
Lignocellulosic Biomass as Source for Lignin-Based Environmentally Benign Antioxidants

 ,
, 
Abstract

1. Introduction
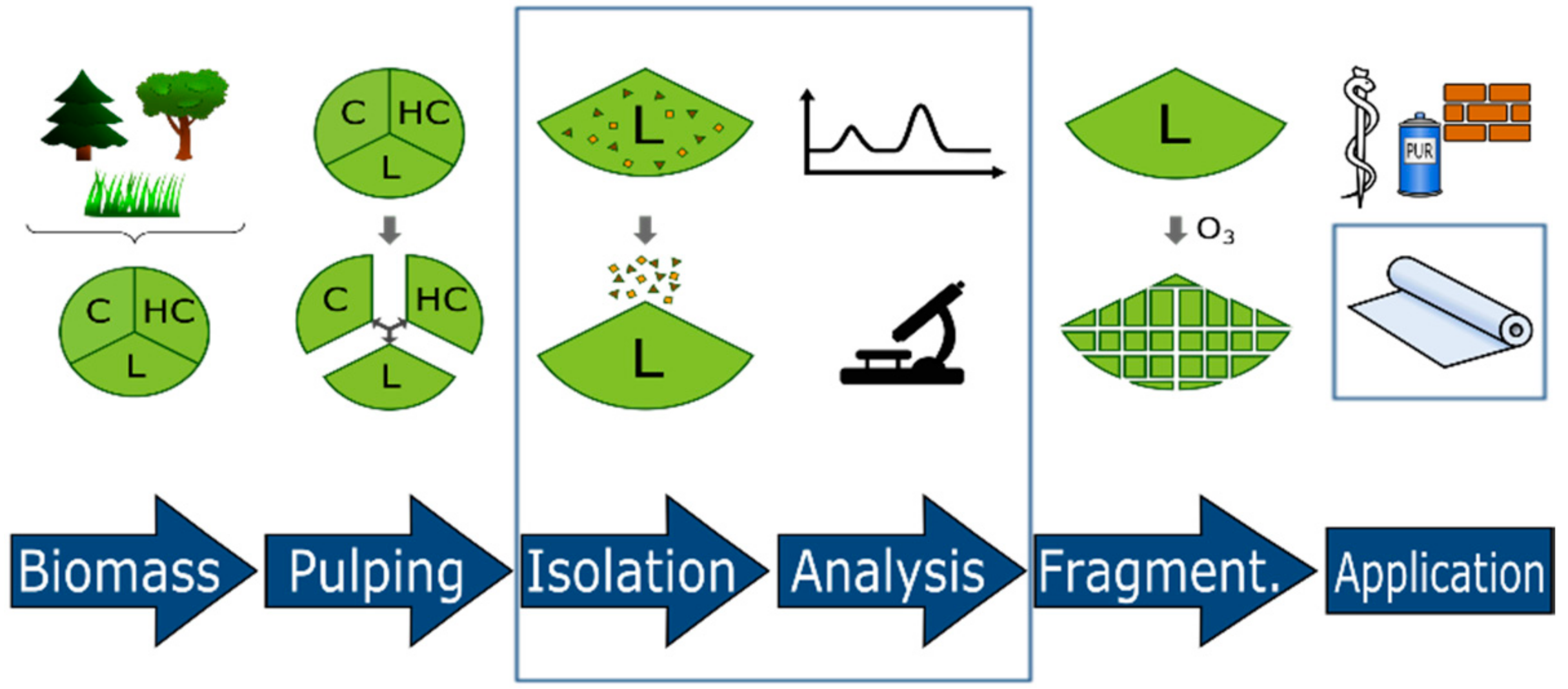
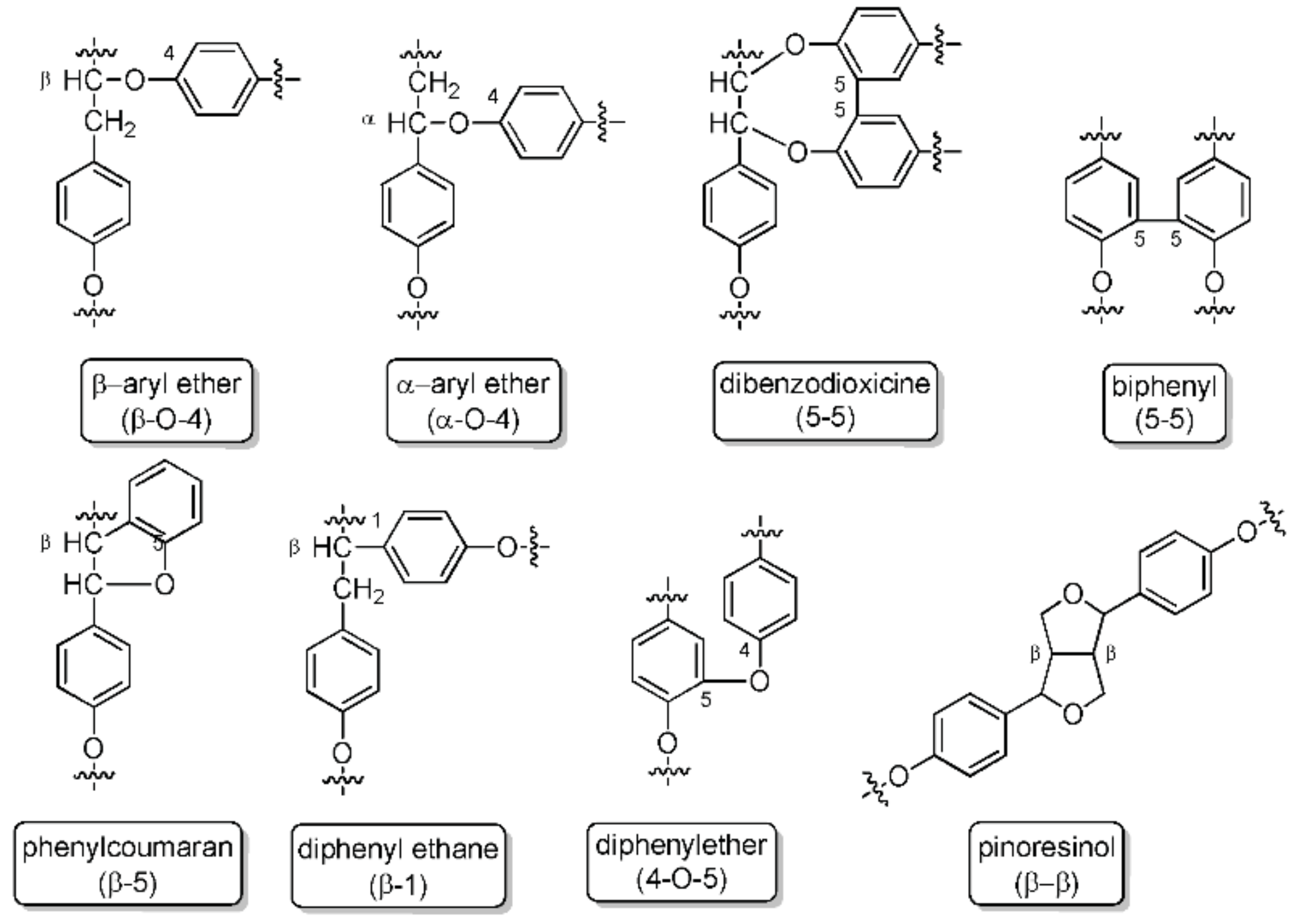
1.1. Lignin Availavility and Structure
1.2. Antioxidant Capacity and Corresponding Assays
2. Results and Discussion
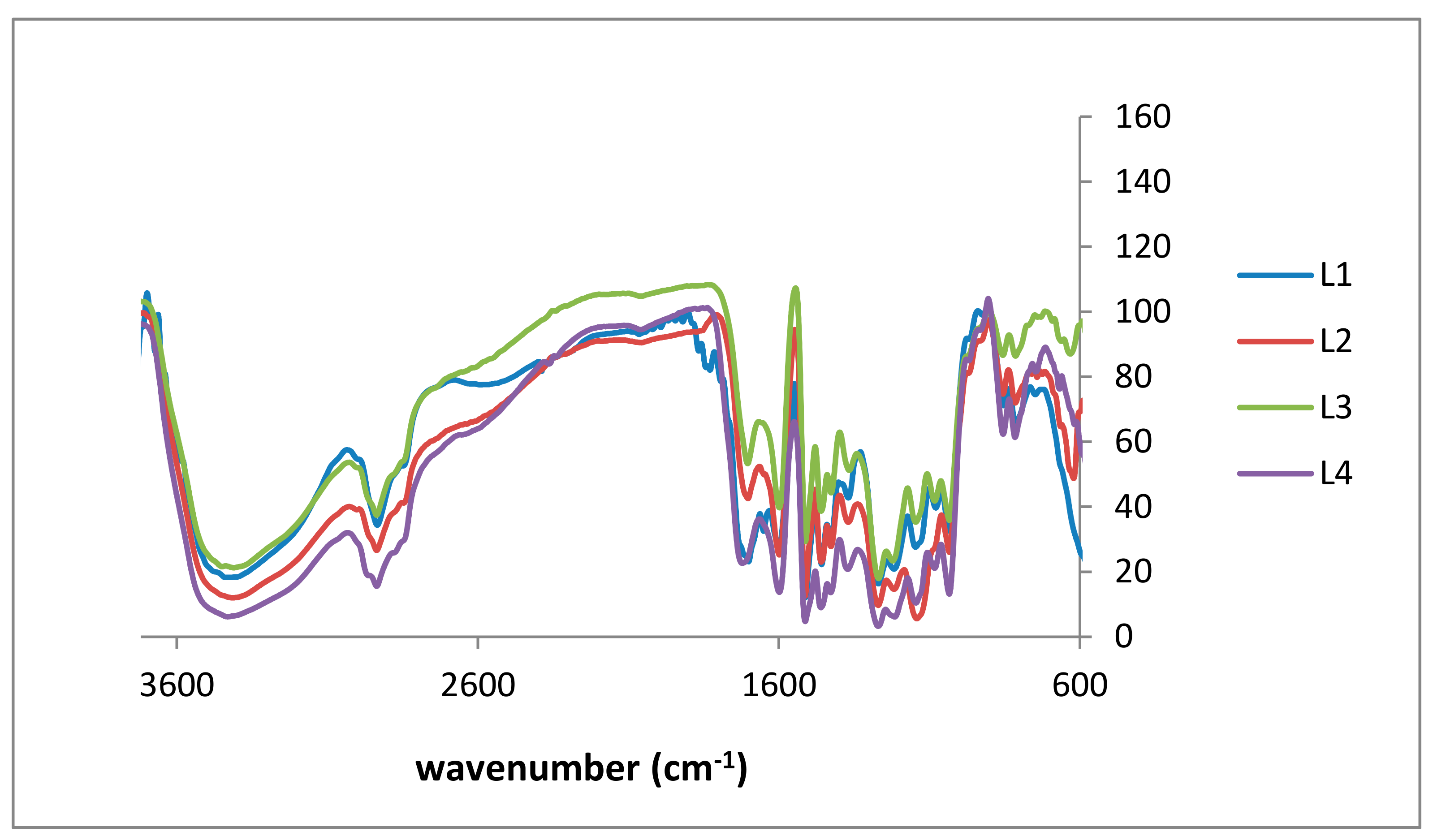
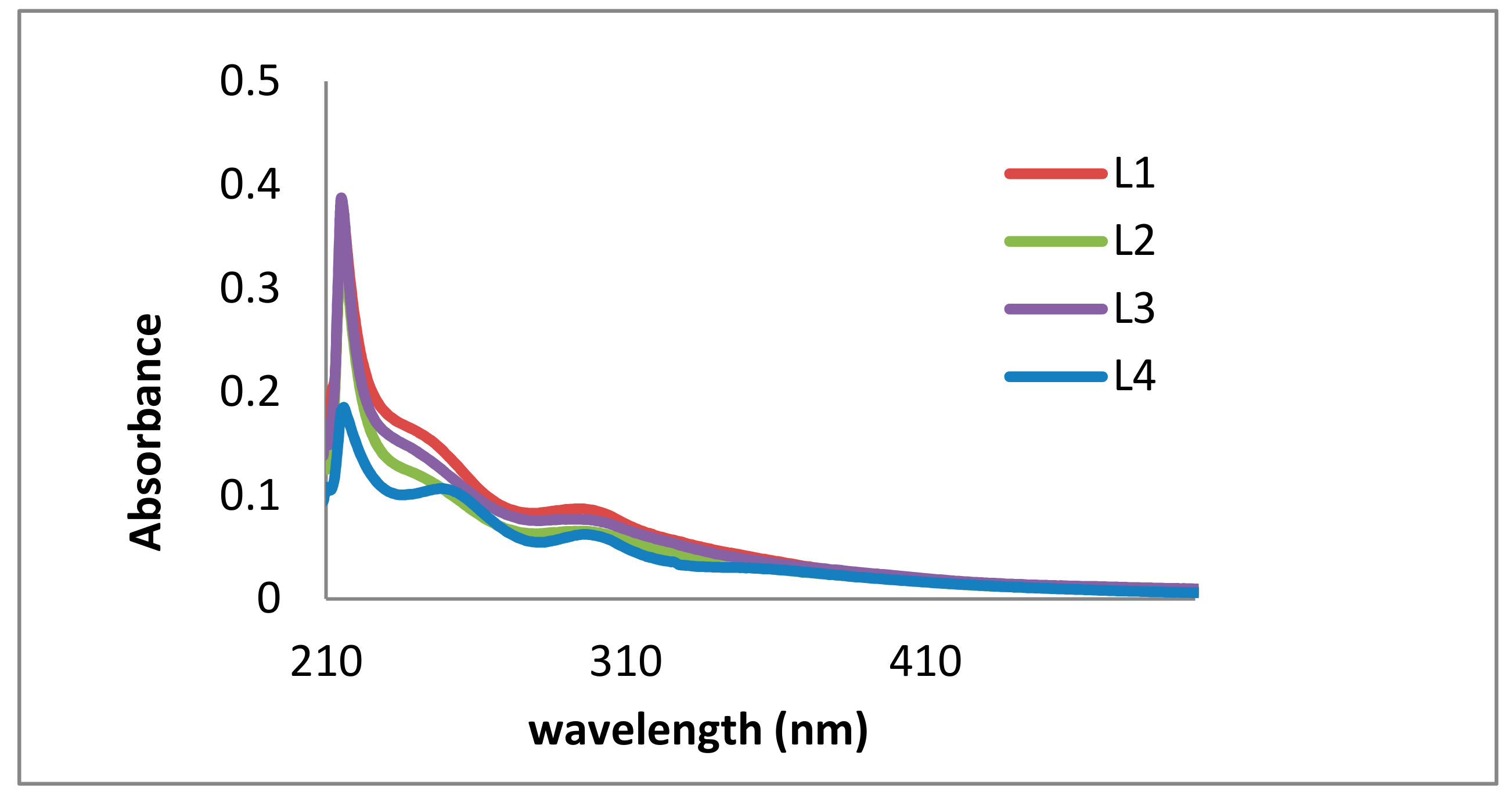
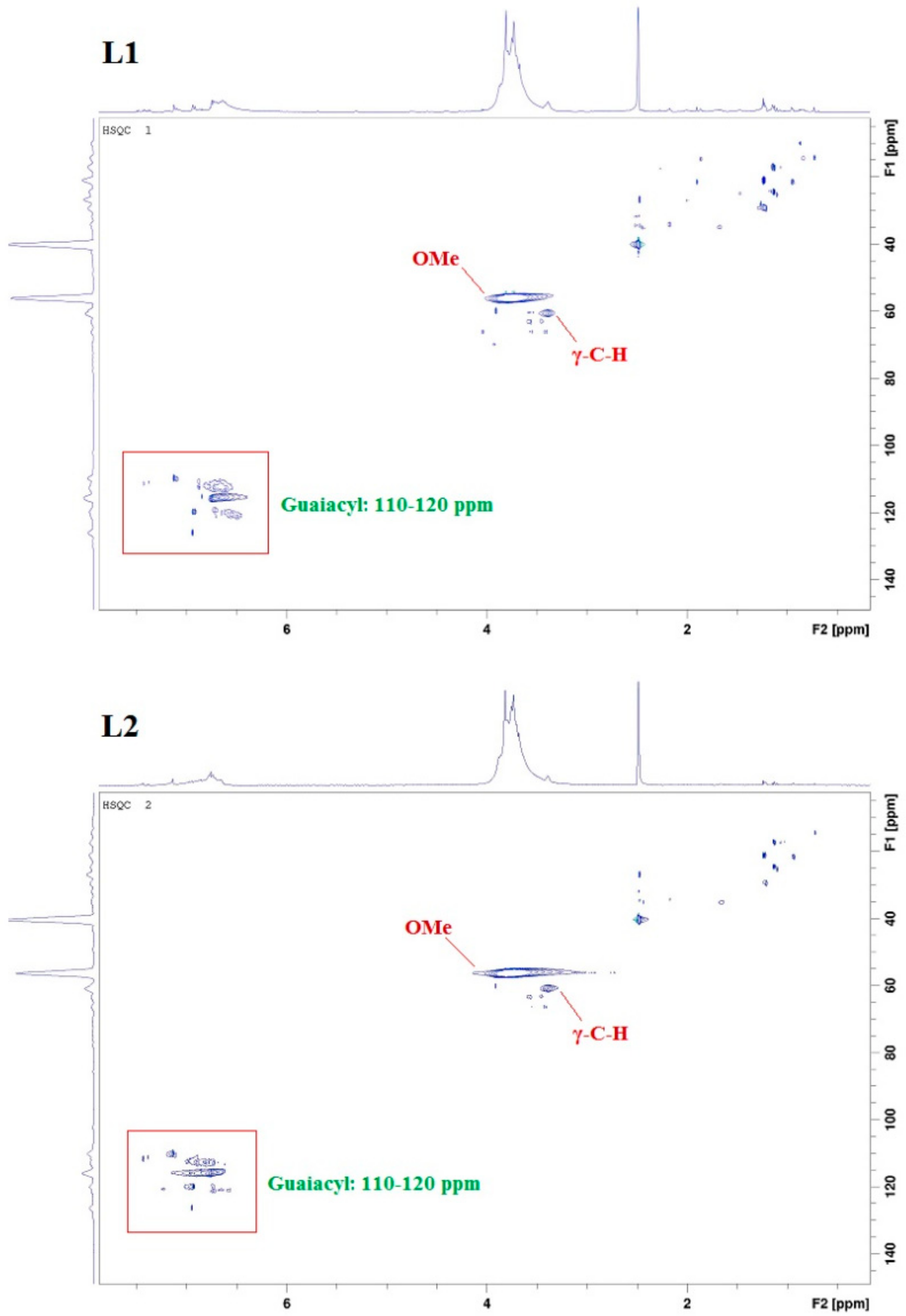
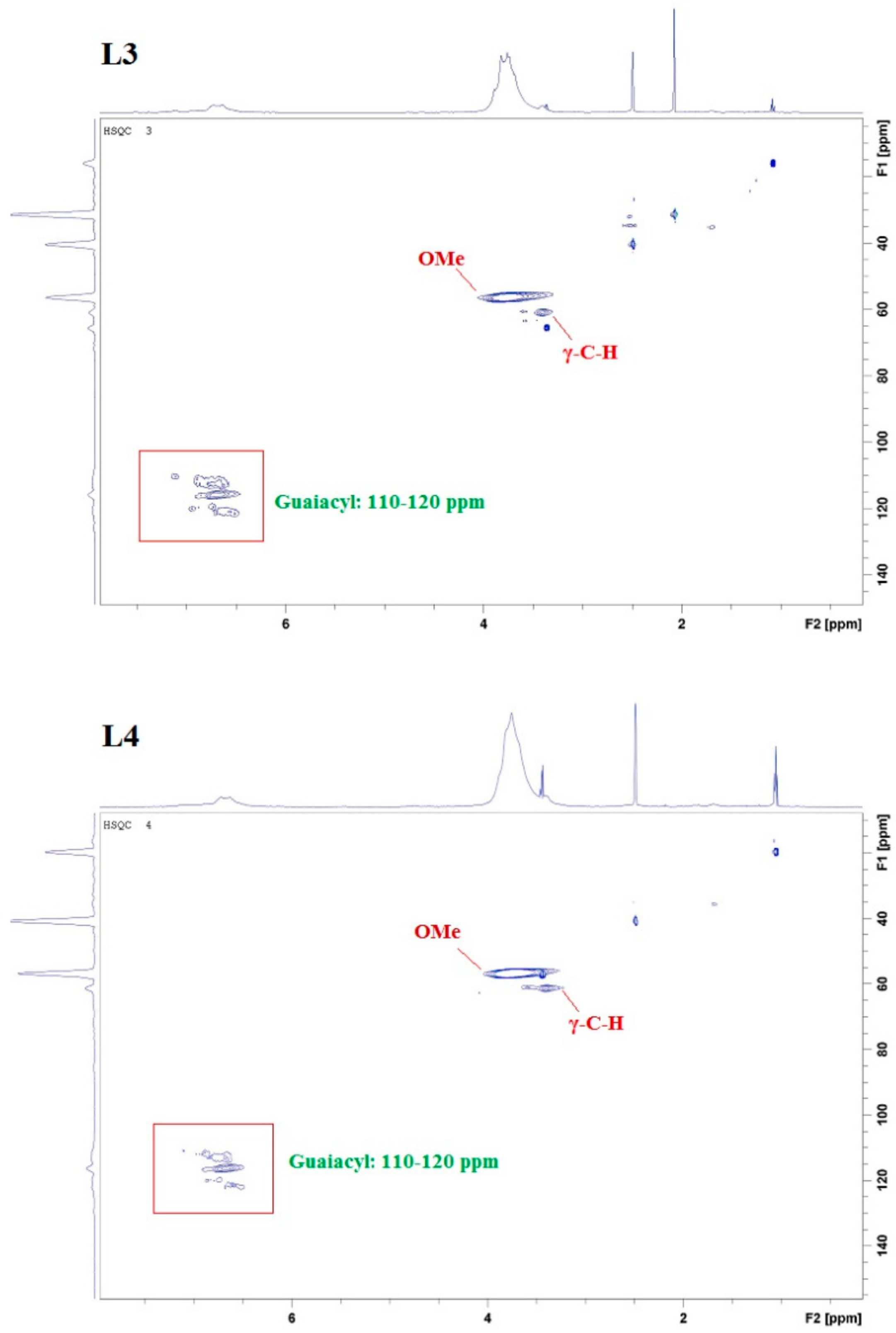
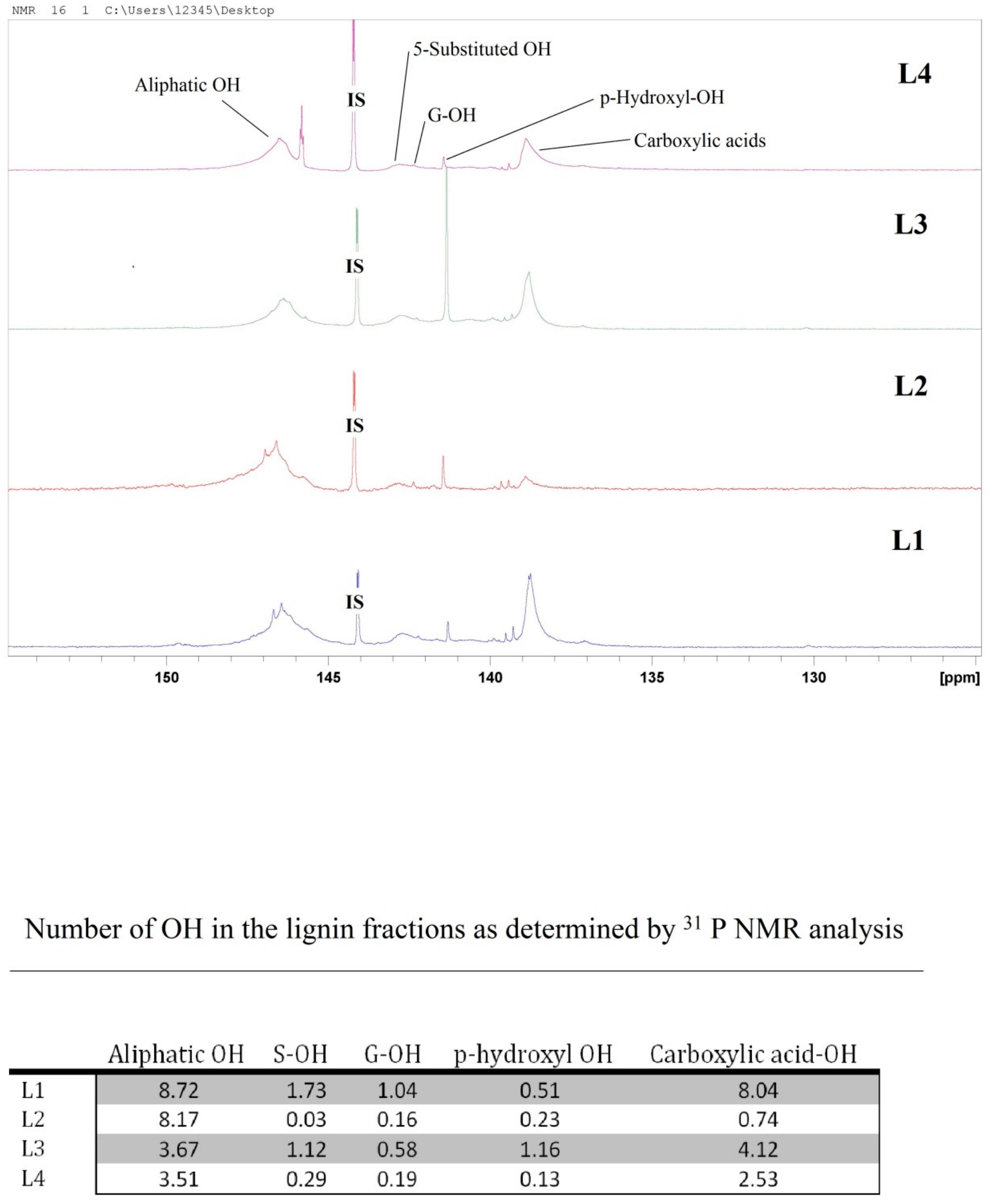
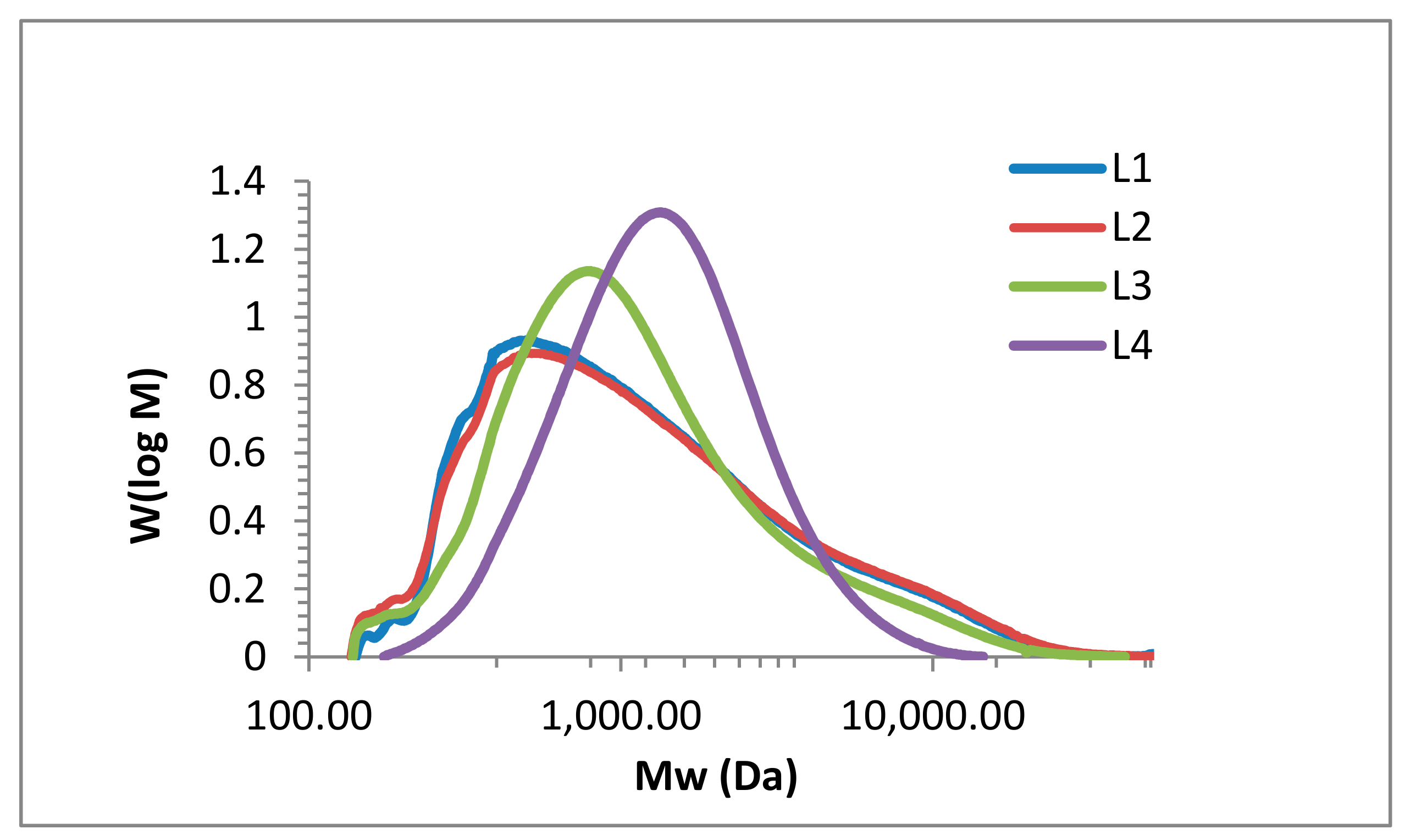
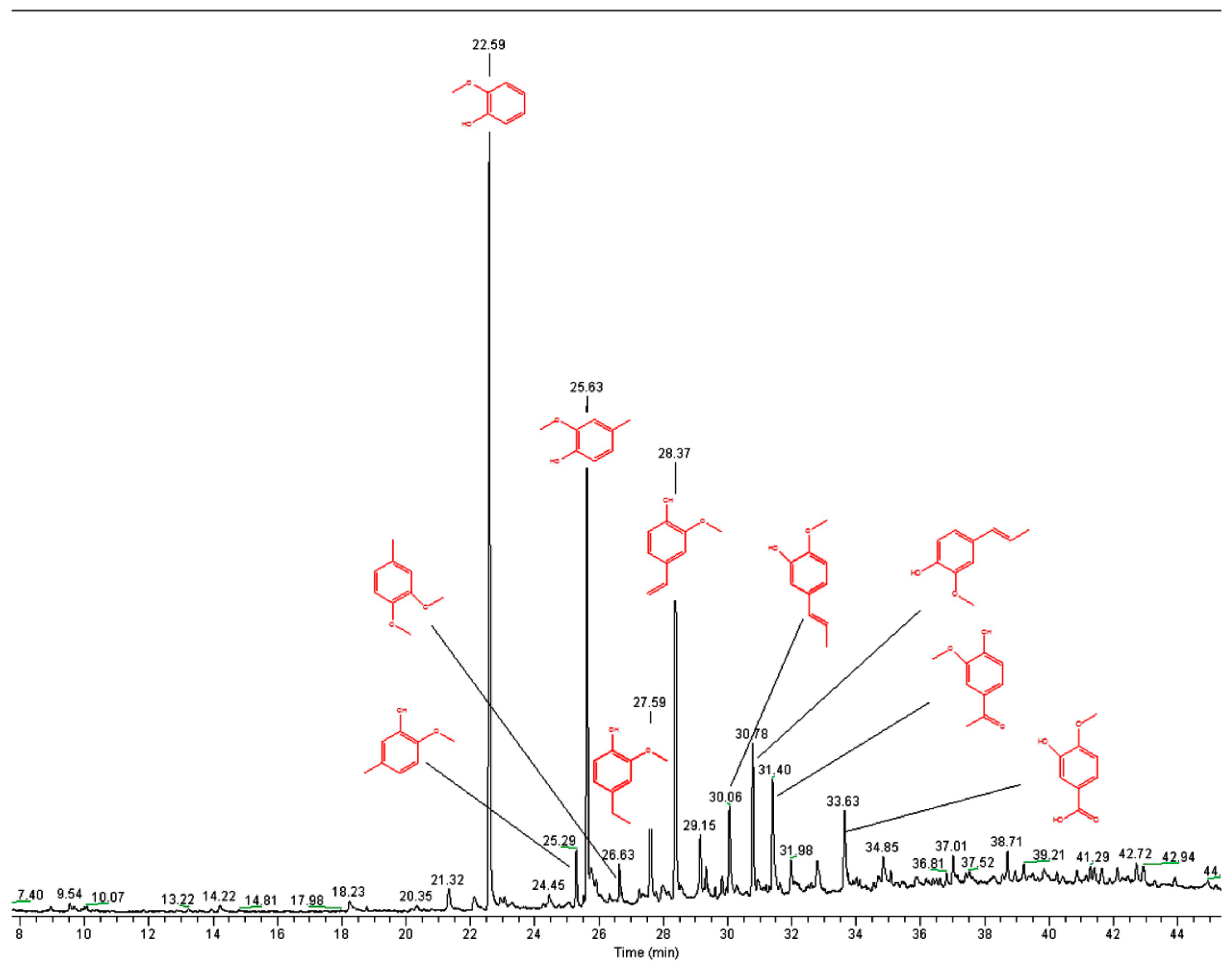
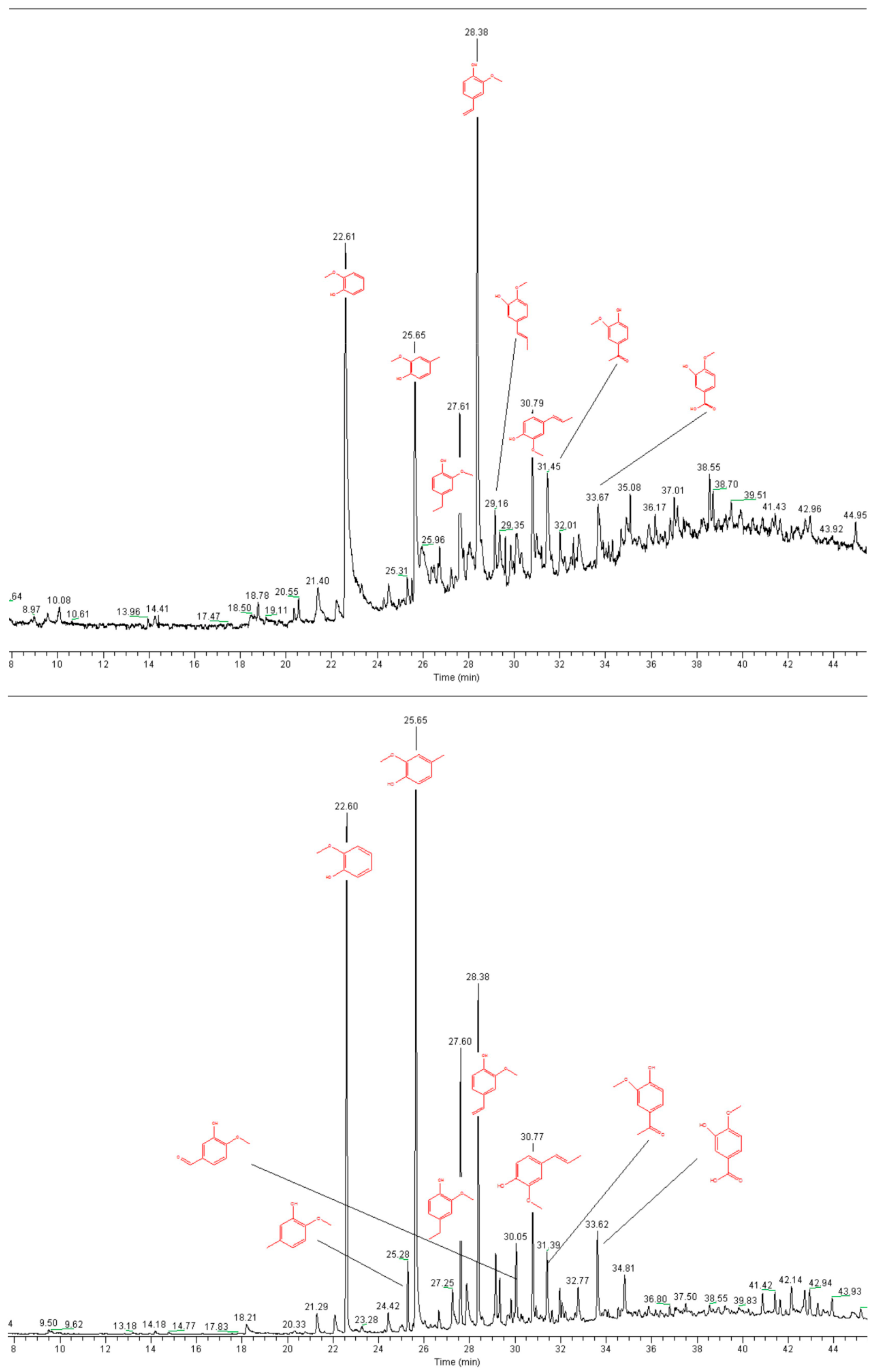
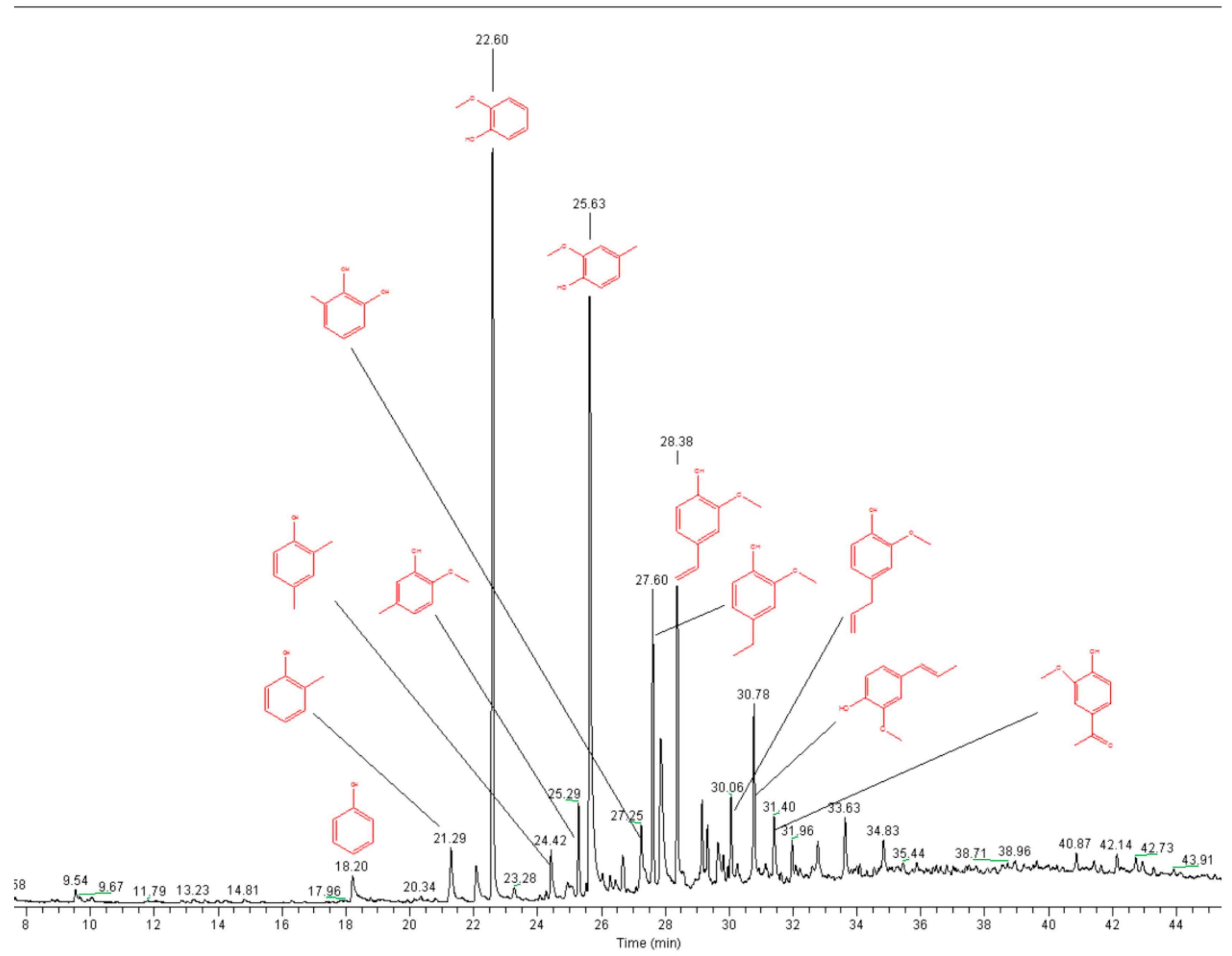
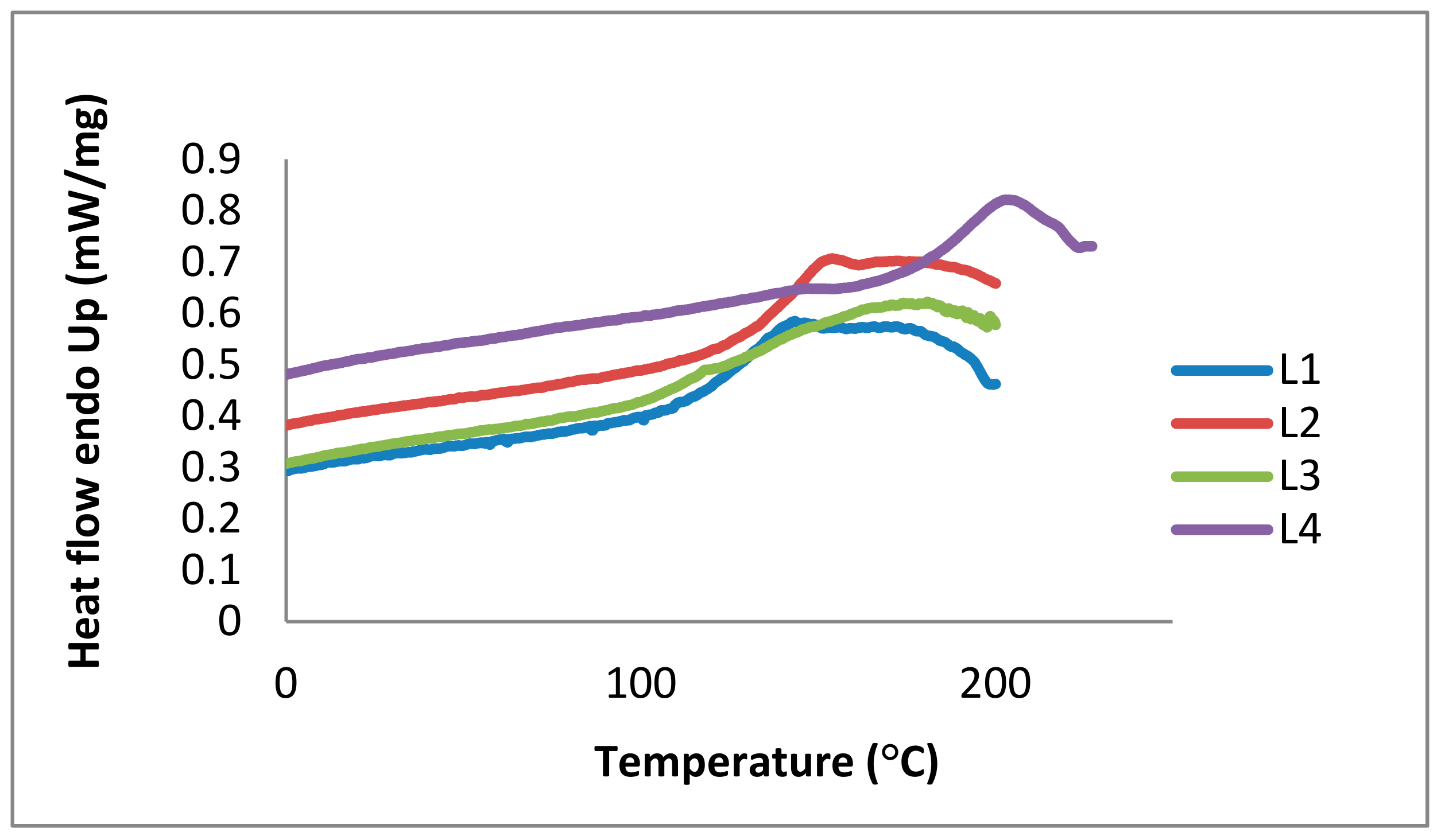
