Novel 5′-Norcarbocyclic Derivatives of Bicyclic Pyrrolo- and Furano[2,3-d]Pyrimidine Nucleosides

 ,
,
Abstract
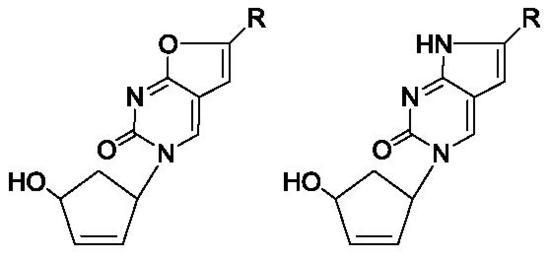
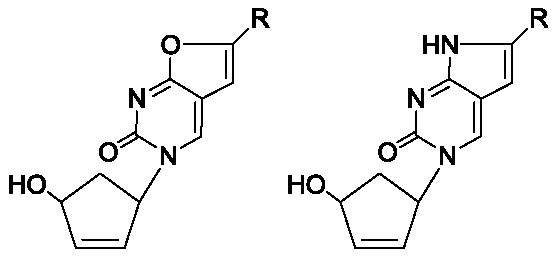
1. Introduction
2. Results and Discussion
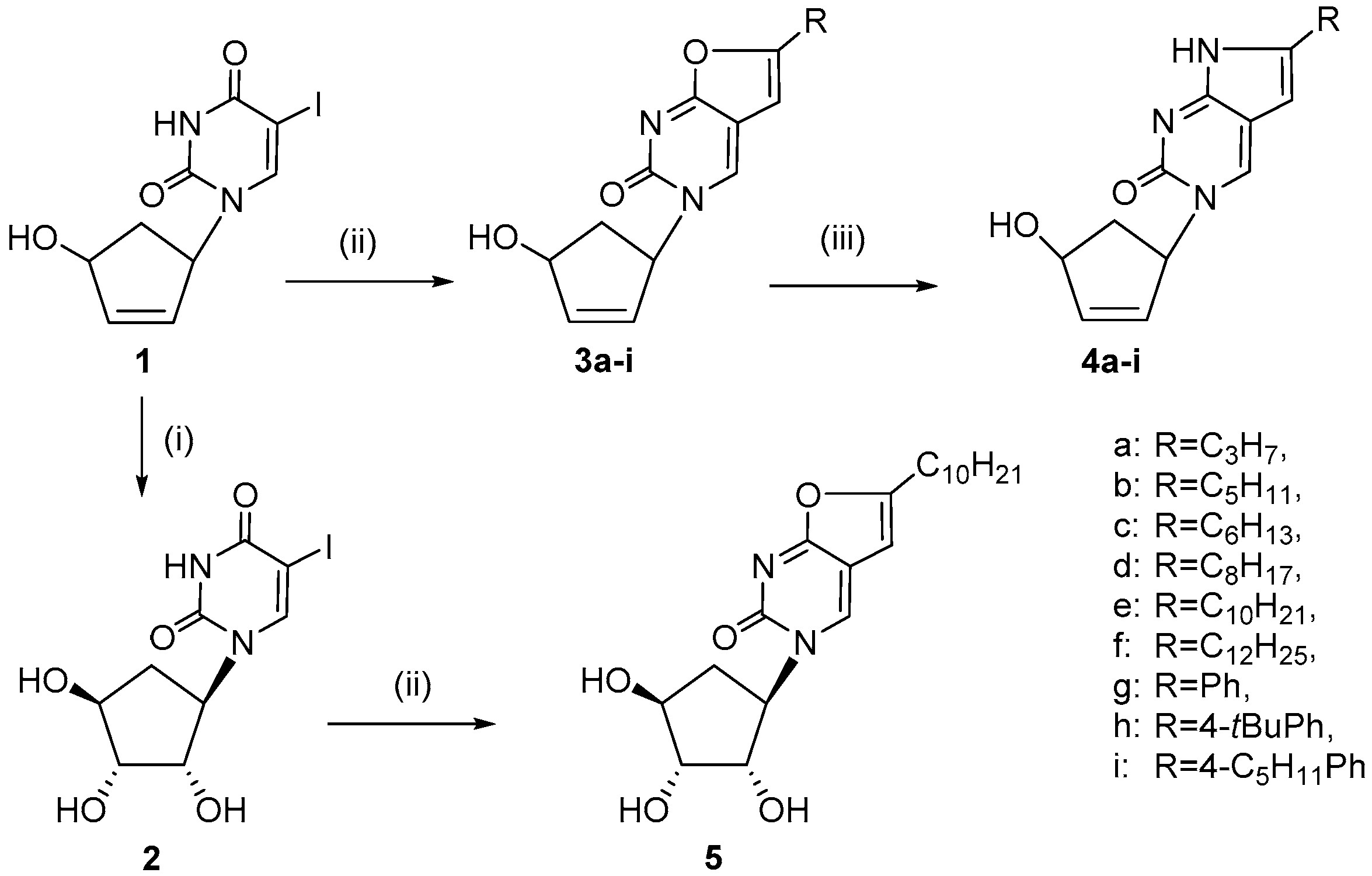
