Hot Spots for Protein Partnerships at the Surface of Cholinesterases and Related α/β Hydrolase Fold Proteins or Domains—A Structural Perspective



Abstract
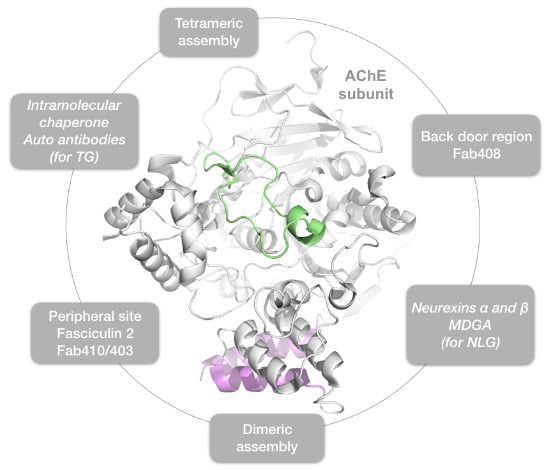
{kind=link}
{kind=link}
{kind=link}
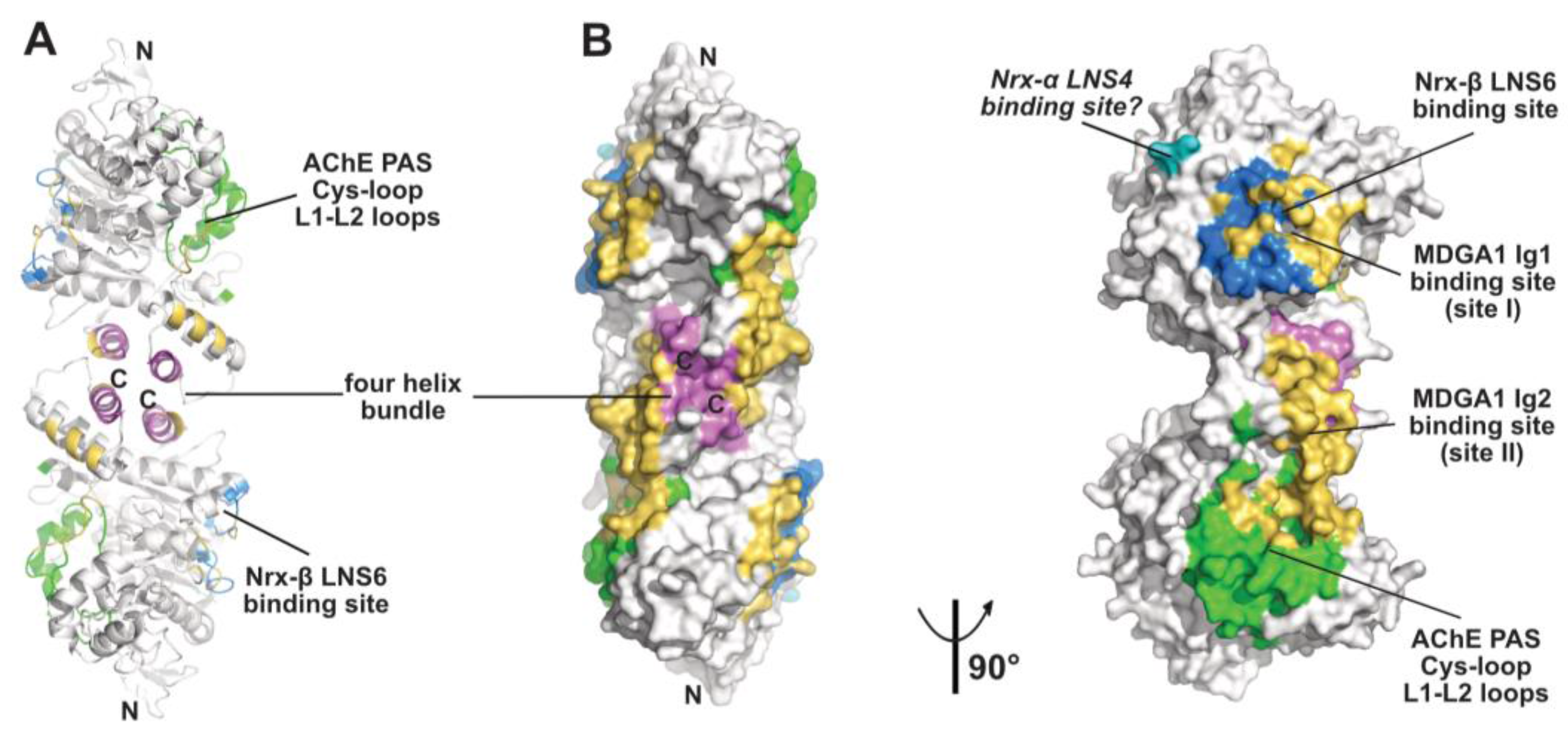{kind=link}
1. Introduction
2. Results
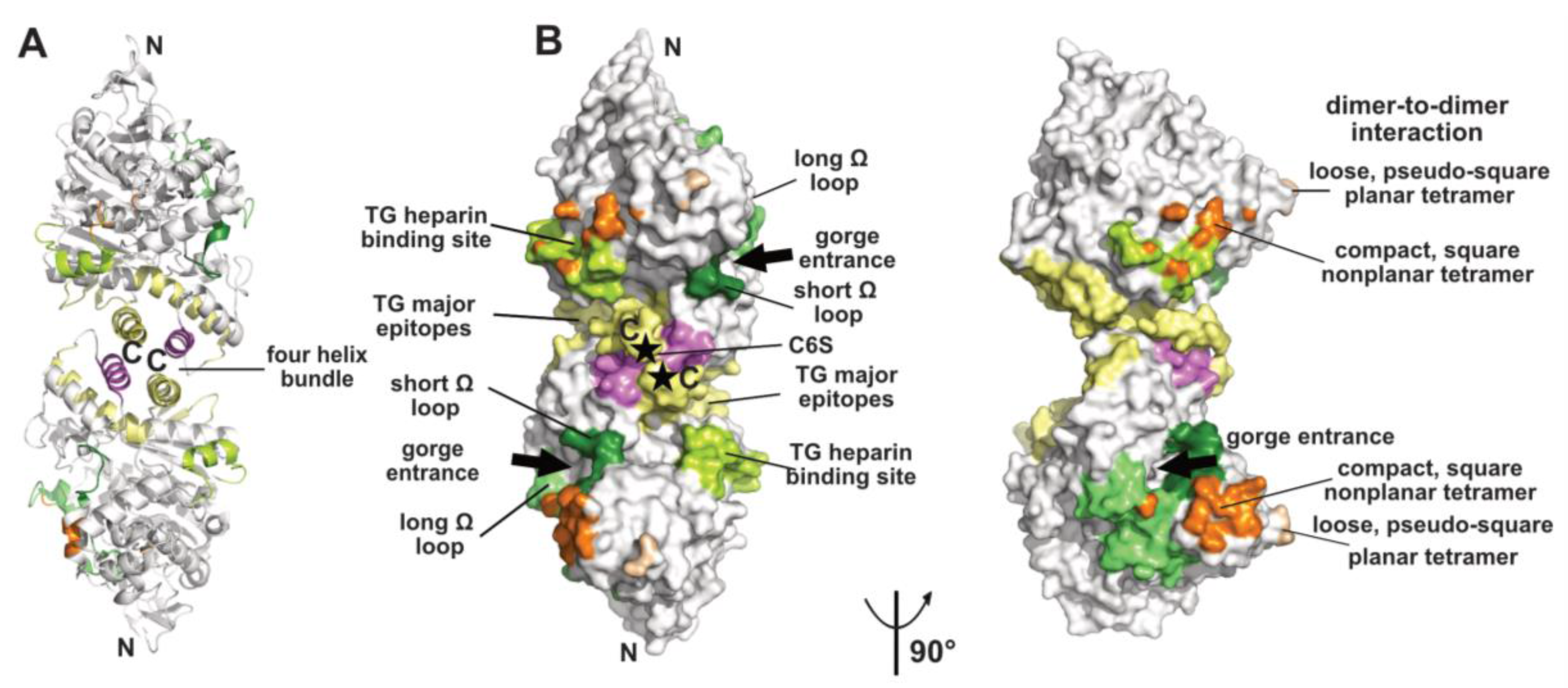
2.1. The Dimer and Tetramer Interfaces
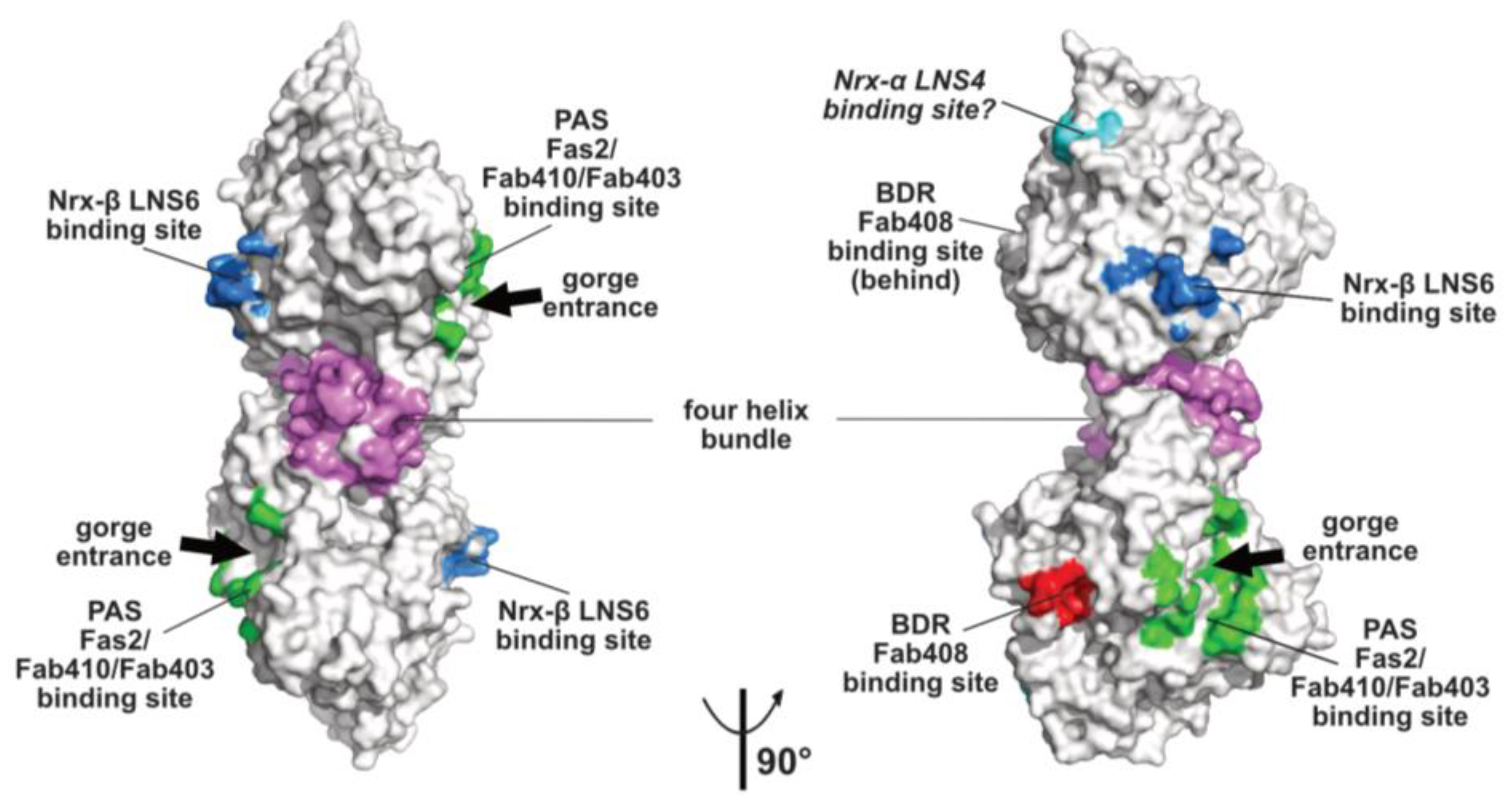
2.2. The Peripheral Anionic Site
2.3. The Neurexin Binding Sites
2.4. The MDGA Binding Interfaces
2.5. The Back-Door Region
3. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AChE | acetylcholinesterase (BfAChE, from Bungarus fasciatus venom; EeAChE; from Electrophorus electricus electroplax; mAChE, recombinant from mouse; TcAChE, from Torpedo californica electroplax); BChE, butyrylcholinesterase (hBChE, recombinant from human) |
| CDR | complementary determining region |
| ChE | cholinesterase |
| EGF | epidermal growth factor |
| GPI | glycosyl-phosphatidylinositol |
| LNS | laminin-neurexin-sex hormone binding protein (domain) |
| MDGA | meprin, A5 protein, and receptor protein-tyrosine phosphatase mu [MAM] domain-containing GPI-anchored (protein) |
| NLG | neuroligin |
| PRAD | proline-rich attachment domain |
| PRiMA | proline-rich membrane-anchoring (domain) |
| WAT | tryptophan amphiphilic tetramerization (domain) |
References
- Massoulié, J.; Sussman, J.; Bon, S.; Silman, I. Structure and functions of acetylcholinesterase and butyrylcholinesterase. Prog. Brain Res. 1993, 98, 139–146. [Google Scholar] [PubMed]
- Taylor, P.; Radić, Z. The cholinesterases: From genes to proteins. Annu. Rev. Pharmacol. Toxicol. 1994, 34, 281–320. [Google Scholar] [CrossRef] [PubMed]
- Silman, I.; Sussman, J.L. Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr. Opin. Pharmacol. 2005, 5, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P. Anticholinesterase agents. In Goodman and Gilman’s the Pharmacological Basis of Therapeutics; Mc-Graw-Hill: New York, NY, USA, 2011; pp. 239–254. [Google Scholar]
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J. The alpha/beta hydrolase fold. Protein Eng. 1992, 5, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Lenfant, N.; Hotelier, T.; Velluet, E.; Bourne, Y.; Marchot, P.; Chatonnet, A. ESTHER, the database of the α/β-hydrolase fold superfamily of proteins: Tools to explore diversity of functions. Nucleic Acids Res. 2013, 41, D423–D429. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; De Jaco, A.; Comoletti, D.; Miller, M.; Camp, S. Cholinesterase confabs and cousins: Approaching forty years. Chem. Biol. Interact. 2013, 203, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Lenfant, N.; Hotelier, T.; Bourne, Y.; Marchot, P.; Chatonnet, A. Tracking the origin and divergence of cholinesterases and neuroligins: The evolution of synaptic proteins. J. Mol. Neurosci. 