
Photophysical Properties of Nitrated and Halogenated Phosphorus Tritolylcorrole Complexes: Insights from Theory

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Computation Details
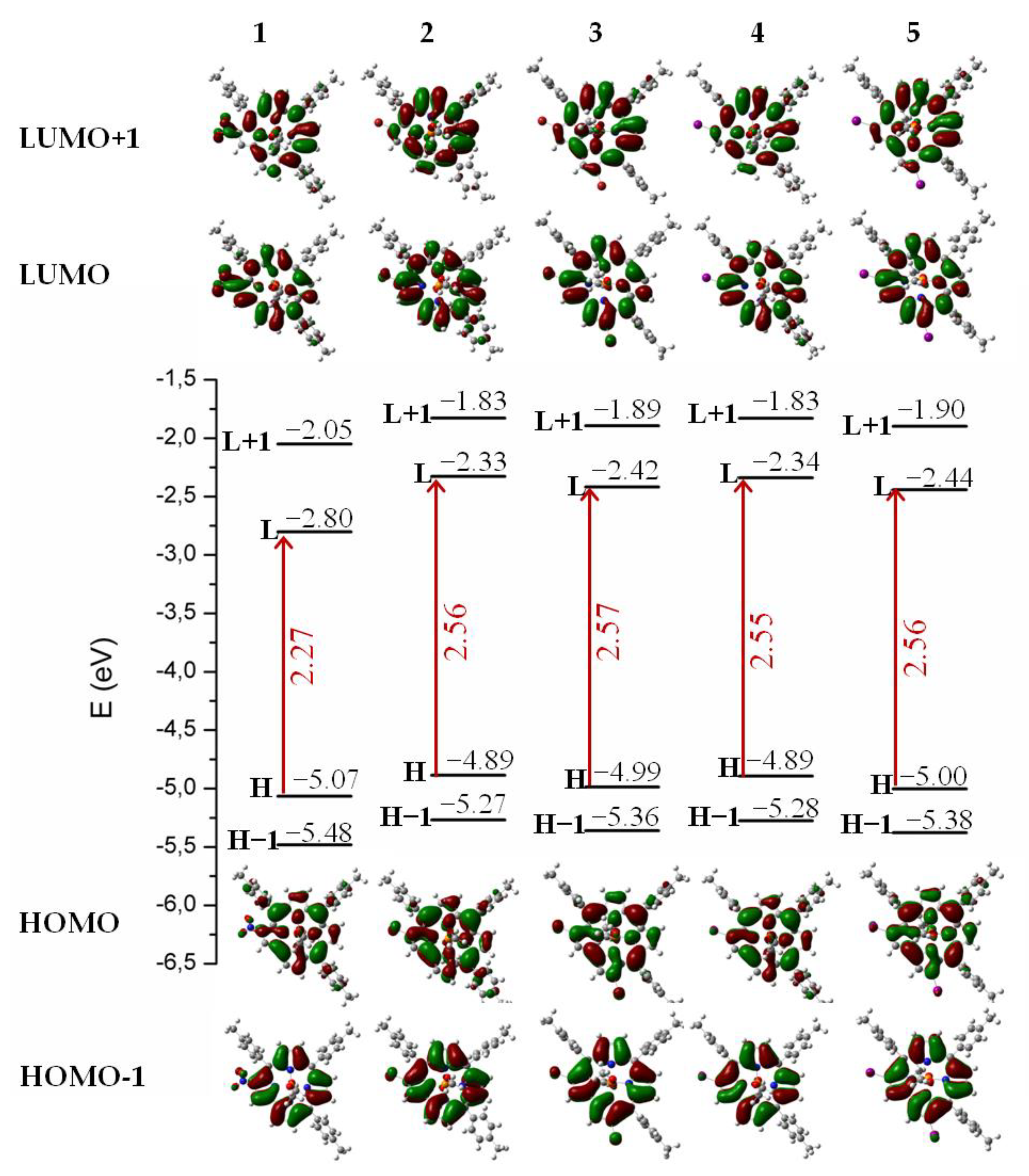
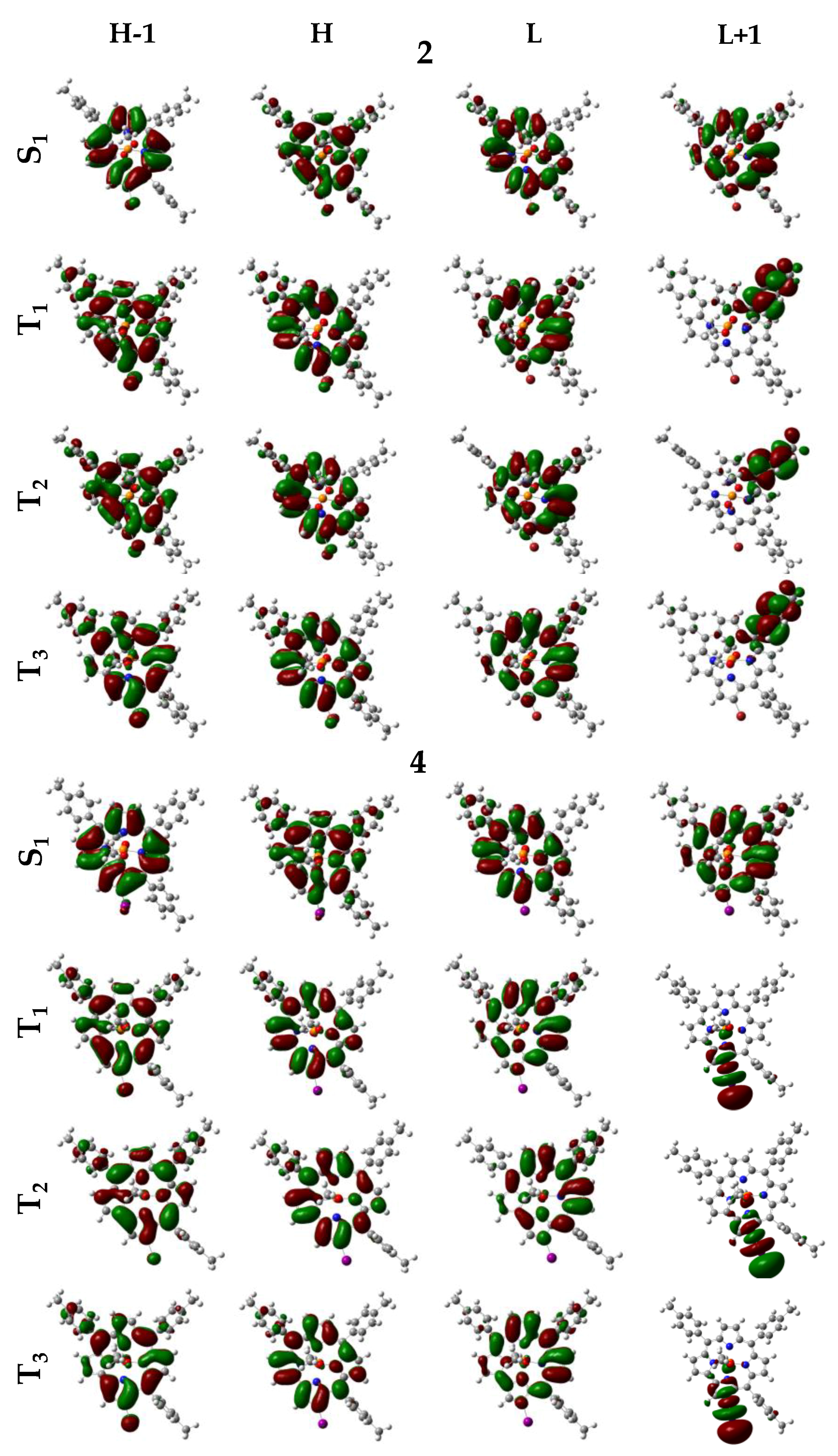
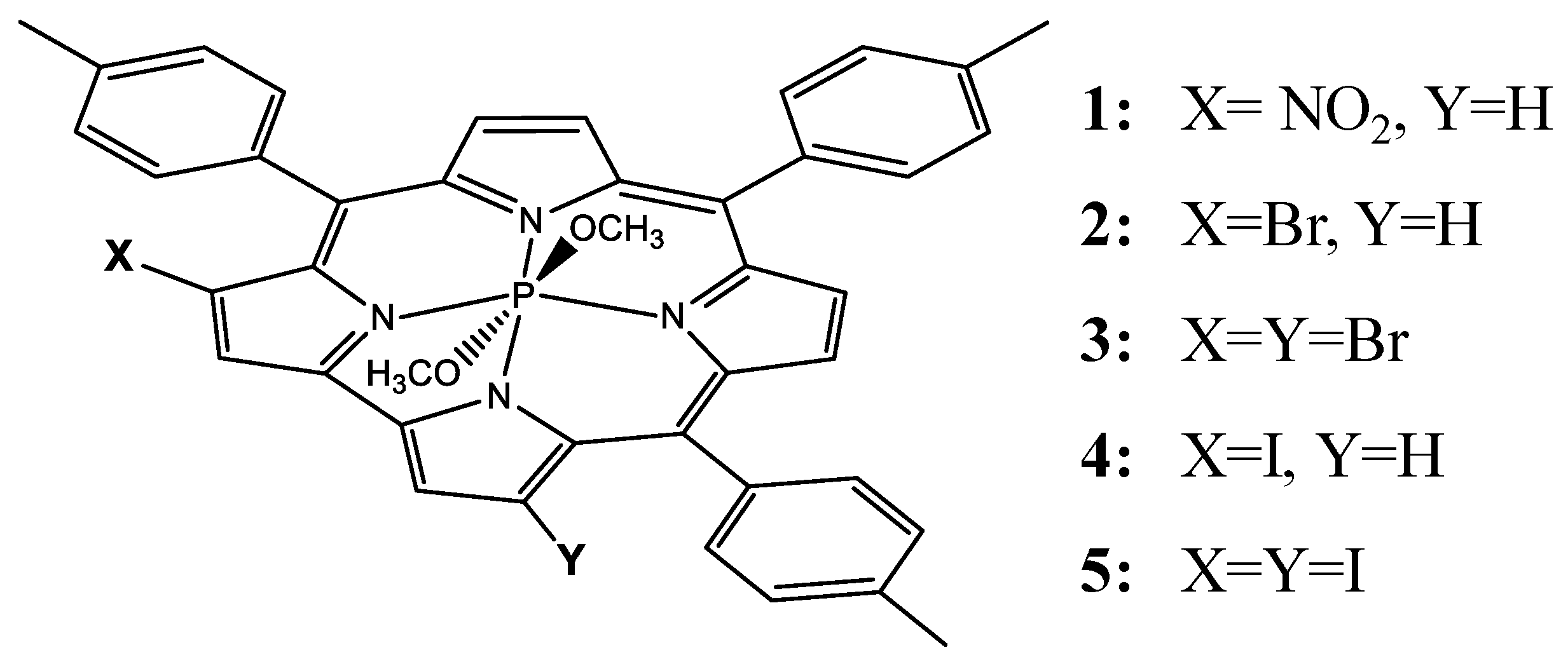
3. Results and Discussion
- (a)
- (b)
- (c)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, A.W.; Kay, I.T. Synthesis of corroles and related ring systems. Proc. R. Soc. Lond. A 1965, 288, 334–341. [Google Scholar] [CrossRef]
- Thomas, K.E.; Alemayehu, A.B.; Conradie, J.; Beavers, C.M.; Ghosh, A. The structural chemistry of metallocorroles: Combined X-ray crystallography and quantum chemistry studies afford unique insights. Acc. Chem. Res. 2012, 45, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Aviv-Harel, I.; Gross, Z. Coordination chemistry of corroles with focus on main group elements. Coord. Chem. Rev. 2011, 255, 717–736. [Google Scholar] [CrossRef]
- Walker, D.C.; Chappel, S.; Mahammed, A.; Brunschwig, B.S.; Winkler, J.R.; Gray, H.B.; Zaban, A.; Gross, Z. The π-system of corroles is much more electron-rich than of porphyrins, and one consequence is that full bromination (17) and chlorination. J. Porphyr. Phthalocyanines 2006, 10, 1259–1262. [Google Scholar] [CrossRef]
- Flamigni, L.; Gryko, D.T. Photoactive corrole-based arrays. Chem. Soc. Rev. 2009, 38, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Agadjanian, H.; Ma, J.; Rentsendorj, A.; Valluripalli, V.; Hwang, J.Y.; Maham-med, A.; Farkas, D.L.; Gray, H.B.; Gross, Z.; Medina-Kauwe, L.K. Tumor detection and elimination by a targeted gallium corrole. Proc. Natl. Acad. Sci. USA 2009, 106, 610–6105. [Google Scholar] [CrossRef] [PubMed]
