1. Introduction
Poria cocos, also named Fuling in Chinese, is the dried sclerotium of
P. cocos (Schw.) Wolf, belonging to the Polyporaceae and grown in China, Japan, Korea, and North America. A well-known traditional Eastern Asian medicine,
P. cocos extracts exert beneficial health effects and fight against illnesses [
1].
P. cocos has important diuretic [
2], antibacterial, antitumor, mitogenic, complement activating, and immune stimulating activities [
3,
4,
5,
6]. However, in the Chinese Pharmacopoeia (2015 edition), information related to the required quality standard of
P. cocos was insufficient, and there has been little research done on
P. cocos.
In order to determine the quality standard of
P. cocos as well as appropriate index standards, we have studied the chemical composition of
P. cocos. An increasing number of chemical investigations on
P. cocos has occurred during the past decades, mainly focused on the isolation of triterpeniods and polysaccharides. Additionally, the lanostane triterpenes from
Poria cocos (Schw.) have been reported to cause cytotoxicity towards several cancer cell lines [
7,
8,
9]. Accordingly, we also expect to find some lead compounds which display cytotoxicity against some cancer cell lines. However, the basic skeletons of these compounds have rarely been determined and there are usually many isomers of them in
P. cocos. In addition, the purification of lanostane triterpenes is very difficult, which makes research on their antitumor activity difficult. In a previous paper, we reported the chemical constituents of a water decoction of
P. cocos [
10]. This inspired us to conduct an additional investigation on this fungus.
Due to the low content of lanostane triterpenes, in this paper, 100 kg dried sclerotia of
P. cocos were extracted with 3 × 1000 L of 95% ethanol, and the chemical constituents and pharmacological characteristics of
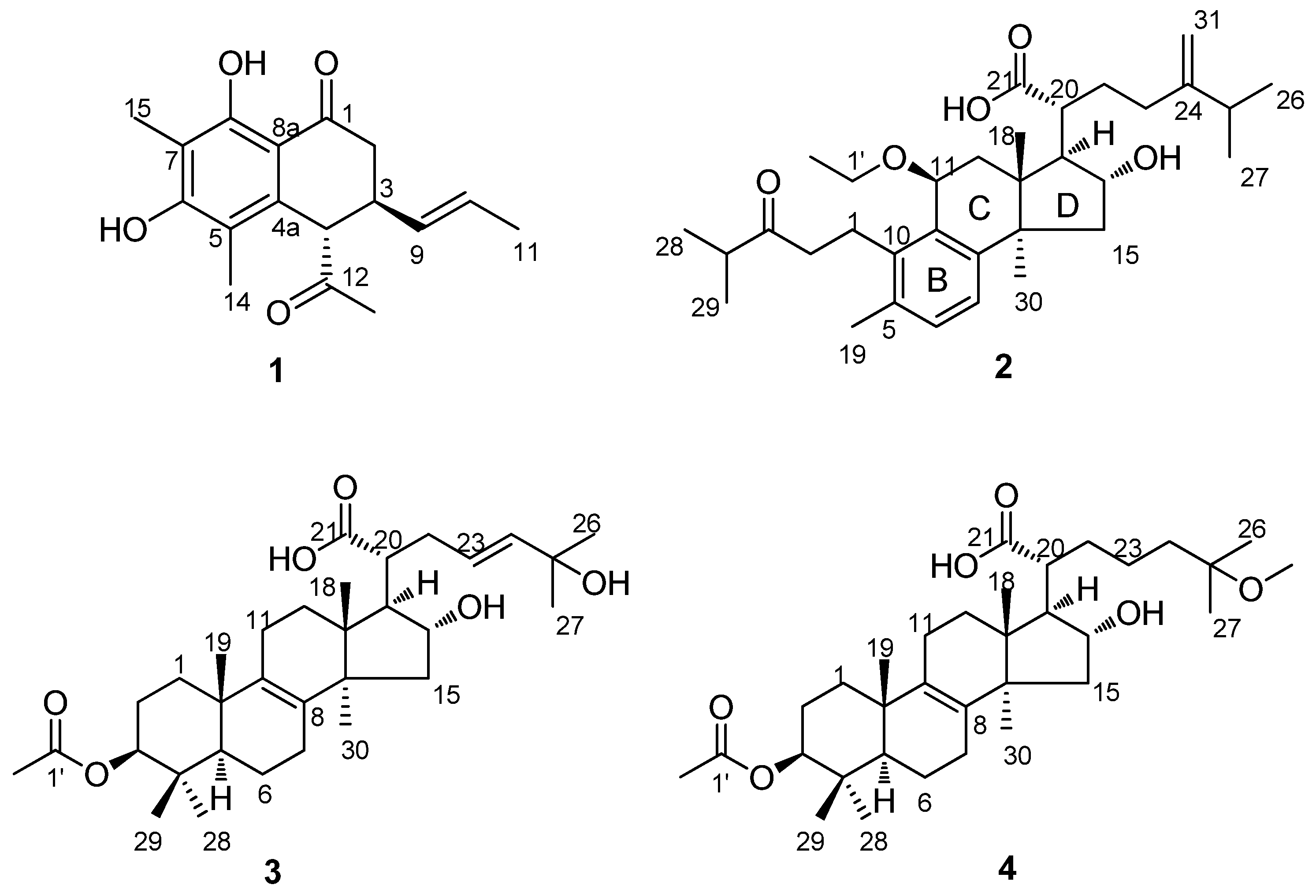
P. cocos were investigated. We obtained 21 compounds, including four new compounds: sohiracillinone (
1), 11
β-ethoxydaedaleanic acid A (
2), ceanphytamic acid A (
3) and ceanphytamic acid B (
4) (
Figure 1). The compounds’ cytotoxic activity was evaluated on 11 human cancer cell lines. Compound
1 showed strong cytotoxicity against the cervical cancer cell line with an IC
50 of 12.93 ± 2.38 μM, and others showed cytotoxicity against a variety of human cancer cell lines to different degrees.
2. Results and Discussion
The EtOH extract of sclerotia of
P. cocos was partitioned with petroleum ether (PE), CH
2Cl
2, and 1-butanol (
n-BuOH). The CH
2Cl
2 layer was chromatographed repeatedly to afford four new compounds (
1–
4 (
Figure 1), along with 17 known compounds: harzianone (
5) [
11], sorbicillin (
6) [
12], 2′,3′-dihydrosorbicillin (
7) [
12], sohirnone A (
8) [
13], pachymic acid (
9) [
14], dehypachymic acid (
10) [
14], trametenolic acid (
11) [
15], deyhdrotrametenolic acid (
12) [
15], polyporenic acid C (
13) [
16], 3
β-p-hydroxybenzoyldehydrotumulosic acid (
14) [
17], daedaleanic acid A (
15) [
18], 3
β,15
α-dihydroxylanosta-7,9(11),24-triene-21-oic acid (
16) [
19], poricoic acid A (
17) [
20], poricoic acid G (
18) [
21], dehydrosulphurenic acid (
19) [
22], eburicoic acid (
20) [
15], and tumulosic acid (
21) [
23]. The structures of the new compounds were established by analysis of spectroscopic data and the known ones were identified by comparison of their NMR data with those reported in the literature.
