A Computational Approach for the Prediction of HIV Resistance Based on Amino Acid and Nucleotide Descriptors




Abstract
1. Introduction
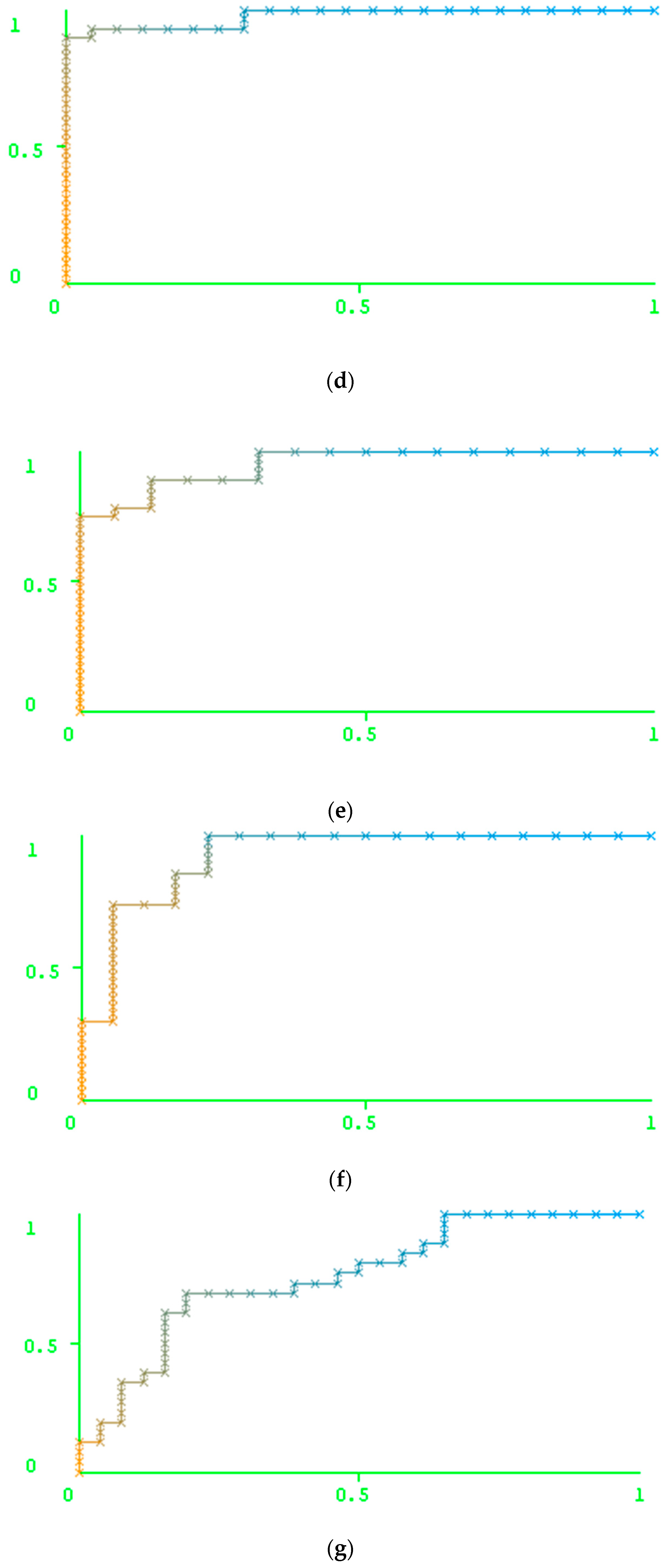
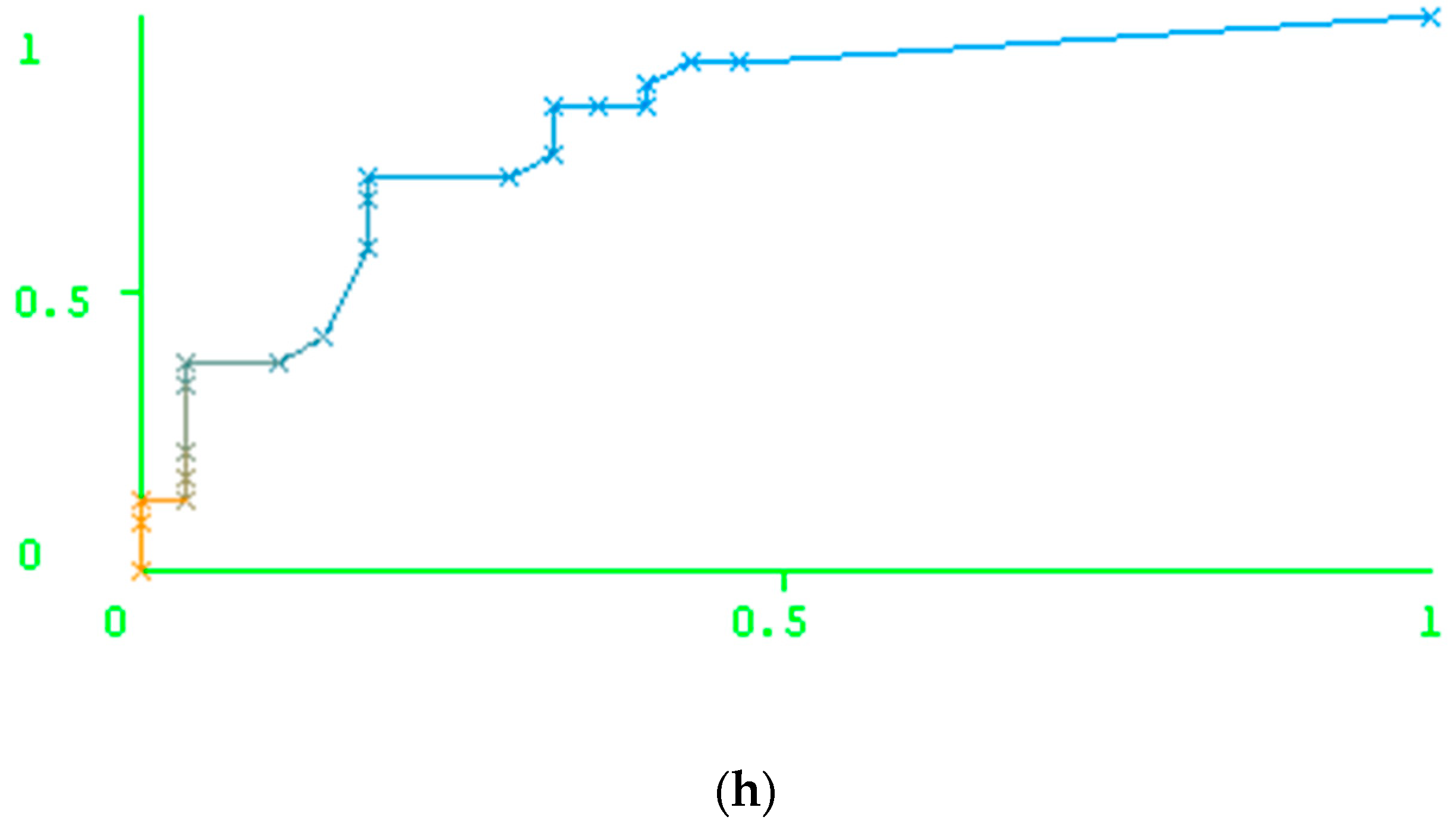
2. Results
Results of the Prediction Based on the Peptide-Based Descriptors and Nucleotide Descriptors
3. Discussion
3.1. Comparison of the Accuracy Obtained Using Particular Types of Descriptors
3.2. Application of the Model to Predict Human Immunodeficiency Virus Type 1 (HIV-1) Resistance to Protease Inhibitors
3.3. Comparison with the Earlier Developed Approaches
4. Materials and Methods
4.1. Datasets
4.2. Descriptors
4.3. Algorithm and Validation
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- The Joint United Nations Programme on HIV/AIDS (UNAIDS). Available online: http://www.unaids.org/ru (accessed on 11 September 2018).