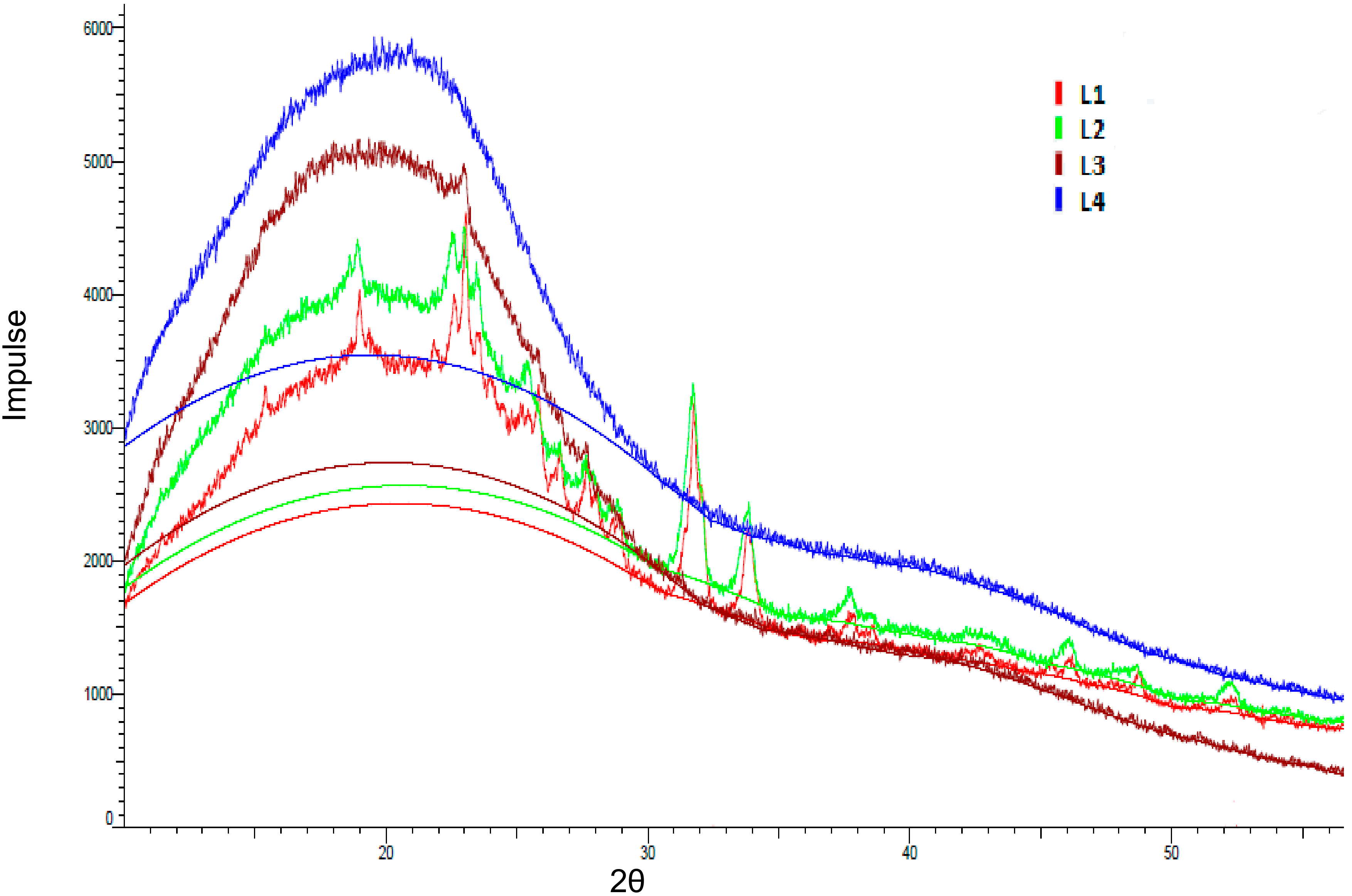
2.1. Lignin Structure Analysis
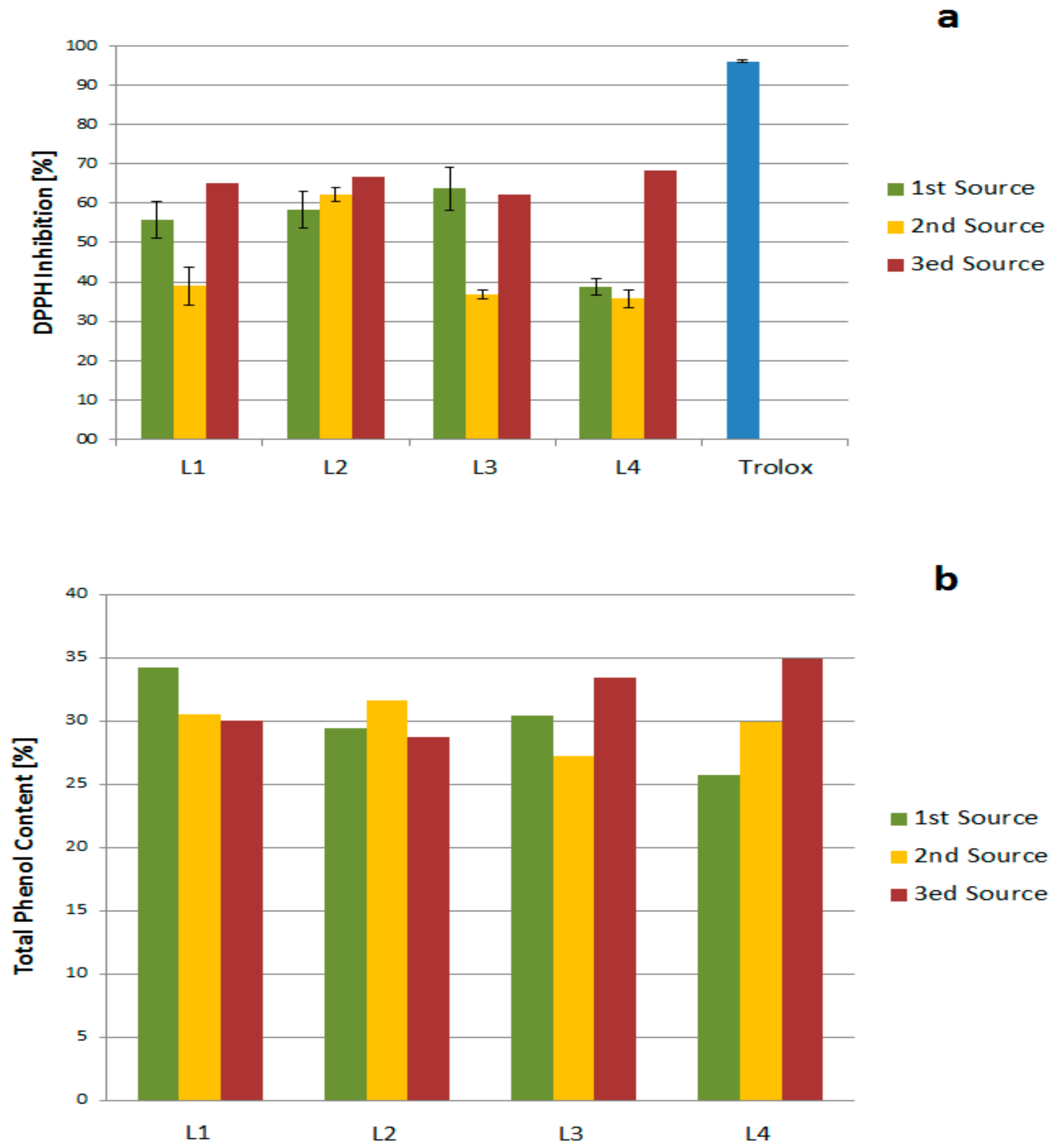
2.2. Antioxidant Activity and Total Phenol Content
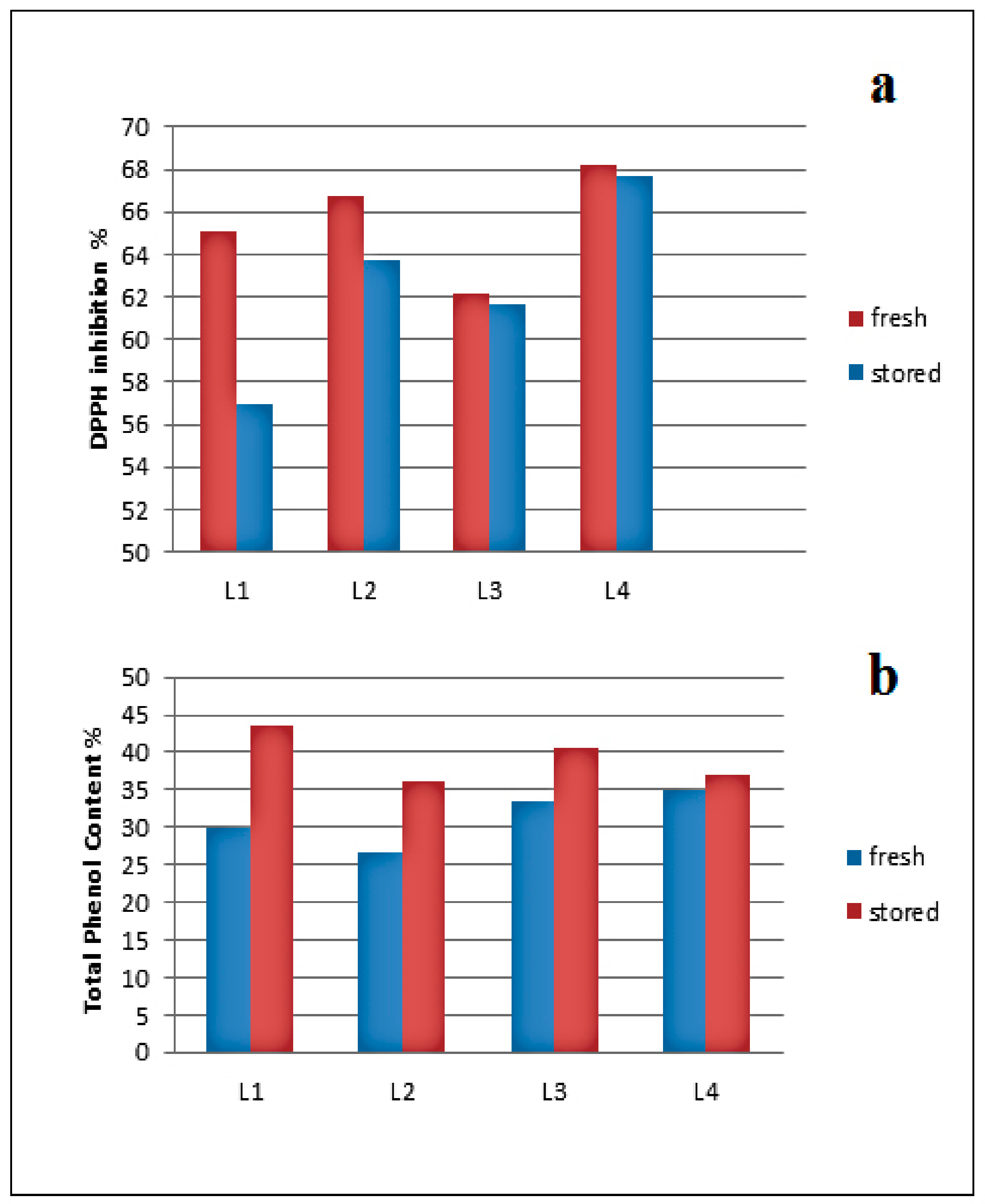
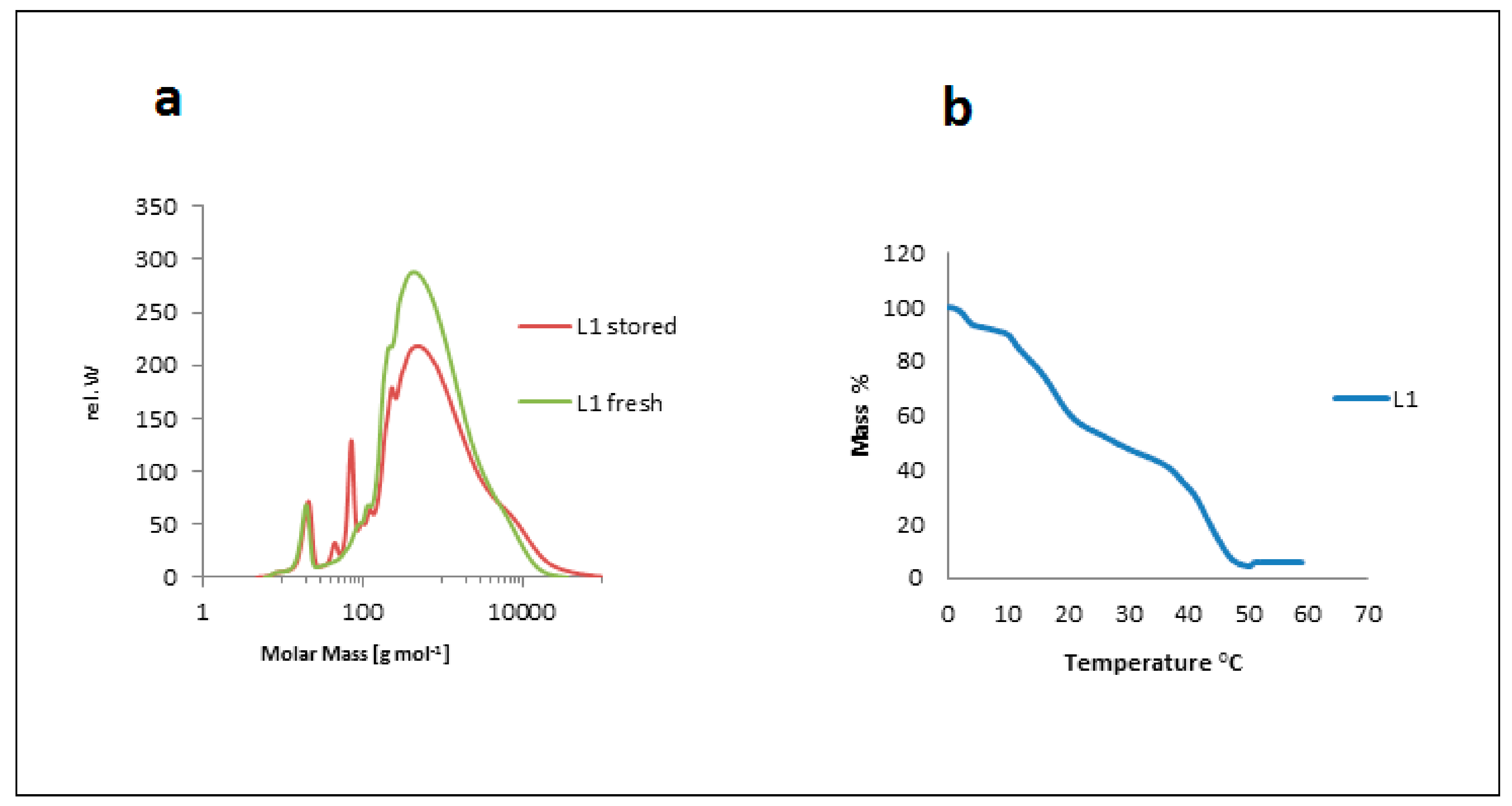
2.2.1. Source, Storage, and Temperature Effects
3. Materials and Methods
3.1. Isolation and Purification
3.2. FTIR Analysis
3.3. UV/Vis Analysis
3.4. 2D HSQC NMR Analysis
3.5. 31P NMR Analysis
3.6. SEC Analysis
3.7. Pyrolysis GC-MS
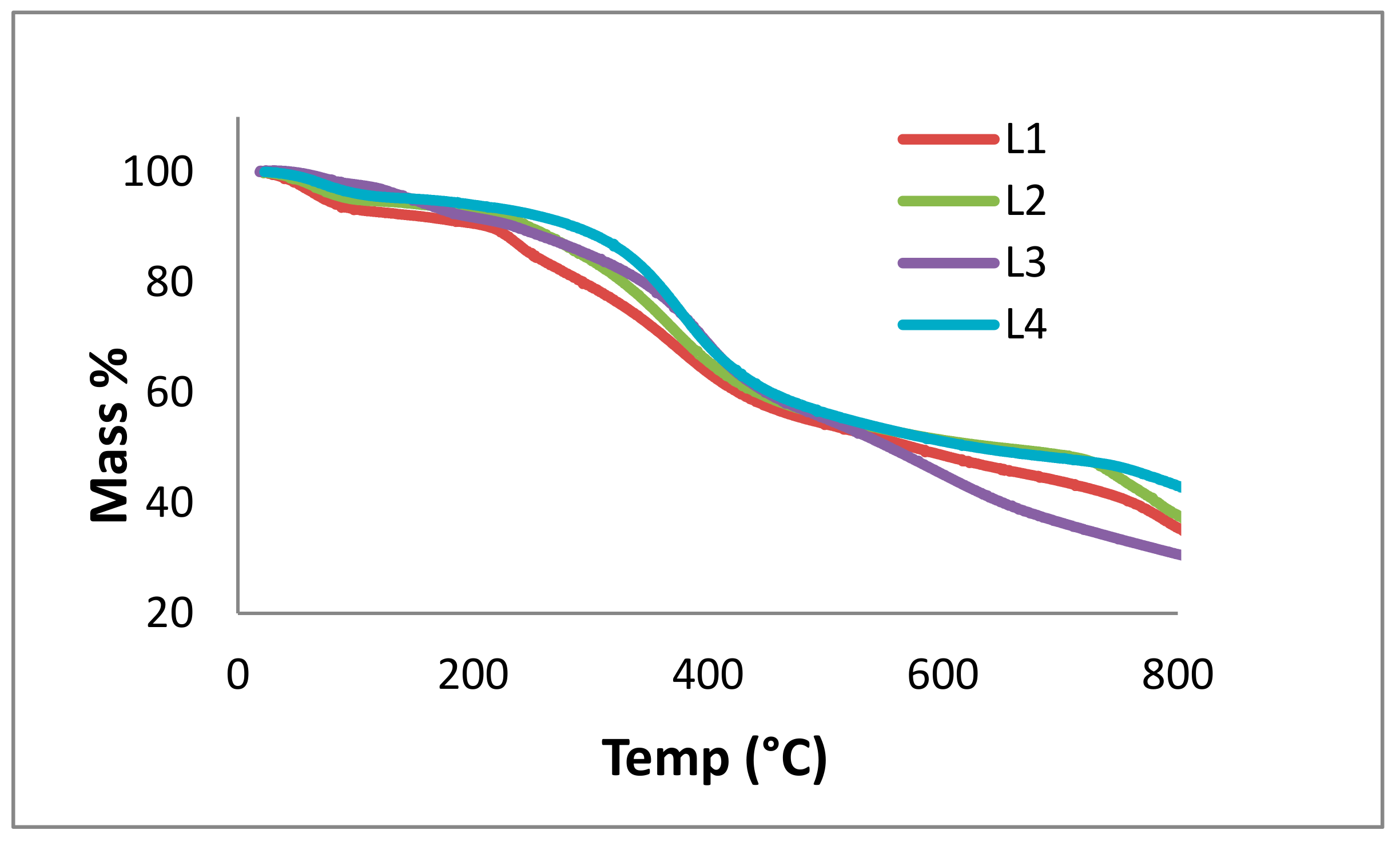
3.8. TGA
3.9. DSC
3.10. X-Ray Diffraction
3.11. Total Phenol Content
3.12. Antioxidant Activity
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kamm, B.; Kamm, M.; Hirth, T.; Schulze, M. Lignocelluloses Based Chemical