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Cell Viability Assay
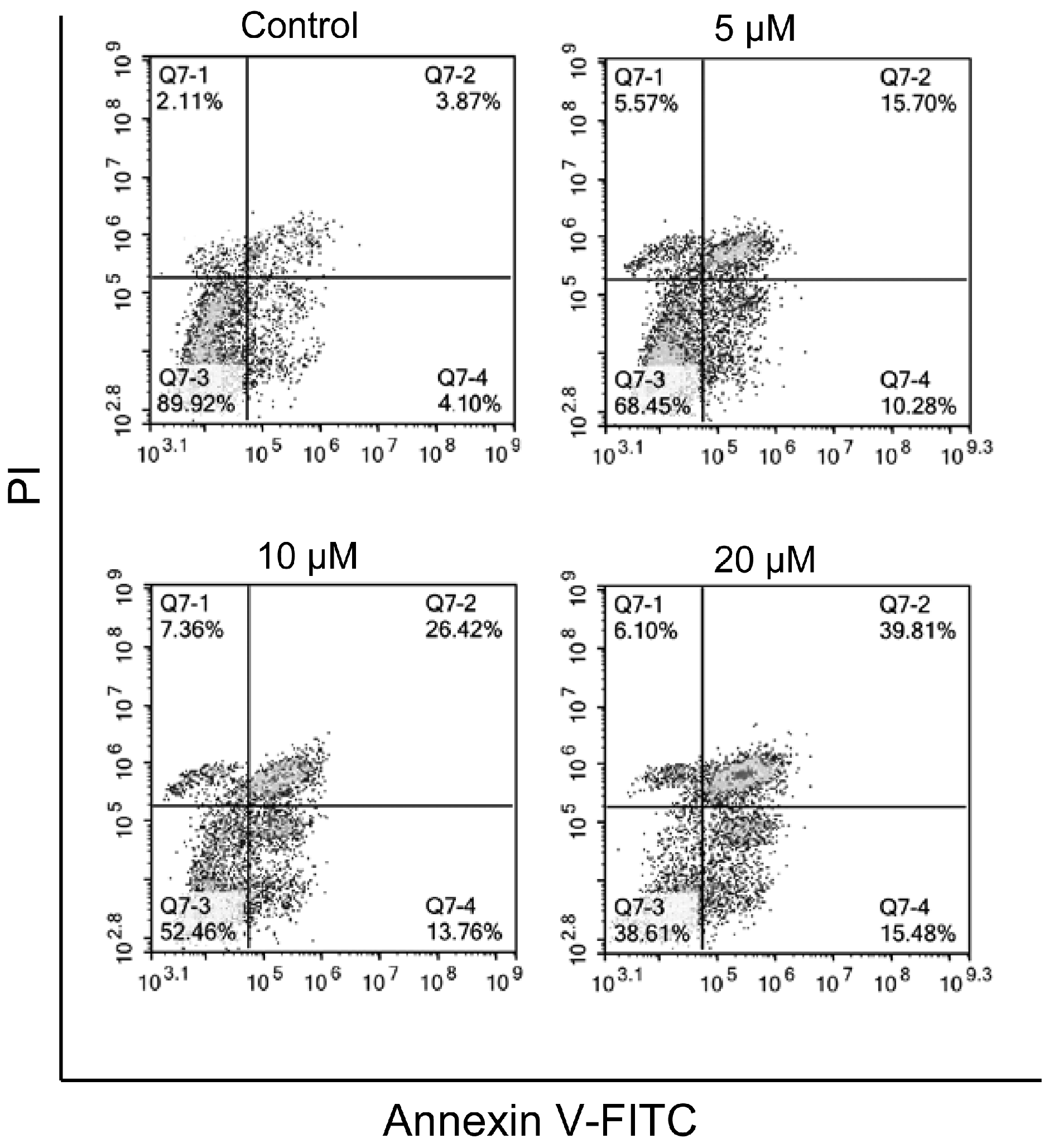
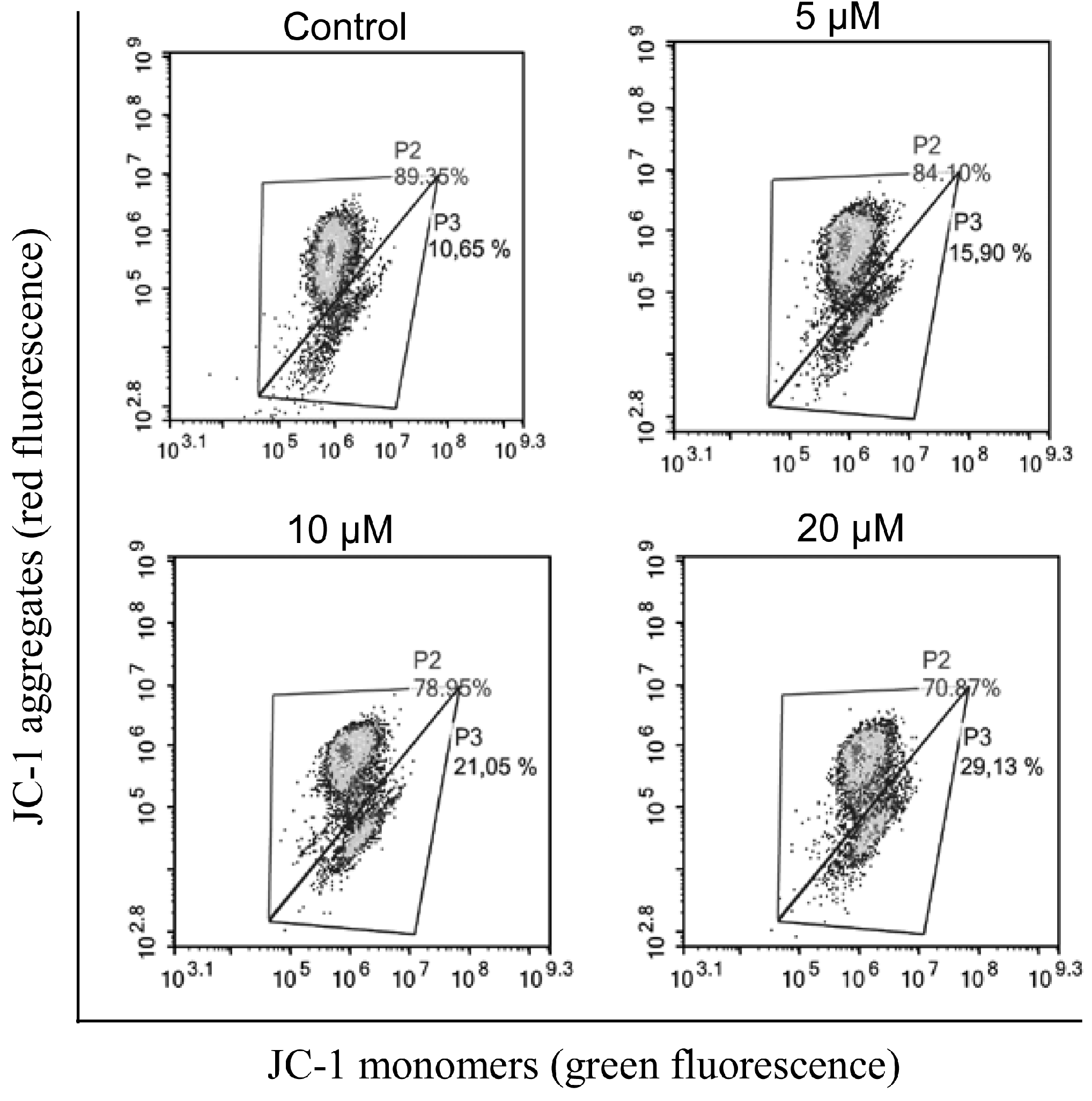
2.2.2. Induction of Apoptosis
3. Materials and Methods
3.1. Chemistry
3.1.1. General Method for the Synthesis of 1-(4′-hydroxy-2′-cyclopenten-1′-yl)-6-alkyl-3H-furano[2,3-d]pyrimidine-2-ones and 1-(4′-hydroxy-2′-cyclopenten-1′-yl)-6-aryl-3H-furano[2,3-d]-pyrimidine-2-ones (3a–i)
3.1.2. General Method for the Synthesis of 1-(4′-hydroxy-2′-cyclopenten-1′-yl)-6-alkyl-3H-pyrrolo[2,3-d]pyrimidine-2-ones and 1-(4′-hydroxy-2′-cyclopenten-1′-yl)-6-aryl-3H-pyrrolo[2,3-d]-pyrimidine-2-ones (4a–i).
