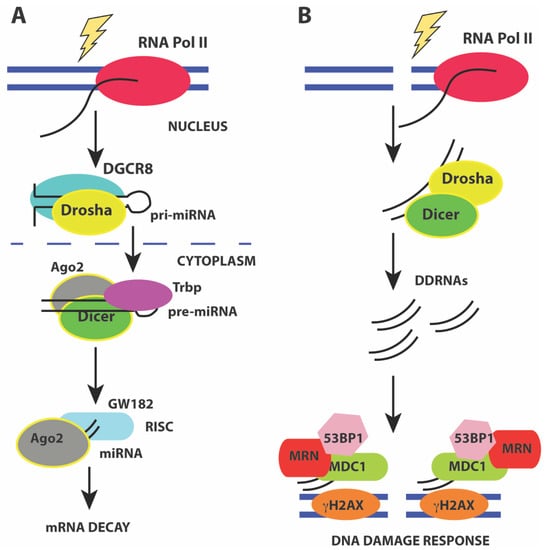
Figure 4.
(A) pathway of miRNA processing to regulate mRNA decay. Pri-miRNA precursors are transcribed and processed before assembling into a macromolecular complex containing the Drosha/DGCR8 heterodimer to release a ~70 nt pre-miRNA hairpin in the nucleus. The pre-miRNA hairpin is exported to the cytoplasm by XPO5 and Ran-GTP. In the cytoplasm, the pre-miRNA hairpin is cleaved by Dicer to generate the mature ~22 nt miRNA duplex that associates with Ago2 leading to the formation of the RNA-induced silencing complex (RISC). Activation of mRNA decay results in base-pairing of the guide strand to the mRNA 3’ UTR and recruitment of mRNA decay factors such as GW182; (B) Activation of the DNA damage response by DDRNAs also involves miRNA processing factors Drosha and Dicer that are required to generate small ~20–35 nt RNAs from the sequences around the site of damage. Formation of DNA damage foci and recruitment of DNA damage factors requires DDRNAs, Drosha, and Dicer.
The miRNA expression profile or “signatures” are predictors of cancer prognosis and are cancer hallmarks [182,183]. miRNA dependent pathways can be regulated at multiple levels upon DNA damage [181,184]. Similar to lncRNAs, the transcription of several miRNA genes, such as the miR-34 family in under the control of p53 and is altered upon DNA-damage [185]. Several miRNAs have been identified that directly regulate the abundance of DSB repair proteins at the posttranscriptional level (Table 3). The up- or down-regulation of miRNAs can affect protein stability of sensors of DNA damage such as γH2AX [186,187], decreasing γH2AX containing damage foci. The kinase ATM, a key regulator of the DNA damage response, is also under the control of several miRNAs (Table 3). Upregulation of miR-18a decreases the DNA damage response due to decreased ATM levels [188] whereas increased levels of miR-421, miR-100 and miR-101 leads to increased radiosensitivity [188,189,190]. The expression of DNA repair effectors is also under microRNA regulation (Table 3). In particular, the tumor suppressor BRCA1 and ssDNA binding protein RAD51, which are important for strand invasion in HR are tightly regulated by a number of miRNAs. MicroRNAs also control protein levels of cell cycle checkpoint factors and apoptosis regulators as has been reviewed [181,184]. Intriguingly, miRNAs can be secreted into body fluids such plasma and urine via exosomes and may be important mediators of DNA damage from radiation-targeted cells to abscopal normal cells, leading to the bystander effect [191]. Exosomes have been reported to contain as many as 764 miRNAs [192,193]. The miR-1246 is an example of a radiation-induced miRNA that is packaged into exosomes and is delivered to non-irradiated cells to decrease NHEJ efficiency by targeting the 3’ UTR of LIG4 [194]. These diverse roles of miRNAs in the DNA damage response and cancer have made them attractive targets for cancer therapy.
Table 3.
miRNAs that directly target genes involved in homologous recombination (HR) or non-homologous end joining (NHEJ).
4.2. Drosha- and Dicer-Dependent Small RNAs (DDRNAs)
Recent studies have shown that small ~21 nt RNAs are induced from the sequences around the site of damage in response to ionizing radiation. These RNAs are called diRNAs in Arabidopsis thaliana [71], qiRNAs in Neurospora crassa [215], endo-siRNAs in Drosophila melanogaster [216], and DDRNAs in human cells [217,218]. These RNAs are generated in a Dicer-dependent manner in all organisms and are distinct from miRNAs (Figure 4B). In mammals, down-regulation or inactivation of Drosha and Dicer impairs formation of DNA damage foci containing phosphor-ATM, MDC1, and 53BP1 in response to ionizing radiation [217,218]. Knockdown of Dicer and Drosha also results in a loss of the G1/S and G2/M checkpoints. Formation of DNA damage foci is RNA-dependent and removal of RNaseA and inhibiting transcription reduces foci formation. Using a chromosomally integrated reporter system and deep sequencing, recent studies show that foci formation requires small RNAs (20–35 nt) called DDRNAs that are generated by Drosha and Dicer from precursor RNAs at the damage site. The precise mechanism by which DDRNAs act is unclear, and it has been proposed that they act later in the DNA damage response and may function to recruit factors such as MDC1 and 53BP1 during HR [217]. A more recent study [219] shows that Drosha has an early role in DSB repair and is required for both HR and NHEJ. Drosha facilitates formation of RNA-DNA hybrids before resection can occur, and may interact directly with BRCA1. However, no DDRNAs were observed in this study [219] that examined endogenous break loci. However, all studies are in agreement for the role of RNA at DNA damage breaks and the requirement for miRNA processing enzymes such as Dicer and Drosha for efficient repair.