2014, 53, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Wang, X.; Di Jeso, B.; Arvan, P. The cholinesterase-like domain, essential in thyroglobulin trafficking for thyroid hormone synthesis, is required for protein dimerization. J. Biol. Chem. 2009, 284, 12752–12761. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Lappi, S. Interaction of fluorescence probes with acetylcholinesterase. The site and specificity of propidium binding. Biochemistry 1975, 14, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Radić, Z.; Pickering, N.A.; Vellom, D.C.; Camp, S.; Taylor, P. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 1993, 32, 12074–12084. [Google Scholar] [CrossRef] [PubMed]
- Haviv, H.; Wong, D.M.; Silman, I.; Sussman, J.L. Bivalent ligands derived from Huperzine A as acetylcholinesterase inhibitors. Curr. Top. Med. Chem. 2007, 7, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.G.; Green, L.G.; Grynszpan, F.; Radić, Z.; Carlier, P.R.; Taylor, P.; Finn, M.G.; Sharpless, K.B. Click chemistry in situ: Acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew. Chem. Int. Ed. 2002, 41, 1053–1057. [Google Scholar] [CrossRef]
- Paraoanu, L.E.; Layer, P.G. Acetylcholinesterase in cell adhesion, neurite growth and network formation. FEBS J. 2008, 275, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cheng, S.; Sussman, J.L.; Silman, I.; Jiang, H. Computational studies on acetylcholinesterases. Molecules 2017, 22, 1324. [Google Scholar] [CrossRef] [PubMed]
- Massoulié, J. The origin of the molecular diversity and functional anchoring of cholinesterases. Neurosignals 2002, 11, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Massoulié, J.; Anselmet, A.; Bon, S.; Krejci, E.; Legay, C.; Morel, N.; Simon, S. Acetylcholinesterase: C-terminal domains, molecular forms and functional localization. J. Physiol. Paris 1998, 92, 183–190. [Google Scholar] [CrossRef]
- Perrier, A.L.; Massoulié, J.; Krejci, E. PRiMA: The membrane anchor of acetylcholinesterase in the brain. Neuron 2002, 33, 275–285. [Google Scholar] [CrossRef]
- Biberoglu, K.; Schopfer, L.M.; Saxena, A.; Tacal, O.; Lockridge, O. Polyproline tetramer organizing peptides in fetal bovine serum acetylcholinesterase. Biochim. Biophys. Acta 2013, 1834, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Krejci, E.; Thomine, S.; Boschetti, N.; Legay, C.; Sketelj, J.; Massoulié, J. The mammalian gene of acetylcholinesterase-associated collagen. J. Biol. Chem. 1997, 272, 22840–22847. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Krejci, E.; Massoulié, J. A four-to-one association between peptide motifs: Four C-terminal domains from cholinesterase assemble with one proline-rich attachment domain (PRAD) in the secretory pathway. EMBO J. 1998, 17, 6178–6187. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Schopfer, L.M.; Masson, P.; Lockridge, O. Lamellipodin proline rich peptides associated with native plasma butyrylcholinesterase tetramers. Biochem. J. 2008, 411, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Schopfer, L.M.; Delacour, H.; Masson, P.; Leroy, J.; Krejci, E.; Lockridge, O. The C5 variant of the butyrylcholinesterase tetramer includes a noncovalently bound 60 kDa lamellipodin fragment. Molecules 2017, 22, 1083. [Google Scholar] [CrossRef] [PubMed]