3.2. Biological Assay
3.2.1. Cell Cultures
3.2.2. Cell Viability Analysis by MTT Assay
3.2.3. Apoptosis Detection by Annexin V Staining
3.2.4. Mitochondria Depolarization Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NMMO | N-methylmorpholine N-oxide |
| JC-1 | 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide |
| PI | propidium iodide |
| PLC | preparative layer chromatography |
| IC50 | the compound concentration that results in 50% cell survival as measured by the MTT assay |
References
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viraldiseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.; Popowycz, F.; Joseph, B. Cytotoxic Nucleoside Analogues: Different Strategies to Improve their Clinical Efficacy. Curr. Med. Chem. 2008, 15, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Yarnold, C.J.; Jones, G.; Velázquez, S.; Barucki, H.; Brancale, A.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J. Potent and selective inhibition of varicella-zoster virus (VZV) by nucleoside analogues with an unusual bicyclic base. J. Med. Chem. 1999, 42, 4479–4484. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; McGuigan, C. Bicyclic pyrimidine nucleoside analogues (BCNAs) as highly selective and potent inhibitors of varicella-zoster virus replication. J. Antimicrob. Chemother. 2002, 50, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Jahnz-Wechmann, Z.; Framski, G.; Januszczyk, P.; Boryski, J. Bioactive fused heterocycles: Nucleoside analogues with an additional ring. Eur. J. Med. Chem. 2015, 97, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Migliore, M.D.; Zonta, N.; McGuigan, C.; Henson, G.; Andrei, G.; Snoeck, R.; Balzarini, J. Synthesis and Antiviral Activity of the Carbocyclic Analogue of the Highly Potent and Selective Anti-VZV Bicyclo Furano Pyrimidines. J. Med. Chem. 2007, 50, 6485–6492. [Google Scholar] [CrossRef] [PubMed]
- Dincer, S.; Cetin, K.T.; Onay-Besikci, A.; Olgen, S. Synthesis, biological evaluation and docking studies of new pyrrolo[2,3-d] pyrimidine derivatives as Src family-selective tyrosine kinase inhibitors. J. Enzyme Inhib. Med. Chem. 2013, 28, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.A.; Serya, R.A.T.; Lasheen, D.S.; Abouzid, K.A.M. Furo[2,3-d]pyrimidine based derivatives as kinase inhibitors and anticancer agents. Future J. Pharm. Sci. 2016, 2, 1–8. [Google Scholar] [CrossRef]
- Romeo, R.; Giofre, S.V.; Garozzo, A.; Bisignano, B.; Corsaro, A.; Chiacchio, M.A. Synthesis and biological evaluation of furopyrimidine N,O-nucleosides. Bioorg. Med. Chem. 2013, 21, 5688–5693. [Google Scholar] [CrossRef] [PubMed]