5. Concluding Remarks
Small RNA and lncRNAs play intricate roles in the DNA damage response, although the mechanisms by which they act remain unclear. Understanding their modes of regulation in DDR provides new opportunities in cancer therapy. lncRNAs can act as guides, scaffolds, decoys in DNA repair. They may also be important for spatial regulation of DNA repair complexes by formation of DNA repair foci. Use of the latest deep sequencing technologies, imaging tools, combined with molecular and structural information on RNA and RNA-protein complexes is needed to understand non-coding RNA function in in DNA repair.
Funding
This work was funded by the M.D. Anderson Cancer Center Knowledge GAP Award. I gratefully acknowledge John A. Tainer for sponsoring our ongoing research efforts (NIH R35CA22043) to understand the roles of lncRNAs in the DNA damage response.
Conflicts of Interest
I declare no conflict of interest.
References
- Lander, E.S. Initial impact of the sequencing of the human genome. Nature 2011, 470, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair. (Amst.) 2014, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Walden, H.; Deans, A.J. The Fanconi anemia DNA repair pathway: Structural and functional insights into a complex disorder. Annu. Rev. Biophys. 2014, 43, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, N.; Daly, M.B.; Feldman, G.L. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet. Med. 2010, 12, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Friedman, E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2007, 96, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Bischof, O.; Kim, S.H.; Irving, J.; Beresten, S.; Ellis, N.A.; Campisi, J. Regulation and localization of the Bloom syndrome protein in response to DNA damage. J. Cell. Biol. 2001, 153, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell. 2009, 36, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, K.A.; Gangloff, S.; Rothstein, R. The RecQ DNA helicases in DNA repair. Annu. Rev. Genet. 2010, 44, 393–417. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell. Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia telangiectasia: A review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- van der Burg, M.; Ijspeert, H.; Verkaik, N.S.; Turul, T.; Wiegant, W.W.; Morotomi-Yano, K.; Mari, P.O.; Tezcan, I.; Chen, D.J.; Zdzienicka, M.Z.; et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin Invest. 2009, 119, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Moshous, D.; Callebaut, I.; de Chasseval, R.; Corneo, B.; Cavazzana-Calvo, M.; Le Deist, F.; Tezcan, I.; Sanal, O.; Bertrand, Y.; Philippe, N.; et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell 2001, 105, 177–186. [Google Scholar] [CrossRef]
- Woodbine, L.; Gennery, A.R.; Jeggo, P.A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair. (Amst.) 2014, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, C.; Mericq, V.; Andrew, S.F.; van Duyvenvoorde, H.A.; Verkaik, N.S.; Losekoot, M.; Porollo, A.; Garcia, H.; Kuang, Y.; Hanson, D.; et al. An XRCC4 splice mutation associated with severe short stature, gonadal failure, and early-onset metabolic syndrome. J. Clin. Endocrinol. Metab. 2015, 100, E789–E798. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Voronova, N.V.; Chistiakov, A.P. Ligase IV syndrome. Eur. J. Med. Genet. 2009, 52, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.S.; Boulton, S.J. Beyond PARP-POLtheta as an anticancer target. Science 2018, 359, 1217–1218. [Google Scholar] [CrossRef] [PubMed]
- Deriano, L.; Roth, D.B. Modernizing the nonhomologous end-joining repertoire: Alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer. Res. 2013, 2, 163–177. [Google Scholar]
- Dutta, A.; Eckelmann, B.; Adhikari, S.; Ahmed, K.M.; Sengupta, S.; Pandey, A.; Hegde, P.M.; Tsai, M.S.; Tainer, J.A.; Weinfeld, M.; et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2017, 45, 2585–2599. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell. 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell. Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Swat, W.; Alt, F.W. The mechanism and regulation of chromosomal V(D)J recombination. Cell 2002, 109, S45–S55. [Google Scholar] [CrossRef]
- Schatz, D.G.; Swanson, P.C. V(D)J recombination: Mechanisms of initiation. Annu. Rev. Genet. 2011, 45, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lees-Miller, S.P. Detection and repair of ionizing radiation-induced DNA double strand breaks: New developments in nonhomologous end joining. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Jette, N.; Lees-Miller, S.P. Non-homologous end joining: Emerging themes and unanswered questions. DNA Repair. (Amst.) 2014, 17, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Duboc, C.; Wu, Q.; Ochi, T.; Liang, S.; Tsutakawa, S.E.; Lees-Miller, S.P.; Nadal, M.; Tainer, J.A.; Blundell, T.L.; et al. Dissection of DNA double-strand-break repair using novel single-molecule forceps. Nat. Struct. Mol. Biol. 2018, 25, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Yu, Y.; Radhakrishnan, S.K.; Chokshi, C.; Tsai, M.S.; Matsumoto, Y.; Kuzdovich, M.; Remesh, S.G.; Fang, S.; Tomkinson, A.E.; et al. An Intrinsically Disordered APLF Links Ku, DNA-PKcs, and XRCC4-DNA Ligase IV in an Extended Flexible Non-homologous End Joining Complex. J. Biol. Chem. 2016, 291, 26987–27006. [Google Scholar] [CrossRef] [PubMed]