- Drăghici, S.; Potter, R.B. Predicting HIV drug resistance with neural networks. Bioinformatics 2003, 19, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Schmidt, B.; Walter, H.; Kaiser, R.; Lengauer, T.; Hoffmann, D.; Korn, K.; Selbig, J. Diversity and complexity of HIV-1 drug resistance: A bioinformatics approach to predicting phenotype from genotype. Proc. Natl. Acad. Sci. USA 2002, 99, 8271–8276. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Däumer, M.; Oette, M.; Korn, K.; Hoffmann, D.; Kaiser, R.; Lengauer, T.; Selbig, J.; Walter, H. Geno2pheno: Estimating phenotypic drug resistance from HIV-1 genotypes. Nucleic Acids Res. 2003, 31, 3850–3855. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Taylor, J.; Wadhera, G.; Ben-Hur, A.; Brutlag, D.L.; Shafer, R.W. Genotypic predictors of human immunodeficiency virus type 1 drug resistance. Proc. Natl. Acad. Sci. USA 2006, 103, 17355–17360. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.J.; Lewis, F.I.; Miller, M.D.; Brown, A.J.L. Genetic basis of variation in tenofovir drug susceptibility in HIV-1. AIDS 2008, 22, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Heider, D.; Verheyen, J.; Hoffmann, D. Machine learning on normalized protein sequences. BMC Res. Notes 2011, 4, 94. [Google Scholar] [CrossRef] [PubMed]
- Van Westen, G.J.P.; Hendriks, A.; Wegner, J.K.; Ijzerman, A.P.; van Vlijmen, H.W.T.; Bender, A. Significantly improved HIV inhibitor efficacy prediction employing proteochemometric models generated from antivirogram data. PLoS Comput. Biol. 2013, 9, e1002899. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.; Senge, R.; Neumann, U.; Hüllermeier, E.; Heider, D. Exploiting HIV-1 protease and reverse transcriptase cross-resistance information for improved drug resistance prediction by means of multi-label classification. BioData Min. 2016, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Amamuddy, O.S.; Bishop, N.T.; Tastan Bishop, Ö. Improving fold resistance prediction of HIV-1 against protease and reverse transcriptase inhibitors using artificial neural networks. BMC Bioinformatics 2017, 18, 369. [Google Scholar]
- Tarasova, O.A.; Filimonov, D.A.; Poroikov, V.V. Computational prediction of human immunodeficiency resistance to reverse transcriptase inhibitors. Biomed. Khim. 2017, 63, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, O.; Filimonov, D.; Poroikov, V. PASS-based approach to predict HIV-1 reverse transcriptase resistance. J. Bioinform. Comput. Biol. 2017, 15, 1650040. [Google Scholar] [CrossRef] [PubMed]
