ROS Modulator Molecules with Therapeutic Potential in Cancers Treatments


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
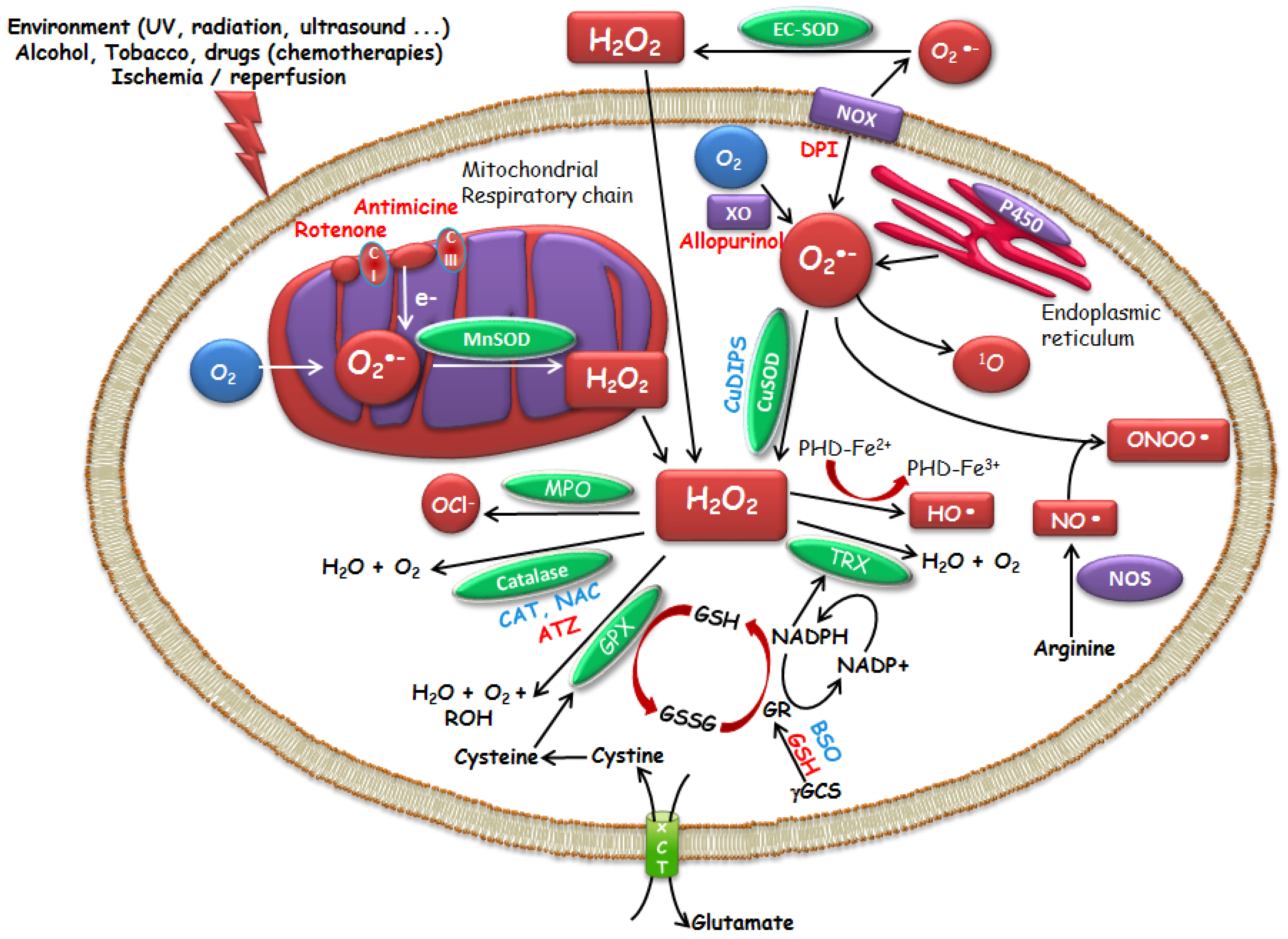
1. Oxidative Stress
1.1. ROS
1.2. Enzymatic and Molecular Controls of ROS Levels
1.3. Redox Equilibrum and Cellular Fate
1.4. Redox Equilibrum and Cancer
2. Therapeutic Potential of Small Molecule Catalysts with ROS Modulating Properties
2.1. Molecules Acting on ROS Production
2.2. Compounds Acting on Nrf2 (Brusatol, DMF)
3. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| 3T3 | mouse fibroblast |
| 5-FU | 5-Fluoro-Uracile |
| A549 | human lung epithelium carcinoma |
| ATZ | aminotriazol |
| BSO | buthionine sulfoximine |
| CAT | catalase |
| CuDIPS | copper (II) (3,5-diisopropylsalicylate) 2 |
| Cu-Zn-SOD | Copper-Zinc-SOD |
| EC-SOD | Zinc-SOD, Zn-SOD |
| GSH | reduced glutathione |
| GSSG | glutathione disulfide, oxidized GSH |
| GST | glutathione S-transferase |
| H2O2 | hydrogen peroxide |
| HO-1 | heme oxygenase 1 |
| HT29 | human colon carcinoma |
| HUVEC | human umbilical vein endothelial cell |
| Keap1 | Kelch-like ECH-associated protein |
| MCF7 | human breast cancer |
| Mn-SOD | Manganese-SOD |
| MnTBAP | Manganese (III) tetrakis (4-benzoic acid) porphyrin |
| NAC | N-acetyl-l-cysteine |
| NOX | NADPH oxidase |
| NQO1 | NAD(P)H dehydrogenase quinone 1 |
| Nrf2 | nuclear erythroid related factor 2 |
| O2•- | superoxide anion |
| •OH | hydroxyl radical |
| ONOOH | nitroperoxide, peroxynitrous acid |
| ONOO- | peroxynitrite |
| NOS | NO synthase |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| TRX | thioredoxin |
| TrxR1 | thioredoxin reductase 1 |
| Xct | cysteine/glutamate exchanger |
References
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.-D.V.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol.-Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.-M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.; Go, Y.-M.; Jones, D.P. Nonequilibrium thermodynamics of thiol/disulfide redox systems: A perspective on redox systems biology. Free Radic. Biol. Med. 2008, 44, 921–937. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. The Utility of Superoxide Dismutase in Studying Free Radical Reactions I. Radicals generated by interaction of sulfite, dimethyl sulfoxide and oxygen. J. Biol. Chem. 1969, 244, 6056–6063. [Google Scholar] [PubMed]
- Nicco, C.; Laurent, A.; Chereau, C.; Weill, B.; Batteux, F. Differential modulation of normal and tumor cell proliferation by reactive oxygen species. Biomed. Pharmacother. 2005, 59, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. Superoxide Dismutase in Redox Biology: The Roles of Superoxide and Hydrogen Peroxide. Available online: http://www.eurekaselect.com/74145/article (accessed on 17 November 2017).