- Sibanda, B.L.; Chirgadze, D.Y.; Ascher, D.B.; Blundell, T.L. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science 2017, 355, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Sibanda, B.L.; Chirgadze, D.Y.; Blundell, T.L. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature 2010, 463, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Pryor, J.M.; Conlin, M.P.; Carvajal-Garcia, J.; Luedeman, M.E.; Luthman, A.J.; Small, G.W.; Ramsden, D.A. Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining. Science 2018, 361, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mut. Res. 2017, 803–805, 51–55. [Google Scholar] [CrossRef] [PubMed]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Lukas, C.; Kitagawa, R.; Melander, F.; Kastan, M.B.; Bartek, J.; Lukas, J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell. Biol. 2006, 173, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Kruhlak, M.J.; Celeste, A.; Dellaire, G.; Fernandez-Capetillo, O.; Muller, W.G.; McNally, J.G.; Bazett-Jones, D.P.; Nussenzweig, A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell. Biol. 2006, 172, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Melander, F.; Stucki, M.; Falck, J.; Bekker-Jensen, S.; Goldberg, M.; Lerenthal, Y.; Jackson, S.P.; Bartek, J.; Lukas, J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004, 23, 2674–2683. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Jackson, S.P. The Mre11 complex: At the crossroads of dna repair and checkpoint signalling. Nat. Rev. Mol. Cell. Biol. 2002, 3, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Lees-Miller, S.P.; Tainer, J.A. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair. (Amst.) 2010, 9, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell. 2018, 71, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Nelms, B.E.; Maser, R.S.; MacKay, J.F.; Lagally, M.G.; Petrini, J.H. In situ visualization of DNA double-strand break repair in human fibroblasts. Science 1998, 280, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Eryilmaz, M.; Schmitt, E.; Krufczik, M.; Theda, F.; Lee, J.H.; Cremer, C.; Bestvater, F.; Schaufler, W.; Hausmann, M.; Hildenbrand, G. Localization Microscopy Analyses of MRE11 Clusters in 3D-Conserved Cell Nuclei of Different Cell Lines. Cancers 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Luijsterburg, M.S.; Acs, K.; Ackermann, L.; Wiegant, W.W.; Bekker-Jensen, S.; Larsen, D.H.; Khanna, K.K.; van Attikum, H.; Mailand, N.; Dantuma, N.P. A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure. EMBO J. 2012, 31, 2511–2527. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S. Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Seeber, A.; Hauer, M.H.; Gasser, S.M. Chromosome Dynamics in Response to DNA Damage. Annu. Rev. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sung, P. Introduction to the Thematic Minireview Series: DNA double-strand break repair and pathway choice. J. Biol. Chem. 2018, 293, 10500–10501. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Misteli, T. Non-coding RNAs in DNA damage and repair. FEBS Lett. 2013, 587, 1832–1839. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ba, Z.; Gao, M.; Wu, Y.; Ma, Y.; Amiard, S.; White, C.I.; Rendtlew Danielsen, J.M.; Yang, Y.G.; Qi, Y. A role for small RNAs in DNA double-strand break repair. Cell 2012, 149, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.G.; Qi, Y. RNA-directed repair of DNA double-strand breaks. DNA Repair. (Amst.) 2015, 32, 82–85. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F. A direct role for small non-coding RNAs in DNA damage response. Trends Cell. Biol. 2014, 24, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Schlackow, M.; Nojima, T.; Gomes, T.; Dhir, A.; Carmo-Fonseca, M.; Proudfoot, N.J. Distinctive Patterns of Transcription and RNA Processing for Human lincRNAs. Mol. Cell. 2017, 65, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Gebhard, C.; Miguel-Escalada, I.; Hoof, I.; Bornholdt, J.; Boyd, M.; Chen, Y.; Zhao, X.; Schmidl, C.; Suzuki, T.; et al. An atlas of active enhancers across human cell types and tissues. Nature 2014, 507, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Hendrich, B.D.; Rupert, J.L.; Lafreniere, R.G.; Xing, Y.; Lawrence, J.; Willard, H.F. The human XIST gene: Analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 1992, 71, 527–542. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Vincent, K.; Pichler, M.; Fodde, R.; Berindan-Neagoe, I.; Slack, F.J.; Calin, G.A. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 2015, 34, 5003–5011. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Guetg, C.; Lienemann, P.; Sirri, V.; Grummt, I.; Hernandez-Verdun, D.; Hottiger, M.O.; Fussenegger, M.; Santoro, R. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J. 2010, 29, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Guetg, C.; Scheifele, F.; Rosenthal, F.; Hottiger, M.O.; Santoro, R. Inheritance of silent rDNA chromatin is mediated by PARP1 via noncoding RNA. Mol. Cell. 2012, 45, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Lanz, R.B.; Razani, B.; Goldberg, A.D.; O’Malley, B.W. Distinct RNA motifs are important for coactivation of steroid hormone receptors by steroid receptor RNA activator (SRA). Proc. Natl. Acad. Sci. USA 2002, 99, 16081–16086. [Google Scholar] [CrossRef] [PubMed]
