
Optimisation of Milk Protein Top-Down Sequencing Using In-Source Collision-Induced Dissociation in the Maxis Quadrupole Time-of-Flight Mass Spectrometer



Abstract
1. Introduction
2. Results and Discussion
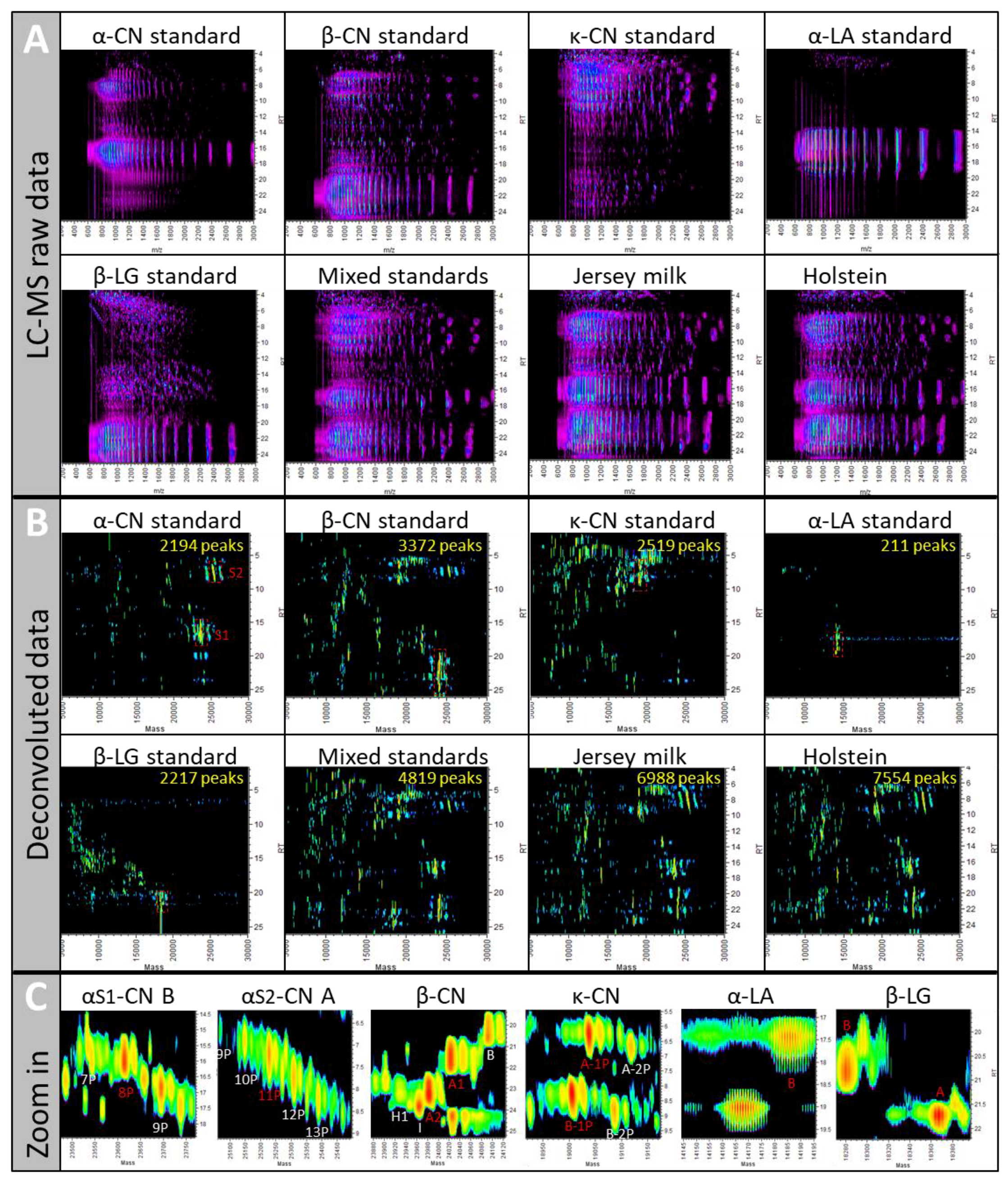
2.1. Sample Complexity
2.1.1. UPLC-ESI-Q-ToF MS Can Handle Complex Samples
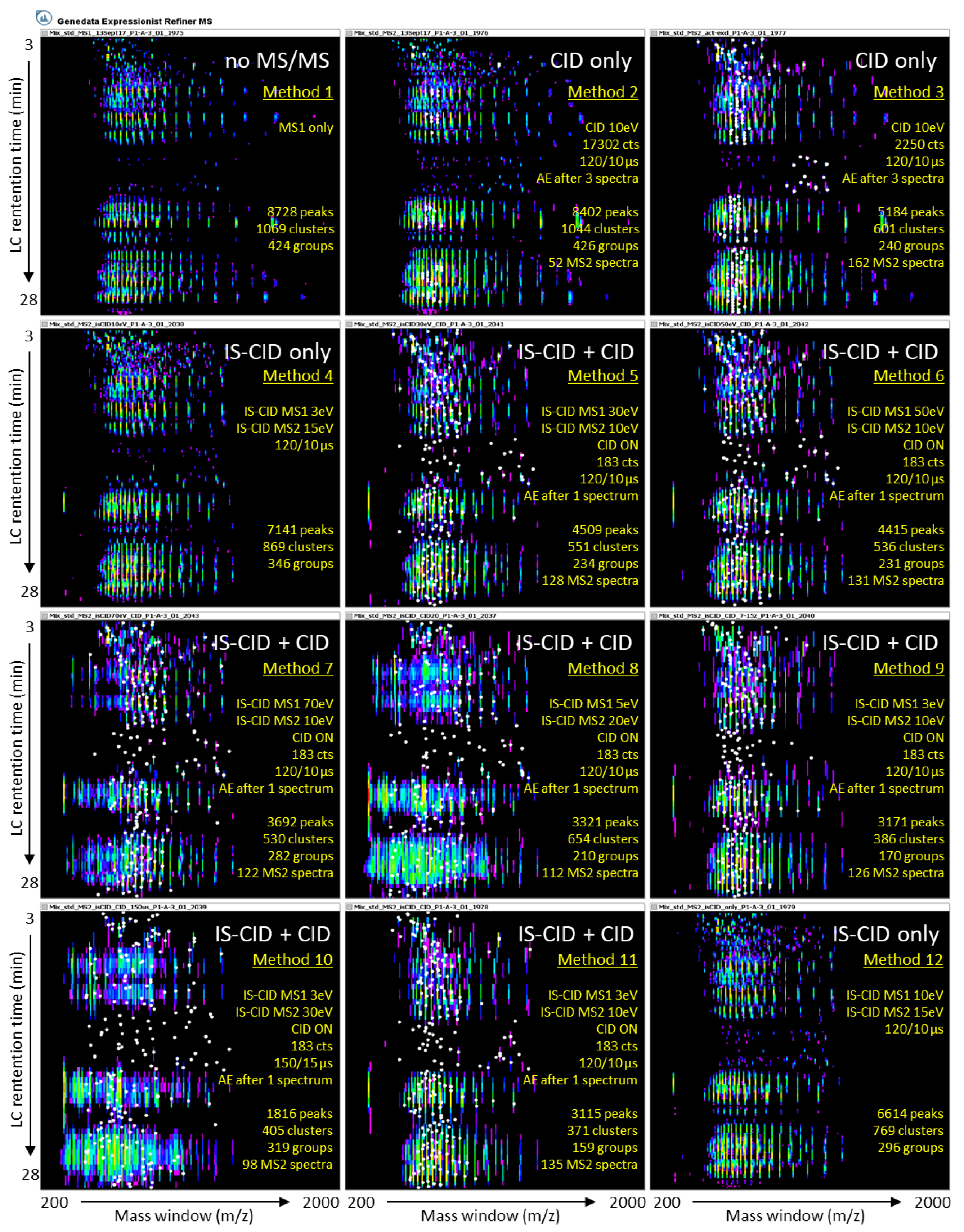
2.1.2. The 11 MS/MS Methods Are Assessed Using a Reference Set of Known Proteins
2.2. Effect of IS-CID and CID on Their Own or Combined on Spectral Data
2.2.1. Precursor Intensity Threshold Is a Key Parameter for CID Fragmentation
2.2.2. Ion Funnel 1 Energy Has Little Impact on IS-CID Fragmentation
2.2.3. Charge State Preference Does Not Influence Precursor Selection
2.2.4. A Long Duty Cycle Minimises MS/MS Events
2.2.5. High Energy Fragmentation Produces Lower m/z Ions
2.2.6. CID Versus IS-CID
2.3. Top-Down Sequencing of Milk Proteins
2.3.1. Reproducibility and Protein Specificity
2.3.2. Low-Energy Fragmentation Drives Top-Down Sequencing Efficacy
2.3.3. AA Position Has No Influence on Fragmentation
2.3.4. Hydrophobicity Affects Fragmentation
2.3.5. High Sequence Coverage Is Critical for Allelic Variants
3. Materials and Methods
3.1. Materials and Sample Preparation
3.2. HPLC Separation of Intact Proteins
3.3. MS1 Analysis
3.4. MS2 Analyses and Top-Down Sequencing
3.5. Protein Annotation in Genedata Expressionist
3.6. Validation and Computational Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| AA | Amino acid |
| ACN | Acetonitrile |
| AE | Active exclusion |
| CAD | Collisionally activated dissociation |
| CID | Collision-induced dissociation |
| CV | Coefficient of variation |
| DAD | Diode array detector |
| DC | Direct current |
| ECD | Electron capture dissociation |
| ESI | Electrospray ionisation |
| ETD | Electron transfer dissociation |
| eV | Electron-volt |
| FA | Formic acid |
| FT | Fourier transform |
| HCD | Higher-energy collisional dissociation |
| HPLC | High-performance liquid chromatography |
| IS-CID | In-source CID |
| kDa | KiloDalton |
| keV | Kiloelectron-volt |
| LC-MS | Liquid chromatography‒mass spectrometry |
| LIT | Linear ion trap |
| m/z | Mass-to-charge ratio |
| MS | Mass spectrometry |
| MS/MS | Tandem MS |
| MS1 | Full scan MS |
| MS2 | Tandem MS |
| MW | Molecular weight |
| ppm | Parts per million |
| PTM | Post-translational modification |
| Q | Quadrupole |
| QQQ | Triple quadrupole |
| Q-ToF | Quadrupole time-of-flight |
| RF | Radiofrequency |
| TFA | Trifluoroacetic acid |
| ToF | Time-of-flight |
| UHT | Ultra-high temperature |
| V | Volt |
| α-LA | Alpha-lactalbumin |
| αS1-CN | Alpha-S1-casein |
| αS2-CN | Alpha-S2-casein |
| β-CN | Beta-casein |
| β-LG | Beta-lactoglobulin |
| κ-CN | Kappa-casein |
References
- Kelleher, N.L.; Lin, H.Y.; Valaskovic, G.A.; Aaserud, D.J.; Fridriksson, E.K.; McLafferty, F.W. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc. 1999, 121, 806–812. [Google Scholar] [CrossRef]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef] [PubMed]
