
Zinc Finger Readers of Methylated DNA

Abstract
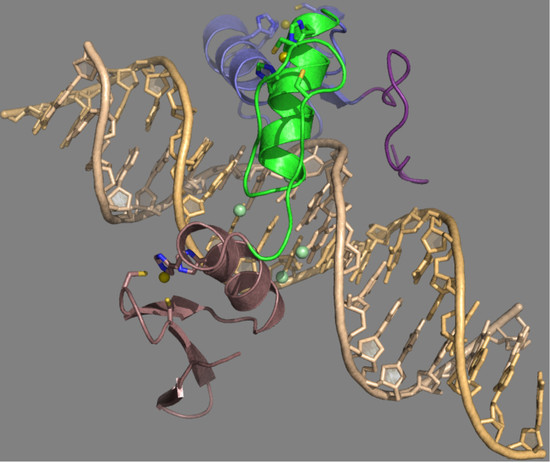
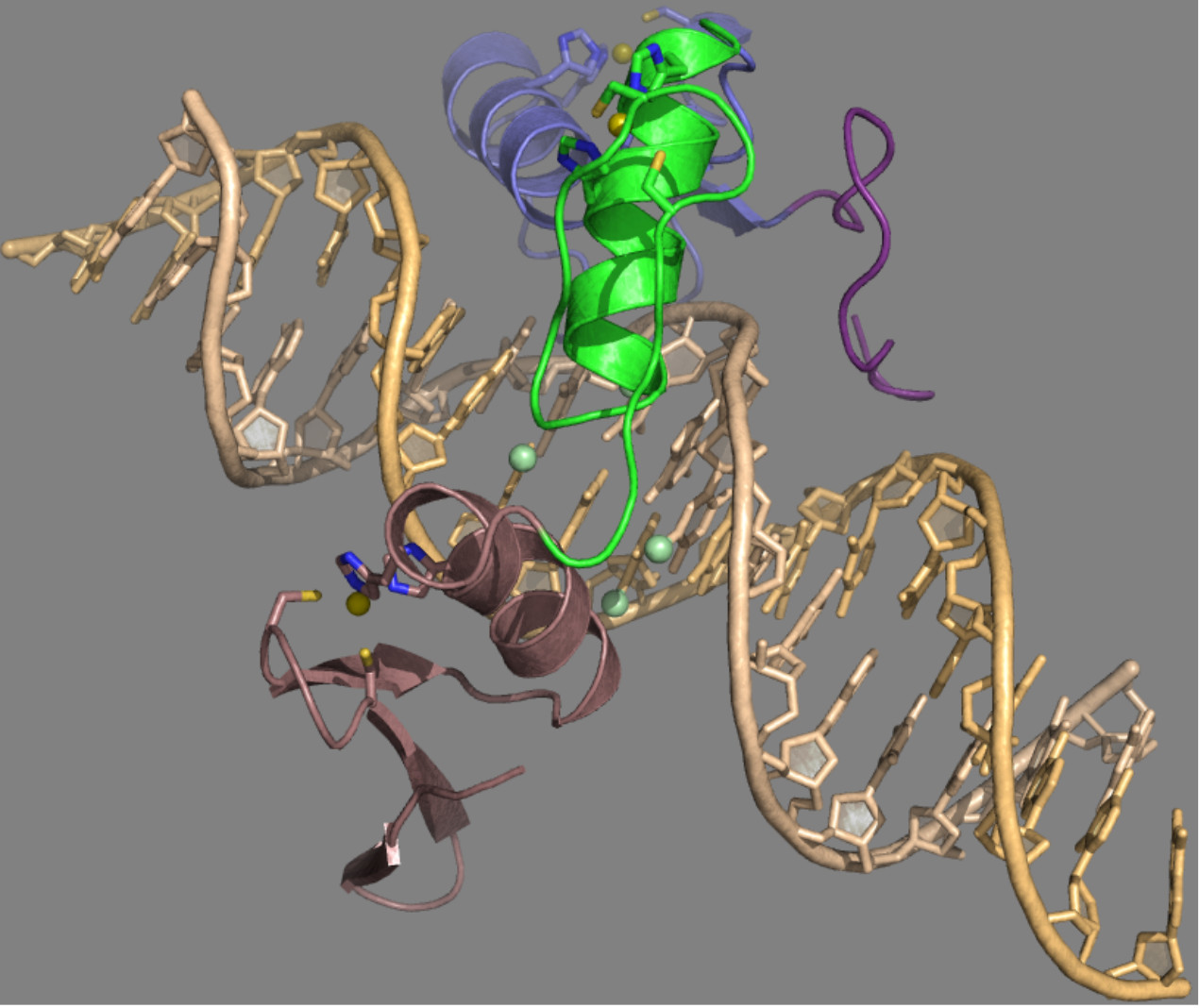{kind=link}
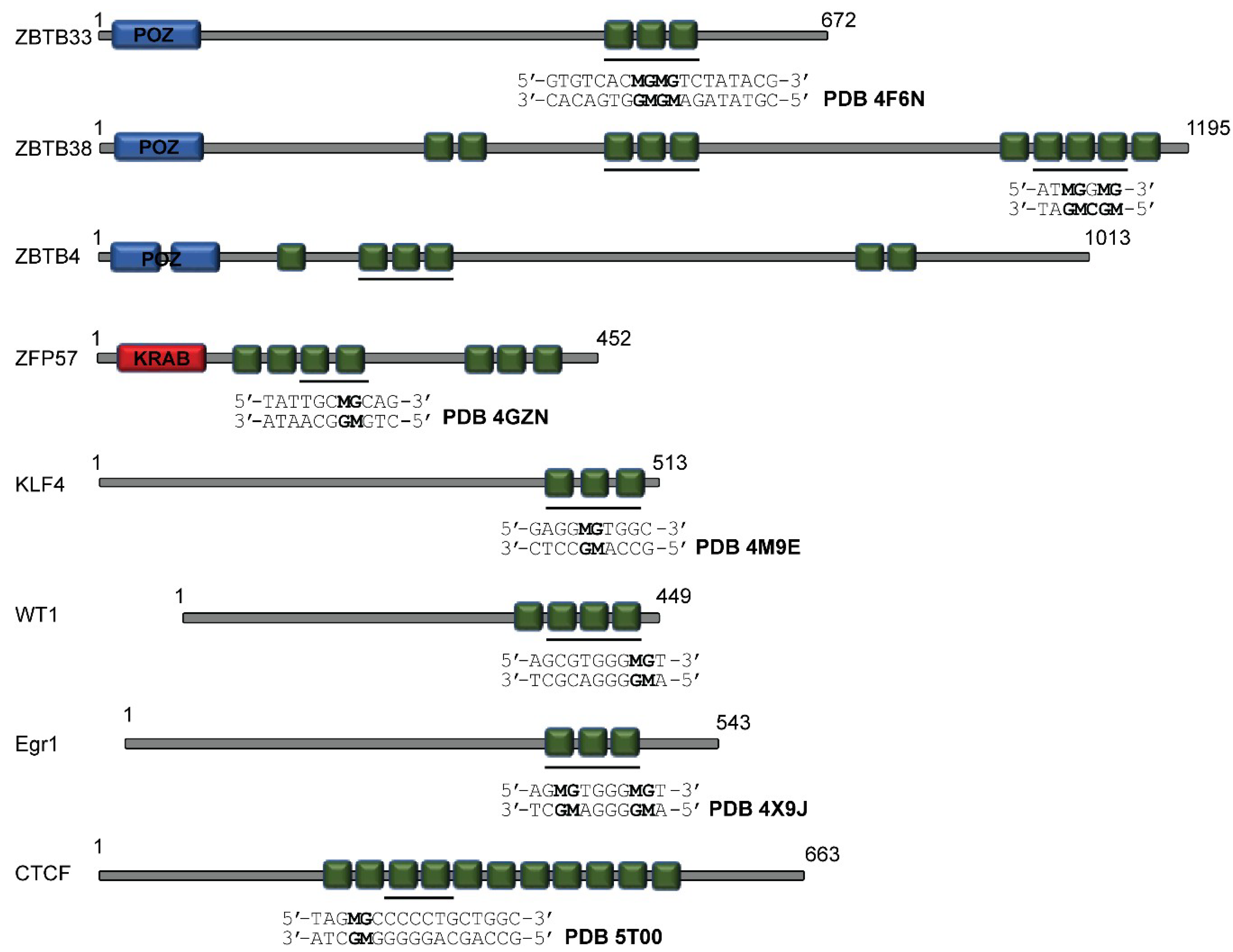{kind=link}
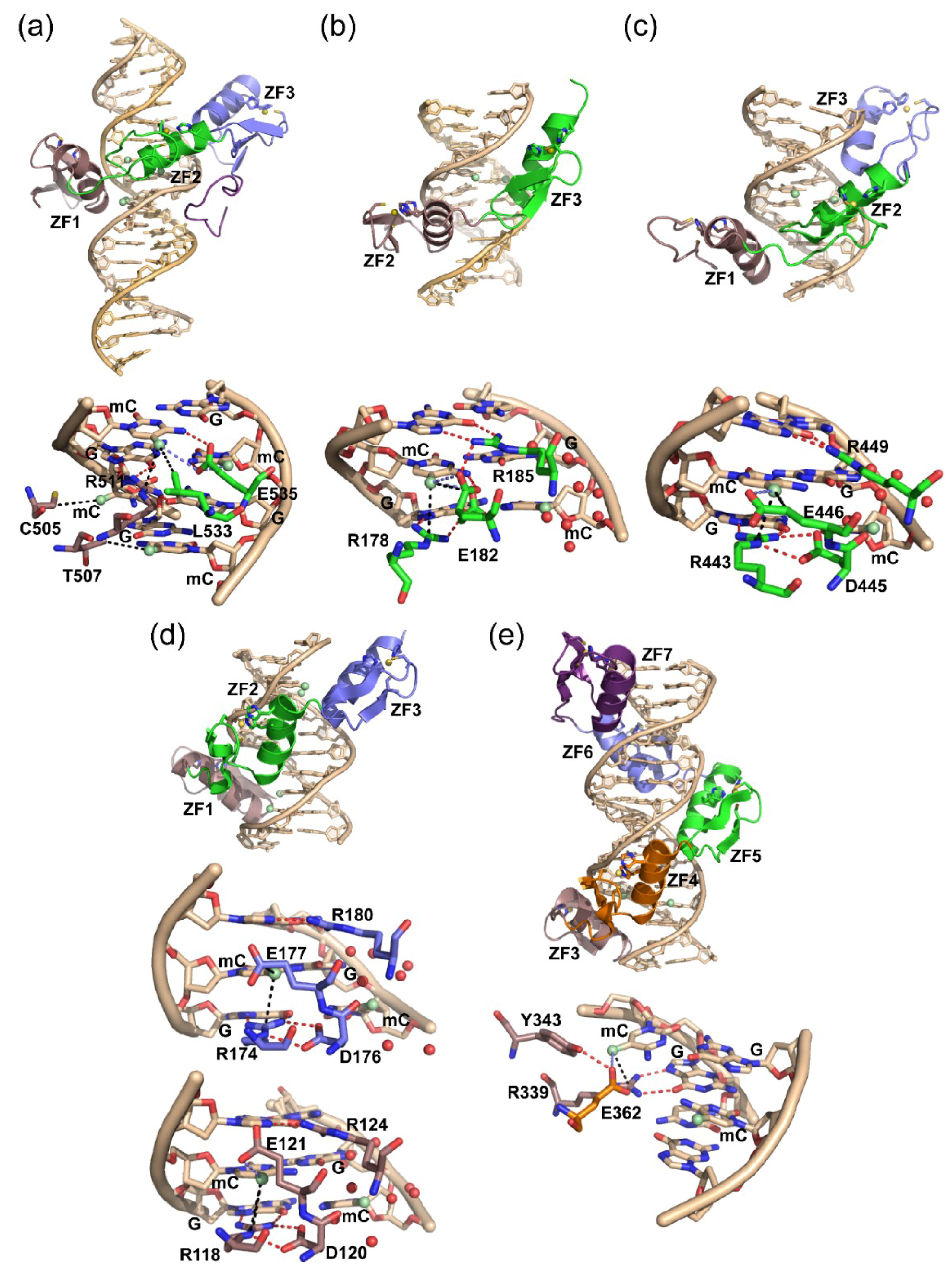{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mechanistic Recognition of mCpG Sites by Zinc Finger Proteins—Structural Perspective
2.1. Structural Insight for ZF Recognition of Methylated DNA
2.2. Additional Methyl-Selective ZF TFs and Alternative Modes of mCpG Recognition
3. Physiological Consequence of Methyl Sensitivity
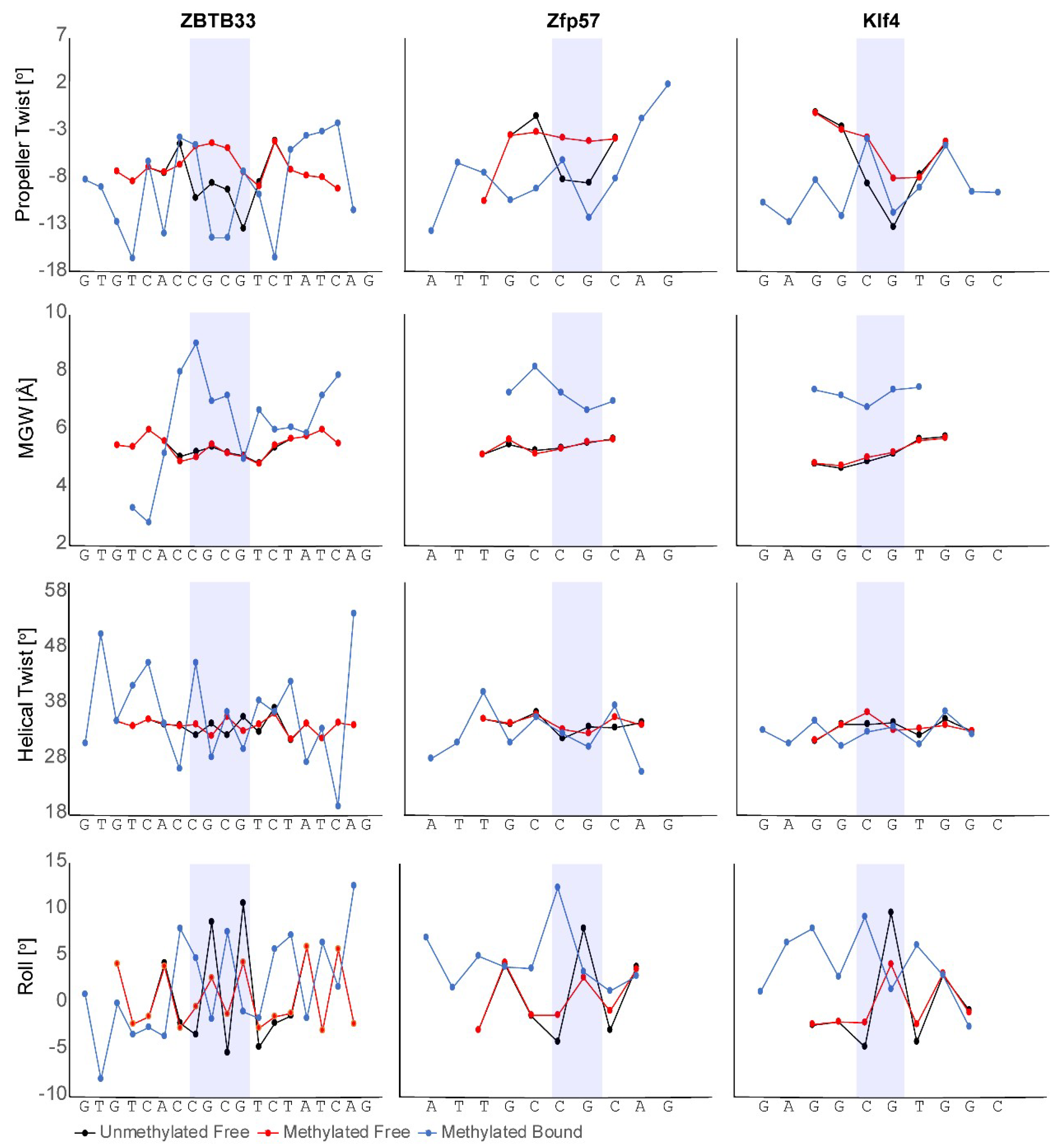
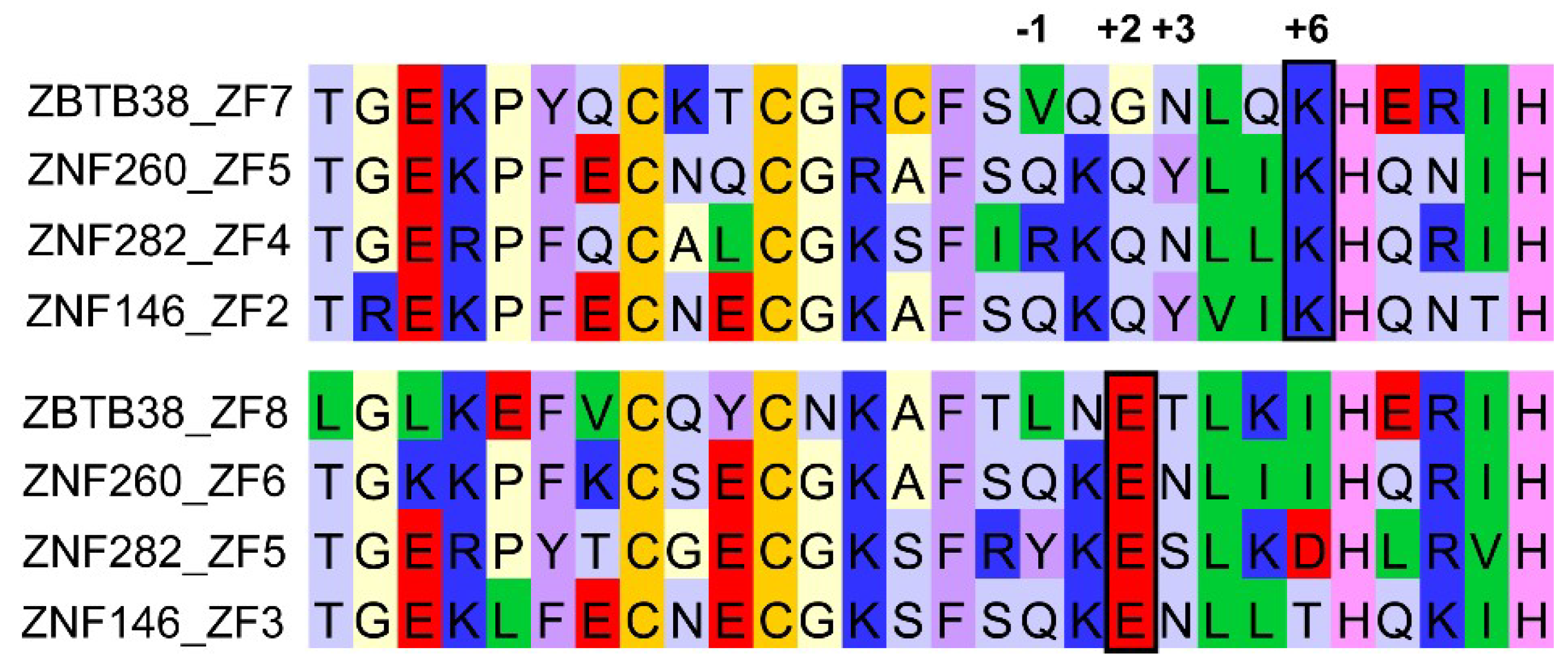
3.1. ZBTB33, ZBTB4, and ZBTB38
3.2. Zfp57
3.3. Klf4
3.4. WT1 and Egr1
3.5. CTCF and Interplay with Other ZF MBPs
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Kim, G.D.; Kelesoglu, N.; Roberts, R.J.; Pradhan, S. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 2002, 21, 4183–4195. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Goll, M.G.; Bestor, T.H. Eukarytic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed]
- Kareta, M.S.; Botello, Z.M.; Ennis, J.J.; Chou, C.; Chédin, F. Reconstitution of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 2006, 281, 25893–25902. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Carninci, P.; Lenhard, B.; Ponjavic, J.; Hayashizaki, Y.; Hume, D.A. Mammalian RNA polymerase II core promoters: Insights from genome-wide studies. Nat. Rev. Genet. 2007, 8, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Augui, S.; Nora, E.P.; Heard, E. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat. Rev. Genet. 2011, 12, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Bartolomei, M.S.; Fergurson-Smith, A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011, 3, a002592. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 587–610. [Google Scholar] [CrossRef] [PubMed]
- Kass, S.U.; Pruss, D.; Wolffe, A.P. How does DNA methylation repress transcription? Trends Genet. 1997, 13, 444–449. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Aberrent DNA methylation as a cancer-inducing mechanism. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 629–656. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.F.; Fruhwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomaki, P.; Lang, J.C.; et al. Aberrant CpG-island methylation has non-random and tumour-type−specific patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Song, J.S.; Costello, J.F. Intratumoral heterogeneity of the epigenome. Cancer Cell 2016, 29, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 2017, 166, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Liu, H.; Su, J.; Wu, X.; Liu, H.; Li, B.; Xiao, X.; Wang, F.; Wu, Q.; Zhang, Y. DiseaseMeth: A human disease methylation database. Nucleic Acids Res. 2011, 40, D1030–D1035. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wei, Y.; Gu, Y.; Zhang, S.; Lyu, J.; Zhang, B.; Chen, C.; Zhu, J.; Wang, Y.; Liu, H.; et al. DiseaseMeth version 2.0 a major expansion and update of the human disease methylation database. Nucleic Acids Res. 2017, 45, D888–D895. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dev. 2018, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Davegårdh, C.; García-Calzón, S.; Bacos, K.; Ling, C. DNA methylation in the pathogenesis of type 2 diabetes in humans. Mol. Metab. 2018, 14, 12–25. [Google Scholar] [CrossRef] [PubMed]
- De Mello, V.D.; Pulkkinen, L.; Lalli, M.; Kolehmainen, M.; Pihlajamäki, J.; Uusitupa, M. DNA methylation in obesity and type 2 diabetes. Ann. Med. 2014, 46, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.M.; Ginbney, E.R. Epigenetic regulation in obesity. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Sayols-Baixeras, S.; Subirana, I.; Fernández-Sanlés, A.; Sentí, M.; Lluís-Ganella, C.; Marrugat, J.; Elosua, R. DNA methylation and obesity traits: An epigenome-wide association study. The REGICOR study. Epigenetics 2017, 12, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Hewagama, A.; Richardson, B. The genetics and epigenetics of autoimune disorders. J. Autoimmun. 2009, 33, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Ronderos, P.; Montoya-Ortiz, G. Epigenetics and Autoimmune Diseases. Autoimmune Dis. 2012, 2012, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Hu, L.; Luo, Z.Y.; Chen, X.P.; Zhou, H.H.; Zhang, W. DNA methylation perspectives in the pathogenesis of autoimmune diseases. Clin. Immunol. 2016, 164, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liu, X.; Deng, Y.; Qing, H. DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci. 2013, 5, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.R.; Guidotti, A. The Dynamics of DNA Methylation in Schizophrenia and Related Psychiatric Disorders. Neuropsychopharmacology 2012, 38, 138. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, M.; Monteggia, L.M. Epigenetics and Psychiatry. Neurotherapeutics 2013, 10, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.-P. Aging and epigenetic drift: A vicious cycle. J. Clin. Investig. 2014, 124, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Cedar, H. DNA methylation and gene expression. Microbiol. Rev. 1991, 55, 451–458. [Google Scholar] [PubMed]