- Lanz, R.B.; McKenna, N.J.; Onate, S.A.; Albrecht, U.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 1999, 97, 17–27. [Google Scholar] [CrossRef]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Carmichael, G.G. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: Functional role of a nuclear noncoding RNA. Mol. Cell. 2009, 35, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell. 2009, 33, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Naganuma, T.; Shioi, G.; Hirose, T. Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J. Cell. Biol. 2011, 193, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Prasanth, K.V.; Tripathi, V.; Colasse, S.; Nakamura, T.; Xuan, Z.; Zhang, M.Q.; Sedel, F.; Jourdren, L.; Coulpier, F.; et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010, 29, 3082–3093. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Nishizawa, M.; Ozaki, T.; Kimura, T.; Hashimoto, I.; Yamada, M.; Kaibori, M.; Kamiyama, Y.; Ito, S.; Okumura, T. Natural antisense transcript stabilizes inducible nitric oxide synthase messenger RNA in rat hepatocytes. Hepatology 2008, 47, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.A.; Assoian, R.K. Transcriptional regulation of the cyclin D1 gene at a glance. J. Cell. Sci. 2008, 121, 3853–3857. [Google Scholar] [CrossRef] [PubMed]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Marin-Bejar, O.; Mas, A.M.; Gonzalez, J.; Martinez, D.; Athie, A.; Morales, X.; Galduroz, M.; Raimondi, I.; Grossi, E.; Guo, S.; et al. The human lncRNA LINC-PINT inhibits tumor cell invasion through a highly conserved sequence element. Genome Biol. 2017, 18, 202. [Google Scholar] [CrossRef] [PubMed]
- Marin-Bejar, O.; Marchese, F.P.; Athie, A.; Sanchez, Y.; Gonzalez, J.; Segura, V.; Huang, L.; Moreno, I.; Navarro, A.; Monzo, M.; et al. Pint lincRNA connects the p53 pathway with epigenetic silencing by the Polycomb repressive complex 2. Genome Biol. 2013, 14, R104. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Garcia, J.T.; Hung, T.; Flynn, R.A.; Shen, Y.; Qu, K.; Payumo, A.Y.; Peres-da-Silva, A.; Broz, D.K.; Baum, R.; et al. An inducible long noncoding RNA amplifies DNA damage signaling. Nat. Genet. 2016, 48, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, Q.; Hu, Z.; Feng, Y.; Fan, L.; Tang, Z.; Yuan, J.; Shan, W.; Li, C.; Hu, X.; et al. Long noncoding RNA LINP1 regulates repair of DNA double-strand breaks in triple-negative breast cancer. Nat. Struct. Mol. Biol. 2016, 23, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, S.; Henriksson, S.; Corcoran, M.; Mendez-Vidal, C.; Wiman, K.G.; Farnebo, M. Wrap53, a natural p53 antisense transcript required for p53 induction upon DNA damage. Mol. Cell. 2009, 33, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, M.; Emadi-Baygi, M.; Hoffmann, M.J.; Schulz, W.A.; Nikpour, P. Altered expression of LINC-ROR in cancer cell lines and tissues. Tumour Biol. 2016, 37, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Zhou, N.; Huang, J.; Liu, Q.; Fukuda, K.; Ma, D.; Lu, Z.; Bai, C.; Watabe, K.; Mo, Y.Y. The human long non-coding RNA-RoR is a p53 repressor in response to DNA damage. Cell. Res. 2013, 23, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Maeda, S.; Liu, C.; Karin, M.; Edgington, T.S. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 2007, 26, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Liu, W.; Zhang, J.; Ohgi, K.A.; Grinstein, J.D.; Dorrestein, P.C.; Rosenfeld, M.G. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 2011, 147, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, J.; Zhou, N.; Zhang, Z.; Zhang, A.; Lu, Z.; Wu, F.; Mo, Y.Y. LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res. 2013, 41, 4976–4987. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Khurana, S.; Kubben, N.; Abdelmohsen, K.; Oberdoerffer, P.; Gorospe, M.; Misteli, T. A BRCA1-interacting lncRNA regulates homologous recombination. EMBO Rep. 2015, 16, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chen, W.; Iyer, M.K.; Cao, Q.; Ma, T.; Han, S.; Sahu, A.; Malik, R.; Wilder-Romans, K.; Navone, N.; et al. PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res. 2014, 74, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chen, W.; Han, S.; Iyer, M.K.; Cao, Q.; Kothari, V.; Evans, J.R.; Knudsen, K.E.; Paulsen, M.T.; Ljungman, M.; et al. The long non-coding RNA PCAT-1 promotes prostate cancer cell proliferation through cMyc. Neoplasia 2014, 16, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D.; et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011, 29, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Hu, X.; Liu, Y.; Han, C.; Sood, A.K.; Calin, G.A.; Zhang, X.; Lu, X. A novel non-coding RNA lncRNA-JADE connects DNA damage signalling to histone H4 acetylation. EMBO J. 2013, 32, 2833–2847. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Mathur, R.; Hu, X.; Liu, Y.; Zhang, X.; Peng, G.; Lu, X. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell. Signal. 2013, 25, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Pilyugin, M.; Irminger-Finger, I. Long non-coding RNA and microRNAs might act in regulating the expression of BARD1 mRNAs. Int. J. Biochem. Cell. Biol. 2014, 54, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Feuerhahn, S.; Lingner, J. TERRA-reinforced association of LSD1 with MRE11 promotes processing of uncapped telomeres. Cell. Rep. 2014, 6, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Maringele, L.; Lydall, D. EXO1-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Delta mutants. Genes Dev. 2002, 16, 1919–1933. [Google Scholar] [CrossRef] [PubMed]
- Gazy, I.; Zeevi, D.A.; Renbaum, P.; Zeligson, S.; Eini, L.; Bashari, D.; Smith, Y.; Lahad, A.; Goldberg, M.; Ginsberg, D.; et al. TODRA, a lncRNA at the RAD51 Locus, Is Oppositely Regulated to RAD51, and Enhances RAD51-Dependent DSB (Double Strand Break) Repair. PLoS ONE 2015, 10, e0134120. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ma, G.; Zhang, Z.; Hua, Q.; Chu, H.; Tong, N.; Yuan, L.; Qin, C.; Yin, C.; Zhang, Z.; et al. A novel antisense long noncoding RNA regulates the expression of MDC1 in bladder cancer. Oncotarget 2015, 6, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Cajigas, I.; Leib, D.E.; Cochrane, J.; Luo, H.; Swyter, K.R.; Chen, S.; Clark, B.S.; Thompson, J.; Yates, J.R., 3rd; Kingston, R.E.; et al. Evf2 lncRNA/BRG1/DLX1 interactions reveal RNA-dependent inhibition of chromatin remodeling. Development 2015, 142, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.J.; Park, J.H.; Park, E.J.; Lee, S.A.; Lee, H.S.; Kang, S.W.; Kwon, J. ATM-mediated phosphorylation of the chromatin remodeling enzyme BRG1 modulates DNA double-strand break repair. Oncogene 2015, 34, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.A.; Moradi Marjaneh, M.; Al-Ejeh, F.; Lim, Y.C.; Shi, W.; Sivakumaran, H.; Tropee, R.; Patch, A.M.; Clark, M.B.; Bartonicek, N.; et al. Long Noncoding RNAs CUPID1 and CUPID2 Mediate Breast Cancer Risk at 11q13 by Modulating the Response to DNA Damage. Am. J. Hum. Genet. 2017, 101, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Segura, V.; Marin-Bejar, O.; Athie, A.; Marchese, F.P.; Gonzalez, J.; Bujanda, L.; Guo, S.; Matheu, A.; Huarte, M. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat. Commun. 2014, 5, 5812. [Google Scholar] [CrossRef] [PubMed]
- Leveille, N.; Melo, C.A.; Rooijers, K.; Diaz-Lagares, A.; Melo, S.A.; Korkmaz, G.; Lopes, R.; Akbari Moqadam, F.; Maia, A.R.; Wijchers, P.J.; et al. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat. Commun. 2015, 6, 6520. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, N.; Zamudio, J.R.; Jong, R.M.; Soukup, D.; Resnick, R.; Sarma, K.; Ward, A.J.; Raj, A.; Lee, J.T.; Sharp, P.A.; et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S. checkpoint. Mol. Cell. 2014, 54, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell. 2012, 47, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gou, H.; Tripathi, B.K.; Huang, J.; Jiang, S.; Dubois, W.; Waybright, T.; Lei, M.; Shi, J.; Zhou, M.; et al. An Apela RNA-Containing Negative Feedback Loop Regulates p53-Mediated Apoptosis in Embryonic Stem Cells. Cell. Stem Cell. 2015, 16, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Farnebo, M. Wrap53, a novel regulator of p53. Cell. Cycle 2009, 8, 2343–2346. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, S.; Henriksson, S.; Corcoran, M.; Mendez-Vidal, C.; Wiman, K.G.; Farnebo, M. Wrap53, a Natural p53 Antisense Transcript Required for p53 Induction upon DNA Damage. Mol. Cell. 2016, 64, 1009. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, S.; Rassoolzadeh, H.; Hedstrom, E.; Coucoravas, C.; Julner, A.; Goldstein, M.; Imreh, G.; Zhivotovsky, B.; Kastan, M.B.; Helleday, T.; et al. The scaffold protein WRAP53beta orchestrates the ubiquitin response critical for DNA double-strand break repair. Genes Dev. 2014, 28, 2726–2738. [Google Scholar] [CrossRef] [PubMed]
- Tycowski, K.T.; Shu, M.D.; Kukoyi, A.; Steitz, J.A. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol. Cell. 2009, 34, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Savage, S.A.; Shkreli, M.; Giri, N.; Jessop, L.; Myers, T.; Chen, R.; Alter, B.P.; Artandi, S.E. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011, 25, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.F.; Ren, L.G.; Xiao, J.Q.; Zhang, Y.; Mao, X.W.; Zhou, L.F. Long non-coding RNA LINP1 promotes the malignant progression of prostate cancer by regulating p53. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4467–4476. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, Y.; Song, X.; Zhang, N.; Sang, Y.; Zhang, H.; Liu, Y.; Chen, B.; Zhao, W.; Wang, L.; et al. Long noncoding RNA LINP1 acts as an oncogene and promotes chemoresistance in breast cancer. Cancer Biol. Ther. 2018, 19, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, H.; Shi, L.; Yu, X.; Gu, Y.; Sun, X. LINP1 facilitates DNA damage repair through non-homologous end joining (NHEJ) pathway and subsequently decreases the sensitivity of cervical cancer cells to ionizing radiation. Cell. Cycle 2018, 17, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Hammerle, M.; Diederichs, S. MALAT1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med. (Berl) 2013, 91, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Lamond, A.I.; Spector, D.L. Nuclear speckles: A model for nuclear organelles. Nat. Rev. Mol. Cell. Biol. 2003, 4, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell. 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3’ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Bulkley, D.; Wang, J.; Valenstein, M.L.; Yario, T.A.; Steitz, T.A.; Steitz, J.A. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 2014, 21, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, Y.; Zhuang, H.; Yang, B.; Hei, K.; Xiao, M.; Hou, C.; Gao, H.; Zhang, X.; Jia, C.; et al. Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation. Nucleic Acids Res. 2017, 45, 9947–9959. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lin, J.; Fang, H.; Fang, J.; Li, C.; Chen, W.; Liu, S.; Ondrejka, S.; Gong, Z.; Reu, F.; et al. Targeting the MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in multiple myeloma. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Blackford, A.N.; Chapman, J.R.; Baskcomb, L.; Gravel, S.; Rusch, A.; Thomas, A.; Blundred, R.; Smith, P.; Kzhyshkowska, J.; et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell. 2012, 45, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. Linking a role of lncRNAs (long non-coding RNAs) with insulin resistance, accelerated senescence, and inflammation in patients with type 2 diabetes. Hum. Genomics 2018, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liang, H.; Yang, H.; Zhou, K.; Xu, L.; Liu, J.; Lai, B.; Song, L.; Luo, H.; Peng, J.; et al. LincRNa-p21: Function and mechanism in cancer. Med. Oncol. (Northwood, London, England) 2017, 34, 98. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, M.; Goff, L.A.; Lodato, S.; Bonev, B.; Groff, A.F.; Gerhardinger, C.; Sanchez-Gomez, D.B.; Hacisuleyman, E.; Li, E.; Spence, M.; et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2013, 2, e01749. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, G.Q.; Chen, H.; Zhao, Z.J.; Chen, H.Z.; Liu, H.; Wang, G.; Jia, Y.H.; Pan, S.H.; Kong, R.; et al. Plasma and tumor levels of Linc-pint are diagnostic and prognostic biomarkers for pancreatic cancer. Oncotarget 2016, 7, 71773–71781. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Gu, H.; Lv, X.; Yuan, C.; Ni, P.; Liu, F. LINC-PINT Activates the Mitogen-Activated Protein Kinase Pathway to Promote Acute Myocardial Infarction by Regulating miR-208a-3p. Circ. J. Off. J. Jpn. Circ. Soc. 2018. [Google Scholar] [CrossRef] [PubMed]
- Permuth, J.B.; Chen, D.T.; Yoder, S.J.; Li, J.; Smith, A.T.; Choi, J.W.; Kim, J.; Balagurunathan, Y.; Jiang, K.; Coppola, D.; et al. Linc-ing Circulating Long Non-coding RNAs to the Diagnosis and Malignant Prediction of Intraductal Papillary Mucinous Neoplasms of the Pancreas. Sci. Rep. 2017, 7, 10484. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Goto, T.; Naemura, M.; Inoue, Y.; Okamoto, H.; Tahara, K. Long Noncoding RNA PANDA Positively Regulates Proliferation of Osteosarcoma Cells. Anticancer Res. 2017, 37, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, C.; Oldoni, E.; Serpente, M.; De Riz, M.A.; Arcaro, M.; D’Anca, M.; Pietroboni, A.M.; Calvi, A.; Lecchi, E.; Goris, A.; et al. LncRNAs expression profile in peripheral blood mononuclear cells from multiple sclerosis patients. J. Neuroimmunol. 2018, 324, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Tsai, S.H.; Cheng, J.C.; Wang, E.Y.; Anglesio, M.S.; Cochrane, D.R.; Fuller, M.; Gibb, E.A.; Wei, W.; Huntsman, D.G.; et al. APELA promotes tumour growth and cell migration in ovarian cancer in a p53-dependent manner. Gynecol. Oncol. 2017, 147, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, J.; Xie, N.; Huang, H.; Xu, S.; Cai, J.; Qi, S. lincROR influences the stemness and crizotinib resistance in EML-ALK(+) non-small-cell lung cancer cells. Onco Targets Ther. 2018, 11, 3649–3657. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.; Zaccagnini, G.; Perfetti, A.; Fuschi, P.; Valaperta, R.; Voellenkle, C.; Castelvecchio, S.; Gaetano, C.; Finato, N.; Beltrami, A.P.; et al. Long noncoding RNA dysregulation in ischemic heart failure. J. Transl. Med. 2016, 14, 183. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, X.; Shen, J. Long non-coding RNAs: Emerging players in osteosarcoma. Tumour Biol. 2016, 37, 2811–2816. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dou, P.; Liu, T.; He, S. Application of Long Noncoding RNAs in Osteosarcoma: Biomarkers and Therapeutic Targets. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, Y.; Zhuang, C.; Xu, W.; Fu, X.; Lv, Z.; Wu, H.; Mou, L.; Zhao, G.; Cai, Z.; et al. Inducing cell growth arrest and apoptosis by silencing long non-coding RNA PCAT-1 in human bladder cancer. Tumour Biol. 2015, 36, 7685–7689. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.C.; Wu, Y.F.; Qu, H.M. Up-regulation of long non-coding RNA PCAT-1 correlates with tumor progression and poor prognosis in gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3021–3027. [Google Scholar] [PubMed]