- Dole, M.; Mack, L.L.; Hines, R.L.; Mobley, R.C.; Ferguson, L.D.; Alice, M.B. Molecular Beams of Macroions. J. Chem. Phys. 1968, 49, 2240–2249. [Google Scholar] [CrossRef]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.A.; Udseth, H.R.; Smith, R.D. Collisional Effects on the Charge Distribution of Ions from Large Molecules, Formed by Electrospray-ionization Mass Spectrometry. Rapid Commun. Mass Spectrom. 1988, 2, 207–210. [Google Scholar] [CrossRef]
- Mann, M.; Meng, C.K.; Fenn, J.B.F. Interpreting Mass Spectra of Multiply Charged Ions. Anal. Chem. 1989, 61, 1702–1708. [Google Scholar] [CrossRef]
- Jones, A.W.; Cooper, H.J. Dissociation techniques in mass spectrometry-based proteomics. Analyst 2011, 136, 3419–3429. [Google Scholar] [CrossRef] [PubMed]
- McLafferty, F.W.; Bryce, T.A. Metastable-ion characteristics: Characterization of isomeric molecules. Chem. Commun. 1967, 0, 1215–1217. [Google Scholar] [CrossRef]
- Jennings, K.R. Collision-induced decompositions of aromatic molecular ions. Int. J. Mass Spectrom. Ion. Phys. 1968, 1, 227–235. [Google Scholar] [CrossRef]
- Garcia, B.A. What does the future hold for Top Down mass spectrometry? J. Am. Soc. Mass Spectrom. 2010, 21, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Pamreddy, A.; Panyala, N.R. Top-down proteomics: Applications, recent developments and perspectives. J. Appl. Bioanal. 2016, 2, 52–75. [Google Scholar] [CrossRef]
- Katta, V.; Chowdhury, S.K.; Chait, B.T. Use of a single-quadrupole mass spectrometer for collision-induced dissociation studies of multiply charged peptide ions produced by electrospray ionization. Anal. Chem. 1991, 63, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Bure, C.; Lange, C. Comparison of dissociation of ions in an electrospray source, or a collision cell in tandem mass spectrometry. Curr. Org. Chem. 2003, 7, 1613–1624. [Google Scholar] [CrossRef]
- Van Dongen, W.D.; van Wijk, J.I.; Green, B.N.; Heerma, W.; Haverkamp, J. Comparison between collision induced dissociation of electrosprayed protonated peptides in the up-front source region and in a low-energy collision cell. Rapid Commun. Mass Spectrom. 1999, 13, 1712–1716. [Google Scholar] [CrossRef]
- Senko, M.W.; Beu, S.C.; McLafferty, F.W. High-resolution tandem mass spectrometry of carbonic anhydrase. Anal. Chem. 1994, 66, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Parcher, J.F.; Wang, M.; Chittiboyina, A.G.; Khan, I.A. In-source collision-induced dissociation (IS-CID): Applications, issues and structure elucidation with single-stage mass analyzers. Drug Test. Anal. 2018, 10, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, N.L. Top-down proteomics. Anal. Chem. 2004, 76, 197A–203A. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.A.; Edmonds, C.G.; Smith, R.D. Primary sequence information from intact proteins by electrospray ionization tandem mass spectrometry. Science 1990, 248, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Loo, J.A.; Quinn, J.P.; Ryu, S.I.; Henry, K.D.; Senko, M.W.; McLafferty, F.W. High-resolution tandem mass spectrometry of large biomolecules. Proc. Natl. Acad. Sci. USA 1992, 89, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Beu, S.C.; Senko, M.W.; Quinn, J.P.; McLafferty, F.W. Improved fourier-transform ion-cyclotron-resonance mass spectrometry of large biomolecules. J. Am. Soc. Mass Spectrom. 1993, 4, 190–192. [Google Scholar] [CrossRef]
- Shevchenko, A.; Chernushevich, I.; Ens, W.; Standing, K.G.; Thomson, B.; Wilm, M.; Mann, M. Rapid ‘de novo’ peptide sequencing by a combination of nanoelectrospray, isotopic labeling and a quadrupole/time-of-flight mass spectrometer. Rapid Commun. Mass Spectrom. 1997, 11, 1015–1024. [Google Scholar] [CrossRef]
- Morris, H.R.; Paxton, T.; Dell, A.; Langhorne, J.; Berg, M.; Bordoli, R.S.; Hoyes, J.; Bateman, R.H. High sensitivity collisionally-activated decomposition tandem mass spectrometry on a novel quadrupole/orthogonal-acceleration time-of-flight mass spectrometer. Rapid Commun. Mass Spectrom. 1996, 10, 889–896. [Google Scholar] [CrossRef]
- Vincent, D.; Elkins, A.; Condina, M.R.; Ezernieks, V.; Rochfort, S. Quantitation and Identification of Intact Major Milk Proteins for High-Throughput LC-ESI-Q-TOF MS Analyses. PLoS ONE 2016, 11, e0163471. [Google Scholar] [CrossRef] [PubMed]
- Domon, B.; Aebersold, R. Mass spectrometry and protein analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.; Kuster, B.; Mann, M. Quadrupole time-of-flight versus triple-quadrupole mass spectrometry for the determination of phosphopeptides by precursor ion scanning. J. Mass Spectrom. 2001, 36, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Nemeth-Cawley, J.F.; Rouse, J.C. Identification and sequencing analysis of intact proteins via collision-induced dissociation and quadrupole time-of-flight mass spectrometry. J. Mass Spectrom. 2002, 37, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Nemeth-Cawley, J.F.; Tangarone, B.S.; Rouse, J.C. “Top Down” characterization is a complementary technique to peptide sequencing for identifying protein species in complex mixtures. J. Proteome Res. 2003, 2, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Ginter, J.M.; Zhou, F.; Johnston, M.V. Generating protein sequence tags by combining cone and conventional collision induced dissociation in a quadrupole time-of-flight mass spectrometer. J. Am. Soc. Mass Spectrom. 2004, 15, 1478–1486. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.J.; Ahmed, T.F.; Miesbauer, L.J.; Edalji, R.; Smith, R.; Harlan, J.; Dorwin, S.; Walter, K.; Holzman, T. Protein fragmentation via liquid chromatography-quadrupole time-of-flight mass spectrometry: The use of limited sequence information in structural characterization. Anal. Biochem. 2005, 341, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Deepalakshmi, P.D. Top-down approach of completely sequencing a 4.9 kDa recombinant peptide using quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 3747–3754. [Google Scholar] [CrossRef] [PubMed]