- Lazarovici, A.; Zhou, T.; Shafer, A.; Dantas Machado, A.C.; Riley, T.R.; Sandstrom, R.; Sabo, P.J.; Lu, Y.; Rohs, R.; Stamatoyannopoulos, J.A. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc. Natl. Acad. Sci. USA 2013, 110, 6376–6381. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Chiu, T.-P.; Kribelbauer, J.F.; Mann, R.S.; Bussemaker, H.J.; Rohs, R. Systematic prediction of DNA shape changes due to CpG methylation explains epigenetic effects on protein-DNA binding. Epigenet. Chromatin 2018, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Dantas Machado, A.C.; Zhou, T.; Rao, S.; Goel, P.; Rastogi, C.; Lazarovici, A.; Bussemaker, H.J.; Rohs, R. Evolving insights on how cytosine methylation affects protein-DNA binding. Brief. Funct. Genom. 2014, 14, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Iguchi-Ariaga, S.M.M.; Schaffner, W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 1989, 3, 612–619. [Google Scholar] [CrossRef]
- Pendergast, G.C.; Ziff, E.B. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science 1991, 251, 186–189. [Google Scholar] [CrossRef]
- Watt, F.; Molloy, P.L. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988, 2, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Comb, M.; Goodman, H.M. CpG methylation inhibits proenkephalin gene expression and binding of the transcription factor AP-2. Nucleic Acids Res. 1990, 18, 3975–3982. [Google Scholar] [CrossRef] [PubMed]
- Gaston, K.; Fried, M. CpG methylation has differential effects on the binding of YY1 and ETS proteins to the bi-directional promoter of the Surf-1 and Surf-2 genes. Nucleic Acids Res. 1995, 23, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Defossez, P.-A.; Stancheva, I. Biological functions of methyl-CpG-binding proteins. Prog. Mol. Biol. Transl. Sci. 2011, 101, 377–398. [Google Scholar] [PubMed]
- Lopez-Serra, L.; Estellar, M. Proteins that bind methylated DNA and human cancer: reading the wrong words. Br. J. Cancer 2008, 98, 1881–1885. [Google Scholar] [CrossRef] [PubMed]
- Parry, L.; Clark, S.J. The roles of the methyl-CpG binding proteins in cancer. Genes Cancer 2011, 2, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Maddison, K.; Clarke, A.R. Mechanism of disease: Methyl-binding domainproteins as potential therapeutic targets in cancer. Nat. Clin. Pract. Oncol. 2007, 4, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Fournier, A.; Sasai, N.; Nakao, M.; Defossez, P.-A. The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Brief. Funct. Genom. 2011, 11, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Joulie, M.; Miotto, B.; Defossez, P.-A. Mammalian methyl-binding proteins: What might they do? Bioessays 2010, 32, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Sasai, N.; Defossez, P.-A. Many paths to one goal? The proteins that recognize methylated DNA in eukaryotes. Int. J. Dev. Biol. 2009, 53, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.; Macleod, D. Reading the DNA methylation signal. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Shimbo, T.; Wade, P.A. Proteins that read DNA methylation. Adv. Exp. Med. Biol. 2016, 945, 303–320. [Google Scholar] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenmann, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MeCP2 recruit histone deactylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Bird, A.; Reinberg, D. Analysis of the NuRD subunits reveals a histone deacetylases core complex and a coneection with DNA methylation. Genes Dev. 1999, 13, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.; Zhang, Y.; Hendrich, B.; Johnson, C.A.; Turner, B.M.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D.; Bird, A. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 1999, 23, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Sasai, N.; Matsuda, E.; Sarashina, E.; Ishada, Y.; Kawaichi, M. Identification of a novel BTB-zinc finger transcriptional repressor, CIBZ, that interacts with CtBP corepressor. Genes Cells 2005, 10, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Marquardt, J.; Elzi, D.; Forster, N.; Starke, S.; Glaum, A.; Yamada, D.; Defossez, P.-A.; Delrow, J.; Eisenman, R.N.; et al. Zbtb4 represses transcription of P21CIP1 and controls the cellular responses to p53 activation. EMBO J. 2008, 27, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.G.; Chan, D.W.; Reynolds, A.B.; Qin, J.; Wong, J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol. Cell 2003, 12, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Lopes, E.C.; Valls, E.; Figueroa, M.E.; Mazur, A.; Meng, F.