- Framski, G.; Wawrzyniak, D.; Jahnz-Wechmann, Z.; Szymanska-Michalak, A.; Barciszewski, J.; Boryski, J.; Kraszewski, A.; Stawinski, J. New Applications of 6-Alkyl-2,3dihydrofurano[2,3-d]pyrimidin-2(1H)-one and 6-Alkyl-2,3-dihydropyrrolo[2,3-d]pyrimidin-2(3H,7H)-one Nucleosides: Anticancer Properties. In Proceedings of the XXI Round Table on Nucleosides, Nucleotides and Nucleic Acids “Chemical Biology of Nucleic Acids”, Poznań, Poland, 24–28 August 2014. Electronic Abstract Book, Poster 13. IS3NA, 2014. [Google Scholar]
- Framski, G.; Wawrzyniak, D.; Jahnz-Wechmann, Z.; Szymanska-Michalak, A.; Kraszewski, A.; Barciszewski, J.; Boryski, J.; Stawinski, J. Searching for anti-glioma activity. Ribonucleoside analogues with modifications in nucleobase and sugar moieties. Acta Biochim. Pol. 2016, 63, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Matyugina, E.; Logashenko, E.; Zenkova, M.; Kochetkov, S.; Khandazhinskaya, A. 5′-Norcarbocyclic analogues of furano[2,3-d] pyrimidine nucleosides. Heterocycl. Commun. 2015, 21, 259–262. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Khandazhinskaya, A.L.; Chernousova, L.N.; Andreevskaya, S.N.; Smirnova, T.G.; Chizhov, A.O.; Karpenko, I.L.; Kochetkov, S.N.; Alexandrova, L.A. The Synthesis and Antituberculosis Activity of 5’-Nor Carbocyclic Uracil Derivatives. Bioorg. Med. Chem. 2012, 20, 6680–6686. [Google Scholar] [CrossRef] [PubMed]
- Khandazhinskaya, A.L.; Shirokova, E.A.; Shipitsin, A.V.; Karpenko, I.L.; Belanov, E.F.; Kukhanova, M.K.; Yasko, M.V. Adenosine N1-oxide analogues as inhibitors of orthopox virus replication. Collect. Czech. Chem. Commun. 2006, 71, 1107–1121. [Google Scholar] [CrossRef]
- Matyugina, E.S.; Khandazhinskaya, A.L.; Kochetkov, S.N. Carbocyclic nucleoside analogues: Classification, target enzymes, mechanisms of action and synthesis. Russ. Chem. Rev. 2012, 81, 729–746. [Google Scholar] [CrossRef]
- Ainai, T.; Wang, Y.-G.; Tokoro, Y.; Kobayashi, Y. Highly Stereoselective Synthesis of Aristeromycin through Dihydroxylation of 4-Aryl-1-azido-2-cyclopentenes. J. Org. Chem. 2004, 69, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.A.; Castilla, J.; Alonso, C.; Pérez, J.M. Novel concepts in the development of platinum antitumor drugs. Curr. Med. Chem. Anticancer Agents 2002, 2, 539–551. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R | HuTu80 | B16 | A549 | KB-3-1 | HeLa | hHFF3 |
|---|---|---|---|---|---|---|---|
| 3a | C3H7 | >100 | >100 | >100 | 40.1 ± 5.2 | 63.4 ± 6.8 | >100 |
| 4a | >100 | >100 | >100 | >100 | >100 | >100 | |
| 3b | C5H11 | >100 | >100 | >100 | >100 | >100 | >100 |
| 4b | 80.4 ± 0.9 | >100 | 45.3 ± 3.9 | 25.2 ± 3.4 | 50.1 ± 4.7 | >100 | |
| 3c | C6H13 | 100 | >100 | 50.3 ± 6.1 | 45.3 ± 5.1 | >100 | >100 |
| 4c | 20.4 ± 3.9 | >100 | 35.2 ± 4.1 | 25.6 ± 2.9 | 50.7 ± 6.2 | >100 | |
| 3d | C8H17 | 46.3 ± 5.7 | 46.2 ± 3.4 | 54.1 ± 6.1 | 47.3 ± 4.2 | 40.6 ± 3.9 | 48.2 ± 5.1 |
| 4d | 30.2 ± 2.9 | >100 | 100 | 25.4 ± 3.1 | 20.1 ± 1.9 | >100 | |
| 3e | C10H21 | 7.3 ± 2.4 | 21.3 ± 3.1 | 36.2 ± 11.3 | 11.2 ± 5.3 | 4.5 ± 0.9 | 11.6 ± 2.1 |
| 4e | 23.4 ± 7.8 | 25.1 ± 2.4 | 36.2 ± 6.2 | 18.4 ± 7.1 | 11 ± 2.1 | 62.5 ± 8.3 | |