- Mahammed, A.; Gross, Z. The importance of developing metal complexes with pronounced catalase-like activity. Catal. Sci. Technol. 2011, 1, 535–540. [Google Scholar] [CrossRef]
- Mahammed, A.; Gross, Z. Metallocorroles as photocatalysts for driving endergonic reactions, exemplified by bromide to bromine conversion. Angew. Chem. Int. Ed. 2015, 54, 12370–12373. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Lubow, J.; Chu, D.; Ma, J.; Agdjanian, H.; Sims, J.; Gray, H.B.; Gross, Z.; Farkas, D.L.; Medina-Kauwe, L.K. A mechanistic study of tumor-targeted corrole toxicity. Mol. Pharm. 2011, 8, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Simkhovich, L.; Mahammed, A.; Goldberg, I.; Gross, Z. Synthesis and characterization of germanium, tin, phosphorus, iron, and rhodium complexes of tris(pentafluorophenyl)corrole, and the utilization of the iron and rhodium corroles as cyclopropanation catalysts. Chem. Eur. J. 2001, 7, 1041–1055. [Google Scholar] [CrossRef]
- Buckley, H.L.; Arnold, J. Recent developments in out-of-plane metallocorrole chemistry across the periodic table. Dalton Trans. 2015, 44, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Furuyama, T.; Satoh, K. Rationally designed phthalocyanines having their main absorption band beyond 1000 nm. J. Am. Chem. Soc. 2011, 133, 19642–19645. [Google Scholar] [CrossRef] [PubMed]
- Pomarico, G.; Tortora, L.; Fronczek, F.R.; Smith, K.M.; Paolesse, R. Selective nitration and bromination of surprisingly ruffled phosphorus corroles. J. Inorg. Biochem. 2016, 158, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Mack, J.; Zheng, L.-M.; Shen, Z.; Kobayashi, N. Phosphorus(V)-corrole: Synthesis, spectroscopic properties, theoretical calculations, and potential utility for in vivo applications in living cells. Inorg. Chem. 2014, 53, 2797–2802. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Lee, W.-Z.; Ravikanth, M. Synthesis, structure and properties of a five-coordinate oxophosphorus(V) meso-triphenylcorrole. Eur. J. Inorg. Chem. 2012, 2012, 4231–4239. [Google Scholar] [CrossRef]