-G.; Chiosis, G.; Laird, P.W.; Schreiber-Agus, N.; Greally, J.M.; Prokhortchouk, E.; et al. Kaiso contributes to DNA methylation-dependent silencing of tumor suppressor genes in colon cancer cell lines. Cancer Res. 2008, 68, 7258–7263. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Serra, L.; Ballestar, E.; Ropero, S.; Setien, F.; Billard, L.-M.; Fraga, M.F.; Lopez-Nieva, P.; Alaminos, M.; Guerrero, D.; Dante, R.; et al. Unmasking of epigenteically silenced candidate tumor suppressor genes by removal of methyl-CpG-binding domain proteins. Oncogene 2008, 27, 3556–3566. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Jin, C.; Long, J.; Fu, D.; Yang, F.; Xu, J.; Yu, X.; Chen, W.; Ni, Q. RNA interference of MBD1 in BxPC-3 human pancreatic cells delivered by PLGA-poloxamer nanoparticles. Cancer Biol. Ther. 2009, 8, 1–5. [Google Scholar] [CrossRef]
- Fukushige, S.; Kondo, E.; Horii, A. Methyl-CpG targeted transcriptional activation allows re-expression of tumor suppressor genes in human cancer cells. Biochem. Biophys. Res. Commun. 2008, 377, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Laird, P.W. Cancer epigentics comes of age. Nat. Genet. 1999, 21, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Herman, J.G. DNA hypermethylation in tumorigenesis: Epigenetics joins genetics. Trends Genet. 2000, 16, 168–174. [Google Scholar] [CrossRef]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D’Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylaiton in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Jjingo, D.; Conley, A.B.; Yi, S.V.; Lunyak, V.V.; Jordan, I.K. On the presence and role of human gene-body DNA methylation. Oncotarget 2012, 3, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Ndlovu, M.N.; Denis, H.; Fuks, F. Exposing the DNA methylome iceberg. Trends Biochem. Sci. 2011, 36, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Sarda, S.; Das, A.; Vinson, C.; Hannenhalli, S. Distal CpG islands can serve as alternative promoters to transcribe genes with silenced proximal promoters. Gen. Res. 2017, 27, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sarda, S.; Hannenhalli, S. Orphan CpG islands as alternative promoters. Transcription 2018, 9, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Maor, G.L.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.T.; Gonzales, F.A.; Jones, P.A. Altered chromatin structure associated with methylation-induced gene silencing in cancer cells: Correlation of accessibility, methylation, MeCP2 binding and acetylation. Nucleic Acids Res. 2001, 29, 4598–4606. [Google Scholar] [CrossRef] [PubMed]
- Baubec, T.; Ivanek, R.; Lienert, F.; Schubeler, D. Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell 2013, 153, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Kemme, C.A.; Marquez, R.; Luu, R.H.; Iwahara, J. Potential role of DNA methylation as a facilitator of target search processes for transcription factors through interplay with methyl-CpG-binding proteins. Nucleic Acids Res. 2017, 45, 7751–7759. [Google Scholar] [CrossRef] [PubMed]
- Kemme, C.A.; Nguyen, D.; Chattopadhyay, A.; Iwahara, J. Regulation of transcription factors via natural decoys in genomic DNA. Transcription 2016, 7, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wan, J.; Su, Y.; Song, Q.; Zeng, Y.; Nguyen, H.N.; Shin, J.; Cox, E.; Rho, H.S.; Woodard, C.; et al. DNA methylation presents distinct binding sites for human transcription factors. eLife 2013, 2, e00726. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Münzel, M.; Wagner, M.; Müller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356, eaaj2239. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Wang, D.; Horton, J.R.; Zhang, X.; Speck, S.H.; Blumenthal, R.M.; Cheng, X. Methyl-dependent and spatial-specific DNA recognition by the orthologous transcription factors human AP-1 and Epstein-Barr virus Zta. Nucleic Acids Res. 2017, 45, 2503–2515. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Bird, A. Identification of a family of methyl-CpG binding proteins. Mol. Cell. Biol. 1998, 18, 6538–6547. [Google Scholar] [CrossRef] [PubMed]
- Meehan, R.R.; Lewis, J.D.; McKay, S.; Kleiner, E.L.; Bird, A.P. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 1989, 58, 499–507. [Google Scholar] [CrossRef]
- Bogdanovic, O.; Veenstra, G.J.C. DNA methylation and methyl-CpG binding proteins: Developmental requirements and function. Chromosoma 2009, 118, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Luu, P.-L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, M.; Wade, P.A. MBD family proteins: Reading the epigenetic code. J. Cell Sci. 2006, 119, 3033–3037. [Google Scholar] [CrossRef] [PubMed]
- Ginder, G.D.; Williams, D.C.J. Readers of DNA methylation, the MBD family as potential therapeutic targets. Pharmacol. Ther. 2018, 184, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Tweedie, S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003, 19, 269–277. [Google Scholar] [CrossRef]