- Kierczak, M.; Ginalski, K.; Dramiński, M.; Koronacki, J.; Rudnicki, W.; Komorowski, J. A Rough Set-Based Model of HIV-1 Reverse Transcriptase Resistome. Bioinform. Biol. Insights 2009, 3, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Bozek, K.; Lengauer, T.; Sierra, S.; Kaiser, R.; Domingues, F.S. Analysis of physicochemical and structural properties determining HIV-1 coreceptor usage. PLoS Comput. Biol. 2013, 9, e1002977. [Google Scholar] [CrossRef] [PubMed]
- Dybowski, J.N.; Riemenschneider, M.; Hauke, S.; Pyka, M.; Verheyen, J.; Hoffmann, D.; Heider, D. Improved Bevirimat resistance prediction by combination of structural and sequence-based classifiers. BioData Min. 2011, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, T.M.; Burger, P.B.; Butch, C.J.; Pelly, S.C.; Liotta, D.C. A machine learning approach for predicting HIV reverse transcriptase mutation susceptibility of biologically active compounds. J. Chem. Inf. Model. 2018, 58, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Heider, D.; Hoffmann, D. Interpol: An R package for preprocessing of protein sequences. BioData Min. 2011, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, O.; Rudik, A.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. QNA-based prediction of sites of metabolism. Molecules 2017, 22, 2123. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, O.; Poroikov, V. HIV resistance prediction to reverse transcriptase inhibitors: Focus on open data. Molecules 2018, 23, 956. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.; Heider, D. Current approaches in computational drug resistance prediction in HIV. Curr. HIV Res. 2016, 14, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Geretti, A.M. Antiretroviral Resistance in Clinical Practice; Mediscript: London, UK, 2006; pp. 145–153. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Drug | Peptide Descriptors | Nucleotide Descriptors | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reverse Transcriptase Inhibitors | ||||||||||
| Sns | Spc | PPV | MCC | AUC | Sns | Spc | PPV | MCC | AUC | |
| 3TC | 0.98 | 0.68 | 0.95 | 0.74 | 0.96 | 0.99 | 0.63 | 0.93 | 0.75 | 0.97 |
| ABC | 0.98 | 0.74 | 0.94 | 0.70 | 0.91 | 0.98 | 0.72 | 0.89 | 0.72 | 0.92 |
| AZT | 0.91 | 0.76 | 0.89 | 0.70 | 0.93 | 0.93 | 0.78 | 0.85 | 0.72 | 0.94 |
| D4T | 0.93 | 0.80 | 0.84 | 0.70 | 0.91 | 0.94 | 0.79 | 0.85 | 0.70 | 0.91 |
| DDI | 0.90 | 0.74 | 0.92 | 0.72 | 0.94 | 0.98 | 0.65 | 0.89 | 0.69 | 0.91 |
| EFV | 0.88 | 0.76 | 0.91 | 0.70 | 0.91 | 0.87 | 0.89 | 0.81 | 0.69 | 0.91 |
| ETR | 0.92 | 0.74 | 0.94 | 0.70 | 0.92 | 0.87 | 0.99 | 0.88 | 0.78 | 0.93 |
| NVP | 0.96 | 0.80 | 0.90 | 0.72 | 0.94 | 0.92 | 0.89 | 0.87 | 0.77 | 0.96 |
| TDF | 0.88 | 0.69 | 0.86 | 0.70 | 0.91 | 0.60 | 0.95 | 0.91 | 0.69 | 0.97 |
| Avg * | 0.93 | 0.75 | 0.91 | 0.71 | 0.93 | 0.90 | 0.81 | 0.88 | 0.72 | 0.94 |
| Protease Inhibitors | ||||||||||
| Sns | Spc | PPV | MCC | AUC | Sns | Spc | PPV | MCC | AUC | |
| FPV | 0.96 | 0.68 | 0.89 | 0.69 | 0.91 | 0.94 | 0.61 | 0.88 | 0.69 | 0.91 |