Products and Product Family Trees. In Biorefineries-Industrial Processes and Products; Kamm, M., Kamm, B., Gruber, P.C., Eds.; Wiley-VCH: Weinheim, Germany, 2006; pp. 97–150. ISBN 3-527-31027-4. [Google Scholar]
- Kromus, S.; Wachter, B.; Koschuh, W.; Mandl, M.; Krotscheck, C.; Narodoslawsky, M. The green biorefinery Austria development of an integrated system for green biomass utilization. Chem. Biochem. Eng. 2004, 8, 8–12. [Google Scholar]
- Kamm, B.; Gruber, P.R.; Kamm, M. Biorefineries-Industrial Processes and Products. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2016; ISBN 9783527306732. [Google Scholar]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. Int. Ed. 2016, 55, 2–54. [Google Scholar] [CrossRef] [PubMed]
- Sipponen, M.H.; Farooq, M.; Koivisto, J.; Pellis, A.; Seitsonen, J.; Österberg, M. Spatially confined lignin nanospheres for biocatalytic ester synthesis in aqueous media. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Alzagameem, A.; El Khaldi-Hansen, B.; Kamm, B.; Schulze, M. Lignocellulosic biomass for energy, biofuels, biomaterials, and chemicals. In Biomass and Green Chemistry, 1st ed.; Vaz, S., Jr., Ed.; Springer International Publishing: Basel, Switzerland, 2018; pp. 95–132. ISBN 978-3-319-66736-2. [Google Scholar]
- Liu, Q.; Luo, L.; Zheng, L. Lignins: Biosynthesis and Biological Functions in Plants. Int. J. Mol. Sci. 2018, 19, 335. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, K.; Kuitunen, S.; Paananen, M.; Sixta, H. Novel Insight into Lignin Degradation during Kraft Cooking. Ind. Eng. Chem. Res. 2014, 53, 2614–2624. [Google Scholar] [CrossRef]
- Løhre, C.; Halleraker, H.V.; Barth, T. Composition of Lignin-to-Liquid Solvolysis Oils from Lignin Extracted in a Semi-Continuous Organosolv Process. Int. J. Mol. Sci. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Hundt, M.; Engel, N.; Schnitzlein, K.; Schnitzlein, M.G. The AlkaPolP process: Fractionation of variouslignocelluloses and continuous pulping within anintegrated biorefinery concept. Chem. Eng. Res. Des. 2016, 107, 13–23. [Google Scholar] [CrossRef]
- Pye, E.K. Industrial lignin production and applications. In Biorefineries–Industrial Processes and Products; Kamm, B., Gruber, P.R., Kamm, M., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; Volume 2, pp. 165–200. ISBN 978-3-527-31027-2. [Google Scholar]
- Gou, M.; Ran, X.; Martin, D.W.; Liu, C.J. The scaffold proteins of lignin biosynthetic cytochrome P450 enzymes. Nat. Plants 2018, 4, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Lupoi, J.S.; Singh, S.; Parthasarathi, R.; Simmons, B.A.; Henry, R.J. Recent innovations in analytical methods for the qualitative and quantitative assessment of lignin. Renew. Sustain. Energy. Rev. 2015, 49, 871–906. [Google Scholar] [CrossRef]
- Determination of Structural Carbohydrates and Lignin in Biomass. Available online: https://www.nrel.gov/docs/gen/fy13/42618.pdf (accessed on 10 September 2018).