- Wade, P.A. Methyl CpG-binding proteins and transcriptional repression. BioEssays 2001, 23, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.H.; Zhou, Z. Emerging molecular and biological functions of MBD2, a reader of DNA methyaltion. Front. Genet. 2016, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Dhasarathy, A.; Wade, P.A. The MBD protein family-reading and epigenetic mark? Mutat. Res. 2008, 647, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Klug, A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010, 79, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, S.A.; Nekludova, L.; Pabo, C.O. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 1999, 3, 183–212. [Google Scholar]
- Buck-Koehntop, B.A.; Stanfield, R.L.; Ekiert, D.C.; Martinez-Yamout, M.A.; Dyson, H.J.; Wilson, I.A.; Wright, P.E. Molecular Basis for recognition of methylated and specific DNA sequences by the zinc finger protein Kaiso. Proc. Natl. Acad. Sci. USA 2012, 109, 15229–15234. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, E.N.; Stanfield, R.L.; Dyson, H.J.; Wright, P.E. CH···O hydrogen bonds mediate highly specific recognition of methylated CpG sites by the zinc finger protein Kaiso. Biochemistry 2018, 57, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Toh, H.; Sasaki, H.; Zhang, X.; Cheng, X. An atomic model of Zfp57 recognition of CpG methylation within a specific DNA sequence. Genes Dev. 2012, 26, 2374–2379. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Wang, D.; Steves, A.N.; Jin, P.; Blumenthal, R.M.; Zhang, X.; Cheng, X. Distinctive Klf4 mutants determine preference for DNA methylation status. Nucleic Acids Res. 2016, 44, 10177–10185. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Olanrewaju, Y.O.; Zheng, Y.; Hashimoto, H.; Blumenthal, R.M.; Zhangg, X.; Cheng, X. Structural Basis for Klf4 recognition of methylated DNA. Nucleic Acids Res. 2014, 42, 4859–4867. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Olanrewaju, Y.O.; Zheng, Y.; Wilson, G.G.; Zhang, X.; Cheng, X. Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. 2014, 28, 2304–2313. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; White, M.A.; Esadze, A.; Iwahara, J. Structural impact of complete CpG methylation within target DNA on specific complex formation of the inducible transcription factoer Egr-1. FEBS Lett. 2015, 589, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Wang, D.; Horton, J.R.; Zhang, X.; Corces, V.G.; Cheng, X. Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol. Cell 2017, 66, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Abramoff, M.D.; Magalhaes, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Buck-Koehntop, B.A.; Defossez, P.-A. On how mammalian transcription factors recognize methylated DNA. Epigenetics 2013, 8, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Blumenthal, R.M.; Cheng, X. A common mode of recognition for methylated CpG. Trends Biochem. Sci. 2013, 38, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Horton, J.R.; Zhang, X.; Blumenthal, R.M.; Cheng, X. Detecting and interpreting DNA methylation marks. Curr. Opin. Struct. Biol. 2018, 53, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Ma, W.; Solov’yov, I.A.; Chipot, C.; Schulten, K. Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Res. 2012, 40, 2747–2758. [Google Scholar] [CrossRef] [PubMed]
- Buck-Koehntop, B.A.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Kaiso uses all three zinc fingers and adjacent sequene motifs for high affinity binding to sequence-specific and methyl-CpG DNA targets. FEBS Lett. 2012, 586, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.M.; Spring, C.M.; Crawford, H.C.; Reynolds, A.B.; Baig, A. The p120ctn-binding partner Kaiso is a bi-modal DNA-binding protein that recognizes both a sequence-specific consensus and methylated CpG dinucleotides. Nucleic Acids Res. 2002, 30, 2911–2919. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.S.; Quigley, G.J.; Tilton, R.F.J.; Rich, A. Hydration of methylated and nonmethylated B-DNA and Z-DNA. J. Phys. Chem. 1988, 92, 939–945. [Google Scholar] [CrossRef]
- Mayer-Jung, C.; Moras, D.; Timsit, Y. Hydration and regulation of methylated CpG steps in DNA. EMBO J. 1998, 17, 2709–2718. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.L.; McNae, I.W.; Schmiedeberg, L.; Klose, R.J.; Bird, A.P.; Walkinshaw, M.D. MeCP2 binding to DNA depends upon hydration of methyl-CpG. Mol. Cell 2008, 29, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Otani, J.; Arita, K.; Kato, T.; Kinoshita, M.; Kimura, H.; Suetake, I.; Tajima, S.; Ariyoshi, M.; Shirakawa, M. Structural basis of the versatile DNA recognition ability of the methyl-CpG binding domain of methyl-CpG binding domain protein 4. J. Biol. Chem. 2013, 288, 6351–6362. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of specificty in protein-DNA recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef] [PubMed]