- Armirotti, A.; Benatti, U.; Damonte, G. Top-down proteomics with a quadrupole time-of-flight mass spectrometer and collision-induced dissociation. Rapid Commun. Mass Spectrom. 2009, 23, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jiang, J.; Wu, Z.; Xie, F. Antimicrobial peptides from the skin of the Asian frog, Odorrana jingdongensis: De novo sequencing and analysis of tandem mass spectrometry data. J. Proteomics 2012, 75, 5807–5821. [Google Scholar] [CrossRef] [PubMed]
- Boeri Erba, E. Investigating macromolecular complexes using top-down mass spectrometry. Proteomics 2014, 14, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Raynes, J.K.; Vincent, D.; Zawadzki, J.L.; Savin, K.; Logan, A.; Mertens, D.; Williams, R.P.W. Investigation of age gelation UHT Milk. Beverages 2018. submitted for publication. [Google Scholar]
- Vincent, D.; Ezernieks, V.; Elkins, A.; Nguyen, N.; Moate, P.J.; Cocks, B.G.; Rochfort, S. Milk Bottom-Up Proteomics: Method Optimization. Front. Genet. 2015, 6, 360. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, N.; Kane, M.; Joshi, L.; Hickey, R.M. Structural and functional characteristics of bovine milk protein glycosylation. Glycobiology 2014, 24, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Farrell, H.M., Jr.; Jimenez-Flores, R.; Bleck, G.T.; Brown, E.M.; Butler, J.E.; Creamer, L.K.; Hicks, C.L.; Hollar, C.M.; Ng-Kwai-Hang, K.F.; Swaisgood, H.E. Nomenclature of the proteins of cows’ milk—Sixth revision. J. Dairy Sci. 2004, 87, 1641–1674. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode | Step | Parameter | Method 1 | Method 2 | Method 3 | Method 4 | Method 5 | Method 6 | Method 7 | Method 8 | Method 9 | Method 10 | Method 11 | Method 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MS | Source | End plate offset | 500 V | 500 V | 500 V | 500 V | 500 V | 500 V | 500 V | 500 V | 500 V | 500 V | 500 V | 500 V |
| MS | Source | Capillary | 4500 V | 4500 V | 4500 V | 4500 V | 4500 V | 4500 V | 4500 V | 4500 V | 4500 V | 4500 V | 4500 V | 4500 V |
| MS | Source | Nebuliser | 1.5 bar | 1.5 bar | 1.5 bar | 1.5 bar | 1.5 bar | 1.5 bar | 1.5 bar | 1.5 bar | 1.5 bar | 1.5 bar | 1.5 bar | 1.5 bar |
| MS | Source | Dry gas | 8 L/min | 8 L/min | 8 L/min | 8 L/min | 8 L/min | 8 L/min | 8 L/min | 8 L/min | 8 L/min | 8 L/min | 8 L/min | 8 L/min |
| MS | Source | Temperature | 190 °C | 190 °C | 190 °C | 190 °C | 190 °C | 190 °C | 190 °C | 190 °C | 190 °C | 190 °C | 190 °C | 190 °C |
| MS | Source | Waste (min) | 0–2.5 | 0–2.5 | 0–2.5 | 0–2.5 | 0–2.5 | 0–2.5 | 0–2.5 | 0–2.5 | 0–2.5 | 0–2.5 | 0–2.5 | 0–2.5 |
| MS | Source | Source (min) | 2.5–40 | 2.5–40 | 2.5–40 | 2.5–40 | 2.5–40 | 2.5–40 | 2.5–40 | 2.5–40 | 2.5–40 | 2.5–40 | 2.5–40 | 2.5–40 |
| MS | Transfer | Funnel RF | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp |
| MS | Transfer | IS-CID energy | 0 eV | 0 eV | 0 eV | 3 eV | 30 eV | 50 eV | 70 eV | 5 eV | 0 eV | 3 eV | 0 eV | 10 eV |
| MS | Transfer | Transfer multipole RF | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp | 400 Vpp |
| MS | Quadrupole | Ion energy | 5 eV | 5 eV | 5 eV | 5 eV | 5 eV | 5 eV | 5 eV | 5 eV | 5 eV | 5 eV | 5 eV | 5 eV |
| MS | Quadrupole | Low mass | 300 m/z | 300 m/z | 300 m/z | 300 m/z | 300 m/z | 300 m/z | 300 m/z | 300 m/z | 300 m/z | 300 m/z | 300 m/z | 300 m/z |
| MS | Collision cell | Collision energy | 10 eV | 10 eV | 10 eV | 10 eV | 10 eV | 10 eV | 10 eV | 10 eV | 10 eV | 10 eV | 10 eV | 10 eV |
| MS | Collision cell | Collision RF (Vpp) | 1800 | 1800 | 1800 | 1800 | 1800 | 1800 | 1800 | 1800 | 1800 | 1800 | 1800 | 1800 |
| MS | Collision cell | Transfer time | 120 μs | 120 μs | 120 μs | 120 μs | 120 μs | 120 μs | 120 μs | 120 μs | 120 μs | 150 μs | 120 μs | 120 μs |
| MS | Collision cell | Pre-pulse storage | 10 μs | 10 μs | 10 μs | 10 μs | 10 μs | 10 μs | 10 μs | 10 μs | 10 μs | 15 μs | 10 μs | 10 μs |
| MS | MS1 | Ion polarity | positive | positive | positive | positive | Positive | Positive | positive | positive | positive | positive | positive | positive |
| MS | MS1 | Mass range (m/z) | 600–3000 | 600–3000 | 600–3000 | 200–2000 | 200–2000 | 200–2000 | 200–2000 | 200–2000 | 200–2000 | 200–2000 | 200–2000 | 200–2000 |
| MS | MS1 | Summation | 15,000 | 15,000 | 15,000 | 18,321 | 18,321 | 18,321 | 18,321 | 18,321 | 18,321 | 18,321 | 18,321 | 18,321 |
| MS | MS1 | Rolling average mode | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| in source | IS-CID | IS-CID | NO | NO | NO | YES | YES | YES | YES | YES | YES | YES | YES | YES |
| in source | IS-CID | IS-CID MS (funnel 1 exit) | 3 eV | 30 eV | 50 eV | 70 eV | 5 eV | 3 eV | 3 eV | 3 eV | 10 eV | |||
| in source | IS-CID | IS-CID MS/MS (funnel 2 entry) | 15 eV | 10 eV | 10 eV | 10 eV | 20 eV | 10 eV | 30 eV | 10 eV | 15 eV | |||
| in source | IS-CID | Acquisition time factor | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |||
| MS/MS | MS2 | Auto MS/MS mode | OFF | ON | ON | OFF | ON | ON | ON | ON | ON | ON | ON | OFF |
| MS/MS | CID | Acquisition | NO | YES | YES | NO | YES | YES | YES | YES | YES | YES | YES | NO |
| MS/MS | CID | MS/MS low (per 1000 sum.) | 104 cts | 104 cts | 104 cts | 104 cts | 104 cts | 104 cts | 104 cts | 104 cts | 104 cts | |||
| MS/MS | CID | MS/MS high (per 1000 sum.) | 106 cts | 106 cts | 106 cts | 106 cts | 106 cts | 106 cts | 106 cts | 106 cts | 106 cts | |||
| MS/MS | MS2 | Preference activated | NO | NO | NO | NO | NO | NO | NO | NO | YES | NO | NO | NO |
| MS/MS | MS2 | Charge state preferred range | 7–15 | |||||||||||
| MS/MS | MS2 | Exclude singly | YES | |||||||||||
| MS/MS | Precursor ions | Exclude (m/z) | 100–600 | 100–600 | 100–600 | 100–600 | 100–600 | 100–600 | 100–600 | 100–600 | 100–600 | |||
| MS/MS | Precursor ions | No. of precursors | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 4 | 4 | |||