- Antonyuk, S.V.; Strange, R.W.; Marklund, S.L.; Hasnain, S.S. The Structure of Human Extracellular Copper–Zinc Superoxide Dismutase at 1.7 Å Resolution: Insights into Heparin and Collagen Binding. J. Mol. Biol. 2009, 388, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Luo, J.L. Glutathione therapy: From prodrugs to genes. Semin. Liver Dis. 1998, 18, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.Q.; Aw, T.Y.; Jones, D.P. Glutathione-dependent protection against oxidative injury. Pharmacol. Ther. 1990, 47, 61–71. [Google Scholar] [CrossRef]
- Korkina, L.G.; Luca, C.D.; Pastore, S.; Kostyuk, V.A. Plant Polyphenols and Tumors: From Mechanisms to Therapies, Prevention, and Protection Against Toxicity of Anti-Cancer Treatments. Available online: http://www.eurekaselect.com/70155/article (accessed on 28 November 2017).
- Mao, X.-Y.; Jin, M.-Z.; Chen, J.-F.; Zhou, H.-H.; Jin, W.-L. Live or let die: Neuroprotective and anti-cancer effects of nutraceutical antioxidants. Pharmacol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Villeneuve, N.F.; Sun, Z.; Wong, P.K.; Zhang, D.D. Dual roles of Nrf2 in cancer. Pharmacol. Res. 2008, 58, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Homma, S.; Ishii, Y.; Morishima, Y.; Yamadori, T.; Matsuno, Y.; Haraguchi, N.; Kikuchi, N.; Satoh, H.; Sakamoto, T.; Hizawa, N.; et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3423–3432. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Boldin-Adamsky, S.; Thimmulappa, R.K.; Rath, S.K.; Ashush, H.; Coulter, J.; Blackford, A.; Goodman, S.N.; Bunz, F.; Watson, W.H.; et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008, 68, 7975–7984. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Dayal, R.; Singh, A.; Pandey, A.; Mishra, K.P. Reactive oxygen species as mediator of tumor radiosensitivity. J. Cancer Res. Ther. 2014, 10, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of reactive oxygen species in biological processes. Klin. Wochenschr. 1991, 69, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.A. Oxidative Stress, Antioxidant Defenses, and Damage Removal, Repair, and Replacement Systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cui, Y.; Niedernhofer, L.J.; Wang, Y. Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage. Chem. Res. Toxicol. 2016, 29, 2008–2039. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Bellon, S.; Berger, M.; Bourdat, A.