- Rohs, R.; West, S.M.; Sosinsky, A.; Liu, P.; Mann, R.S.; Honig, B. The role of DNA shape in protein-DNA recognition. Nature 2009, 461, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Slatttery, M.; Zhou, T.; Yang, L.; Dantas Machado, A.C.; Gordan, R.; Rohs, R. Abscence of a simple code: how transcription factors read the genome. Trends Biochem. Sci. 2014, 39, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Lavery, R.; Moakher, M.; Maddocks, J.H.; Petkeviciute, D.; Zakrzewska, K. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009, 37, 5917–5929. [Google Scholar] [CrossRef] [PubMed]
- Filion, G.J.P.; Zhenilo, S.; Salozhin, S.; Yamada, D.; Prokhortchouk, E.; Defossez, P.-A. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell. Biol. 2006, 26, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Sasai, N.; Nakao, M.; Defossez, P.-A. Sequence-specific recognition of methylated DNA by human zinc-finger proteins. Nucleic Acids Res. 2010, 38, 5015–5022. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, Y.; Matsuda, E.; Nishil, T.; Ishida, Y.; Kawaichi, M. Down-regulation of CIBZ, a novel substrate of Caspase-3, induces apoptosis. J. Biol. Chem. 2008, 283, 14242–14247. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, Y.; Omon, R.; Nishii, T.; Ishida, Y.; Kawaich, M.; Matsuda, E. The methyl-CpG-binding protein CIBZ suppresses myogenic differentiation by directly inhibiting myogenin expression. Cell Res. 2011, 21, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Pozner, A.; Hudson, N.O.; Trewhella, J.; Terooatea, T.W.; Miller, S.A.; Buck-Koehntop, B.A. The C-terminal zinc fingers of ZBTB38 are novel selective readers of DNA methylation. J. Mol. Biol. 2018, 430, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, H.; Chatail-Hermitte, F.; Ravassard, P.; Bayard, E.; Brunet, I.; Mallet, J. ZENON, a novel POZ Kruppel-like DNA binding protein associated with differentiaon and/or survivial of late postmitotic neurons. Mol. Cell. Biol. 2005, 25, 1713–1729. [Google Scholar] [CrossRef] [PubMed]
- Prokhortchouk, A.; Hendrich, B.; Jorgensen, H.R.A.; Wilm, M.; Georgiev, G.; Bird, A.; Prokhortchouk, E. The p120 catenin partner Kaiso is a DNA methylation-dependent transricptional repressor. Genes Dev. 2001, 15, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Prokhortchouk, A.V.; Aitkhozhina, D.S.; Sablina, A.A.; Ruzov, A.S.; Prokhortchouk, E.B. Kaiso, a new protein of the BTB/POZ family, specifcally binds to methylated DNA sequences. Russ. J. Genet. 2001, 37, 603–609. [Google Scholar] [CrossRef]
- Donaldson, N.S.; Pierre, C.C.; Antsey, M.I.; Robinson, S.C.; Weerawardane, S.M.; Daniel, J.M. Kaiso represses the cell cycle gene cyclin D1 via sequence-specific and methyl-CpG-dependent mechanisms. PLoS ONE 2012, 7, e50398. [Google Scholar] [CrossRef] [PubMed]
- Pozner, A.; Terooatea, T.W.; Buck-Koehntop, B.A. Cell specific Kaiso (ZBTB33) regulation of cell cycle through cyclin D1 and cyclin E1. J. Biol. Chem. 2016, 291, 24538–24550. [Google Scholar] [CrossRef] [PubMed]
- Rodova, M.; Kelly, K.F.; VanSuan, M.; Daniel, J.M.; Werle, M.J. Regulation of the Rapsyn promoter by Kaiso and d-Catenin. Mol. Cell. Biol. 2004, 24, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Raghav, S.K.; Waszak, S.M.; Krier, I.; Gubelmann, C.; Isakova, A.; Mikkelsen, T.S.; Deplancke, B. Integrative genomics identifies the corepressor SMRT as a gatekeeper of adipogenesis through the transcription factors C/EBPbeta and KAISO. Mol. Cell 2012, 46, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Cofre, J.; Menezes, J.R.; Pizzatti, L.; Abdelhay, E. Knock-down of Kaiso induces proliferation and blocks granulocytic differentiation in blast crisis of chronic myeloid leukemia. Cancer Cell Int. 2012, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Han, L.-H.; Su, Q.-X. Kaiso affects cell proliferation and cycle of lung cancer by inhibiting cyclin D1 transcript. Prog. Anatom. Sci. 2014, 20, 257–261. [Google Scholar]
- Koh, D.-I.; Han, D.; Ryu, H.; Choi, W.-I.; Jeon, B.-N.; Kim, M.-K.; Kim, Y.; Kim, J.Y.; Parry, L.; Clarke, A.R.; et al. KAISO, a critical regulator of p53-mediated transcription of CDKN1A and apoptotic genes. Proc. Natl. Acad. Sci. USA 2014, 111, 15078–15083. [Google Scholar] [CrossRef] [PubMed]