| ATV | 0.98 | 0.68 | 0.90 | 0.70 | 0.92 | 0.97 | 0.61 | 0.90 | 0.70 | 0.91 |
| IDV | 0.97 | 0.74 | 0.89 | 0.72 | 0.93 | 0.98 | 0.86 | 0.92 | 0.77 | 0.96 |
| LPV | 0.96 | 0.76 | 0.86 | 0.70 | 0.92 | 0.92 | 0.71 | 0.87 | 0.74 | 0.93 |
| NFV | 0.96 | 0.89 | 0.88 | 0.77 | 0.96 | 0.95 | 0.84 | 0.91 | 0.77 | 0.94 |
| SQV | 0.94 | 0.79 | 0.89 | 0.75 | 0.93 | 0.96 | 0.83 | 0.91 | 0.77 | 0.94 |
| TPV | 0.96 | 0.74 | 0.86 | 0.72 | 0.94 | 0.92 | 0.64 | 0.89 | 0.70 | 0.92 |
| DRV | 0.96 | 0.78 | 0.88 | 0.72 | 0.91 | 0.98 | 0.82 | 0.84 | 0.72 | 0.92 |
| Avg | 0.96 | 0.76 | 0.88 | 0.72 | 0.93 | 0.95 | 0.74 | 0.89 | 0.73 | 0.93 |
| Drug | Nr * | Ns | Sns | Spc | PPV | MCC | AUC |
|---|---|---|---|---|---|---|---|
| FPV | 65 | 340 | 0.43 | 0.94 | 0.51 | 0.40 | 0.91 |
| ATV | 96 | 271 | 0.84 | 0.96 | 0.82 | 0.81 | 0.98 |
| IDV | 214 | 184 | 0.78 | 0.76 | 0.69 | 0.90 | 0.92 |
| LPV | 145 | 142 | 0.79 | 0.94 | 0.77 | 0.70 | 0.93 |
| NFV | 248 | 168 | 0.94 | 0.98 | 0.88 | 0.96 | 0.97 |
| SQV | 192 | 223 | 0.83 | 0.80 | 0.80 | 0.71 | 0.94 |
| TPV | 78 | 118 | 0.52 | 0.96 | 0.50 | 0.50 | 0.96 |
| DRV | 64 | 124 | 0.65 | 0.94 | 0.59 | 0.76 | 0.96 |
| Avg | 0.725 | 0.91 | 0.70 | 0.72 | 0.95 |
| Drug | BA (Our) | BA [3] | BA [6] | AUC (Our) | AUC [7] | MCR * (Our) | MCR [10] |
|---|---|---|---|---|---|---|---|
| 3TC | 0.81 | 0.89 | 0.9 | 0.97 | 0.94 | 7.29 | 3.87 |
| ABC | 0.85 | 0.85 | 0.69 | 0.92 | 0.92 | 6.8 | 6.53 |
| AZT | 0.86 | 0.89 | 0.70 | 0.94 | 0.91 | 13.96 | 36.19 |
| D4T | 0.87 | 0.75 | 0.76 | 0.94 | 0.90 | 10.01 | 7.31 |
| DDI | 0.82 | 0.68 | 0.75 | 0.91 | 0.85 | 10.90 | 8.05 |
| EFV | 0.88 | 0.902 | 0.84 | 0.96 | 0.93 | 18.08 | 16.08 |
| ETR | 0.93 | N/D | N/D | 0.93 | N/D | 10.01 | 6.58 |
| NVP | 0.91 | 0.91 | 0.91 | 0.94 | 0.92 | 12.7 | 24.87 |
| RPV | N/D | 0.89 | N/D | N/D | N/D | N/D | 1.55 |
| TDF | 0.78 | N/D | N/D | 0.92 | 0.83 | 12.3 | 5.39 |
| FPV | 0.78 | N/D | N/D | 0.92 | N/D | 15.8 | 16.08 |
| ATV | 0.79 | 0.87 | 0.71 | 0.93 | 0.93 | 26.2 | 26.69 |
| IDV | 0.92 | 0.89 | 0.75 | 0.98 | 0.97 | 8.2 | 34.29 |
| LPV | 0.82 | N/D | 0.77 | 0.94 | 0.96 | 23.8 | 9.79 |
| NFV | 0.90 | 0.89 | 0.76 | 0.96 | 0.94 | 7.15 | 25.23 |
| SQV | 0.90 | 0.88 | 0.75 | 0.96 | 0.96 | 11.15 | 30.37 |
| TPV | 0.78 | N/D | N/D | 0.87 | N/D | 4.77 | 9.07 |
| DRV | 0.79 | N/D | N/D | 0.92 | N/D | 2.38 | 2.98 |
| Avg | 0.854 | 0.857 | 0.78 | 0.94 | 0.92 | 11.85 | 15.05 |
| Drug | Random Forest (Our) | Decision Trees [3,4] | R * | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sns | Spc | PPV | BA | AUC | Sns | Spc | PPV | BA | ||
| FPV | 0.41 | 0.97 | 0.89 | 0.69 | 0.94 | 0.99 | 0.34 | 0.52 | 0.675 | 32 |
| ATV | 0.69 | 0.99 | 0.99 | 0.84 | 0.97 | 0.86 | 0.72 | 0.70 | 0.91 | 89 |
| IDV | 0.99 | 0.96 | 0.92 | 0.98 | 0.98 | 0.91 | 0.66 | 0.89 | 0.785 | 190 |