-G.; Douki, T.; Duarte, V.; Frelon, S.; Gasparutto, D.; Muller, E.; Ravanat, J.-L.; et al. Recent aspects of oxidative DNA damage: Guanine lesions, measurement and substrate specificity of DNA repair glycosylases. Biol. Chem. 2002, 383, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.J.; Skouras, C.; Haugk, B.; Charnley, R.M. Keap1-Nrf2 signalling in pancreatic cancer. Int. J. Biochem. Cell Biol. 2015, 65, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Gañán-Gómez, I.; Wei, Y.; Yang, H.; Boyano-Adánez, M.C.; García-Manero, G. Oncogenic functions of the transcription factor Nrf2. Free Radic. Biol. Med. 2013, 65, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.-L.; Schreiber, S.; Schäfer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.-Q.; Yi, Y.W.; Kang, H.J.; Hong, Y.B.; Tang, W.; Wang, A.; Seong, Y.-S.; Bae, I. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2014, 44, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Hileman, E.O.; Liu, J.; Albitar, M.; Keating, M.J.; Huang, P. Intrinsic oxidative stress in cancer cells: A biochemical basis for therapeutic selectivity. Cancer Chemother. Pharmacol. 2004, 53, 209–219. [Google Scholar] [CrossRef] [PubMed]
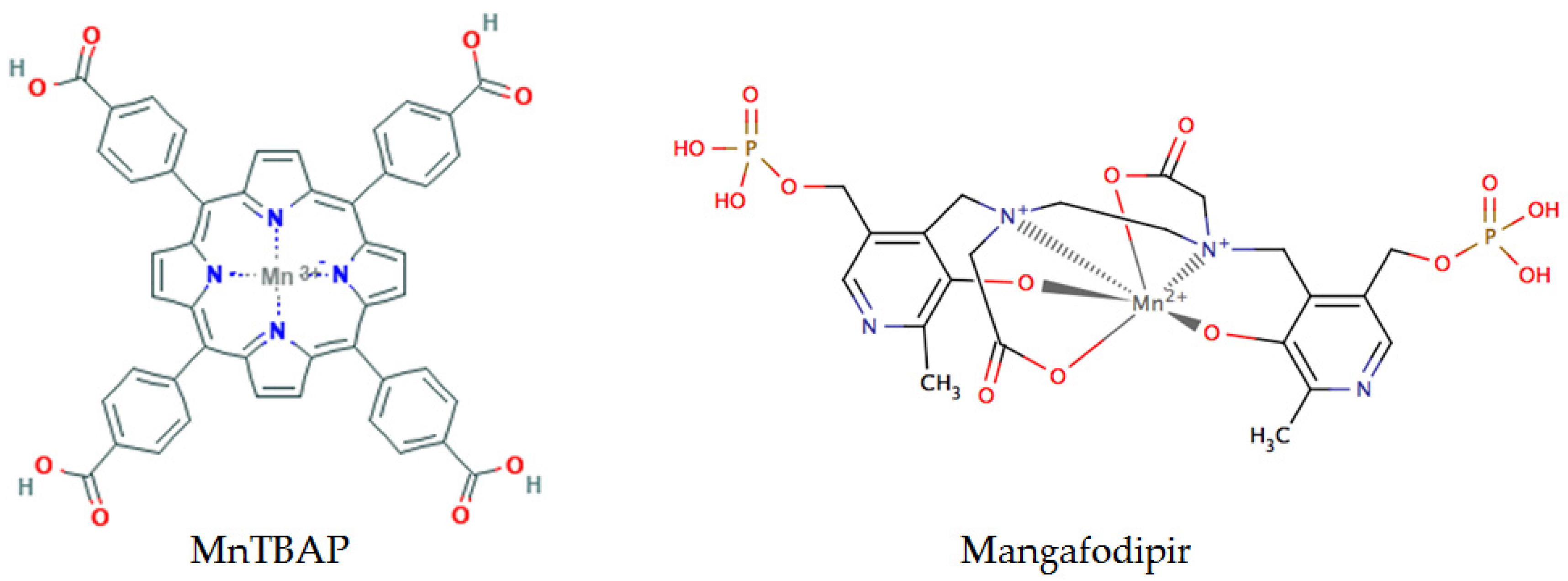
- Asplund, A.; Grant, D.; Karlsson, J.O. Mangafodipir (MnDPDP)-and MnCl2-induced endothelium-dependent relaxation in bovine mesenteric arteries. J. Pharmacol. Exp. Ther. 1994, 271, 609–614. [Google Scholar] [PubMed]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St. Clair, D.; Batinic-Haberle, I. Manganese superoxide dismutase, MnSOD and its mimics. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2012, 1822, 794–814. [Google Scholar] [CrossRef] [PubMed]
- Batinić-Haberle, I.; Rebouças, J.S.; Spasojević, I. Superoxide Dismutase Mimics: Chemistry, Pharmacology, and Therapeutic Potential. Antioxid. Redox Signal. 2010, 13, 877–918. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.O.G.; Ignarro, L.J.; Lundström, I.; Jynge, P.; Almén, T. Calmangafodipir [Ca4Mn(DPDP)5], mangafodipir (MnDPDP) and MnPLED with special reference to their SOD mimetic and therapeutic properties. Drug Discov. Today 2015, 20, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Varbiro, G.; Veres, B.; Gallyas, F.; Sumegi, B. Direct effect of Taxol on free radical formation and mitochondrial permeability transition. Free Radic. Biol. Med. 2001, 31, 548–558. [Google Scholar] [CrossRef]
- Baker, D. Oxaliplatin: A New Drug for the Treatment of Metastatic Carcinoma of the Colon or Rectum. Available online: Https://www.ncbi.nlm.nih.gov/pubmed/12684591 (accessed on 1 December 2017).