- Aguilo, F.; Di Cecilia, S.; Walsh, M.J. Long Non-coding RNA ANRIL and Polycomb in Human Cancers and Cardiovascular Disease. Curr. Top. Microbiol. Immunol. 2016, 394, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Simion, V.; Haemmig, S.; Feinberg, M.W. LncRNAs in vascular biology and disease. Vasc. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B. Telomerase and cancer therapeutics. Nat. Rev. Cancer 2008, 8, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhuang, S.; Wen, J.; Zheng, K. Long non-coding RNA MDC1-AS inhibits human gastric cancer cell proliferation and metastasis through an MDC1-dependent mechanism. Exp. Ther. Med. 2018, 15, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Zhu, J.; Xie, S.; Li, F.; Xu, Q. MDC1-AS, an antisense long noncoding RNA, regulates cell proliferation of glioma. Biomed. Pharmacother. 2016, 81, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Cajigas, I.; Chakraborty, A.; Swyter, K.R.; Luo, H.; Bastidas, M.; Nigro, M.; Morris, E.R.; Chen, S.; VanGompel, M.J.W.; Leib, D.; et al. The Evf2 Ultraconserved Enhancer lncRNA Functionally and Spatially Organizes Megabase Distant Genes in the Developing Forebrain. Mol. Cell. 2018, 71, 956–972. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.M.; Vangompel, M.J.; Sametsky, E.A.; Clark, M.F.; Savage, J.C.; Disterhoft, J.F.; Kohtz, J.D. Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci. 2009, 12, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.J.; Phan, A.T.; Kuryavyi, V. Human telomere, oncogenic promoter and 5’-UTR G-quadruplexes: Diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007, 35, 7429–7455. [Google Scholar] [CrossRef] [PubMed]
- Cusanelli, E.; Chartrand, P. Telomeric repeat-containing RNA TERRA: A noncoding RNA connecting telomere biology to genome integrity. Front. Genet. 2015, 6, 143. [Google Scholar] [CrossRef] [PubMed]
- Azzalin, C.M.; Lingner, J. Telomere functions grounding on TERRA firma. Trends Cell. Biol. 2015, 25, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Centore, R.C.; O’Sullivan, R.J.; Rai, R.; Tse, A.; Songyang, Z.; Chang, S.; Karlseder, J.; Zou, L. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 2011, 471, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Bottai, G.; Pasculli, B.; Calin, G.A.; Santarpia, L. Targeting the microRNA-regulating DNA damage/repair pathways in cancer. Expert Opin. Biol. Ther. 2014, 14, 1667–1683. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Pan, Y.; Navarro, F.; Dykxhoorn, D.M.; Moreau, L.; Meire, E.; Bentwich, Z.; Lieberman, J.; Chowdhury, D. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat. Struct. Mol. Biol. 2009, 16, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.W.; Li, M.; Cavenee, W.K.; Mitchell, P.S.; Zhou, X.; Tewari, M.; Furnari, F.B.; Taniguchi, T. MicroRNA-138 modulates DNA damage response by repressing histone H2AX expression. Mol. Cancer Res. 2011, 9, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Tessitore, A.; Cicciarelli, G.; Del Vecchio, F.; Gaggiano, A.; Verzella, D.; Fischietti, M.; Vecchiotti, D.; Capece, D.; Zazzeroni, F.; Alesse, E. MicroRNAs in the DNA Damage/Repair Network and Cancer. Int J. Genomics 2014, 2014, 820248. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Du, L.; Nagabayashi, G.; Seeger, R.C.; Gatti, R.A. ATM is down-regulated by N-Myc-regulated microRNA-421. Proc. Natl. Acad. Sci. USA 2010, 107, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Ng, W.L.; Zhang, X.; Wang, P.; Zhang, Z.; Mo, Y.Y.; Mao, H.; Hao, C.; Olson, J.J.; Curran, W.J.; et al. Targeting DNA-PKcs and ATM with miR-101 sensitizes tumors to radiation. PLoS ONE 2010, 5, e11397. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.Y. Secreted microRNAs: A new form of intercellular communication. Trends Cell. Biol. 2012, 22, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gonzalez, R.; Gauthier, S.A.; Kumar, A.; Saito, M.; Saito, M.; Levy, E. A Method for Isolation of Extracellular Vesicles and Characterization of Exosomes from Brain Extracellular Space. Methods Mol. Biol. 2017, 1545, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Beach, A.; Zhang, H.G.; Ratajczak, M.Z.; Kakar, S.S. Exosomes: An overview of biogenesis, composition and role in ovarian cancer. J. Ovarian Res. 2014, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Mo, L.J.; Song, M.; Huang, Q.H.; Guan, H.; Liu, X.D.; Xie, D.F.; Huang, B.; Huang, R.X.; Zhou, P.K. Exosome-packaged miR-1246 contributes to bystander DNA damage by targeting LIG4. Br. J. Cancer 2018, 119, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, A.; De Pitta, C.; Moret, F.; Barbieri, V.; Celotti, L.; Mognato, M. The DNA-damage response to gamma-radiation is affected by miR-27a in A549 cells. Int. J. Mol. Sci. 2013, 14, 17881–17896. [Google Scholar] [CrossRef] [PubMed]