- Bassey-Archibong, B.I.; Rayner, L.G.A.; Hercules, S.M.; Aarts, C.W.; Dvorkin-Gheva, A.; Bramson, J.L.; Hassell, J.A.; Daniel, J.M. Kaiso depletion attenuates the growth and survival of triple negative breast cancer cells. Cell Death Dis. 2017, 8, e2689. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Zhang, J.; Lan, H.; Xu, Y.; Wang, H. Kaiso protects human umbilical vein endothelial cells against apoptosis by differentially regulating the expression of B-cell CLL/lymphoma 2 family members. Sci. Rep. 2017, 7, 7116. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Wang, H.; Zhou, J.; Hardy, S.; Turner, T.; Austin, D.; He, Q.; Wells, A.; Grizzle, W.E.; Yates, C. Nuclear Kaiso indicates aggressive prostate cancers and promotes migration and invasiveness of prostate cancer cells. Am. J. Pathol. 2012, 181, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Kwiecien, J.M.; Bassey-Archibong, B.I.; Dabrowski, W.; Rayner, L.G.A.; Lucas, A.R.; Daniel, J.M. Loss of Kaiso expression in breast cancer cells prevents intra-vascular invasion in the lung and secondary metastasis. PLoS ONE 2017, 12, e0183883. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, W.; Black, S.; Turner, O.; Daniel, J.M.; Dean-Colomb, W.; He, Q.P.; Davis, M.; Yates, C. Kaiso, a transcriptional repressor, promotes cell migration and invasion of prostate cancer cells through regulation of miR-31 expression. Oncotarget 2016, 7, 5677–5689. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ma, J.; Wang, X.; Peng, F.; Chen, X.; Zheng, B.; Wang, C.; Dai, Z.; Ai, J.; Zhao, S. Kaiso (ZBTB33) downregulation by mirna-181a inhibits cell proliferation, invasion, and the epithelial–mesenchymal transition in glioma cells. Cell Physiol. Biochem. 2018, 48, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Wang, H.; Karanam, B.; Theodore, S.; Dean-Colomb, W.; Welch, D.R.; Grizzle, W.; Yates, C. Nuclear localization of Kaiso promotes the poorly differentiated phenotype and EMT in infiltrating ductal carcinomas. Clin. Exp. Metastasis 2014, 31, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, J.F.; van de Ven, R.A.H.; Ercan, C.; van der Groep, P.; van der Wall, E.; Bult, P.; Christgen, M.; Lehmann, U.; Daniel, J.; van Diest, P.J.; et al. Nuclear Kaiso Expression is Associated with High Grade and Triple-Negative Invasive Breast Cancer. PLoS ONE 2012, 7, e37864. [Google Scholar] [CrossRef] [PubMed]
- Bassey-Archibong, B.I.; Kwiecien, J.M.; Milosavljevic, S.B.; Hallett, R.M.; Rayner, L.G.; Erb, M.J.; Crawford-Brown, C.J.; Stephenson, K.B.; Bédard, P.A.; Hassell, J.A.; et al. Kaiso depletion attenuates transforming growth factor-β signaling and metastatic activity of triple-negative breast cancer cells. Oncogenesis 2016, 5, e208. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Chadalapaka, G.; Lee, S.-O.; Yamada, D.; Sastre-Garau, X.; Defossez, P.-A.; Park, Y.-Y.; Lee, J.-S.; Safe, S. Identification of oncogenic microRNA-17-92/ZBTB4/specificty protein axis in breast cancer. Oncogene 2011, 31, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Chadalapaka, G.; Pathi, S.S.; Jin, U.-H.; Lee, J.-S.; Park, Y.-Y.; Cho, S.-G.; Chintharlapalli, S.; Safe, S. Induction of the transcriptional repressor ZBTB4 in prostate cancer cells by drug-induced targeting of microRNA-17-92/106b-25 clusters. Mol. Cancer Ther. 2012, 11, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.W.; Chadalapaka, G.; Cho, S.-G.; Lee, S.; Jin, U.-H.; Jutooru, I.; Choi, K.; Leung, Y.-K.; Ho, S.-M.; Safe, S.; et al. The transcriptional repressor ZBTB4 regulates EZH2 through microRNA-ZBTB4-Specificity protein signaling axis. Neoplasia 2014, 16, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Roussel-Gervais, A.; Naciri, I.; Kirsh, O.; Kasprzyk, L.; Velasco, G.; Grillo, G.; Dubus, P.; Defossez, P.-A. Loss of the methyl-CpG-binding protein ZBTB4 alters mitotic checkpoint, inreases aneuploidy and promotes tumorigenesis. Cancer Res. 2017, 77, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Li, J.; Yang, S.; Li, P.; Zhang, X.; Liu, H. CIBZ, a novel BTB domain-containing protein, is involved in mouse spinal cord injury via mitochondrial pathway independent of p53 gene. PLoS ONE 2012, 7, e33156. [Google Scholar] [CrossRef] [PubMed]
- Nishii, T.; Oikawa, Y.; Ishida, Y.; Kawaichi, M.; Matsuda, E. CtBP-interacting BTB zinc finger protein (CIBZ) promotes proliferation and G1/S transition in embryonic stem cells via Nanog. J. Biol. Chem. 2012, 287, 12417–12424. [Google Scholar] [CrossRef] [PubMed]
- Kotoku, T.; Kosaka, K.; Nishio, M.; Ishida, Y.; Kawaichi, M.; Matsuda, E. CIBZ regulates mesodermal and cardiac differentiation of by suppressing T and Mesp1 expression in mouse embryonic stem cells. Sci. Rep. 2016, 6, 34188. [Google Scholar] [CrossRef] [PubMed]
- Blattler, A.; Yao, L.; Wang, Y.; Ye, Z.; Jin, V.X.; Farnham, P.J. ZBTB33 binds unmethylated regions of the genome associated with actively expressed genes. Epigenet. Chromatin 2013, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Zhang, B.; Tian, W.; Gu, L.; Lu, Z.; Deng, D. Kaiso mainly locates in the nucleus in vivo and binds to methylated, but not hydroxymethylated DNA. Chin. J. Cancer Res. 2015, 27, 148–155. [Google Scholar] [PubMed]