- Araç, D.; Boucard, A.A.; Ozkan, E.; Strop, P.; Newell, E.; Südhof, T.C.; Brunger, A.T. Structures of neuroligin-1 and the neuroligin-1/neurexin-1 beta complex reveal specific protein-protein and protein-Ca2+ interactions. Neuron 2007, 56, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Fabrichny, I.P.; Leone, P.; Sulzenbacher, G.; Comoletti, D.; Miller, M.T.; Taylor, P.; Bourne, Y.; Marchot, P. Structural analysis of the synaptic protein neuroligin and its beta-neurexin complex: Determinants for folding and cell adhesion. Neuron 2007, 56, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Comoletti, D.; Ferracci, G.; Conrod, S.; Garcia, S.U.; Taylor, P.; Bourne, Y.; Marchot, P. Structural insights into the exquisite selectivity of neurexin/neuroligin synaptic interactions. EMBO J. 2010, 29, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Kim, D.; Won, S.Y.; Han, K.A.; Park, D.; Cho, E.; Yun, N.; An, H.J.; Um, J.W.; Kim, E.; et al. Structural insights into modulation of neurexin-neuroligin trans-synaptic adhesion by MDGA1/Neuroligin-2 complex. Neuron 2017, 94, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, S.P.; Zhong, X.; Seshadrinathan, S.; Chen, H.; Machius, M.; Rudenko, G. Molecular mechanism of MDGA1: Regulation of neuroligin 2:neurexin trans-synaptic bridges. Neuron 2017, 94, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Elegheert, J.; Cvetkovska, V.; Clayton, A.J.; Heroven, C.; Vennekens, K.M.; Smukowski, S.N.; Regan, M.C.; Jia, W.; Smith, A.C.; Furukawa, H.; et al. Structural mechanism for modulation of synaptic neuroligin-neurexin signaling by MDGA proteins. Neuron 2017, 95, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Thoumine, O.; Marchot, P. A Triad of crystals sheds light on MDGA interference with neuroligation. Neuron 2017, 95, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Marchot, P. The neuroligins and their ligands: From structure to function at the synapse. J. Mol. Neurosci. 2014, 53, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Pinan-Lucarré, B.; Ji, T.; Jospin, M.; Bessereau, J.L. C. elegans punctin clusters GABA(A) receptors via neuroligin binding and UNC-40/DCC recruitment. Neuron 2015, 86, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Maro, G.S.; Gao, S.; Olechwier, A.M.; Hung, W.L.; Liu, M.; Özkan, E.; Zhen, M.; Shen, K. MADD-4/Punctin and neurexin organize C. elegans GABAergic postsynapses through neuroligin. Neuron 2015, 86, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, W.; Pelkey, K.A.; Duan, J.; Mao, X.; Wang, Y.X.; Craig, M.T.; Dong, L.; Petralia, R.S.; McBain, C.J.; et al. Molecular dissection of neuroligin 2 and Slitrk3 reveals an essential framework for GABAergic synapse development. Neuron 2017, 96, 808–826. [Google Scholar] [CrossRef] [PubMed]