| MS/MS | Threshold | Absolute (per 1000 sum.) | 1153 cts | 170 cts | 10 cts | 10 cts | 10 cts | 10 cts | 10 cts | 10 cts | 10 cts | |||
| MS/MS | Threshold | Absolute | 17,302 cts | 2550 cts | 183 cts | 183 cts | 183 cts | 183 cts | 183 cts | 183 cts | 183 cts | |||
| MS/MS | Active exclusion | ON/OFF | ON | ON | ON | ON | ON | ON | ON | ON | ON | |||
| MS/MS | Active exclusion | Exclude after | 3 spectra | 3 spectra | 1 spectra | 1 spectra | 1 spectra | 1 spectra | 1 spectra | 1 spectra | 1 spectra | |||
| MS/MS | Active exclusion | Release after | 5 min | 5 min | 5 min | 5 min | 5 min | 5 min | 5 min | 5 min | 5 min |
| Method | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LC separation (min) | 22.5 | 22.5 | 22.5 | 22.5 | 22.5 | 22.5 | 22.5 | 22.5 | 22.5 | 22.5 | 22.5 | 22.5 |
| LC separation (s) | 1350 | 1350 | 1350 | 1350 | 1350 | 1350 | 1350 | 1350 | 1350 | 1350 | 1350 | 1350 |
| Duty cycle (s) | 2.7 | 13.7 | 13.7 | 5.6 | 13.7 | 13.7 | 13.8 | 19.1 | 24.6 | 24.6 | 24.6 | 5.5 |
| No. of full MS1 scans | 493 | 99 | 99 | 243 | 99 | 99 | 98 | 71 | 55 | 55 | 55 | 247 |
| No. of precursors | 2 | 2 | 2 | 2 | 2 | 3 | 4 | 4 | 4 | |||
| (a) Theor. no. of MS2 events | 0 | 197 | 198 | 0 | 197 | 198 | 196 | 212 | 219 | 219 | 220 | 0 |
| (b) MS1 peaks (300–3000 m/z) | 8728 | 8402 | 5184 | 7141 | 4509 | 4415 | 3692 | 3321 | 3171 | 1816 | 3115 | 6614 |
| (c) MS1 peaks (700–3000 m/z) | 8711 | 8387 | 5184 | 7124 | 4503 | 4415 | 3599 | 3040 | 3169 | 1628 | 3113 | 6599 |
| (d) MS1 peaks (300–700 m/z) | 17 | 15 | 0 | 17 | 6 | 0 | 93 | 281 | 2 | 188 | 2 | 15 |
| (d)/(b) (%) | 0.2 | 0.2 | 0.0 | 0.2 | 0.1 | 0.0 | 2.5 | 8.5 | 0.1 | 10.4 | 0.1 | 0.2 |
| MS1 clusters | 1069 | 1044 | 601 | 869 | 551 | 536 | 530 | 654 | 386 | 405 | 371 | 769 |
| MS1 groups | 424 | 426 | 240 | 346 | 234 | 231 | 282 | 487 | 170 | 319 | 159 | 296 |
| (e) Observed No. of MS2 spectra | 0 | 56 | 170 | 0 | 196 | 196 | 196 | 210 | 218 | 218 | 218 | 0 |
| (e)/(a) (%) | 28 | 86 | 99 | 99 | 100 | 99 | 99 | 99 | 99 | |||
| (e)/(b) (%) | 0.7 | 3.3 | 4.3 | 4.4 | 5.3 | 6.3 | 6.9 | 12.0 | 7.0 | |||
| MS2 m/z range | 730–1636 | 644–1854 | 643–1955 | 703–1955 | 657–1966 | 609–1913 | 607–1774 | 609–1966 | 634–1928 | |||
| 600–700 m/z | 1 | 3 | 2 | 18 | 3 | 20 | 4 | |||||
| 700–800 m/z | 2 | 2 | 5 | 5 | 5 | 14 | 4 | 19 | 8 | |||
| 800–900 m/z | 4 | 12 | 20 | 20 | 15 | 22 | 26 | 27 | 29 | |||
| 900–1000 m/z | 20 | 53 | 45 | 44 | 21 | 34 | 56 | 36 | 54 | |||
| 1000–1100 m/z | 28 | 77 | 53 | 51 | 47 | 23 | 63 | 24 | 57 | |||
| 1100–1200 m/z | 5 | 29 | 26 | 35 | 24 | 30 | 18 | 23 | ||||
| 1200–1300 m/z | 2 | 19 | 21 | 26 | 26 | 20 | 29 | 18 | ||||
| 1300–1400 m/z | 2 | 3 | 7 | 17 | 14 | 8 | 7 | 2 | ||||
| 1400–1500 m/z | 2 | 2 | 5 | 4 | 11 | 2 | 10 | 5 | ||||
| 1500–1600 m/z | 2 | 3 | 4 | 5 | 8 | 3 | 9 | 3 | ||||
| 1600–1700 m/z | 2 | 6 | 4 | 4 | 7 | 7 | 1 | 8 | 7 | |||
| 1700–1800 m/z | 3 | 4 | 3 | 3 | 1 | 2 | 5 | 5 | ||||