Compound
1 was obtained as a yellow oil. Its high resolution electrospray ionization mass spectrum (HRESIMS) exhibited a molecular ion peak at
m/z 288.1364, corresponding to the molecular formula of C
17H
20O
4 (calcd. 288.1362), and requiring eight degrees of unsaturation. The
13C-NMR data showed 17 carbon signals. Analyses of the
1H-NMR,
13C-NMR (
Table 1), and the HSQC spectra of
1 revealed the presence of two methyl signals in a single peak (
δH 2.06,
δC 11.7;
δH 2.13,
δC 7.5), one methyl signal in a double peak (
δH 1.57,
J = 6.5 Hz,
δC 7.5), one carbonyl group (
δC 201.6), one acetyl group (
δH 2.24,
δC 29.5, 207.6), and one 1,2-disubstituted double bond (
δH 5.37,
J = 15.0, 6.2, 0.9 Hz,
δC 131.6;
δH 5.52,
δC 127.4) with a set of aromatic carbon signals for quaternary bonds (
δC 161.6;
δC 159.3;
δC 136.7;
δC 114.0;
δC 111.5;
δC 109.1). Additionally, the data showed a methylene group (
δH 2.53,
J = 17.8 Hz,
δH 2.77,
J = 17.8, 5.3 Hz,
δC 39.6) and two methyne groups (
δH 3.22,
δC 38.6;
δH 4.03,
δC 55.6). These spectroscopic features together with the formula indicated that
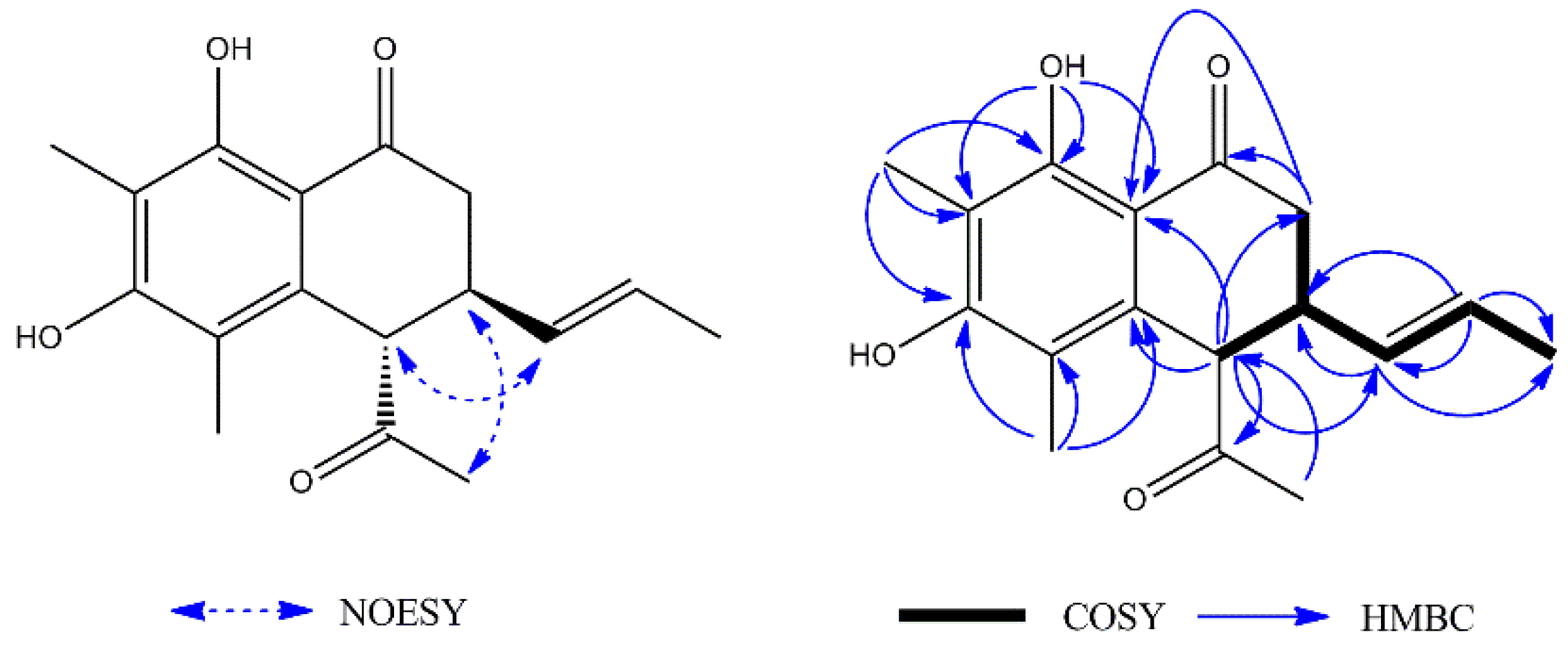
1 was a naphthalenone derivative. The difference between
1 and naphthalenone was the substituent groups on the aromatic ring and cyclohexanone. The
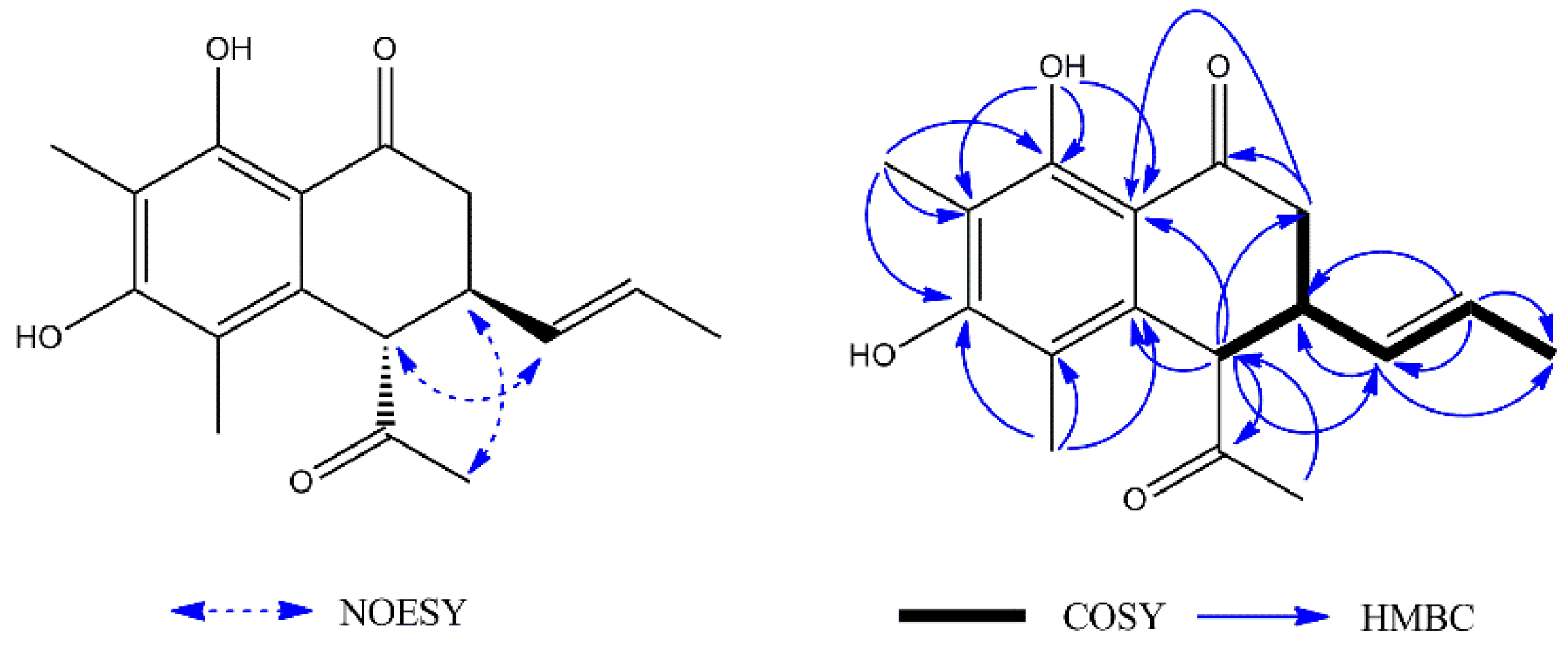
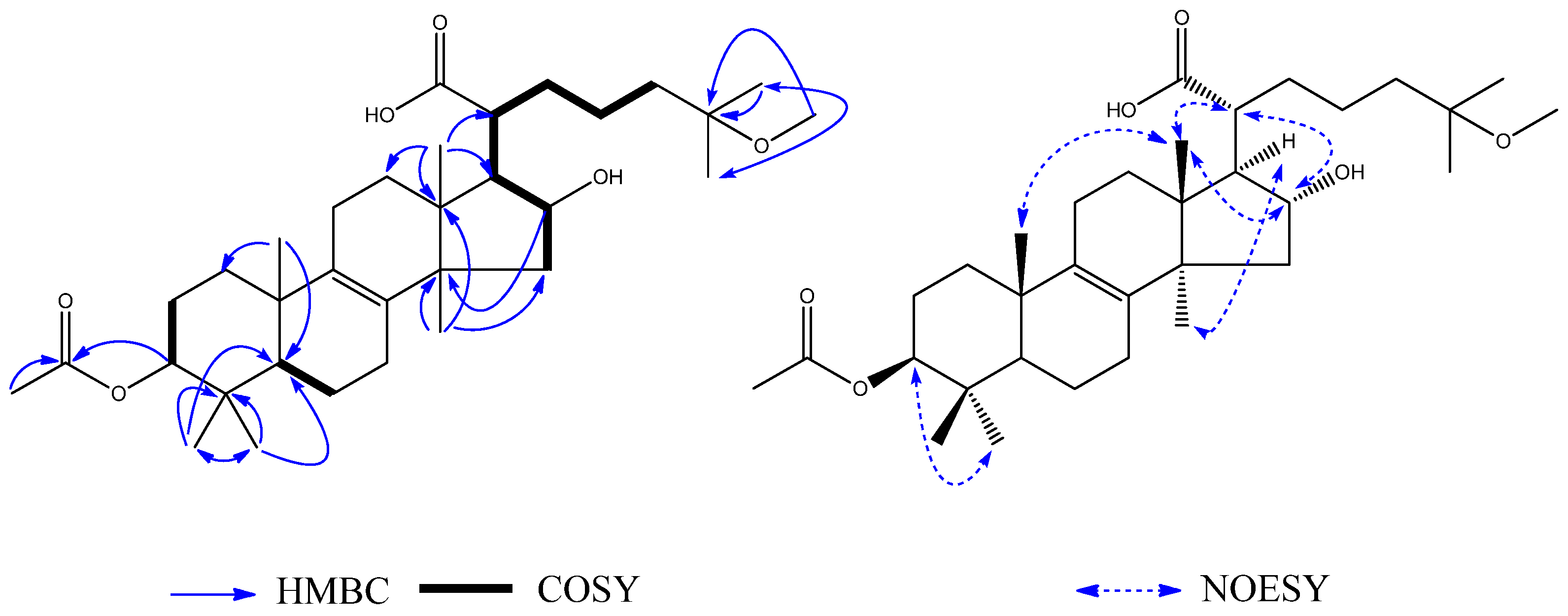
1H–
1H COSY spectrum (
Figure 2) showed the following two fragments: –CH
2(C-2)–CH(C-3)–CH(C-4)– and –CH(C-9)=CH(C-10)–CH
3(C-11). In addition, in the HMBC spectrum (
Figure 2), the hydrogen signal of olefin H-9 (
δH 5.37)/H-10 (
δH 5.52) was correlated with C-3 (