- Paolesse, R.; Boschi, T.; Licoccia, S.; Khoury, R.G.; Smith, K.M. Phosphorus complex of corrole. Chem. Commun. 1998, 1119–1120. [Google Scholar] [CrossRef]
- Kadish, K.M.; Ou, Z.; Adamian, V.A.; Guilard, R.; Gros, C.P.; Erben, C.; Will, S.; Vogel, E. Corroles with group 15 ions. 2. synthesis and characterization of octaethylcorroles containing a phosphorus central atom. Inorg. Chem. 2000, 39, 5675–5682. [Google Scholar] [CrossRef] [PubMed]
- Vestfrid, J.; Goldberg, I.; Gross, Z. Tuning the photophysical and redox properties of metallocorroles by iodination. Inorg. Chem. 2014, 53, 10536–10542. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, W.; Mackeyev, Y.; Lee, G.-S.; Kim, H.-J.; Tachikawa, T.; Hong, S.; Lee, S.; Kim, J.; Wilson, L.J.; et al. Selective oxidative degradation of organic pollutants by singlet oxygen-mediated photosensitization: Tin porphyrin versus C60 aminofullerene systems. Environ. Sci. Technol. 2012, 46, 9606–9613. [Google Scholar] [CrossRef] [PubMed]
- Han, S.K.; Sik, R.H.; Motten, A.G.; Chignell, C.F.; Bilski, P.J. Photosensitized oxidation of tetrabromobisphenol a by humic acid in aqueous solution. Photochem. Photobiol. 2009, 85, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Kohn, T.; Nelson, K.L. Sunlight-mediated inactivation of MS2 coliphage via exogenous singlet oxygen produced by sensitizers in natural waters. Environ. Sci. Technol. 