- Cygler, M.; Schrag, J.D.; Sussman, J.L.; Harel, M.; Silman, I.; Gentry, M.K.; Doctor, B.P. Relationship between sequence conservation and three-dimensional structure in a large family of esterases, lipases, and related proteins. Protein Sci. 1993, 2, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Taylor, P.; Marchot, P. Acetylcholinesterase inhibition by fasciculin: Crystal structure of the complex. Cell 1995, 83, 503–512. [Google Scholar] [CrossRef]
- Marchot, P.; Ravelli, R.B.; Raves, M.L.; Bourne, Y.; Vellom, D.C.; Kanter, J.; Camp, S.; Sussman, J.L.; Taylor, P. Soluble monomeric acetylcholinesterase from mouse: Expression, purification, and crystallization in complex with fasciculin. Protein Sci. 1996, 5, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Renault, L.; Marchot, P. Crystal structure of snake venom acetylcholinesterase in complex with inhibitory antibody fragment Fab410 bound at the peripheral site: Evidence for open and closed states of a back door channel. J. Biol. Chem. 2015, 290, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Massoulié, J.; Pezzementi, L.; Bon, S.; Krejci, E.; Vallette, F.M. Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 1993, 41, 31–91. [Google Scholar] [CrossRef]
- Brazzolotto, X.; Wandhammer, M.; Ronco, C.; Trovaslet, M.; Jean, L.; Lockridge, O.; Renard, P.Y.; Nachon, F. Human butyrylcholinesterase produced in insect cells: Huprine-based affinity purification and crystal structure. FEBS J. 2012, 279, 2905–2916. [Google Scholar] [CrossRef] [PubMed]
- Brazzolotto, X.; Igert, A.; Guillon, V.; Santoni, G.; Nachon, F. Bacterial expression of human butyrylcholinesterase as a tool for nerve agent bioscavengers development. Molecules 2017, 22, 1828. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Asojo, O.A.; Borgstahl, G.E.; Masson, P.; Lockridge, O. Role of water in aging of human butyrylcholinesterase inhibited by echothiophate: The crystal structure suggests two alternative mechanisms of aging. Biochemistry 2005, 44, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Ngamelue, M.N.; Homma, K.; Lockridge, O.; Asojo, O.A. Crystallization and X-ray structure of full-length recombinant human butyrylcholinesterase. Acta Crystallogr. Sect. F 2007, 63, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Comoletti, D.; Grishaev, A.; Whitten, A.E.; Tsigelny, I.; Taylor, P.; Trewhella, J. Synaptic arrangement of the neuroligin/beta-neurexin complex revealed by X-ray and neutron scattering. Structure 2007, 15, 693–705. [Google Scholar] [CrossRef] [PubMed]