δC 38.6), suggesting that a propenyl moiety was located at C-3. The correlations of H-13 (
δH 2.24) with C-4 (
δC 55.6) and H-4 (
δH 4.03) with C-12 (
δC 207.6) showed that an acetyl group was located at C-4. The correlations of the hydroxy group (
δH 13.29, s, 1H) with C-7 (
δC 109.1) and C-8 (
δC 161.6), C-8a (
δC 111.5) showed it was located at C-8; the correlations of H-15 (
δH 2.13) with C-6 (
δC 159.3), C-7 (
δC 109.1), and C-8 (
δC 161.6) and the correlations of H-14 (
δH 2.06) with C-4a (
δC 136.7), C-5 (
δC 114.0), and C-6 (
δC 159.3) showed that two methyl groups were situated at C-5 and C-7 on the aromatic ring. The NOESY analysis (
Figure 2) showed correlations between H-3 (
δH 3.22) and H-13 (
δH 2.24), H-4 (
δH 4.03) and H-9 (
δH 5.37) which indicated the H-3 might be
α-oriented and H-4 was
β-oriented. According to the established relative configuration, the pair of enantiomers of
1 are (3
S,4
S)-
1a and (3
R,4
R)-
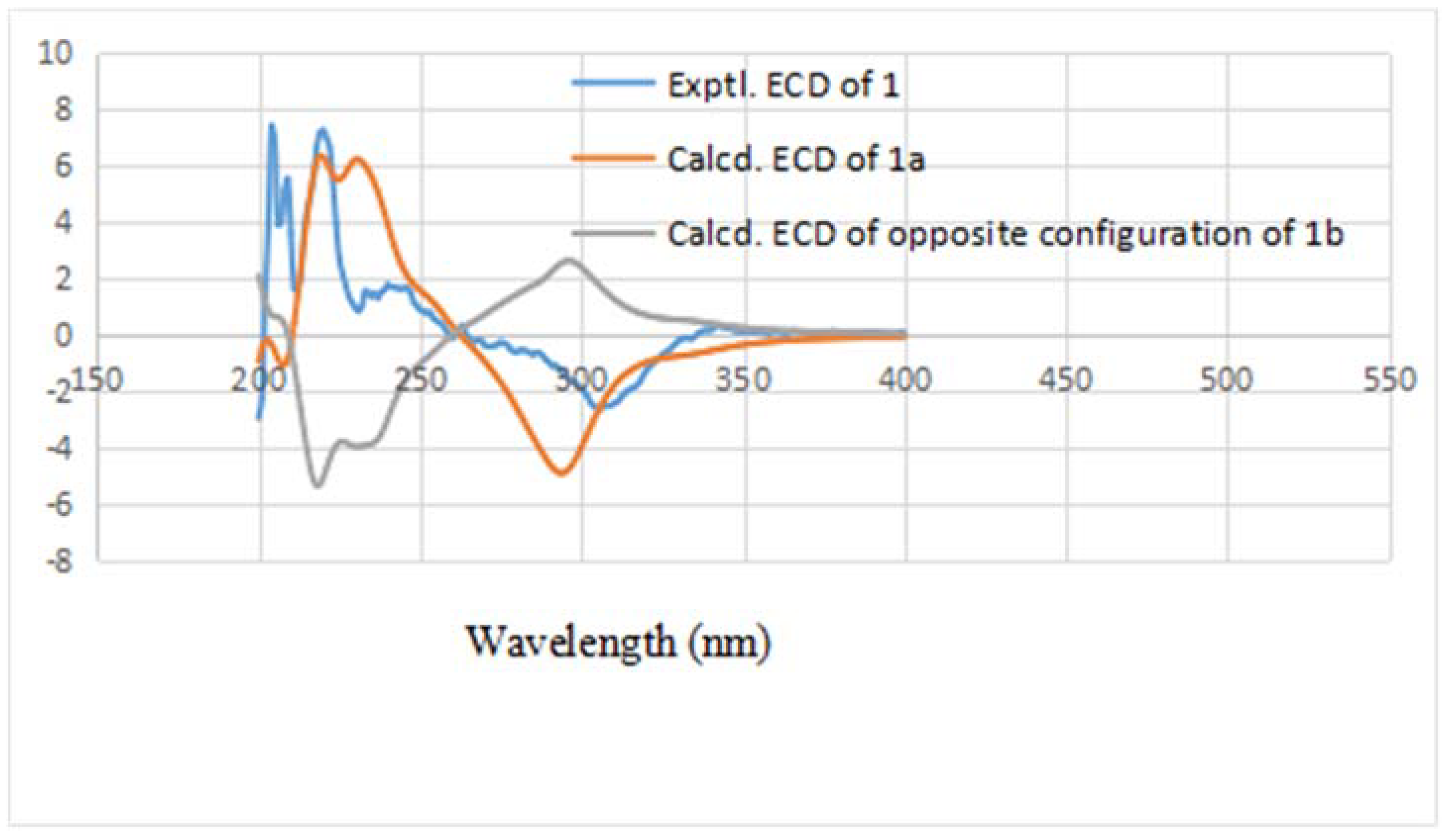
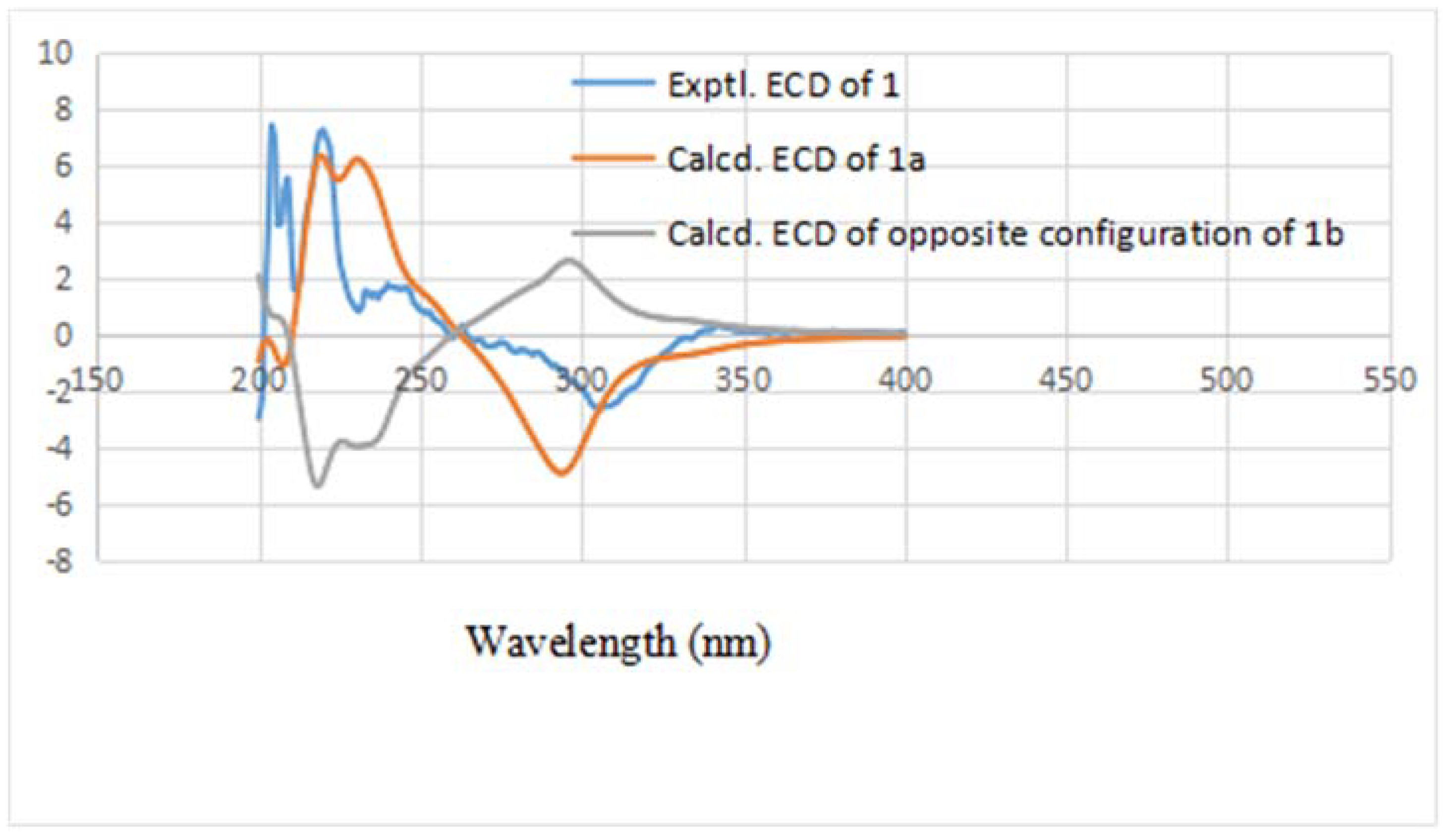
1b, respectively. In order to establish the absolute configuration of
1, the electronic circular dichroism (ECD) spectrum (
Figure 3) was determined, which exhibited a negative Cotton effect at 294 nm (n → π* transition). The calculated data indicated the 3
S and 4
S absolute configurations. Using the NMR and the CD data, the structure of
1 was determined to be (3
S,4
S)-4-acetyl-6,8-dihydroxy-5,7-dimethyl-3-((
E)-prop-1-en-1-yl)-3,4-dihydronaphthalen-1(2
H)-one, and it was named sohiracillinone.
Compound
2 was obtained as a white amorphous powder. Based on the HRESIMS, the molecular formula of
2 was determined to be C
33H
50O