- Hwang, P.M.; Bunz, F.; Yu, J.; Rago, C.; Chan, T.A.; Murphy, M.P.; Kelso, G.F.; Smith, R.A.J.; Kinzler, K.W.; Vogelstein, B. Ferredoxin reductase affects p53-dependent, 5-fluorouracil–induced apoptosis in colorectal cancer cells. Nat. Med. 2001, 7, 1111. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.; Nicco, C.; Chéreau, C.; Goulvestre, C.; Alexandre, J.; Alves, A.; Lévy, E.; Goldwasser, F.; Panis, Y.; Soubrane, O.; et al. Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 2005, 65, 948–956. [Google Scholar] [PubMed]
- Alexandre, J.; Nicco, C.; Chéreau, C.; Laurent, A.; Weill, B.; Goldwasser, F.; Batteux, F. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J. Natl. Cancer Inst. 2006, 98, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Vorotnikova, E.; Rosenthal, R.A.; Tries, M.; Doctrow, S.R.; Braunhut, S.J. Novel Synthetic SOD/Catalase Mimetics Can Mitigate Capillary Endothelial Cell Apoptosis Caused by Ionizing Radiation. Radiat. Res. 2010, 173, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Coriat, R.; Leconte, M.; Kavian, N.; Bedda, S.; Nicco, C.; Chereau, C.; Goulvestre, C.; Weill, B.; Laurent, A.; Batteux, F. Mangafodipir protects against hepatic ischemia-reperfusion injury in mice. PLoS ONE 2011, 6, e27005. [Google Scholar] [CrossRef] [PubMed]
- Bedda, S.; Laurent, A.; Conti, F.; Chéreau, C.; Tran, A.; Tran-Van Nhieu, J.; Jaffray, P.; Soubrane, O.; Goulvestre, C.; Calmus, Y.; et al. Mangafodipir prevents liver injury induced by acetaminophen in the mouse. J. Hepatol. 2003, 39, 765–772. [Google Scholar] [CrossRef]
- McDonald, M. A Superoxide Dismutase Mimetic with Catalase Activity (EUK-8) Reduces the Organ Injury in Endotoxic Shock. Available online: http://www.sciencedirect.com.frodon.univ-paris5.fr/science/article/pii/S0014299903015383?via%3Dihub (accessed on 28 November 2017).
- Rong, Y.; Doctrow, S.R.; Tocco, G.; Baudry, M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc. Natl. Acad. Sci. USA 1999, 96, 9897–9902. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, S.; Shang, D.-J.; Wang, X.-L.; You, Z.-L.; Li, H.-B. Antihyperglycemic and neuroprotective effects of one novel Cu–Zn SOD mimetic. Bioorg. Med. Chem. Lett. 2011, 21, 4320–4324. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.; Nicco, C.; Tran Van Nhieu, J.; Borderie, D.; Chéreau, C.; Conti, F.; Jaffray, P.; Soubrane, O.; Calmus, Y.; Weill, B.; et al. Pivotal role of superoxide anion and beneficial effect of antioxidant molecules in murine steatohepatitis. Hepatology 2004, 39, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Serena, C.; Calvo, E.; Clares, M.P.; Diaz, M.L.; Chicote, J.U.; Beltrán-Debon, R.; Fontova, R.; Rodriguez, A.; García-España, E.; García-España, A. Significant In Vivo Anti-Inflammatory Activity of Pytren4Q-Mn a Superoxide Dismutase 2 (SOD2) Mimetic Scorpiand-Like Mn (II) Complex. PLoS ONE 2015, 10, e0119102. [Google Scholar] [CrossRef] [PubMed]
- Salvemini, D.; Muscoli, C.; Riley, D.P.; Cuzzocrea, S. Superoxide Dismutase Mimetics. Pulm. Pharmacol. Ther. 2002, 15, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Tovmasyan, A.; Reboucas, J.S.; Benov, L. Simple Biological Systems for Assessing the Activity of Superoxide Dismutase Mimics. Antioxid. Redox Signal. 2013, 20, 2416–2436. [Google Scholar] [CrossRef] [PubMed]