- De Jaco, A.; Dubi, N.; Camp, S.; Taylor, P. Congenital hypothyroidism mutations affect common folding and trafficking in the α/β-hydrolase fold proteins. FEBS J. 2012, 279, 4293–4305. [Google Scholar] [CrossRef] [PubMed]
- Di Jeso, B.; Arvan, P. Thyroglobulin from molecular and cellular biology to clinical endocrinology. Endocr. Rev. 2016, 37, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Erregragui, K.; Prato, S.; Miquelis, R.; Barrande, C.; Daniel, C.; Fert, V. Antigenic mapping of human thyroglobulin—Topographic relationship between antigenic regions and functional domains. Eur. J. Biochem. 1997, 244, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Van de Graaf, S.A.; Ris-Stalpers, C.; Pauws, E.; Mendive, F.M.; Targovnik, H.M.; de Vijlder, J.J. Up to date with human thyroglobulin. J. Endocrinol. 2001, 170, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Conte, M.; Arcaro, A.; D’Angelo, D.; Gnata, A.; Mamone, G.; Ferranti, P.; Formisano, S.; Gentile, F. A single chondroitin 6-sulfate oligosaccharide unit at Ser-2730 of human thyroglobulin enhances hormone formation and limits proteolytic accessibility at the carboxyl terminus. Potential insights into thyroid homeostasis and autoimmunity. J. Biol. Chem. 2006, 281, 22200–22211. [Google Scholar] [CrossRef] [PubMed]
- Raves, M.L.; Giles, K.; Schrag, J.D.; Schmid, M.F.; Phillips, G.N., Jr.; Chiu, W.; Howard, A.J.; Silman, I.; Sussman, J.L. Quaternary structure of tetrameric acetylcholinesterase. In Structure and Function of Cholinesterases and Related Proteins; Rotundo, R.L., Doctor, B.P., Taylor, P., Gentry, M.K., Quinn, D.M., Eds.; Plenum Publishing Corp.: New York, NY, USA, 1998; pp. 351–356. [Google Scholar]
- Bourne, Y.; Grassi, J.; Bougis, P.E.; Marchot, P. Conformational flexibility of the acetylcholinesterase tetramer suggested by X-ray crystallography. J. Biol. Chem. 1999, 274, 30370–30376. [Google Scholar] [CrossRef] [PubMed]
- Dvir, H.; Harel, M.; Bon, S.; Liu, W.Q.; Vidal, M.; Garbay, C.; Sussman, J.L.; Massoulié, J.; Silman, I. The synaptic acetylcholinesterase tetramer assembles around a polyproline II helix. EMBO J. 2004, 23, 4394–4405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; McCammon, J.A. The association of tetrameric acetylcholinesterase with ColQ tail: A block normal mode analysis. PLoS Comput. Biol. 2005, 1, e62. [Google Scholar] [CrossRef] [PubMed]
- Gorfe, A.A.; Chang, C.E.; Ivanov, I.; McCammon, J.A. Dynamics of the acetylcholinesterase tetramer. Biophys. J. 2008, 94, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Taylor, P.; Bougis, P.E.; Marchot, P. Crystal structure of mouse acetylcholinesterase. A peripheral site-occluding loop in a tetrameric assembly. J. Biol. Chem. 1999, 274, 2963–2970. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Schalk, I.; Ehret-Sabatier, L.; Bouet, F.; Goeldner, M.; Hirth, C.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 9031–9035. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Taylor, P.; Radić, Z.; Marchot, P. Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. EMBO J. 2003, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.E.; Harel, M.; Silman, I.; Sussman, J.L. Structure of a complex of the potent and specific inhibitor BW284C51 with Torpedo californica acetylcholinesterase. Acta Crystallogr. D 2002, 58, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Kleywegt, G.J.; Ravelli, R.B.; Silman, I.; Sussman, J.L. Crystal structure of an acetylcholinesterase-fasciculin complex: Interaction of a three-fingered toxin from snake venom with its target. Structure 1995, 3, 1355–1366. [Google Scholar] [CrossRef]
- Remy, M.H.; Frobert, Y.; Grassi, J. Characterization of monoclonal antibodies that strongly inhibit Electrophorus electricus acetylcholinesterase. Eur. J. Biochem. 1995, 231, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Le Goff, A.; Frobert, Y.; Grassi, J.; Massoulie, J. The binding sites of inhibitory monoclonal antibodies on acetylcholinesterase. Identification of a novel regulatory site at the putative “back door”. J. Biol. Chem. 1999, 274, 27740–27746. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Renault, L.; Essono, S.; Mondielli, G.; Lamourette, P.; Boquet, D.; Grassi, J.; Marchot, P. Molecular characterization of monoclonal antibodies that inhibit acetylcholinesterase by targeting the peripheral site and backdoor region. PLoS ONE 2013, 8, e77226. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.; Moore, S.W. The Leu-Arg-Glu (LRE) adhesion motif in proteins of the neuromuscular junction with special reference to proteins of the carboxylesterase/cholinesterase family. Comp. Biochem. Physiol. Part D 2013, 8, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.; Moore, S.W. Investigations into the development of catalytic activity in anti-acetylcholinesterase idiotypic and anti-idiotypic antibodies. J. Mol. Recognit. 2009, 22, 188–196. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A structural motif of acetylcholinesterase that promotes amyloid beta- peptide fibril formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.L.; Brazzolotto, X.; Macdonald, I.R.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: A crystallographic, kinetic and calorimetric study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef] [PubMed]
- Lockridge, O.; Norgren, R.B.; Johnson, R.C.; Blake, T.A. Naturally occurring genetic variants of human acetylcholinesterase and butyrylcholinesterase and their potential impact on the risk of toxicity from cholinesterase inhibitors. Chem. Res. Toxicol. 2016, 29, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Brimijoin, S.; Chen, V.P.; Pang, Y.P.; Geng, L.; Gao, Y. Physiological roles for butyrylcholinesterase: A BChE-ghrelin axis. Chem. Biol. Interact. 