5, giving a quasi-molecular ion peak at
m/z 525.3557 [M − H]
− (calcd. 525.3580). The
1H-NMR data (
Table 2) showed signals for three methyls at
δH 0.91 (3H, s, H
3-18),
δH 2.26 (3H, s, H
3-19),
δH 1.18 (3H, s, H
3-30), and two mutually-coupled aromatic protons at
δH 7.03 (1H, d,
J = 6.2 Hz, H-6),
δH 6.77 (1H, d,
J = 6.2 Hz, H-7). Additionally, two pairs of equivalent secondary methyl signals were identified at
δH 1.08 (3H, s, H
3-28),
δH 1.10 (3H, s, H
3-29) and
δH 1.05 (6H, overlap (ov.), H
3-26 and H
3-27). The 32 carbon signals observed in the
13C-NMR spectrum were sorted into eight methyls, indicating the presence of two isopropyl groups, eight methyl groups, seven methylene groups, and six methine groups, suggesting that
2 is a lanostane triterpene that is similar to daedaleanic acid A [
17]. The difference between the planar structure of
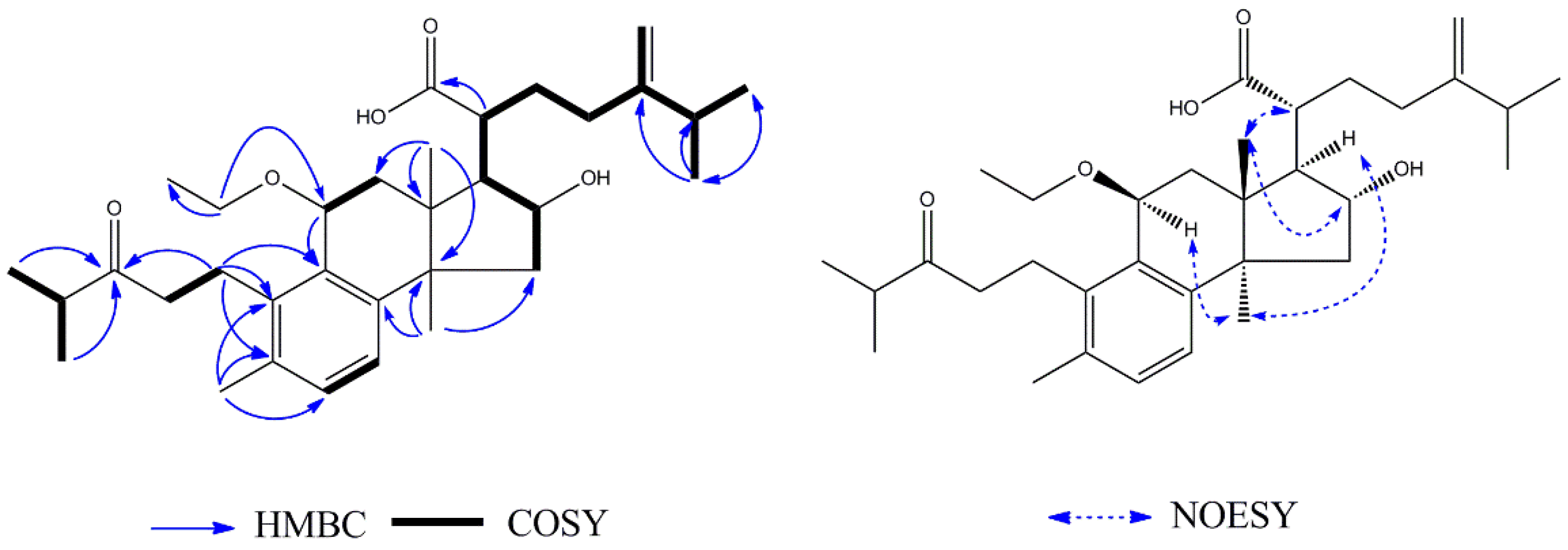
2 and daedaleanic acid A is the presence of an ethoxy group at C-11. In the HMBC spectrum, H-1′ (
δH 3.56, 1H, m;
δH 3.38, 1H, m) was correlated with C-11 (
δC 73.3) and CH
3-2′ (
δC 15.9), suggesting that the oxethyl group was located at C-11. The relative configuration of
2 was established by the NOESY experiment. Significant NOE correlations between H
3-18 (
δH 0.91, 3H, s) and H-16 (
δH 4.33, 1H, t) and H-20 (
δH 2.57, 1H, m), and between H
3-30 (
δH 1.18, 3H, s) and H-17 (
δH 2.30, 1H, m) and H-11 (
δH 4.47, 1H, d) indicated that OH-16 and H-17 have
α-orientations. From the analysis of all of these data and from a biosynthetic point of view, the structure of
2 was assigned as shown in
Figure 4 and named 11
β-ethoxydaedaleanic acid A.