2007, 41, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Romero, O.C.; Straub, A.P.; Kohn, T.; Nguyen, T.H. Role of temperature and suwannee river natural organic matter on inactivation kinetics of rotavirus and bacteriophage MS2 by solar irradiation. Environ. Sci. Technol. 2011, 45, 10385–10393. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, M.C.; Crutchley, R.J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233–234, 351–371. [Google Scholar] [CrossRef]
- Yano, S.; Hirohara, S.; Obata, M.; Hagiya, Y.; Ogura, S.; Ikeda, I.; Kataoka, H.; Tanaka, M.; Joh, T. Current states and future views in photodynamic therapy. J. Photochem. Photobiol. C 2011, 12, 46–67. [Google Scholar] [CrossRef]
- Allison, R.R.; Bagnato, V.S.; Sibata, C.H. Future of oncologic photodynamic therapy. Future Oncol. 2010, 6, 929–940. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, I.J.; Dougherty, T.J. Basic principles of photodynamic therapy. J. Porphyr. Phthalocyan. 2001, 5, 105–129. [Google Scholar] [CrossRef]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Gattuso, H.; Dumont, E.; Marazzi, M.; Monari, A. Two-photon-absorption DNA sensitization via solvated electron production: Unraveling photochemical pathways by molecular modeling and simulation. Phys. Chem. Chem. Phys. 2016, 18, 18598–18606. [Google Scholar] [CrossRef] [PubMed]
- Herzberg, G. Spectra of Diatomic Molecules, 2nd ed.; Van Nostrand Reinhold: New York, NY, USA, 1950; pp. 344–346. [Google Scholar]
- Marian, C.M. Spin-orbit coupling and intersystem crossing in molecules. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 187–203. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photoradiation therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Mazzone, G.; Alberto, M.E.; De Simone, B.C.; Marino, T.; Russo, N. Can expanded bacteriochlorins act as photosensitizers in photodynamic therapy? Good news from density functional theory computations. Molecules 2016, 21, 288. [Google Scholar] [CrossRef] [PubMed]