| 1800–1900 m/z | 3 | 4 | 4 | 7 | 7 | 4 | 1 | |||||
| 1900–2000 m/z | 2 | 2 | 2 | 1 | 2 | 2 |
| Protein | Variant | PTM | Code | Mo. Mass (Da) | Length (AAs) | RT (min) | Sample 1 | Method 2 | Method 3 | Method 4 | Method 5 | Method 6 | Method 7 | Method 8 | Method 9 | Method 10 | Method 11 | Method 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| αS1-CN | B | 8P | αS1-CN B 8P | 23,600.3 | 199 | 15–18 | I | 196 | 196 | 195 | 194 | 195 | 190 | 70 | 195 | 7 | 194 | 195 |
| αS2-CN | A | 11P | αS2-CN A 11P | 25,213.0 | 207 | 6.8–8.3 | I | 14 | 6 | 83 | 6 | 6 | 6 | 6 | 6 | 0 | 6 | 83 |
| β-CN | A1 | 5P | β-CN A1 5P | 24,008.2 | 209 | 21–22 | I | 204 | 204 | 197 | 205 | 205 | 203 | 197 | 205 | 95 | 205 | 195 |
| β-CN | A2 | 5P | β-CN A2 5P | 23,968.2 | 209 | 21.5–23 | I | 202 | 202 | 201 | 203 | 203 | 201 | 186 | 203 | 92 | 203 | 200 |
| κ-CN | A | 1P | κ-CN A 1P | 19,026.5 | 169 | 6.5–8.5 | I | 160 | 160 | 165 | 157 | 157 | 156 | 119 | 157 | 31 | 157 | 165 |
| κ-CN | B | 1P | κ-CN B 1P | 18,993.6 | 169 | 8.5–10 | I | 162 | 162 | 157 | 160 | 159 | 157 | 133 | 161 | 59 | 161 | 154 |
| α-LA | B | α-LA B | 14,176.8 | 123 | 15–18 | I | 98 | 97 | 84 | 94 | 100 | 98 | 90 | 97 | 86 | 97 | 83 | |
| β-LG | A | β-LG A | 18,355.5 | 162 | 22–24 | I | 162 | 162 | 161 | 155 | 155 | 153 | 153 | 162 | 151 | 162 | 161 | |
| β-LG | B | β-LG B | 18,269.4 | 162 | 20–22 | I | 159 | 158 | 159 | 153 | 154 | 149 | 155 | 159 | 146 | 159 | 158 | |
| αS1-CN | B | 8P | αS1-CN B 8P | 23,600.3 | 199 | 15–18 | M | 178 | 178 | 192 | 179 | 177 | 173 | 91 | 178 | 52 | 181 | 192 |
| αS2-CN | A | 11P | αS2-CN A 11P | 25,213.0 | 207 | 6.8–8.3 | M | 121 | 123 | 150 | 111 | 116 | 108 | 52 | 115 | 8 | 115 | 151 |
| β-CN | A1 | 5P | β-CN A1 5P | 24,008.2 | 209 | 21–22 | M | 171 | 172 | 168 | 162 | 167 | 156 | 79 | 163 | 27 | 165 | 174 |
| β-CN | A2 | 5P | β-CN A2 5P | 23,968.2 | 209 | 21.5–23 | M | 201 | 201 | 201 | 203 | 201 | 200 | 182 | 202 | 43 | 203 | 201 |
| κ-CN | A | 1P | κ-CN A 1P | 19,026.5 | 169 | 6.5–8.5 | M | 108 | 109 | 122 | 106 | 109 | 93 | 41 | 106 | 14 | 107 | 124 |
| κ-CN | B | 1P | κ-CN B 1P | 18,993.6 | 169 | 8.5–10 | M | 122 | 125 | 112 | 120 | 120 | 110 | 33 | 122 | 10 | 121 | 116 |
| α-LA | B | α-LA B | 14,176.8 | 123 | 15–18 | M | 86 | 85 | 96 | 83 | 82 | 77 | 37 | 83 | 21 | 86 | 94 | |
| β-LG | A | β-LG A | 18,355.5 | 162 | 22–24 | M | 158 | 157 | 156 | 158 | 158 | 157 | 135 | 157 | 62 | 157 | 155 | |
| β-LG | B | β-LG B | 18,269.4 | 162 | 20–22 | M | 111 | 112 | 126 | 106 | 107 | 96 | 42 | 106 | 20 | 111 | 125 | |
| αS1-CN | B | 8P | αS1-CN B 8P | 23,600.3 | 199 | 15–18 | J | 194 | 195 | 198 | 195 | 195 | 189 | 137 | 194 | 97 | 195 | 198 |
| αS2-CN | A | 11P | αS2-CN A 11P | 25,213.0 | 207 | 6.8–8.3 | J | 92 | 140 | 177 | 122 | 110 | 97 | 18 | 114 | 20 | 126 | 177 |
| β-CN | A1 | 5P | β-CN A1 5P | 24,008.2 | 209 | 21–22 | J | 206 | 206 | 205 | 206 | 206 | 205 | 191 | 206 | 110 | 206 | 204 |