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.R.; Morrison, G.; Dolan, M.E.; Fleming, G.F. Chemotherapy-induced peripheral neuropathy: Current status and progress. Gynecol. Oncol. 2016, 140, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-T.; Wang, S.-J. Pyridoxine inhibits depolarization-evoked glutamate release in nerve terminals from rat cerebral cortex: A possible neuroprotective mechanism? J. Pharmacol. Exp. Ther. 2009, 331, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Coriat, R.; Alexandre, J.; Nicco, C.; Quinquis, L.; Benoit, E.; Chéreau, C.; Lemaréchal, H.; Mir, O.; Borderie, D.; Tréluyer, J.-M.; et al. Treatment of oxaliplatin-induced peripheral neuropathy by intravenous mangafodipir. J. Clin. Investig. 2014, 124, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Son, A.-R.; Ahn, J.; Song, J.-Y. Niclosamide enhances ROS-mediated cell death through c-Jun activation. Biomed. Pharmacother. 2014, 68, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, P.-K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Cerles, O.; Benoit, E.; Chéreau, C.; Chouzenoux, S.; Morin, F.; Guillaumot, M.-A.; Coriat, R.; Kavian, N.; Loussier, T.; Santulli, P.; et al. Niclosamide Inhibits Oxaliplatin Neurotoxicity while Improving Colorectal Cancer Therapeutic Response. Mol. Cancer Ther. 2017, 16, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Arteel, G.E.; Kanda, T.; Engman, L.; Sies, H. Water-Soluble Organotellurium Compounds: Catalytic Protection against Peroxynitrite and Release of Zinc from Metallothionein. Chem. Res. Toxicol. 2000, 13, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Marut, W.K.; Kavian, N.; Servettaz, A.; Nicco, C.; Ba, L.A.; Doering, M.; Chéreau, C.; Jacob, C.; Weill, B.; Batteux, F. The Organotelluride Catalyst (PHTE)2NQ Prevents HOCl-Induced Systemic Sclerosis in Mouse. J. Investig. Dermatol. 2012, 132, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Coriat, R.; Marut, W.; Leconte, M.; Ba, L.B.; Vienne, A.; Chéreau, C.; Alexandre, J.; Weill, B.; Doering, M.; Jacob, C.; et al. The organotelluride catalyst LAB027 prevents colon cancer growth in the mice. Cell Death Dis. 2011, 2, e191. [Google Scholar] [CrossRef] [PubMed]
- Mecklenburg, S.; Shaaban, S.; Ba, L.A.; Burkholz, T.; Schneider, T.; Diesel, B.; Kiemer, A.K.; Röseler, A.; Becker, K.; Reichrath, J.; et al. Exploring synthetic avenues for the effective synthesis of selenium- and tellurium-containing multifunctional redox agents. Org. Biomol. Chem. 2009, 7, 4753–4762. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Liu, L.; Xue, H.; Li, L.; Han, C.; Wang, L.; Chen, Z.; Liu, G. Chemical library and structure-activity relationships of 11-demethyl-12-oxo calanolide A analogues as anti-HIV-1 agents. J. Med. Chem. 2008, 51, 1432–1446. [Google Scholar] [CrossRef] [PubMed]
- Kidane, A.G.; Salacinski, H.; Tiwari, A.; Bruckdorfer, K.R.; Seifalian, A.M. Anticoagulant and antiplatelet agents: Their clinical and device application(s) together with usages to engineer surfaces. Biomacromolecules 2004, 5, 798–813. [Google Scholar] [CrossRef] [PubMed]
- Whang, W.K.; Park, H.S.; Ham, I.; Oh, M.; Namkoong, H.; Kim, H.K.; Hwang, D.W.; Hur, S.Y.; Kim, T.E.; Park, Y.G.; et al. Natural compounds, fraxin and chemicals structurally related to fraxin protect cells from oxidative stress. Exp. Mol. Med. 2005, 37, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Kumar, A.; Kumar, M.; Sarkar, J.; Sinha, S. Synthesis and in vitro evaluation of novel coumarin–chalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 7205–7211. [Google Scholar] [CrossRef] [PubMed]
- Jamier, V.; Marut, W.; Valente, S.; Chereau, C.; Chouzenoux, S.; Nicco, C.; Lemarechal, H.; Weill, B.; Kirsch, G.; Batteux, F.; et al. Chalcone-Coumarin Derivatives as Potential Anti-Cancer Drugs: An in Vitro and in Vivo Investigation. Available online: http://www.eurekaselect.com/119149/article (accessed on 15 November 2017).