- De Simone, B.C.; Mazzone, G.; Russo, N.; Sicilia, E.; Toscano, M. Excitation energies, singlet–triplet energy gaps, spin–orbit matrix elements and heavy atom effects in BOIMPYs as possible photosensitizers for photodynamic therapy: A computational investigation. Phys. Chem. Chem. Phys. 2018, 20, 2656–2661. [Google Scholar] [CrossRef] [PubMed]
- Noimark, S.; Salvadori, E.; Gómez-Bombarelli, R.; MacRobert, A.J.; Parkina, I.P.; Kay, C.W.M. Comparative study of singlet oxygen production by photosensitiser dyes encapsulated in silicone: Towards rational design of anti-microbial surfaces. Phys. Chem. Chem. Phys. 2016, 18, 2810. [Google Scholar] [CrossRef] [PubMed]
- Quartarolo, A.D.; Sicilia, E.; Russo, N. On the potential use of squaraine derivatives as photosensitizers in photodynamic therapy: A TDDFT and RICC2 survey. J. Chem. Theory Comput. 2009, 5, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V. Solvent effect on vertical electronic transitions by the polarizable continuum model. J. Chem. Phys. 2000, 112, 2427–2435. [Google Scholar] [CrossRef]
- Tomasi, J.; Menucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Rinkevicius, Z.; Tunell, I.; Sałek, P.; Vahtras, O.; Ågren, H. Restricted density functional theory of linear time-dependent properties in open-shell molecules. J. Chem. Phys. 2003, 119, 34–46. [Google Scholar] [CrossRef]
- Ågren, H.; Vahtras, O.; Minaev, B. Response theory and calculations of spin-orbit coupling phenomena in molecules. Adv. Quantum Chem. 1996, 27, 71–162. [Google Scholar]
- Dalton. A Molecular Electronic Structure Program. Release Dalton 2011. Available online: http://daltonprogram.org/ (accessed on 24 February 2016).