Compound
3 was obtained as a white amorphous powder. The HRESIMS showed a
m/z at 530.3616 calcd. for C
32H
49O
6, 530.3607 [M − H]
− indicated a molecular formula of C
32H
50O
6. Its
1H-NMR (
Table 2) spectrum showed the signals of eight methyl signals at
δH 0.79 (3H, s, H
3-18),
δH 1.02 (3H, s, H
3-19),
δH 1.24 (3H, s, H
3-26),
δH 1.24 (3H, s, H
3-27),
δH 0.89 (3H, s, H
3-28),
δH 0.91 (3H, s, H
3-29),
δH 1.13 (3H, s, H
3-30) and
δH 2.03 (3H, s, H
3-2′) and two oxygen-bearing methynes at
δH 4.45 (1H, t, H-3) and
δH 4.05 (1H, t, H-16). Its
13C-NMR (
Table 2) and HSQC spectra proved the existence of eight methyl groups, as well as an acetyl carbon at
δ 172.9 (C, C-1′) and a pair of tetrasubstituted olefinic carbonds at
δ 135.6 (C, C-8) and
δ 136.0 (C, C-9) and three oxygenated carbons at
δ 82.5 (CH, C-3),
δ 77.9 (CH, C-16),
δ 71.1 (C, C-25). In addition, the
13C NMR and HSQC spectra also exhibited two olefinic signals at
δC 126.4 (
δH 5.62, 1H, ov., C-23) and
δC 140.2 (
δH 5.62, 1H, ov., C-24). There were
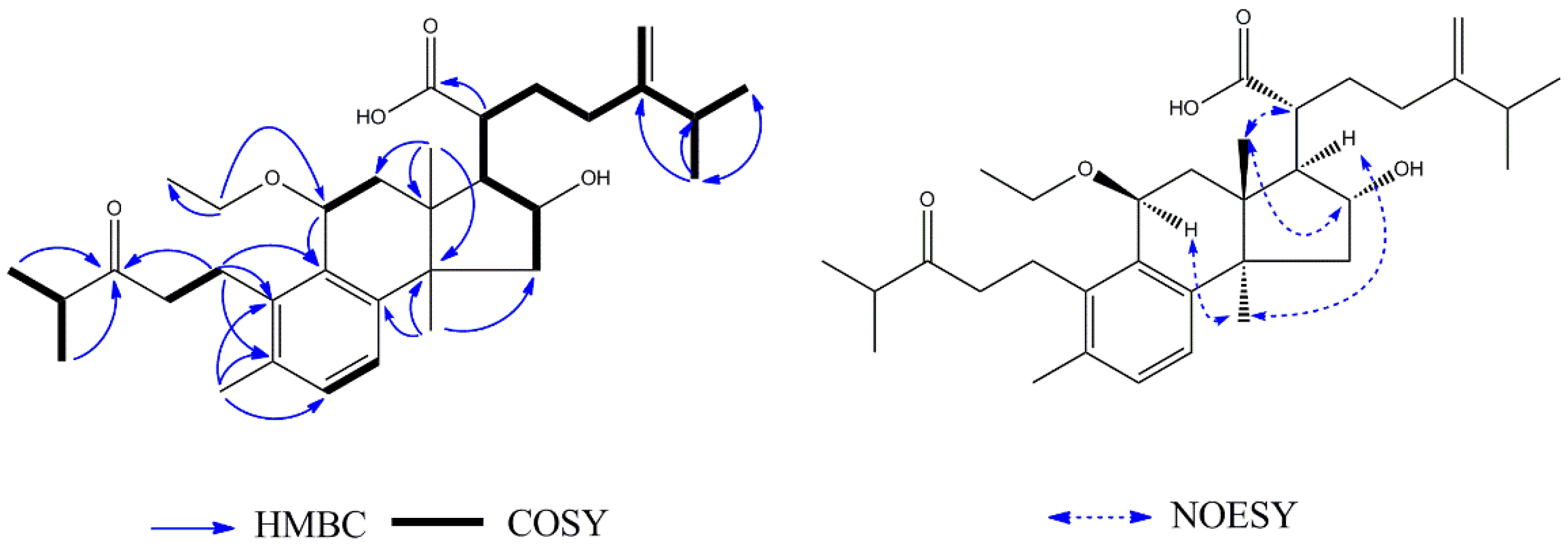
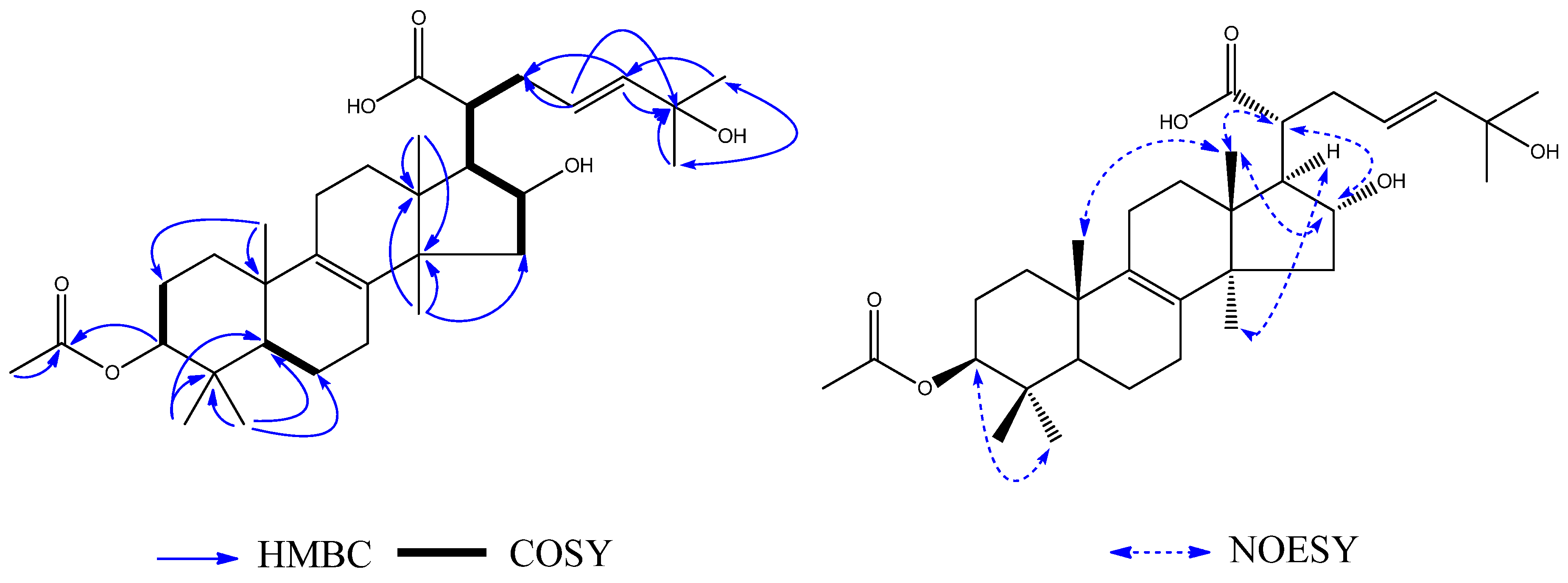
1H–
1H COSY (
Figure 5) correlations between H-2 (
δH 1.66) and H-3 (
δH 4.45), between H-5 (
δH 1.16) and H-6 (
δH 1.66), between H-15 (