- Lignin Market Size Growth/Global Industry Analysis Report, 2018–2025. Available online: https://www.grandviewresearch.com/industry-analysis/lignin-market (accessed on 10 September 2018).
- Research and Markets: ‘Global Lignin Market 2015–2021—Products (Lignosulfonate, Kraft Lignin and Others) & Applications (Concrete Additives, Animal Feed, Dyestuff, Others). Available online: https://www.businesswire.com/news/home/20150925005744/en/Research-Markets-Global-Lignin-Market-2015-2021 (accessed on 10 September 2018).
- Beisl, S.; Miltner, A.; Friedl, A. Lignin from Micro- to Nanosize: Production Methods. Int. J. Mol. Sci. 2017, 18, 1244. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Apak, R.; Capanoglu, E.; Shahidi, F. (Eds.) Measurement of Antioxidant Activity & Capacity, 1st ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2017; ISBN 9781119135388. [Google Scholar]
- Garrett, A.R.; Murray, B.K.; Robison, R.A.; O’Neill, K.L. Measuring antioxidant capacity using the orac and tosc assays. In Advanced Protocols in Oxidative Stress II. Methods in Molecular Biology (Methods and Protocols); Armstrong, D., Ed.; Humana Press: Totowa, NJ, USA, 2010; Volume 594, ISBN 978-1-60761-411-1. [Google Scholar]
- Tai, A.; Sawano, T.; Yazama, F.; Ito, H. Evaluation of antioxidant activity of vanillin by using multiple antioxidant assays. Biochim. Biophys. Acta 2011, 1810, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Dizhbite, T.; Lauberts, M.; Viksna, A.; Dobele, G.; Bikovens, O.; Telysheva, G. Characterization of softwood and hardwood lignoboost kraft lignins with emphasis on their antioxidant activity. Bioresources 2014, 9, 2051–2068. [Google Scholar] [CrossRef]
- Benzie, I.F.; Devaki, M. The ferric reducing/antioxidant power (FRAP) assay for non-enzymatic antioxidant capacity: Concepts, procedures, limitations and applications. In Measurement of Antioxidant Activity & Capacity, 1st ed.; Apak, R., Capanoglu, E., Shahidi, F., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2017; Chapter 5; ISBN 9781119135388. [Google Scholar]
- Özyürek, M.; Güclü, K.; Tütem, E.; Başkan, K.S.; Erçağ, E.; Çelik, S.E.; Baki, S.; Yıldız, L.; Karaman, S.; Apak, R. A comprehensive review of CUPRAC methodology. Anal. Methods 2011, 3, 2439–2453. [Google Scholar] [CrossRef]
- Cano, A.; Arnao, M.B. ABTS/TEAC (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)/Trolox®-Equivalent Antioxidant Capacity) radical scavenging mixed-mode assay. In Measurement of Antioxidant Activity & Capacity, 1st ed.; Apak, R., Capanoglu, E., Shahidi, F., Eds.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2017; Chapter 7; ISBN 9781119135388. [Google Scholar]
- Mishra, K.; Ojha, H.; Chaudhury, N.K. Estimation of antiradical properties of antioxidants using DPPH_ assay: A critical review and results. Food Chem. 2011, 130, 1036–1043. [Google Scholar] [CrossRef]
- Sanchez-Rangel, J.C.; Benavides, J.; Heredia, J.B.; Cisneros-Zevallosc, L.; Jacobo-Velázquez, D.A. The Folin–Ciocalteu assay revisited: Improvement of its specificity for total phenolic content determination. Anal. Methods 2013, 5, 5990–5999. [Google Scholar] [CrossRef]
- Witzler, M.; Alzagameem, A.; Bergs, M.; El Khaldi-Hansen, B.; Klein, S.E.; Hielscher, D.; Kamm, B.; Kreyenschmidt, J.; Tobiasch, E.; Schulze, M. Lignin-Derived Biomaterials for Drug Release and Tissue Engineering. Molecules 2018, 23, 1885. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, A.; Kumar, M.S.Y. Critical Review on the Analytical Mechanistic Steps in the Evaluation of Antioxidant Activity. Crit. Rev. Anal. Chem. 2018, 48, 214–236. [Google Scholar] [CrossRef] [PubMed]
- Dizhbite, T.; Telysheva, G.; Jurkjane, V.; Viesturs, U. Characterization of the radical scavenging activity of lignins––natural antioxidants. Bioresour. Technol. 2004, 95, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.; Erdocia, X.; Gatto, D.A.; Labidi, J. Characterisation of Kraft lignin separated by gradient acid precipitation. Ind. Crops Prod. 2014, 55, 149–154. [Google Scholar] [CrossRef]
- Hansen, B.; Kamm, B.; Schulze, M. Qualitative and quantitative analysis of lignin produced from beech wood by different conditions of the Organosolv process. J. Polym. Environ. 2016, 24, 85–97. [Google Scholar] [CrossRef]
- Hansen, B.; Kamm, B.; Schulze, M. Qualitative and quantitative analysis of lignins from different sources and isolation methods for an application as a biobased chemical resource and polymeric material. In Analytical Techniques and Methods for Biomass Products; Vaz, S., Jr., Seidl, P., Eds.; Springer: Berlin, Germany, 2017; pp. 15–44. ISBN 978-3-319-41414-0. [Google Scholar]
- Constant, S.; Wienk, H.L.J.; Frissen, A.E.; de Peinder, P.; Boelens, R.; van Es, D.S.; Grisel, R.H.-J.; Weckhuysen, B.M.; Huijgen, W.J.J.; Gosselink, R.J.A.; et al. New insights into the structure and composition of technical lignins: A comparative characterization study. Green Chem. 2016, 18, 2651. [Google Scholar] [CrossRef]
- Fiţigău, I.F.; Peter, F.; Boeriu, C.G. Structural Analysis of Lignins from Different Sources. World Acad. Sci. Eng. Technol. 2013, 76, 107–112. [Google Scholar]
- Boeriu, C.G.; Bravo, D.; Gosselink, R.J.A.; van Dam, J.E.G. Characterisation of structure-dependent functional properties of Lignin with infrared spectroscopy. Ind. Crops Prod. 2004, 20, 205–218. [Google Scholar] [CrossRef]
- Do, X.T.; Nöster, J.; Weber, M.; Nietsch, A.; Jung, C.; Witzleben, S.; Schulze, M. Comparative Studies of Lignin Depolymerisation: Photolysis versus Ozonolysis in Alkaline Medium. In Proceedings of the Annual Conference of the GDCh Division Sustainable Chemistry, Aachen, Germany, 17–19 September 2018. [Google Scholar]
- Vivekanand, V.; Chawade, A.; Larsson, M.; Larsson, A.; Olsson, O. Identification and qualitative characterization of high and low lignin lines from an oat TILLING population. Ind. Crops Prod. 2014, 59, 1–8. [Google Scholar] [CrossRef]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Lauberts, M.; Dizhbite, T.; Lauberte, L.; Jurkjane, V.; Telysheva, G. Antioxidant activity of various lignins and lignin-related phenylpropanoid units with high and low molecular weight. Holzforschung 2015, 69, 1–12. [Google Scholar] [CrossRef]
- Gilca, I.A.; Ghitescu, R.E.; Puitel, A.C.; Popa, V.I. Preparation of lignin nanoparticles by chemical modification. Iran. Polym. J. 2014, 23, 355–363. [Google Scholar] [CrossRef]
- Argyropoulos, D.S. Quantitative phosphorus 31 NMR analysis of lignins: A new tool for the lignin chemist. J Wood Chem. Technol. 1994, 14, 45–63. [Google Scholar] [CrossRef]
- Sun, S.-N.; Cao, X.-F.; Xu, F.; Sun, R.-C.; Jones, G.L. Structural Features and Antioxidant Activities of Lignins from Steam-Exploded Bamboo (Phyllostachys pubescens). J. Agric. Food Chem. 2014, 62, 5939–5947. [Google Scholar] [CrossRef] [PubMed]
- Aminzadeh, S.; Lauberts, M.; Dobele, G.; Ponomarenko, J.; Mattsson, T.; Lindstroem, M.E.; Sevastyanova, O. Membrane filtration of kraft lignin: Structural characteristics and antioxidant activity of the low-molecular-weight fraction. Ind. Crops Prod. 2018, 112, 200–209. [Google Scholar] [CrossRef]
- Monakhova, Y.; Diehl, B.W.K.; Do, X.T.; Witzleben, S.; Schulze, M. Novel method for the determination of average molecular weight of natural polymers based on 2D DOSY NMR and chemometrics: Example of heparin. J. Pharm. Biomed. Anal. 2018, 149, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Sulaeva, I.; Zinovyev, G.; Plankeele, J.M.; Sumerskii, I.; Rosenau, T.; Potthast, A. Fast Track to Molar Mass Distributions of Technical Lignins. ChemSusChem 2017, 10, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Acosta, J.L.; Torres-Chavez, P.I.; Ramirez-Womg, B.; Lopez-Saiz, C.M.; Montano-Leyva, B. Antioxidant, Antimicrobial, and Antimutagenic Properties of Technical Lignins and Their Applications. Bioresources 2016, 11, 5452–5481. [Google Scholar] [CrossRef]
- Sadeghifar, H.; Argyropoulos, D.S. Correlations of the Antioxidant Properties of Softwood Kraft Lignin Fractions with the Thermal Stability of its Blends with Polyethylene. ACS Sustain. Chem. Eng. 2015, 3, 349–356. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, M.; Zhao, L.; Zhu, L. Influence of Interactions among Three Biomass Components on the Pyrolysis Behavior. Ind. Eng. Chem. Res. 2018, 57, 5241–5249. [Google Scholar] [CrossRef]
- Vallejos, M.E.; Felissia, F.E.; Curvelo, A.A.S.; Zambon, M.D.; Ramos, L.; Area, M.C. Chemical and physico-chemical characterization of lignins obtained from ethanol-water fractionation of bagasse. BioResources 2011, 6, 1158–1171. [Google Scholar]