- Koseki, S.; Schmidt, M.W.; Gordon, M.S. Effective nuclear charges for the first- through third-row transition metal elements in spin–orbit calculations. J. Phys. Chem. A 1998, 102, 10430–10436. [Google Scholar] [CrossRef]
- Alberto, M.E.; Pirillo, J.; Russo, N.; Adamo, C. A theoretical exploration of TypeI/TypeII dual photoreactivity of new promising Ru(II)-dyads for PDT approach. Inorg. Chem. 2016, 55, 11185–11192. [Google Scholar] [CrossRef] [PubMed]
- De Simone, B.C.; Mazzone, G.; Russo, N.; Sicilia, E.; Toscano, M. Computational investigation of the influence of halogen atoms on the photophysical properties of tetraphenylporphyrin and its Zinc(II) complexes. J. Phys. Chem. A 2018, 122, 2809–2815. [Google Scholar] [CrossRef] [PubMed]
- De Simone, B.C.; Mazzone, G.; Pirillo, J.; Russo, N.; Sicilia, E. Halogen atom effect on the photophysical properties of substituted aza-BODIPY derivatives. Phys. Chem. Chem. Phys. 2017, 19, 2530–2536. [Google Scholar] [CrossRef] [PubMed]
- Ruud, K.; Schimmelpfennig, B.; Ågren, H. Internal and external heavy-atom effects on phosphorescence radiative lifetimes calculated using a mean-field spin-orbit Hamiltonian. Chem. Phys. Lett. 1999, 310, 215–221. [Google Scholar] [CrossRef]
- Alberto, M.E.; De Simone, B.C.; Mazzone, G.; Sicilia, E.; Russo, N. The heavy atom effect on Zn(II) phthalocyanine derivatives: A theoretical exploration of the photophysical properties. Phys. Chem. Chem. Phys. 2015, 17, 23595–23601. [Google Scholar] [CrossRef] [PubMed]
- Alberto, M.E.; Iuga, C.; Quartarolo, A.D.; Russo, N. Bisanthracene bis(dicarboxylic imide)s as potential photosensitizers in photodynamic therapy. A theoretical investigation. J. Chem. Inf. Model. 2013, 53, 2334–2340. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Liu, H.Y.; Shen, H.; Hu, J.; Zhang, G.L.; Wang, H.; Ji, L.N.; Chang, C.K.; Jiang, H.F. Fluorescence properties of halogenated mono-hydroxyl corroles: The heavy-atom effects. J. Porphyr. Phthalocyanines 2009, 13, 1221–1226. [Google Scholar] [CrossRef]
- El-Sayed, M.A. Triplet state. Its radiative and nonradiative properties. Acc. Chem. Res. 1968, 1, 8–16. [Google Scholar] [CrossRef]
- Peterson, K.A.; Shepler, B.C.; Figgen, D.; Stoll, H. On the spectroscopic and thermochemical properties of ClO, BrO, IO, and their anions. J. Phys. Chem. A 2006, 110, 13877–13883. [Google Scholar] [CrossRef] [PubMed]
- Alberto, M.E.; Marino, T.; Quartarolo, A.D.; Russo, N. Photophysical origin of the reduced photodynamic therapy activity of temocene compared to Foscan®: Insights from theory. Phys. Chem. Chem. Phys. 2013, 15, 16167–16171. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cmpd. | State | ΔE | λ | f | Transitions | λexp |
|---|---|---|---|---|---|---|
| 1 | S1 | 1.84 | 673 | 0.275 | H→L (93%) | 635 |
| S2 | 2.19 | 566 | 0.161 | H − 1→L (75%) | 590 | |
| S3 | 2.68 | 463 | 0.439 | H→L + 1 (64%) | 447 | |
| S4 | 3.18 | 390 | 0.559 | H − 1→L + 1 (29%), H − 4→L (29%) | 428 | |
| T1 | 1.18 | 1050 | H→L (96%) | |||