- Blattler, A.; Farnham, P.J. Cross-talk between Site-specific Transcription Factors and DNA Methylation States. J. Biol. Chem. 2013, 288, 34287–34294. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Negre, N.; Li, Q.; Mieczkowska, J.O.; Slattery, M.; Liu, T.; Zhang, Y.; Kim, T.-K.; He, H.H.; Zieba, J.; et al. Systematic evaluation of factors influencing ChIP-seq fidelity. Nat. Methods 2012, 9, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Head, S.R.; Komori, H.K.; LaMere, S.A.; Whisenant, T.; Van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library construction ofr next-generation sequencing: Overviews and challenges. BioTechniques 2014, 56, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Schmidl, C.; Renderio, A.F.; Sheffield, N.C.; Bock, C. ChIPmentation: Fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods 2015, 12, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Wallerman, O.; Nord, H.; Bysani, M.; Borghini, L.; Wadelius, C. lobChIP: From cells to sequencing ready ChIP libraries in a single day. Epigenet. Chromatin 2015, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, S. A simple method for generating high-resolution maps of genome-wide protein binding. eLife 2015, 4, e09225. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 2017, 6, e21856. [Google Scholar] [CrossRef] [PubMed]
- Strogantsev, R.; Ferguson-Smith, A.C. Proteins involved in establishment and maintenance of imprinted methylation marks. Brief. Funct. Genom. 2012, 11, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Leder, P. Identifying genes preferentially expressed in undifferentiated embryonic stem cells. BMC Cell Biol. 2007, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ito, M.; Zhou, F.; Youngson, N.; Zuo, X.; Leder, P.; Fergurson-Smith, A.C. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev. Cell 2008, 15, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Quenneville, S.; Verde, G.; Corsinotti, A.; Kapopoulou, A.; Jakobsson, J.; Offner, S.; Baglivo, I.; Pedone, P.V.; Grimaldi, G.; Riccio, A.; et al. In Embryonic Stem Cells, ZFP57/KAP1 Recognize a Methylated Hexanucleotide to Affect Chromatin and DNA Methylation of Imprinting Control Regions. Mol. Cell 2011, 44, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Sheng, J.; Lau, H.-T.; McDonald, C.M.; Andrade, M.; Cullen, D.E.; Bell, F.T.; Iacovino, M.; Kyba, M.; Xu, G.; et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J. Biol. Chem. 2012, 287, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Yamaguchi, Y.; Kinjo, T.; Song, X.; Akagi, T.; Takamura, H.; Ohta, T.; Yokota, T.; Koide, H. The stem cell transcription factor ZFP57 induces IGF2 expression to promote anchorage-independent growth in cancer cells. Oncogene 2015, 34, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.M.; Christy, R.J.; Yang, V.W. Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. J. Biol. Chem. 1996, 271, 20009–20017. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotentstem cells from mouse embryonic and adult fibroblastcultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.T.; Pevsner, J.; Yang, V.W. The biology of the mammalian Krüppel-like family of transcription factors. Int. J. Biochem. Cell. Biol. 2000, 32, 1103–1121. [Google Scholar] [CrossRef]
- Evans, P.M.; Liu, C. Role of Krüppel-like factor 4 in normal homeostasis, cancer, and stem cells. Acta Biochim. Biophys. Sin. 2008, 40, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, M.K.; Vanderbilt, D.B.; Salkeni, M.A.; Ruppert, J.M. Kruppel-like Pluripotency Factors as Modulators of Cancer Cell Therapeutic Responses. Cancer Res. 2016, 76, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; Yang, V.W. Krüppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Rowland, B.D.; Pepper, D.S. KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer 2006, 6, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Su, Y.; Song, Q.; Tung, B.; Oyinlade, O.; Liu, S.; Ying, M.; Ming, G.-L.; Song, H.; Qian, J.; et al. Methylated cis-regulatory elements mediate KLF4-dependent gene transactivation and cell migration. eLife 2017, 6, e20068. [Google Scholar] [CrossRef] [PubMed]
- Pagel, J.I.; Deindl, E. Early growth response 1—A transcription factor in the crossfire of signal transcduction cascades. Indian J. Biochem. Biophys. 2011, 48, 226–235. [Google Scholar] [PubMed]
- Bozon, B.; Davis, S.; Laroche, S. A requirement for the immediate early gene zif268 in reconsolidation of recognition memory after retrieval. Neuron 2003, 40, 695–701. [Google Scholar] [CrossRef]
- Khachigian, L.M.; Lindner, V.; Williams, A.J.; Collins, T. Egr-1-induced endothelial gene expression: A common theme in vascular injury. Science 1996, 271, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.L.; Everitt, B.J.; Thomas, K.L. Independent cellular processes for hippocampal memory consolidation and reconsolidation. Science 2004, 304, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Fujita, T.; Lu, J.; Okada, K.; Shan Zou, Y.; Mackman, N.; Pinsky, D.J.; Stern, D.M. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat. Med. 2000, 6, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Haber, D.A. Wilms tumor and the WT1 gene. Exp. Cell Res. 2001, 264, 74–99. [Google Scholar] [CrossRef] [PubMed]