- Ramezani, N.; Sain, M. Thermal and Physiochemical Characterization of Lignin Extracted from Wheat Straw by Organosolv Process. J. Polym. Environ. 2018, 26, 3109–3116. [Google Scholar] [CrossRef]
- Reda, S.Y. Evaluation of antioxidants stability by thermal analysis and its protective effect in heated edible vegetable oil. Food Sci Technol. 2011, 31, 475–480. [Google Scholar] [CrossRef]
- Dos Santos, P.S.B.; Fuentes da Silva, S.H.; Dos Reis Paganotto, G.F.; Labidi, J. Characterization of kraft lignin with XRD. In Proceedings of the 3rd International Conference on Processing Technologies for the Forest and Bio-based Products Industries PTF BPI 2014, Kuchl, Austria, 10–12 September 2014. [Google Scholar] [CrossRef]
- Garcia, A.; Toledano, A.; Serrano, L.; Egues, I.; Gonzalez, M.; Marin, F.; Labidi, J. Characterization of lignins obtained by selective precipitation. Sep. Purif. Technol. 2009, 68, 193–198. [Google Scholar] [CrossRef]
- Singh, M.; Jha, A.; Kumar, A.; Hettiarachchy, N.; Rai, A.K.; Sharma, D. Influence of the solvents on the extraction of major phenolic compounds (punicalagin, ellagic acid and gallic acid) and their antioxidant activities in pomegranate aril. J. Food Sci. Technol. 2014, 51, 2070–2077. [Google Scholar] [CrossRef] [PubMed]
- Akowuah, G.A.; Ismail, Z.; Norhayati, I.; Sadikun, A. The effects of different extraction solvents of varying polarities on polyphenols of Orthosiphon stamineus and evaluation of the free radical-scavenging activity. Food Chem. 2005, 93, 311–317. [Google Scholar] [CrossRef]
- Felıcio, C.M.; da Hora Machadoa, A.E.; Castellan, A. Routes of degradation of β-O-4 syringyl and guaiacyl lignin model compounds during photobleaching processes. J. Photochem. Photobiol. A Chem. 2003, 156, 253–265. [Google Scholar] [CrossRef]
- García, A.; González, M.; Spigno, G.; Labidi, J. Lignin as natural radical scavenger. Effect of the obtaining and purification processes on the antioxidant behaviour of lignin. Biochem. Eng. J. 2012, 67, 173–185. [Google Scholar] [CrossRef]
- Garcia, A.; Amendola, D.; González, M.; Spigno, G.; Labidi, J. Lignin as natural radical scavenger. Study of the antioxidant capacity of apple tree pruning lignin obtained by different methods. Chem. Eng. Trans. 2011, 24, 925–931. [Google Scholar]
- Kaur, R.; Uppal, S.K. Structural characterization and antioxidant activity of lignin from sugarcane bagasse. Colloid Polym. Sci. 2015, 293, 2585–2592. [Google Scholar] [CrossRef]
- Sun, S.L.; Wen, J.L.; Ma, M.G.; Sun, R.C.; Jones, G.L. Structural features and antioxidant activities of degraded lignins from steam exploded bamboo stem. Ind Crop Prod 2014, 56, 128–136. [Google Scholar] [CrossRef]
- Gadioli, R.; Waldman, W.R.; De Paoli, M.A. Lignin as a green primary antioxidant for polypropylene. J. Appl. Polym. Sci. 2016, 133, 43558. [Google Scholar] [CrossRef]
- Do, X.T.; Nietzsch, A.; Jung, C.; Witzleben, S.; Schulze, M. Lignin-Depolymerisation via UV-Photolysis and Titanium Dioxide Photocatalysis. Preprints 2017, 2017100128. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antioxidant Assay | Mechanism | Advantages | Disadvantages | References |
|---|---|---|---|---|
| ORAC (Oxygen Radical Absorbance Capacity) | Hydrogen Atom Transfer | • can be adapted to detect both hydrophilic and hydrophobic antioxidants by altering the radical source and solvent • ORAC values account for lag-time, initial rate and total extent of inhibition in a single value • automation is possible | • fluorescence quenching is very sensitive, so any impurity has to be avoided • to achieve reproducible results, constant reaction conditions are required (temperature, pH, oxygen and reagent concentrations etc.) • detection requires fluorometer (fluorescence easy to be quenched) • analysis time about one hour • measurement is limited to peroxyl-radicals as oxidants | [20] |
| FRAP (Ferric Reducing Antioxidant Power) | Single Electron Transfer | • simple, quick, inexpensive, robust, does not require special equipment • direct method to measure the combined activity of multiple, reductive antioxidants in a sample • automation is possible | • no exact reaction time/reactivity is varying for different samples • thiol-containing antioxidants like glutathione are not detected | [23] |
| CUPRAC (Cupric Reduction Antioxidant Capacity) | Single Electron Transfer | • simple, quick, inexpensive, robust, does not require special equipment • all classes of antioxidants are detected, including thiols • applicable to both, hydrophilic and lipophilic antioxidants | • no exact reaction time/reactivity is strongly varying for different samples | [24] |
| ABTS (2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) | mixture of HAT/SET | • simple, quick, wide pH-range, often used • soluble in aqueous and organic solvents and not affected by ionic strength -> applicable to a wide range of hydrophilic and lipophilic antioxidants • several wavelenghts are available for photometric detection of the ABTS-radical | • no exact reaction time/reactivity is strongly varying for different samples • the bulky ABTS-radical is not a good model for small, biologically more relevant radicals like HO • etc. | [25] |
| DPPH (2,2-diphenyl-1-picrylhydrazyl) | mixture of HAT/SET | • simple, quick, often used, no special equipment needed • DPPH-radical is commercially available; no in situ-generation necessary | • no exact reaction time/reactivity is varying for different samples • DPPH-radical may have a poor reactivity with antioxidants due to its stability an sterical hindrance | [26] |
| FC/TPC (Folin-Ciocalteu-Assay or Total-Phenolics-Assay) | mixture of HAT/SET | • simple, often used, does not require special equipment | • no exact reaction time/reactivity is varying for different samples • interferences with other reductive substances may influence the results | [27] |
| L1 (cm−1) | L2 (cm−1) | L3 (cm−1) | L4 (cm−1) | KL Lit. [34,35] (cm−1) | Signal Assignment |
|---|---|---|---|---|---|
| 3396 | 3408 | 3414 | 3396 | 3415 | O–H stretching |
| 2931 | 2931 | 2926 | 2925 | 2935 | C–H stretching |
| 2834 | 2814 | 2834 | 2833 | 2843 | tertiary C–H group |
| 1695 | 1695 | 1700 | 1702 | 1660 | carbonyl-carboxyl stretching |
| 1577 | 1583 | 1593 | 1595 | 1505 | aromatic/carbonyl stretching |
| 1452 | 1449 | 1455 | 1459 | 1451 | C–H deformation |
| 1263 | 1262 | 1265 | 1262 | 1265 | C–O stretching, aromatic (phenyl) |
| 1028 | 1028 | 1026 | 1028 | 1029 | C–O deformation (methoxy group) |
| 810 | 810 | 807 | 807 | 814 | C–H out-of-plane in m-position of guaicyl units |
| 848 | 848 | 848 | 848 | - | C–H out-of-plane in m-position of guaicyl units |
| λ exp. (nm) | λ Lit. (nm) | Functional Group | Intensity | Excitation | Reference |
|---|---|---|---|---|---|
| 215–222 | 279–280 | Non-conjugated phenolic groups (G/S rich) | high | π–π* | Azadi et al. [39] |
| 296–303 | 316–320 | Conjugated phenolic groups (p-coumaric acid, ferulic acid) | low | n–π* | Vivekanand et al. [38] |
| Fraction | Mn (g mol −1) | Mw (g·mol−1) | PD |
|---|---|---|---|
| L1 | 720 | 2108 | 2.9 |
| L2 | 706 | 2226 | 3.2 |
| L3 | 757 | 1816 | 2.4 |
| L4 | 1043 | 1690 | 1.6 |
| L1 | L2 | L3 | L4 | DL | OLSW | Lit. [30] | |
|---|---|---|---|---|---|---|---|
| DPPH inhibition (%) | 65.1 ± 3.7 | 66.8 ± 6.6 | 62.2 ± 9.5 | 68.2 ± 3.6 | 64 ± 2.6 | 42 ± 1.9 | 54.76 |
| TPC (%) | 30 ± 1.2 | 26.8 ± 0.5 | 33.5 ± 0.9 | 35 ± 1.0 | 33.3 ± 1.6 | 34.1 ± 1.0 | 29.61 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzagameem, A.; Khaldi-Hansen, B.E.; Büchner, D.; Larkins, M.; Kamm, B.; Witzleben, S.; Schulze, M. Lignocellulosic Biomass as Source for Lignin-Based Environmentally Benign Antioxidants. Molecules 2018, 23, 2664. https://doi.org/10.3390/molecules23102664
Alzagameem A, Khaldi-Hansen BE, Büchner D, Larkins M, Kamm B, Witzleben S, Schulze M. Lignocellulosic Biomass as Source for Lignin-Based Environmentally Benign Antioxidants. Molecules. 2018; 23(10):2664. https://doi.org/10.3390/molecules23102664
Chicago/Turabian StyleAlzagameem, Abla, Basma El Khaldi-Hansen, Dominik Büchner, Michael Larkins, Birgit Kamm, Steffen Witzleben, and Margit Schulze. 2018. "Lignocellulosic Biomass as Source for Lignin-Based Environmentally Benign Antioxidants" Molecules 23, no. 10: 2664. https://doi.org/10.3390/molecules23102664
APA StyleAlzagameem, A., Khaldi-Hansen, B. E., Büchner, D., Larkins, M., Kamm, B., Witzleben, S., & Schulze, M. (2018). Lignocellulosic Biomass as Source for Lignin-Based Environmentally Benign Antioxidants. Molecules, 23(10), 2664. https://doi.org/10.3390/molecules23102664