2016, 259, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Comoletti, D.; Taylor, P.; Bourne, Y.; Marchot, P. Structure-function relationships of the alpha/beta-hydrolase fold domain of neuroligin: A comparison with acetylcholinesterase. Chem. Biol. Interact. 2010, 187, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, H.; Shim, A.H.; Focia, P.J.; He, X. Structural basis for synaptic adhesion mediated by neuroligin-neurexin interactions. Nat. Struct. Mol. Biol. 2008, 15, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Miyazaki, N.; Matoba, K.; Nogi, T.; Iwasaki, K.; Takagi, J. Higher-order architecture of cell adhesion mediated by polymorphic synaptic adhesion molecules neurexin and neuroligin. Cell Rep. 2012, 2, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Nogi, T.; Yasui, N.; Iwasaki, K.; Takagi, J. Structural Basis for Variant-Specific Neuroligin-Binding by α-Neurexin. PLoS ONE 2011, 6, e19411. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.T.; Mileni, M.; Comoletti, D.; Stevens, R.C.; Harel, M.; Taylor, P. The crystal structure of the α-neurexin-1 extracellular region reveals a hinge point for mediating synaptic adhesion and function. Structure 2011, 19, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Venugopal, V.; Murray, B.; Rudenko, G. The structure of neurexin 1α reveals features promoting a role as synaptic organizer. Structure 2011, 19, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Marinò, M.; Zhao, J.; McCluskey, R.T. Megalin (gp330): A putative endocytic receptor for thyroglobulin (Tg). Endocrinology 1998, 139, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Marinò, M.; Friedlander, J.A.; McCluskey, R.T.; Andrews, D. Identification of a heparin-binding region of rat thyroglobulin involved in megalin binding. J. Biol. Chem. 1999, 274, 30377–30386. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; Pinchera, A.; McCluskey, R.T.; Chiovato, L.; Marinò, M. Binding of heparin to human thyroglobulin (Tg) involves multiple binding sites including a region corresponding to a binding site of rat Tg. Eur. J. Endocrinol. 2002, 146, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, D.R.; Faerman, C.H.; Axelsen, P.H.; Silman, I.; Sussman, J.L. An electrostatic mechanism for substrate guidance down the aromatic gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA 1993, 90, 5128–5132. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Straatsma, T.P.; McCammon, J.A.; Ripoll, D.R.; Faerman, C.H.; Axelsen, P.H.; Silman, I.; Sussman, J.L. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science 1994, 263, 1276–1278. [Google Scholar] [CrossRef] [PubMed]