δH 2.18), H-16 (
δH 4.05), H-17 (
δH 2.06), H-20 (
δH 2.32), H-22 (
δH 2.50), and H-23 (
δH 5.62). Therefore, it can be concluded that
3 was a lanost-8-ene triterpenoid. On the basis of the HMBC (
Figure 5), correlations from H
3-26 (
δH 1.24) to CH
3-27 (
δC 29.8), H
3-27 (
δH 1.24) to CH
3-26 (
δC 29.8), H
3-27 (
δH 1.24) to C-25 (
δC 71.1), and H
3-26 (
δH 1.24) to C-24 (
δC 140.2) indicated the presence of a hydroxy group at C-25. The correlation from H-3 (
δH 4.45) to C-1′ (
δC 172.9) showed that the acetyl group was located at C-3. The NOE correlations between H-16 (
δH 4.05) and H-20 (
δH 2.32), and between H
3-30 (
δH 1.13) and H-17 (
δH 2.06) indicated
α-orientations for OH-16 and H-17. The correlation between H
3-18 (
δH 0.79) and H
3-19 (
δH 1.02), H-16 (
δH 4.05) and H-20 (
δH 2.32) showed that CH
3-18, CH
3-19 and H-20 have
β-orientations. Thus, the structure of
3 was determined to be 3
β-acetoxy-16
α,25-dihydroxylanost-8,23-dien-21-oic acid, and it was named ceanphytamic acid A (
Figure 5).
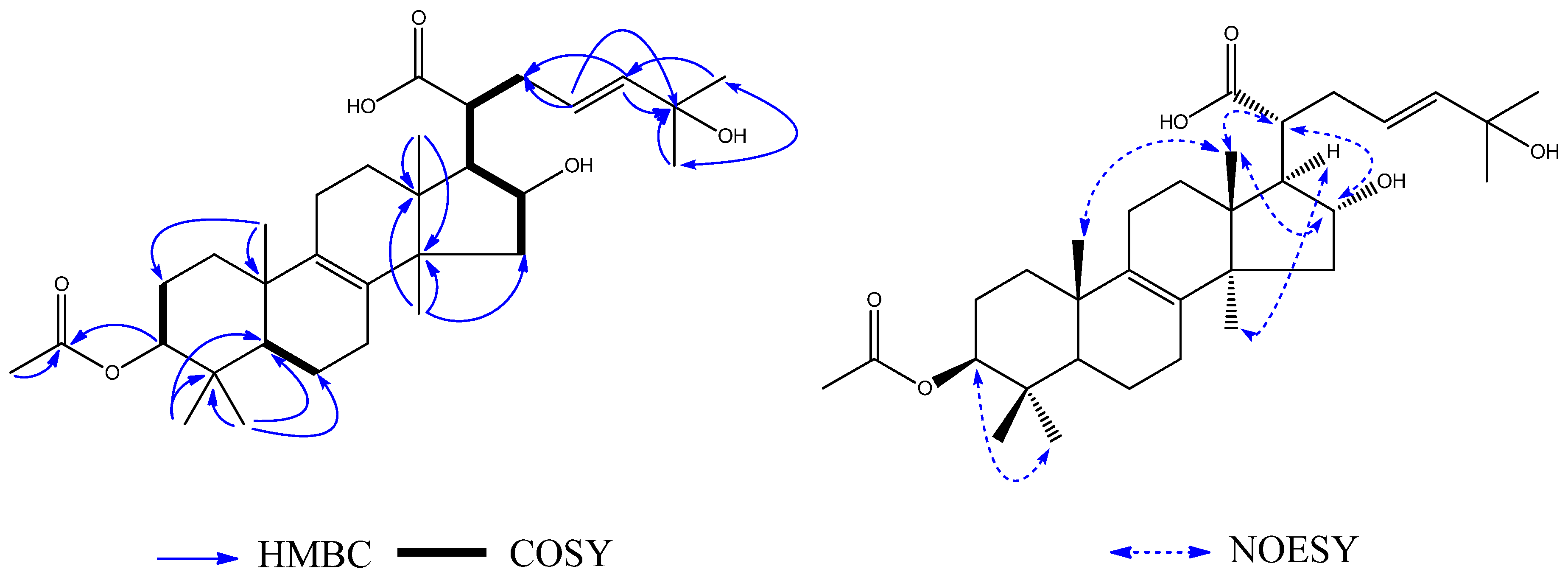
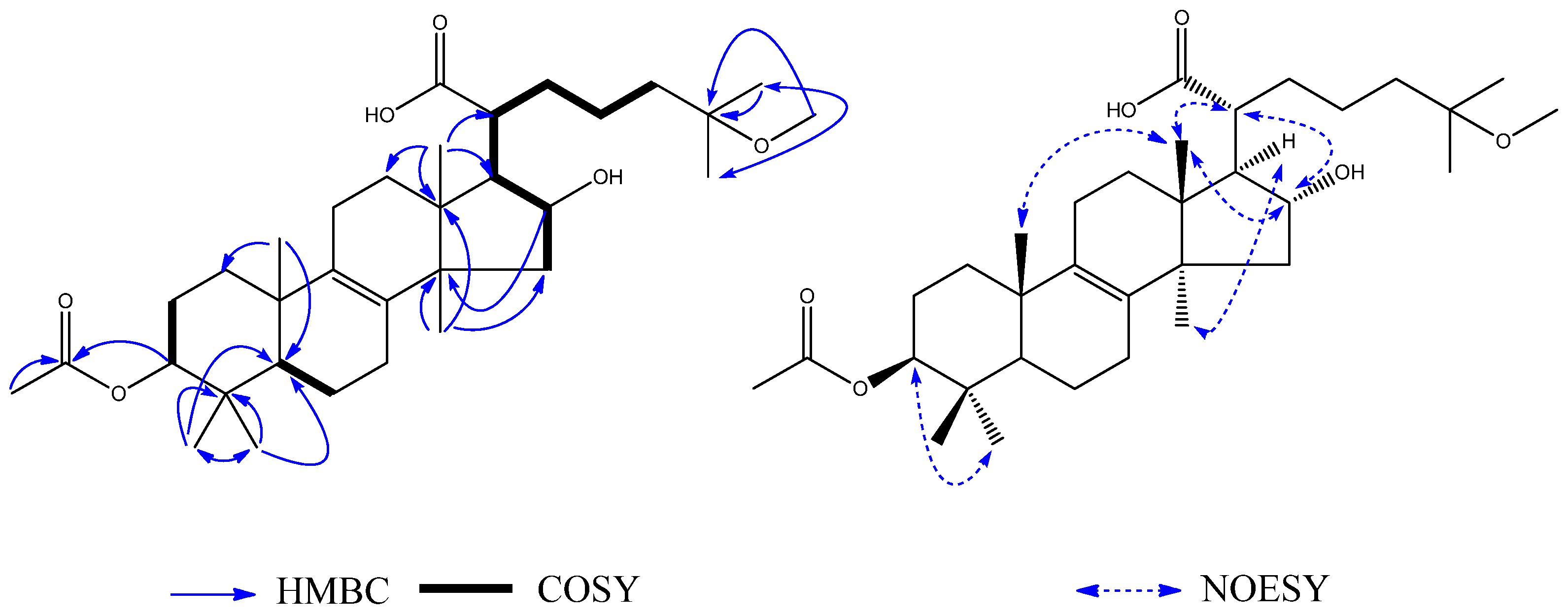
Compound
4 was obtained as a white amorphous powder. The HRESIMS showed a
m/z at 546.3913 calcd. for C
33H
54O
6, 546.3920 [M + H]
+.