| β-CN | A2 | 5P | β-CN A2 5P | 23,968.2 | 209 | 21.5–23 | J | 199 | 199 | 201 | 200 | 200 | 199 | 188 | 199 | 106 | 199 | 201 |
| κ-CN | A | 1P | κ-CN A 1P | 19,026.5 | 169 | 6.5–8.5 | J | 82 | 132 | 151 | 121 | 107 | 104 | 12 | 110 | 30 | 119 | 154 |
| κ-CN | B | 1P | κ-CN B 1P | 18,993.6 | 169 | 8.5–10 | J | 164 | 164 | 157 | 164 | 163 | 164 | 146 | 164 | 78 | 163 | 157 |
| α-LA | B | α-LA B | 14,176.8 | 123 | 15–18 | J | 120 | 120 | 121 | 120 | 120 | 111 | 62 | 120 | 38 | 120 | 121 | |
| β-LG | A | β-LG A | 18,355.5 | 162 | 22–24 | J | 154 | 154 | 153 | 154 | 154 | 154 | 139 | 154 | 73 | 154 | 153 | |
| β-LG | B | β-LG B | 18,269.4 | 162 | 20–22 | J | 158 | 156 | 156 | 158 | 156 | 154 | 134 | 156 | 76 | 156 | 155 | |
| αS1-CN | B | 8P | αS1-CN B 8P | 23,600.3 | 199 | 15–18 | H | 191 | 191 | 197 | 183 | 184 | 177 | 133 | 185 | 89 | 183 | 197 |
| αS2-CN | A | 11P | αS2-CN A 11P | 25,213.0 | 207 | 6.8–8.3 | H | 172 | 181 | 192 | 179 | 179 | 172 | 65 | 178 | 88 | 180 | 193 |
| β-CN | A1 | 5P | β-CN A1 5P | 24,008.2 | 209 | 21–22 | H | 204 | 204 | 200 | 204 | 204 | 204 | 181 | 204 | 170 | 204 | 201 |
| β-CN | A2 | 5P | β-CN A2 5P | 23,968.2 | 209 | 21.5–23 | H | 206 | 207 | 203 | 207 | 207 | 207 | 198 | 207 | 168 | 207 | 203 |
| κ-CN | A | 1P | κ-CN A 1P | 19,026.5 | 169 | 6.5–8.5 | H | 147 | 150 | 165 | 147 | 153 | 133 | 54 | 151 | 72 | 150 | 165 |
| κ-CN | B | 1P | κ-CN B 1P | 18,993.6 | 169 | 8.5–10 | H | 89 | 97 | 118 | 91 | 86 | 82 | 11 | 89 | 17 | 95 | 118 |
| α-LA | B | α-LA B | 14,176.8 | 123 | 15–18 | H | 118 | 119 | 119 | 106 | 105 | 94 | 52 | 107 | 23 | 104 | 119 | |
| β-LG | A | β-LG A | 18,355.5 | 162 | 22–24 | H | 157 | 159 | 156 | 158 | 158 | 155 | 136 | 156 | 97 | 157 | 155 | |
| β-LG | B | β-LG B | 18,269.4 | 162 | 20–22 | H | 162 | 162 | 159 | 162 | 162 | 160 | 138 | 162 | 128 | 162 | 158 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincent, D.; Mertens, D.; Rochfort, S. Optimisation of Milk Protein Top-Down Sequencing Using In-Source Collision-Induced Dissociation in the Maxis Quadrupole Time-of-Flight Mass Spectrometer. Molecules 2018, 23, 2777. https://doi.org/10.3390/molecules23112777
Vincent D, Mertens D, Rochfort S. Optimisation of Milk Protein Top-Down Sequencing Using In-Source Collision-Induced Dissociation in the Maxis Quadrupole Time-of-Flight Mass Spectrometer. Molecules. 2018; 23(11):2777. https://doi.org/10.3390/molecules23112777
Chicago/Turabian StyleVincent, Delphine, Dominik Mertens, and Simone Rochfort. 2018. "Optimisation of Milk Protein Top-Down Sequencing Using In-Source Collision-Induced Dissociation in the Maxis Quadrupole Time-of-Flight Mass Spectrometer" Molecules 23, no. 11: 2777. https://doi.org/10.3390/molecules23112777
APA StyleVincent, D., Mertens, D., & Rochfort, S. (2018). Optimisation of Milk Protein Top-Down Sequencing Using In-Source Collision-Induced Dissociation in the Maxis Quadrupole Time-of-Flight Mass Spectrometer. Molecules, 23(11), 2777. https://doi.org/10.3390/molecules23112777