- Bisso, A.; Faleschini, M.; Zampa, F.; Capaci, V.; De Santa, J.; Santarpia, L.; Piazza, S.; Cappelletti, V.; Daidone, M.; Agami, R.; et al. Oncogenic miR-181a/b affect the DNA damage response in aggressive breast cancer. Cell. Cycle 2013, 12, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Liu, J.L.; Li, J.P.; Xiao, F.; Zhang, Z.X.; Zhang, L. MicroRNA-124 (miR-124) regulates Ku70 expression and is correlated with neuronal death induced by ischemia/reperfusion. J. Mol. Neurosci. 2014, 52, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.E.; Meghani, K.; Brault, M.E.; Leclerc, L.; He, Y.J.; Day, T.A.; Elias, K.M.; Drapkin, R.; Weinstock, D.M.; Dao, F.; et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell. Rep. 2016, 14, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Fu, S.L.; Xu, S.Q.; Zhou, X.; Liu, X.S.; Xu, Y.J.; Zhao, J.P.; Wei, S. By downregulating Ku80, hsa-miR-526b suppresses non-small cell lung cancer. Oncotarget 2015, 6, 1462–1477. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Zhang, Z.Y.; Fu, S.L.; Xie, J.G.; Liu, X.S.; Xu, Y.J.; Zhao, J.P.; Xiong, W.N. Hsa-miR-623 suppresses tumor progression in human lung adenocarcinoma. Cell. Death Dis. 2016, 7, e2388. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.; Steptoe, A.L.; Martin, H.C.; Wani, S.; Nones, K.; Waddell, N.; Mariasegaram, M.; Simpson, P.T.; Lakhani, S.R.; Gabrielli, B.; et al. MicroRNA-182-5p targets a network of genes involved in DNA repair. RNA 2013, 19, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M.; et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell. 2011, 41, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Ambrosone, C.B.; DiCioccio, R.A.; Odunsi, K.; Lele, S.B.; Zhao, H. A functional polymorphism in the miR-146a gene and age of familial breast/ovarian cancer diagnosis. Carcinogenesis 2008, 29, 1963–1966. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.E.; Pan, Y.; Park, E.; Konstantinopoulos, P.; De, S.; D’Andrea, A.; Chowdhury, D. MicroRNAs down-regulate homologous recombination in the G1 phase of cycling cells to maintain genomic stability. Elife 2014, 3, e02445. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Clark, P.; Quann, K.; Zhou, H.; Londin, E.; Jing, Y.; Rigoutsos, I. Post-transcriptional Regulation of BRCA2 through Interactions with miR-19a and miR-19b. Front. Genet. 2016, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.W.; Calses, P.; Kemp, C.J.; Taniguchi, T. MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res. 2012, 72, 4037–4046. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Wang, Q.; Liu, R.; Chen, Z.; Zhang, X.; Zhou, P.; Wang, Z. LncRNA lnc-RI regulates homologous recombination repair of DNA double-strand breaks by stabilizing RAD51 mRNA as a competitive endogenous RNA. Nucleic Acids Res. 2018, 46, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xue, F.; Zhang, W. miR-506: A regulator of chemo-sensitivity through suppression of the RAD51-homologous recombination axis. Chin. J. Cancer 2015, 34, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Lovat, F.; Fassan, M.; Casadei, L.; Cascione, L.; Jacob, N.K.; Carasi, S.; Palmieri, D.; Costinean, S.; Shapiro, C.L.; et al. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc. Natl. Acad. Sci. USA 2014, 111, 4536–4541. [Google Scholar] [CrossRef] [PubMed]
- Neijenhuis, S.; Bajrami, I.; Miller, R.; Lord, C.J.; Ashworth, A. Identification of miRNA modulators to PARP inhibitor response. DNA Repair. (Amst.) 2013, 12, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.W.; Wang, Y.; Dhillon, K.K.; Calses, P.; Villegas, E.; Mitchell, P.S.; Tewari, M.; Kemp, C.J.; Taniguchi, T. Systematic screen identifies miRNAs that target RAD51 and RAD51D to enhance chemosensitivity. Mol. Cancer Res. 2013, 11, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Santarpia, L.; Kim, J.; Esteva, F.J.; Moretti, E.; Buzdar, A.U.; Di Leo, A.; Le, X.F.; Bast, R.C., Jr.; Park, S.T.; et al. Plasma microRNA 210 levels correlate with sensitivity to trastuzumab and tumor presence in breast cancer patients. Cancer 2012, 118, 2603–2614. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Nakamura, K.; Davies, R.; Nahas, S.A.; Brown, C.; Tunuguntla, R.; Gatti, R.A.; Hu, H. ATM-dependent MiR-335 targets CtIP and modulates the DNA damage response. PLoS Genet. 2013, 9, e1003505. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Chang, S.S.; Choudhary, S.; Aalto, A.P.; Maiti, M.; Bamford, D.H.; Liu, Y. qiRNA is a new type of small interfering RNA induced by DNA damage. Nature 2009, 459, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Michalik, K.M.; Bottcher, R.; Forstemann, K. A small RNA response at DNA ends in Drosophila. Nucleic Acids Res. 2012, 40, 9596–9603. [Google Scholar] [CrossRef] [PubMed]
- Francia, S.; Cabrini, M.; Matti, V.; Oldani, A.; d’Adda di Fagagna, F. DICER, DROSHA and DNA damage response RNAs are necessary for the secondary recruitment of DNA damage response factors. J. Cell. Sci. 2016, 129, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; de Hoon, M.; Anelli, V.; Mione, M.; Carninci, P.; d’Adda di Fagagna, F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012, 488, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.T.; Hawley, B.R.; Skalka, G.L.; Baldock, R.A.; Smith, E.M.; Bader, A.S.; Malewicz, M.; Watts, F.Z.; Wilczynska, A.; Bushell, M. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 2018, 9, 532. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).