- Colletier, J.P.; Royant, A.; Specht, A.; Sanson, B.; Nachon, F.; Masson, P.; Zaccai, G.; Sussman, J.L.; Goeldner, M.; Silman, I.; et al. Use of a ‘caged’ analogue to study the traffic of choline within acetylcholinesterase by kinetic crystallography. Acta Crystallogr. D 2007, 63, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Colletier, J.P.; Weik, M.; Qin, G.; Jiang, H.; Silman, I.; Sussman, J.L. Long route or shortcut? A molecular dynamics study of traffic of thiocholine within the active-site gorge of acetylcholinesterase. Biophys. J. 2010, 99, 4003–4011. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Stojan, J.; Fournier, D. Insights into substrate and product traffic in the Drosophila melanogaster acetylcholinesterase active site gorge by enlarging a back channel. FEBS J. 2008, 275, 2659–2664. [Google Scholar] [CrossRef] [PubMed]
- Sanson, B.; Colletier, J.P.; Xu, Y.; Lang, P.T.; Jiang, H.; Silman, I.; Sussman, J.L.; Weik, M. Backdoor opening mechanism in acetylcholinesterase based on X-ray crystallography and molecular dynamics simulations. Protein Sci. 2011, 20, 1114–1118. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Radić, Z.; Sulzenbacher, G.; Kim, E.; Taylor, P.; Marchot, P. Substrate and product trafficking through the active center gorge of acetylcholinesterase analyzed by crystallography and equilibrium binding. J. Biol. Chem. 2006, 281, 29256–29267. [Google Scholar] [CrossRef] [PubMed]
- De la Escalera, S.; Bockamp, E.O.; Moya, F.; Piovant, M.; Jiménez, F. Characterization and gene cloning of neurotactin, a Drosophila transmembrane protein related to cholinesterases. EMBO J. 1990, 9, 3593–3601. [Google Scholar] [PubMed]
- Yan, J.; Oliveira, G.; Coutinho, A.; Yang, C.; Feng, J.; Katz, C.; Sram, J.; Bockholt, A.; Jones, I.R.; Craddock, N.; et al. Analysis of the Neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 2005, 10, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Vellom, D.C.; Radić, Z.; Li, Y.; Pickering, N.A.; Camp, S.; Taylor, P. Amino acid residues controlling acetylcholinesterase and butyrylcholinesterase specificity. Biochemistry 1993, 32, 12–17. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourne, Y.; Marchot, P. Hot Spots for Protein Partnerships at the Surface of Cholinesterases and Related α/β Hydrolase Fold Proteins or Domains—A Structural Perspective. Molecules 2018, 23, 35. https://doi.org/10.3390/molecules23010035
Bourne Y, Marchot P. Hot Spots for Protein Partnerships at the Surface of Cholinesterases and Related α/β Hydrolase Fold Proteins or Domains—A Structural Perspective. Molecules. 2018; 23(1):35. https://doi.org/10.3390/molecules23010035
Chicago/Turabian StyleBourne, Yves, and Pascale Marchot. 2018. "Hot Spots for Protein Partnerships at the Surface of Cholinesterases and Related α/β Hydrolase Fold Proteins or Domains—A Structural Perspective" Molecules 23, no. 1: 35. https://doi.org/10.3390/molecules23010035
APA StyleBourne, Y., & Marchot, P. (2018). Hot Spots for Protein Partnerships at the Surface of Cholinesterases and Related α/β Hydrolase Fold Proteins or Domains—A Structural Perspective. Molecules, 23(1), 35. https://doi.org/10.3390/molecules23010035