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
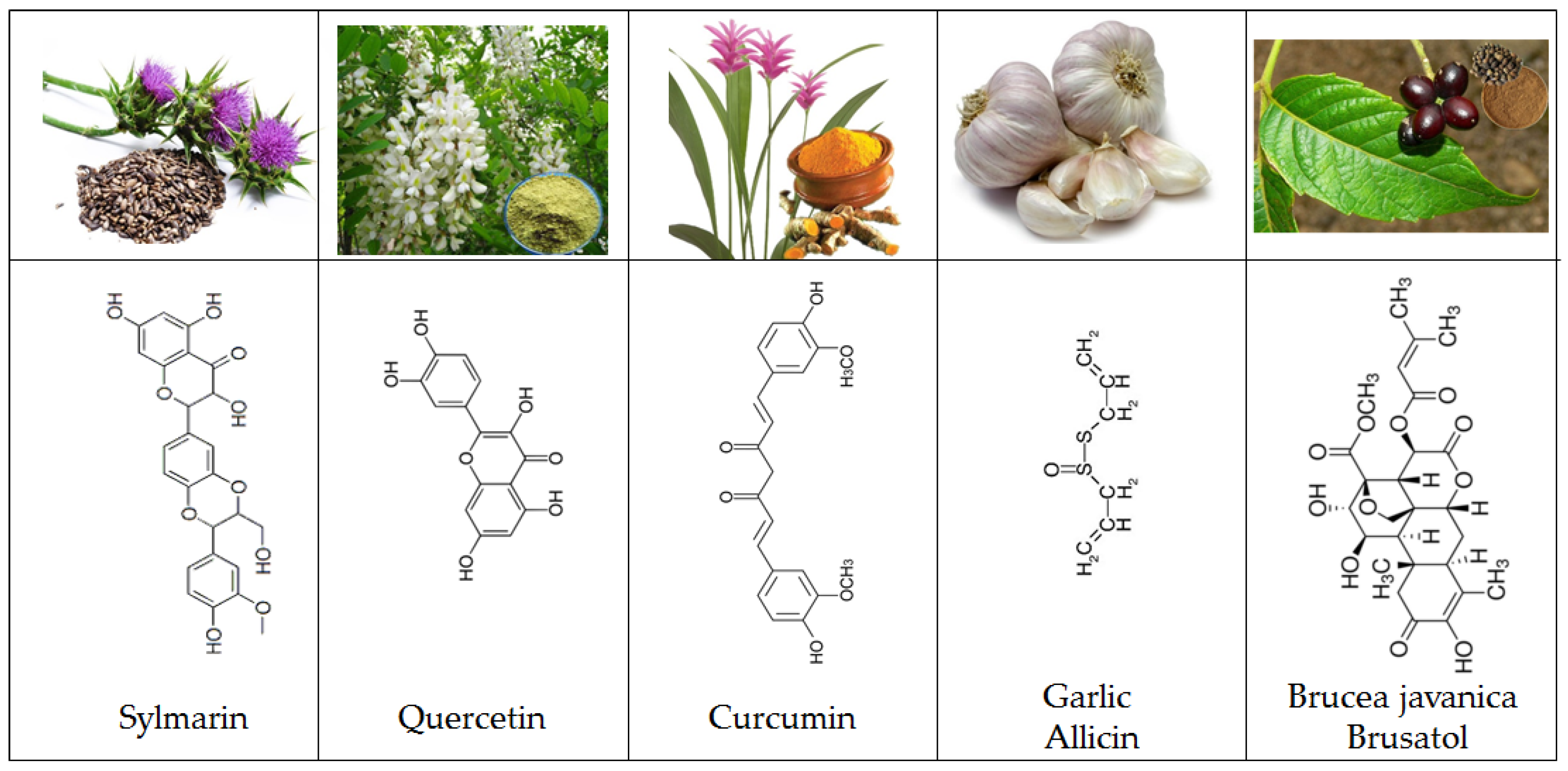
- Dajas, F. Life or death: Neuroprotective and anticancer effects of quercetin. J. Ethnopharmacol. 2012, 143, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, J.; Ji, C. Quercetin: A potential drug to reverse multidrug resistance. Life Sci. 2010, 87, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Shen, C.; Li, C.; Liu, Y.; Gao, D.; Shi, C.; Peng, F.; Liu, Z.; Zhao, B.; Zheng, Z.; et al. Inhibition of autophagy induced by quercetin at a late stage enhances cytotoxic effects on glioma cells. Tumor Biol. 2016, 37, 3549–3560. [Google Scholar] [CrossRef] [PubMed]
- Dutta, K.R.; Banerjee, S.; Mitra, A. Medicinal plants of west Midnapore, India: Emphasis on phytochemical containment having role on oral cancer. Int. J. Phytopharmacol. 2012, 3, 198–208. [Google Scholar] [CrossRef]
- Hu, X.-T.; Ding, C.; Zhou, N.; Xu, C. Quercetin protects gastric epithelial cell from oxidative damage in vitro and in vivo. Eur. J. Pharmacol. 2015, 754, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, K. Quercetin induces cytochrome-c release and ROS accumulation to promote apoptosis and arrest the cell cycle in G2/M, in cervical carcinoma: Signal cascade and drug-DNA interaction. Cell Prolif. 2013, 46, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Borska, S.; Chmielewska, M.; Wysocka, T.; Drag-Zalesinska, M.; Zabel, M.; Dziegiel, P. In vitro effect of quercetin on human gastric carcinoma: Targeting cancer cells death and MDR. Food Chem. Toxicol. 2012, 50, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Kunnumakkara, A.B.; Anand, P.; Aggarwal, B.B. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008, 269, 199–225. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y. Involvement of ROS in Curcumin-Induced Autophagic Cell Death. Available online: http://www.kjpp.net/journal/viewJournal.html?year=2011&vol=15&page=1 (accessed on 17 November 2017).