| LPV | 0.99 | 0.83 | 0.92 | 0.92 | 0.92 | 0.90 | 0.90 | 0.99 | 0.90 | 96 |
| NFV | 0.97 | 0.97 | 0.95 | 0.99 | 0.97 | 0.86 | 0.50 | 0.86 | 0.68 | 215 |
| SQV | 0.91 | 0.82 | 0.91 | 0.92 | 0.96 | 0.83 | 0.49 | 0.82 | 0.66 | 162 |
| TPV | 0.10 | 0.99 | 0.09 | 0.76 | 0.78 | 0.54 | 0.89 | 0.53 | 0.715 | 16 |
| DRV | 0.20 | 0.99 | 0.16 | 0.76 | 0.80 | 0.75 | 0.88 | 0.75 | 0.815 | 24 |
| Drug | FR * | Total | Susceptible | Resistant |
|---|---|---|---|---|
| 3TC | 1.5 | 1727 | 635 | 1092 |
| ABC | 4.5 | 1655 | 1494 | 161 |
| AZT | 2.2 | 1747 | 1002 | 745 |
| D4T | 1.7 | 1755 | 1632 | 123 |
| DDI | 1.7 | 1756 | 1034 | 722 |
| EFV | 2.5 | 1378 | 1278 | 100 |
| ETR | 2.9 | 1836 | 1754 | 82 |
| NVP | 2.5 | 1844 | 962 | 882 |
| RPV ** | N/D | N/D | N/D | N/D |
| TDF | 1.5 | 1378 | 1218 | 160 |
| FPV | 20 | 1965 | 1614 | 351 |
| ATV | 2.2 | 1309 | 714 | 595 |
| IDV | 2.4 | 2007 | 1036 | 971 |
| LPV | 6.7 | 1693 | 917 | 717 |
| NFV | 3.6 | 2102 | 954 | 1148 |
| SQV | 2.07 | 2012 | 925 | 1087 |
| TPV | 1.2 | 1060 | 477 | 583 |
| DRV | 5.5 | 734 | 147 | 582 |
| Drug | FR * | Total | Susceptible | Resistant |
|---|---|---|---|---|
| 3TC | 1.5 | 720 | 74 | 646 |
| ABC | 4.5 | 740 | 181 | 563 |
| AZT | 2.2 | 718 | 272 | 446 |
| D4T | 1.7 | 723 | 258 | 465 |
| DDI | 1.7 | 720 | 123 | 597 |
| EFV | 2.5 | 744 | 353 | 391 |
| ETR | 2.9 | 193 | 57 | 136 |
| NVP | 2.5 | 756 | 316 | 440 |
| RPV | N/D | N/D | N/D | N/D |
| TDF | 1.5 | 423 | 234 | 189 |
| FPV | 20 | 774 | 666 | 108 |
| ATV | 2.2 | 352 | 150 | 202 |
| IDV | 2.4 | 795 | 367 | 428 |
| LPV | 6.7 | 614 | 332 | 282 |
| NFV | 3.6 | 833 | 342 | 491 |
| SQV | 2.07 | 827 | 445 | 382 |
| TPV | 1.2 | 196 | 101 | 96 |
| DRV | 5.5 | 165 | 139 | 26 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarasova, O.; Biziukova, N.; Filimonov, D.; Poroikov, V. A Computational Approach for the Prediction of HIV Resistance Based on Amino Acid and Nucleotide Descriptors. Molecules 2018, 23, 2751. https://doi.org/10.3390/molecules23112751
Tarasova O, Biziukova N, Filimonov D, Poroikov V. A Computational Approach for the Prediction of HIV Resistance Based on Amino Acid and Nucleotide Descriptors. Molecules. 2018; 23(11):2751. https://doi.org/10.3390/molecules23112751
Chicago/Turabian StyleTarasova, Olga, Nadezhda Biziukova, Dmitry Filimonov, and Vladimir Poroikov. 2018. "A Computational Approach for the Prediction of HIV Resistance Based on Amino Acid and Nucleotide Descriptors" Molecules 23, no. 11: 2751. https://doi.org/10.3390/molecules23112751
APA StyleTarasova, O., Biziukova, N., Filimonov, D., & Poroikov, V. (2018). A Computational Approach for the Prediction of HIV Resistance Based on Amino Acid and Nucleotide Descriptors. Molecules, 23(11), 2751. https://doi.org/10.3390/molecules23112751