- Niaudet, P.; Gubler, M.C. WT1 and glomerular diseases. Pediatric. Nephrol. 2006, 21, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.D.; Wagner, N.; Schedl, A. The complex life of WT1. J. Cell Sci. 2003, 116, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Hastie, N.D. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development 2017, 144, 2862–2872. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, P.; Hastie, N.D. The many facets of the Wilms’ tumor gene, WT1. Hum. Mol. Genet. 2006, 15, R196–R201. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.; Schug, J.; Lefterova, M.; Cronican, A.A.; Fitz, N.F.; Davenport, F.A.; Carter, A.; Castranio, E.L.; Lefterov, I. Genome-wide approaches reveal EGR1-controlled regulatory networks associated with neurodegeneration. Neurobiol. Dis. 2014, 63, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Kubosaki, A.; Tomaru, Y.; Tagami, M.; Arner, E.; Miura, H.; Suzuki, T.; Suzuki, M.; Suzuki, H.; Hayashizaki, Y. Genome-wide investigation of in vivo EGR-1 binding sites in monocytic differentiation. Genome Biol. 2009, 10, R41. [Google Scholar] [CrossRef] [PubMed]
- Kemme, C.A.; Esadze, A.; Iwahara, J. Influence of quasi-specific sites on kinetics of target DNA search by a sequence-specific DNA-binding protein. Biochemistry 2015, 54, 6684–6691. [Google Scholar] [CrossRef] [PubMed]
- Kann, M.; Ettou, S.; Jung, Y.L.; Lenz, M.O.; Taglienti, M.E.; Park, P.J.; Schermer, B.; Benzing, T.; Kreidberg, J.A. Genome-wide analysis of Wilms’ tumor 1-Controlled gene expression in podocytes reveals key regulatory mechanisms. J. Am. Soc. Nephrol. 2015, 26, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Motamedi, F.J.; Badro, D.A.; Clarkson, M.; Rita Lecca, M.; Bradford, S.T.; Buske, F.A.; Saar, K.; Hübner, N.; Brändli, A.W.; Schedl, A. WT1 controls antagonistic FGF and BMP-pSMAD pathways in early renal progenitors. Nat. Commun. 2014, 5, 4444. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylsytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.F.; Yue, C.; Zhou, Y.; Houghton, P.J.; Soboloff, J. Wilms tumor suppressor 1 (WT1) and early growth response 1 (EGR1) are regulators of STIM1 expression. J. Biol. Chem. 2010, 285, 10591–10596. [Google Scholar] [CrossRef] [PubMed]
- Nakahashi, H.; Kwon, K.-R.K.; Resch, W.; Vian, L.; Dose, M.; Stavreva, D.; Hakim, O.; Pruett, N.; Nelson, S.; Yamane, A.; et al. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Reports 2013, 3, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.S.; Pugh, B.J. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 2011, 147, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Terooatea, T.W.; Pozner, A.; Buck-Koehntop, B.A. PAtChCap: Input Strategy for Improving Analysis of ChIP-exo Data and Beyond. Nucleic Acids Res. 2016, 44, e159. [Google Scholar] [PubMed]
- Wang, H.; Maurano, M.T.; Qu, H.; Varley, K.E.; Gertz, J.; Pauli, F.; Lee, K.; Canfield, T.; Weaver, M.; Sanstrom, R.; et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012, 22, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- De La Rosa-Velazquez, I.A.; Rincon-Arano, H.; Benitez-Bribiesca, L.; Recillas-Targa, F. Epigenetic regulation of the human retinoblastoma tumor suppressor gene promoter by CTCF. Cancer Res. 2007, 67, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Defossez, P.-A.; Kelly, K.F.; Filion, G.J.P.; Perez-Torrado, R.; Magdinier, F.; Menoni, H.; Nordgaard, C.L.; Daniel, J.M.; Gilson, E. The human enhancer blocker CTC-binding factor interacts with the transcription factor Kaiso. J. Biol. Chem. 2005, 280, 43017–43023. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y. Recognition of DNA methylation by zinc fingers. Nat. Struct. Biol. 1998, 5, 264–265. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y.; Klug, A. Physical basis of a protein-DNA recognition code. Curr. Opin. Struct. Biol. 1997, 7, 117–125. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hudson, N.O.; Buck-Koehntop, B.A. Zinc Finger Readers of Methylated DNA. Molecules 2018, 23, 2555. https://doi.org/10.3390/molecules23102555
Hudson NO, Buck-Koehntop BA. Zinc Finger Readers of Methylated DNA. Molecules. 2018; 23(10):2555. https://doi.org/10.3390/molecules23102555
Chicago/Turabian StyleHudson, Nicholas O., and Bethany A. Buck-Koehntop. 2018. "Zinc Finger Readers of Methylated DNA" Molecules 23, no. 10: 2555. https://doi.org/10.3390/molecules23102555
APA StyleHudson, N. O., & Buck-Koehntop, B. A. (2018). Zinc Finger Readers of Methylated DNA. Molecules, 23(10), 2555. https://doi.org/10.3390/molecules23102555