1H–
1H COSY (
Figure 6) correlations were shown between H-2 (
δH 1.72) and H-3 (
δH 4.59), between H-5 (
δH 1.04) and H-6 (
δH 1.61), and between H-15 (
δH 2.34), H-16 (
δH 4.47), H-17 (
δH 2.30), H-20 (
δH 2.86), H-22 (
δH 1.24), H-23 (
δH 2.33), and H-24 (
δH 1.41). The
1H-NMR showed nine methyl groups at H
3-18 (
δH 1.05, 3H, s), H
3-19 (
δH 0.89, 3H, s), H
3-26 (
δH 1.01, 3H, s), H
3-27 (
δH 1.00, 3H, s), H
3-28 (
δH 0.84, 3H, s), H
3-29 (
δH 0.86, 3H, s), H
3-30 (
δH 1.40, 3H, s), H
3-2′ (
δH 2.02, 3H, s), and H
3-OMe (
δH 3.08, 3H, s). The
13C-NMR (
Table 2) and HSQC spectrum proved the existence of nine methyl groups, as well as an acetyl carbon at C-1′ (
δC 170.8) and three oxygenated carbons at C-3 (
δC 80.7), C-16 (
δC 76.6), and C-25 (
δC 74.4). The signals observed in the
1H NMR and
13C NMR spectra (
Table 2) closely resembled those of
3 except for the partial signals of the C-17 side-chain and the presence of an additional methyl group
3, indicating that
4 also had the same lanostane skeleton as
3. On the basis of the HMBC (
Figure 6) correlations between H
3-26 (
δH 1.01) and CH
3-27 (
δC 25.0), H
3-27 (
δH 1.00) and CH
3-26 (
δC 25.0), H
3-26 (
δH 1.01) and C-25 (
δC 74.4), and H
3-OMe (δ
H 3.08) and CH
3-25 (
δC 74.4), the presence of a methoxy group at C-25 was indicated. The NOE correlations between H
3-30 (
δH 1.40) and H-17 (
δH 2.30) indicated
α-orientations for H-17. The correlations between H
3-18 (
δH 1.05) and H
3-19 (
δH 0.89), and between H-16 (
δH 4.47) and H-20 (
δH 2.86) indicated
β-orientations for CH
3-18, CH
3-19 and H-20. Therefore, the structure of
4 (
Figure 6) was determined to be 3
β-acetoxy-16
α-hydroxylanost-25-methoxyl-8-en-21-oic acid, and it was named ceanphytamic acid B.
All of the isolated compounds were evaluated for cytotoxicity against eleven human cancer cell lines: A549 (adenocarcinomic human alveolar basal epithelial cells, ATCC: CCL-185), Calu-1 (human lung cancer cells, ATCC: HTB-54), HeLa (human cervical cancer cells, ATCC: CCL-2), MDA-MB-231 (human breast invasive ductal carcinoma cells, ATCC: HTB-26), SW579 (human thyroid squamous cell carcinoma cells, ATCC: HTB-107), SK-OV-3 (human ovarian cancer cells, ATCC: HTB-77), T84 (human lung cancer cells, ATCC: CCL-248), Hep G2 (human liver cancer cells, ATCC: HB-8065), Hep 3B2.1-7 (human liver cancer cells, ATCC: HB-8064), KATO III (human gastric carcinoma cells, ATCC: HTB-103), MCF-7 (human breast adenocarcinoma cell, ATCC: HTB-22) by the CCK-8 method. None of compounds exhibited cytotoxicity against T84 cells, Hep G2 cells, Hep 3B2.1-7 cells, KATO III cells, or MCF-7 cells. Compound
1 showed stronger cytotoxicity against HeLa cells than the other tested compounds, and
2–
4 showed different degree cytotoxicity against A549, SK-OV-3 and SW579 cells (
Table 3). Compounds
5–
6,
9–
10 and
12–
14 showed selective inactive cytotoxicity against these cancer cell lines. However, the cytotoxic activities were far less potent compared to those of the positive controls.
3. Experimental Section
3.1. General Information
Optical rotations were obtained with an Autopol VI polarimeter (No. 90079, Rudolph Research Analytical, Hackettstown, NewJersey, USA). IR spectra were recorded with a Perkin Elmer FT-IR spectrometer (Beijing Purkinje General Istrument Co., Ltd., Beijing, People’s Republic of China). One-dimensional and two-dimensional NMR spectra were run on a AVANCE-III instrument (Bruker, Bremen, Germany) operating at 600/400 MHz for 1H, 125/100 MHz for 13C, respectively, with tetramethylsilane as an internal standard. HRESIMS spectra were obtained on a UPLC Premier QTOF spectrometer (Waters, Milford, Massachusetts, USA). Reversed phase medium pressure liquid chromatography (RP-MPLC) was performed on a GRACE system (Grace, Columbia, MD, USA). Preparative HPLC was performed on an 1260 series instrument (Agilent, Santa Clara, CA, USA) equipped with a Capcellpak C18 column (250 × 20 mm, 5 μm, flow rate: 16 mL/min, Shiseido, Tokyo, Japan). Column chromatography were performed with silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China), Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Stockholm, Sweden), and YMC gel ODS-AQ (50 µm, YMC Co., Ltd., Kyoto, Japan). Thin-layer chromatography (TLC) and preparative TLC were carried out on HSGF254 plates (Yantai Jiangyou Silica Gel Development Co., Ltd., Yantai, China).
3.2. Plant Material
The sclerotia of Poria cocos was purchased from Shanghai Kang Qiao Herbal Pieces Co., Ltd. (Shanghai, China), and identified by Professor Hong Xu from Shanghai University of Traditional Chinese Medicine. A voucher specimen (150910) was deposited at Shanghai R&D Center for standardization of Chinese Medicines, Shanghai, China.
3.3. Extraction and Isolation
Dried sclerotia of P. cocos (100 kg) was extracted three times, each time with 95% EtOH (1000 L) to give a crude extract (2300 g), which was suspended in water and successively partitioned with petroleum ether (PE), CH2Cl2 and n-BuOH. The CH2Cl2 extracts (804 g) were loaded into silica gel column chromatography (200–300 mesh) and eluted with a stepwise gradient of PE-EtOAc (100:1 to 1:1) and EtOAc-MeOH (20:1 to 1:1) to obtain 13 fractions (Fr. A-Fr. M) on the basis of their TLC characteristics.
Fraction B (18.4 g) was repeatedly chromatographed on silica gel (PE/EtOAc = 50:1 to 1:1) and Sephadex LH-20 (MeOH) columns to obtain six fractions (Fr. B-1 to Fr. B-6). Fr. B-2 (1.7 g) was subjected to an ODS preparative MPLC column eluted with MeOH-H2O (3:7 to 8:2, v/v), followed by Sephadex LH-20 (MeOH) and preparative HPLC (86% CH3CN in H2O, 16 mL/min) to give harzianone (5, 2.1 mg). Fr. B-3 (2.2 g) was chromatographed by preparative HPLC (82% CH3CN in H2O, 16 mL/min) to yield sorbicillin (6, 4.5 mg), 2′,3′-dihydrosorbicillin (7, 3.7 mg), sohirnone A (8, 6.2 mg). Fr. B-5 (4.0 g) was eluted repeatedly with MeOH-H2O (1:7 to 9:1, v/v) and preparative HPLC to yield sohiracillinone (1, 1.9 mg), 11β-ethoxydaedaleanic acid A (2, 1.6 mg), and daedaleanic acid A (15, 2.0 mg), respectively.