| 3f | C12H25 | 7.1 ± 0.8 | 4.5 ± 0.3 | 10.5 ± 1.9 | 10.2 ± 4.1 | 2.5 ± 0.3 | 10.2 ± 1.8 |
| 4f | 3.1 ± 0.4 | 3.1 ± 0.3 | 4.5 ± 0.2 | 4.5 ± 0.9 | 3.2 ± 0.4 | 3.4 ± 0.7 | |
| 3g | Ph | >100 | >100 | >100 | 30.1 ± 4.1 | 45.3 ± 5.1 | >100 |
| 4g | >100 | >100 | 50.2 ± 6.7 | 45.4 ± 5.3 | 90.2 ± 9.6 | >100 | |
| 3h | 4-tBuPh | 9.9 ± 1.7 | 10.1 ± 2.3 | 7.2 ± 0.8 | 1.7 ± 0.2 | 2.3 ± 0.3 | 5.1 ± 0.7 |
| 4h | 25.4 ± 3.1 | 35.6 ± 4.2 | 25.3 ± 2.9 | 15.3 ± 1.1 | 25.8 ± 4.6 | 70.9 ± 9.1 | |
| 3i | 4-C5H11Ph | 5.1 ± 0.6 | 21.3 ± 2.9 | 47.5 ± 5.8 | 8.2 ± 1.3 | 6.5 ± 0.9 | >100 |
| 4i | 8.5 ± 1.1 | 13.4 ± 3.2 | 15.6 ± 4.1 | 9.8 ± 0.7 | 11.1 ± 0.9 | 21.9 ± 4.8 | |
| 5 | C10H21 | >100 | >100 | >100 | 50.3 ± 9.8 | 70.2 ± 8.9 | >100 |
| Compound | R | HuTu80 | B16 | A549 | KB-3-1 | HeLa |
|---|---|---|---|---|---|---|
| 3a | C3H7 | 1 | 1 | 1 | >2.5 | >1.6 |
| 4a | 1 | 1 | 1 | 1 | 1 | |
| 3b | C5H11 | 1 | 1 | 1 | 1 | 1 |
| 4b | >1.2 | 1 | >2.2 | >3.9 | >1.9 | |
| 3c | C6H13 | 1 | 1 | >1.9 | >2.2 | 1 |
| 4c | >4.9 | 1 | >2.8 | >3.9 | >1.9 | |
| 3d | C8H17 | 1 | 1 | 0.9 | 1 | 1.2 |
| 4d | >3.3 | 1 | 1 | >3.9 | >4.9 | |
| 3e | C10H21 | 1.6 | 0.5 | 0.3 | 1 | 2.6 |
| 4e | 2.6 | 2.5 | 1.7 | 3.4 | 5.7 | |
| 3f | C12H25 | 1.4 | 2.3 | 1 | 1 | 4.1 |
| 4f | 1.1 | 1.1 | 0.8 | 0.8 | 1 | |
| 3g | Ph | 1 | 1 | 1 | >3.3 | >2.2 |
| 4g | 1 | 1 | >2 | >2.2 | >1.1 | |
| 3h | 4-tBuPh | 0.5 | 0.5 | 0.7 | 3 | 2.2 |
| 4h | 2.8 | 1.9 | 2.8 | 4.6 | 2.7 | |
| 3i | 4-C5H11Ph | >19.6 | >4.7 | >2.1 | >12.2 | >15.3 |
| 4i | 2.6 | 1.6 | 1.4 | 2.2 | 1.9 | |
| 5 | C10H21 | 1 | 1 | 1 | >2 | >1.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimenko, A.A.; Matyugina, E.S.; Logashenko, E.B.; Solyev, P.N.; Zenkova, M.A.; Kochetkov, S.N.; Khandazhinskaya, A.L. Novel 5′-Norcarbocyclic Derivatives of Bicyclic Pyrrolo- and Furano[2,3-d]Pyrimidine Nucleosides. Molecules 2018, 23, 2654. https://doi.org/10.3390/molecules23102654
Klimenko AA, Matyugina ES, Logashenko EB, Solyev PN, Zenkova MA, Kochetkov SN, Khandazhinskaya AL. Novel 5′-Norcarbocyclic Derivatives of Bicyclic Pyrrolo- and Furano[2,3-d]Pyrimidine Nucleosides. Molecules. 2018; 23(10):2654. https://doi.org/10.3390/molecules23102654
Chicago/Turabian StyleKlimenko, Anna A., Elena S. Matyugina, Evgeniya B. Logashenko, Pavel N. Solyev, Marina A. Zenkova, Sergey N. Kochetkov, and Anastasia L. Khandazhinskaya. 2018. "Novel 5′-Norcarbocyclic Derivatives of Bicyclic Pyrrolo- and Furano[2,3-d]Pyrimidine Nucleosides" Molecules 23, no. 10: 2654. https://doi.org/10.3390/molecules23102654
APA StyleKlimenko, A. A., Matyugina, E. S., Logashenko, E. B., Solyev, P. N., Zenkova, M. A., Kochetkov, S. N., & Khandazhinskaya, A. L. (2018). Novel 5′-Norcarbocyclic Derivatives of Bicyclic Pyrrolo- and Furano[2,3-d]Pyrimidine Nucleosides. Molecules, 23(10), 2654. https://doi.org/10.3390/molecules23102654