- Lao, C.D.; Ruffin, M.T.; Normolle, D.; Heath, D.D.; Murray, S.I.; Bailey, J.M.; Boggs, M.E.; Crowell, J.; Rock, C.L.; Brenner, D.E. Dose escalation of a curcuminoid formulation. BMC Complement. Altern. Med. 2006, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, J.; Ankola, D.D.; Beniwal, V.; Singh, D.; Kumar, M.N. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur. J. Pharm. Sci. 2009, 37, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin Inhibits Formation of Amyloid β Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in Vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Gruhlke, M.C.H.; Slusarenko, A.J. The biology of reactive sulfur species (RSS). Plant Physiol. Biochem. 2012, 59, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Choi, Y.J.; Cha, S.H.; Choi, C.H.; Cho, W.H. Allicin inhibits cell growth and induces apoptosis in U87MG human glioblastoma cells through an ERK-dependent pathway. Oncol. Rep. 2012, 28, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Goldstein, D.; Krishnan, A.V.; Lin, C.S.-Y.; Friedlander, M.L.; Cassidy, J.; Koltzenburg, M.; Kiernan, M.C. Chemotherapy-induced peripheral neurotoxicity: A critical analysis. CA. Cancer J. Clin. 2013, 63, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Gao, L.; Pan, J.; Wang, X. Study on the inhibitory effect of allicin on human gastric cancer cell line sgc-7901 and its mechanism. Afr. J. Tradit. Complement. Altern. Med. 2014, 11, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Cho, S.-J.; Kwon, H.; Lee, K.-R.; Rhee, D.-K.; Pyo, S. Caspase-independent cell death by allicin in human epithelial carcinoma cells: Involvement of PKA. Cancer Lett. 2005, 224, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Borlinghaus, J.; Albrecht, F.; Gruhlke, M.C.H.; Nwachukwu, I.D.; Slusarenko, A.J. Allicin: Chemistry and Biological Properties. Molecules 2014, 19, 12591–12618. [Google Scholar] [CrossRef] [PubMed]
- Haghi, A.; Azimi, H.; Rahimi, R. A Comprehensive Review on Pharmacotherapeutics of Three Phytochemicals, Curcumin, Quercetin, and Allicin, in the Treatment of Gastric Cancer. J. Gastrointest. Cancer 2017, 48, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Lau, F.Y.; Chui, C.H.; Gambari, R.; Kok, S.H.L.; Kan, K.L.; Cheng, G.Y.M.; Wong, R.S.M.; Teo, I.T.N.; Cheng, C.H.; Wan, T.S.K.; et al. Antiproliferative and apoptosis-inducing activity of Brucea javanicaitalic extract on human carcinoma cells. Int. J. Mol. Med. 2005, 16, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-H.; Jin, H.-Z.; Zhang, W.-D.; Yan, S.-K.; Shen, Y.-H. Chemical constituents of plants from the genus Brucea. Chem. Biodivers. 2009, 6, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Xie, J.; Xu, S.; Lv, H.; Lin, M.; Yuan, S.; Bai, J.; Hou, Q.; Yu, S. Novel nitric oxide-releasing derivatives of brusatol as anti-inflammatory agents: Design, synthesis, biological evaluation, and nitric oxide release studies. J. Med. Chem. 2014, 57, 7600–7612. [Google Scholar] [CrossRef] [PubMed]
- Turpaev, K.; Welsh, N. Brusatol inhibits the response of cultured beta-cells to pro-inflammatory cytokines in vitro. Biochem. Biophys. Res. Commun. 2015, 460, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Feng, X.; Ma, D.; Yang, J.; Jiang, H.; Zhang, Y.; He, W. Brusatol isolated from Brucea javanica (L.) Merr. induces apoptotic death of insect cell lines. Pestic. Biochem. Physiol. 2013, 107, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, B.; Shi, Q.; Wang, X.; Wang, D.; Zhu, L. Brusatol inhibits HIF-1 signaling pathway and suppresses glucose uptake under hypoxic conditions in HCT116 cells. Sci. Rep. 2016, 6, 39123. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.; Tian, W.; La Clair, J.J.; Tan, A.-C.; Ooi, A.; Chapman, E.; Zhang, D.D. Brusatol overcomes chemoresistance through inhibition of protein translation. Mol. Carcinog. 2017, 56, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, Q.; Wang, Y.; Du, L.; Xu, C.; Liu, Q. Brusatol Enhances the Radiosensitivity of A549 Cells by Promoting ROS Production and Enhancing DNA Damage. Int. J. Mol. Sci. 2016, 17, 977. [Google Scholar] [CrossRef] [PubMed]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.-Q.; Lin, Z.