| T2 | 1.62 | 766 | H − 1→L (92%) | |||
| 2 | S1 | 2.22 | 559 | 0.310 | H→L (86%) | 599 |
| S2 | 2.37 | 522 | 0.007 | H − 1→L (54%), H→L + 1 (45%) | 526 | |
| S3 | 3.03 | 405 | 1.112 | H→L + 1 (52%), H − 1→L (44%) | 421 | |
| S4 | 3.18 | 390 | 1.066 | H − 1→L + 1 (82%) | 410 | |
| T1 | 1.41 | 879 | H→L (99%) | |||
| T2 | 1.76 | 705 | H→L + 1 (55%), H − 1→L (43%) | |||
| T3 | 1.96 | 632 | H − 1→L (56%), H→L + 1 (43%) | |||
| 3 | S1 | 2.24 | 555 | 0.295 | H→L (86%) | 599 |
| S2 | 2.39 | 520 | 0.011 | H − 1→L (56%), H→L + 1 (43%) | 527 | |
| S3 | 3.07 | 404 | 1.176 | H→L + 1 (54%), H − 1→L (41%) | 422 | |
| S4 | 3.21 | 386 | 0.995 | H − 1→L + 1 (83%) | 411 | |
| T1 | 1.40 | 885 | H→L (99%) | |||
| T2 | 1.78 | 697 | H→L + 1 (50%), H − 1→L (48%) | |||
| T3 | 1.97 | 629 | H − 1→L (51%), H→L + 1 (48%) | |||
| 4 | S1 | 2.22 | 559 | 0.318 | H→L, (86%) | |
| S2 | 2.37 | 522 | 0.008 | H − 1→L (54%), H→L + 1 (45%) | ||
| S3 | 3.04 | 407 | 1.101 | H→L + 1 (51%), H − 1→L (42%) | ||
| S4 | 3.18 | 390 | 1.069 | H − 1→L + 1 (82%) | ||
| T1 | 1.41 | 877 | H→L (99%) | |||
| T2 | 1.76 | 706 | H→L + 1 (54%), H − 1→L (44%) | |||
| T3 | 1.96 | 632 | H − 1→L (56%),H→L + 1 (43%) | |||
| 5 | S1 | 2.23 | 556 | 0.311 | H→L (86%) | |
| S2 | 2.39 | 519 | 0.014 | H − 1→L (57%), H→L + 1 (42%) | ||
| S3 | 3.05 | 406 | 1.186 | H→L + 1 (53%), H − 1→L (40%) | ||
| S4 | 3.22 | 385 | 1.018 | H − 1→L + 1 (82%) | ||
| T1 | 1.40 | 887 | H→L (99%) | |||
| T2 | 1.78 | 698 | H − 1→L (50%), H→L + 1 (48%) | |||
| T3 | 1.97 | 629 | H→L + 1 (50%), H − 1→L (49%) |
| Cmpd. | ΔES1–T1 | ΔES1–T2 | ΔES1–T3 | |||
|---|---|---|---|---|---|---|
| 1 | 0.48 | 0.66 | 0.35 | 0.22 | ||
| 2 | 0.28 | 0.81 | 0.67 | 0.46 | 0.32 | 0.26 |
| 3 | 0.26 | 0.84 | 0.76 | 0.46 | 0.27 | 0.27 |
| 4 | 13.10 | 0.81 | 35.0 | 0.46 | 5.4 | 0.26 |
| 5 | 8.54 | 0.83 | 77.27 | 0.45 | 12.65 | 0.26 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alberto, M.E.; De Simone, B.C.; Mazzone, G.; Russo, N.; Toscano, M. Photophysical Properties of Nitrated and Halogenated Phosphorus Tritolylcorrole Complexes: Insights from Theory. Molecules 2018, 23, 2779. https://doi.org/10.3390/molecules23112779
Alberto ME, De Simone BC, Mazzone G, Russo N, Toscano M. Photophysical Properties of Nitrated and Halogenated Phosphorus Tritolylcorrole Complexes: Insights from Theory. Molecules. 2018; 23(11):2779. https://doi.org/10.3390/molecules23112779
Chicago/Turabian StyleAlberto, Marta Erminia, Bruna Clara De Simone, Gloria Mazzone, Nino Russo, and Marirosa Toscano. 2018. "Photophysical Properties of Nitrated and Halogenated Phosphorus Tritolylcorrole Complexes: Insights from Theory" Molecules 23, no. 11: 2779. https://doi.org/10.3390/molecules23112779
APA StyleAlberto, M. E., De Simone, B. C., Mazzone, G., Russo, N., & Toscano, M. (2018). Photophysical Properties of Nitrated and Halogenated Phosphorus Tritolylcorrole Complexes: Insights from Theory. Molecules, 23(11), 2779. https://doi.org/10.3390/molecules23112779