Fr. C (21.0 g) was repeatedly chromatographed on silica gel (PE/EtOAc = 50:1 to 1:1) to obtain seven fractions (Fr. C-1 to Fr. C-7). Fr. C-1 (11.5 g) was subjected to an ODS preparative MPLC column eluted with MeOH-H2O (4:5 to 9:1, v/v), and followed by preparative HPLC to yield pachymic acid (9, 0.6 g), dehypachymic acid (10, 0.5 g), and polyporenic acid C (13, 0.3 g), respectively. Fr. C-2 was also isolated by the same method as Fr.C-1 to yield deyhdrotrametenolic acid (12, 4.5 mg), trametenolic acid (11, 3.2 mg), eburicoic acid (20, 3.7 mg), and 3β-p-hydroxybenzoyl-dehydrotumulosic acid (14, 2.8 mg), respectively.
Fr. D (9.5 g) was repeatedly chromatographed on silica gel (PE/EtOAc = 50:1 to 1:1) to obtain five fractions (Fr. D-1 to Fr. D-5). Fr. D-2 (1.5 g) was subjected to an ODS preparative MPLC column eluted with MeOH-H2O (1:5 to 9:1, v/v), and followed by preparative HPLC (84% CH3CN in H2O, 16 mL/min) to yield ceanphytamic acid A (3, 2.1 mg). Fr. D-4 (2.2 g) was subjected to an ODS preparative MPLC column eluted with MeOH-H2O (1:8 to 9:1, v/v), and followed by preparative HPLC (82% CH3CN in H2O, 16 mL/min) to yield poricoic acid A (17, 3.1 mg), and poricoic acid G (18, 3.7 mg).
Fr. F (12.7 g) was repeatedly chromatographed on silica gel (PE/EtOAc = 50:1 to 1:1) to obtain seven fractions (Fr. F-1 to Fr. F-7). Fr. F-3 (0.3 g) was eluted by HPLC (84% CH3CN in H2O, 16 mL/min) to yield ceanphytamic acid B (4, 2.0 mg). Fr. F-5 (0.8 g) was eluted by HPLC (84% CH3CN in H2O, 16 mL/min) to yield 3β,15α-dihtdroxylanosta-7,9(11),24-trien-21-oic acid (16, 3.1 mg).
Fr. G (7.1 g) was repeatedly chromatographed on silica gel (PE/EtOAc = 50:1 to 1:1) to obtain three fractions (Fr. G-1 to Fr. G-3). Fr. G-1 was eluted by HPLC (78% CH3CN in H2O, 16 mL/min) to yield dehydrosulphurenic acid (19, 3.8 mg).
Fr. H (5.1 g) was repeatedly chromatographed on silica gel (PE/EtOAc = 100:1 to 1:1) to obtain three fractions (Fr. H-1 to Fr. H-3). Fr. H-2 was subjected to an ODS preparative MPLC column eluted with MeOH-H2O (1:9 to 9:1, v/v) to yield tumulosic acid (21, 3.9 mg).
3.4. Spectroscopic Data
Sohiracillinones (
1): Yellow oil;
= −19.2 (
c = 0.033, CHCl
3). IR: 2920, 2852, 1740, 1596, 1413, 1366, 1099, 779, 720, 616 cm
−1.
1H and
13C NMR are shown in
Table 1. Negative HRESIMS: 288.1364 ([M − H]
−, C
17H
19O
4; calcd. 288.1362). More Spectroscropic data can be found in the
Supplementary Materials (Figures S1–S35).
11β-Ethoxydaedaleanic acid A (
2): White amorphous powder;
= +6.7 (
c = 0.1, C
5H
5N). IR: 3395, 2919, 2849, 1645, 1438, 1069, 804, 760, 703 cm
−1.
1H and
13C NMR are shown in
Table 2. Negative HRESIMS: 525.3557 ([M − H]
−, C
33H
49O
5; calcd. 525.3580). More Spectroscropic data can be found in the
Supplementary Materials (Figures S1–S35).
Ceanphytamic acid A (
3): White amorphous powder;
= +16.7 (
c = 0.03, C
5H
5N). IR: 3676, 2988, 2902, 1702, 1635, 1394, 1075, 1066, 1056, 892 cm
−1.
1H and
13C NMR are shown in
Table 2. Negative HRESIMS: 530.3616 ([M − H]
−, C
32H
49O
6; calcd. 530.3607). More Spectroscropic data can be found in the
Supplementary Materials (Figures S1–S35).
Ceanphytamic acid B (
4): White amorphous powder;
= +19.4 (
c = 0.03, C
5H
5N). IR: 3676, 2988, 2902, 1707, 1620, 1407, 1394, 1251, 1066, 1057, 891 cm
−1.
1H and
13C NMR are shown in
Table 2. Positive HRESIMS: 546.3913 ([M + H]
+, C
33H
53O
6; calcd. 546.3920). More Spectroscropic data can be found in the
Supplementary Materials (Figures S1–S35).
3.5. Cell Culture
Nine human cancer cell lines, A549, Calu-1, HeLa, SK-OV-3, T84, Hep G2, Hep 3B2.1-7, KATO III, and MCF-7, were kindly provided by Stem Cell Bank, Chinese Academy of Sciences. Two human cancer cell lines, MDA-MB-231 and SW579, were kindly provided by Professor Hong Xu from Shanghai University of Traditional Chinese Medicine. A Cell Counting Kit-8 was purchased from Shanghai DOJINDO Co., Ltd. (Shanghai, China). Hep G2, Hep 3B2.1-7, HeLa, and MCF-7 were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) and 0.1% (v/v) penicillin-streptomycin in a humidified incubator with 5% CO2 at 37 °C. Calu-1 and SK-OV-3 were maintained in McCoy’s 5A supplemented with 10% (v/v) heat-inactivated FBS and 0.1% (v/v) penicillin-streptomycin in a humidified incubator with 5% CO2 at 37 °C. A549 was maintained in F12K supplemented with 10% (v/v) heat-inactivated FBS and 0.1% (v/v) penicillin-streptomycin in a humidified incubator with 5% CO2 at 37 °C. KATO III, MDA-MB-231, and SW579 were maintained in RPMI 1640 supplemented with 10% (v/v) heat-inactivated FBS and 0.1% (v/v) penicillin-streptomycin in a humidified incubator with 5% CO2 at 37 °C. T84 was maintained in D-MEM/F-12 supplemented with 20% (v/v) heat-inactivated FBS and 0.1% (v/v) penicillin-streptomycin in a humidified incubator with 5% CO2 at 37 °C. All cell culture media and reagents were purchased from Thermo Scientific Hyclone (Logan, UT, USA).
3.6. Cell Assay
The compounds’ cytotoxicity was measured by the CCK-8 method. Compounds were dissolved with dimethylsulfoxide (DMSO). The cells were seeded onto 96-well microplates at a density of 1 × 104 cells per well in 100 μL of medium each. After incubation at 37 °C in a humidified incubator for 24 h, the cells were treated with various concentrations of each compound. After incubation, 10% CCK-8 (10 μL in 90 μL FBS) was added to each well of the plate. After removal from the medium, the cells were incubated at 37 °C for 1 h. Cell viability was calculated as a percentage of viable cells in the compound-treated group vs. the control group by the following equation: Cell viability (%) = [1 − OD (Compound)/OD (Blank)] × 100%.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}