-X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Lai, Z.-Q.; Leung, A.W.N.; Leung, P.S.; Li, Z.-S.; Lin, Z.-X. Exploring brusatol as a new anti-pancreatic cancer adjuvant: Biological evaluation and mechanistic studies. Oncotarget 2017. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Ye, W.; Huang, C.; Lou, B.; Zhang, J.; Yu, D.; Huang, X.; Chen, B.; Zhou, M. Brusatol inhibits growth and induces apoptosis in pancreatic cancer cells via JNK/p38 MAPK/NF-κb/Stat3/Bcl-2 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 487, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Lau, S.T.; Leung, P.S.; Che, C.T.; Lin, Z.X. Seven quassinoids from Fructus Bruceae with cytotoxic effects on pancreatic adenocarcinoma cell lines. Phytother. Res. PTR 2011, 25, 1796–1800. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.T.; Lin, Z.-X.; Zhao, M.; Leung, P.S. Brucea javanica fruit induces cytotoxicity and apoptosis in pancreatic adenocarcinoma cell lines. Phytother. Res. PTR 2008, 22, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.Q.; Huang, X.E.; Wu, X.Y.; Liu, J.; Wang, L.; Tang, J.H. Safety of Brucea javanica and cantharidin combined with chemotherapy for treatment of NSCLC patients. Asian Pac. J. Cancer Prev. 2014, 15, 8603–8605. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 Mutations and Nrf2 Pathway Activation in Epithelial Ovarian Cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Gold, R.; Miller, D.H.; Macmanus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Yang, M.; et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet Lond. Engl. 2008, 372, 1463–1472. [Google Scholar] [CrossRef]
- Saidu, N.E.B.; Noé, G.; Cerles, O.; Cabel, L.; Kavian-Tessler, N.; Chouzenoux, S.; Bahuaud, M.; Chéreau, C.; Nicco, C.; Leroy, K.; et al. Dimethyl Fumarate Controls the NRF2/DJ-1 Axis in Cancer Cells: Therapeutic Applications. Mol. Cancer Ther. 2017, 16, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Johnson, D.A.; Johnson, J.A. Keap1-Nrf2 activation in the presence and absence of DJ-1. Eur. J. Neurosci. 2010, 31, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Zhao, Y.; Ma, C.-Y.; Xu, X.-M.; Zhang, Y.-Q.; Wang, C.-G.; Jin, J.; Shen, X.; Gao, J.-L.; Li, N.; et al. Dimethyl fumarate induces necroptosis in colon cancer cells through GSH depletion/ROS increase/MAPKs activation pathway. Br. J. Pharmacol. 2015, 172, 3929–3943. [Google Scholar] [CrossRef] [PubMed]
- Loewe, R.; Valero, T.; Kremling, S.; Pratscher, B.; Kunstfeld, R.; Pehamberger, H.; Petzelbauer, P. Dimethylfumarate impairs melanoma growth and metastasis. Cancer Res. 2006, 66, 11888–11896. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Soga, T.; Pollard, P.J.; Adam, J. The emerging role of fumarate as an oncometabolite. Front. Oncol. 2012, 2, 85. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.F.; Kirsner, R.S. Mechanisms of Drug Action: The Potential of Dimethylfumarate for the Treatment of Neoplasms. J. Investig. Dermatol. 2011, 131, 1181. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Cardaci, S.; Jerby, L.; MacKenzie, E.D.; Sciacovelli, M.; Johnson, T.I.; Gaude, E.; King, A.; Leach, J.D.G.; Edrada-Ebel, R.; et al. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.C.U.; Listopad, J.J.; Rentzsch, C.U.; Igney, F.H.; von Bonin, A.; Hennekes, H.H.; Asadullah, K.; Docke, W.-D.F. Dimethylfumarate induces immunosuppression via glutathione depletion and subsequent induction of heme oxygenase 1. J. Investig. Dermatol. 2007, 127, 835–845. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicco, C.; Batteux, F. ROS Modulator Molecules with Therapeutic Potential in Cancers Treatments. Molecules 2018, 23, 84. https://doi.org/10.3390/molecules23010084
Nicco C, Batteux F. ROS Modulator Molecules with Therapeutic Potential in Cancers Treatments. Molecules. 2018; 23(1):84. https://doi.org/10.3390/molecules23010084
Chicago/Turabian StyleNicco, Carole, and Frédéric Batteux. 2018. "ROS Modulator Molecules with Therapeutic Potential in Cancers Treatments" Molecules 23, no. 1: 84. https://doi.org/10.3390/molecules23010084
APA StyleNicco, C., & Batteux, F. (2018). ROS Modulator Molecules with Therapeutic Potential in Cancers Treatments. Molecules, 23(1), 84. https://doi.org/10.3390/molecules23010084