icMRCI+Q Study of the Spectroscopic Properties of the 14 Λ-S and 49 Ω States of the SiN− Anion in the Gas Phase

Abstract
:1. Introduction
2. Methodology
3. Results and Discussion
3.1. Electron Affinity of SiN and the Stable States of SiN−
3.1.1. Electron Affinity of SiN
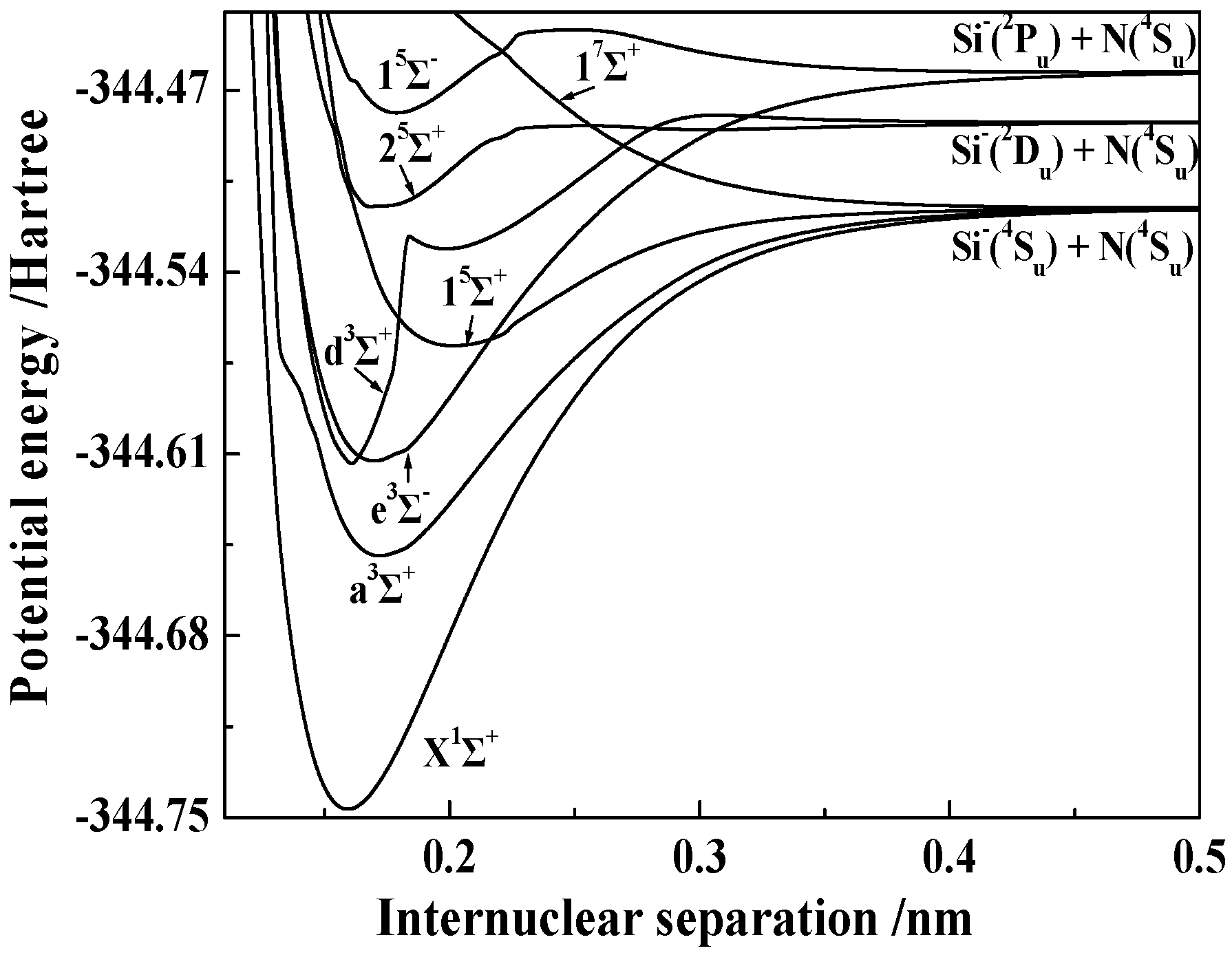
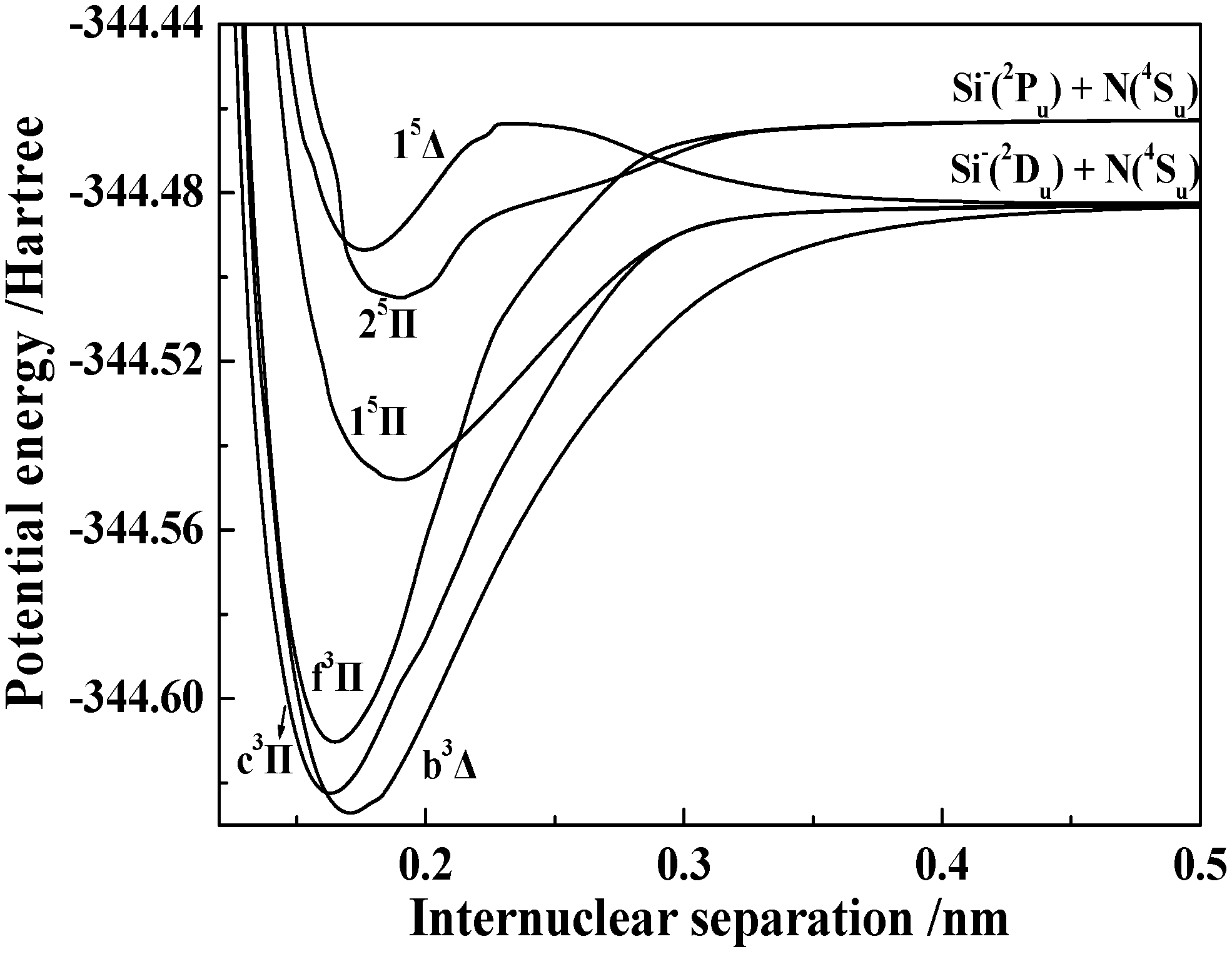
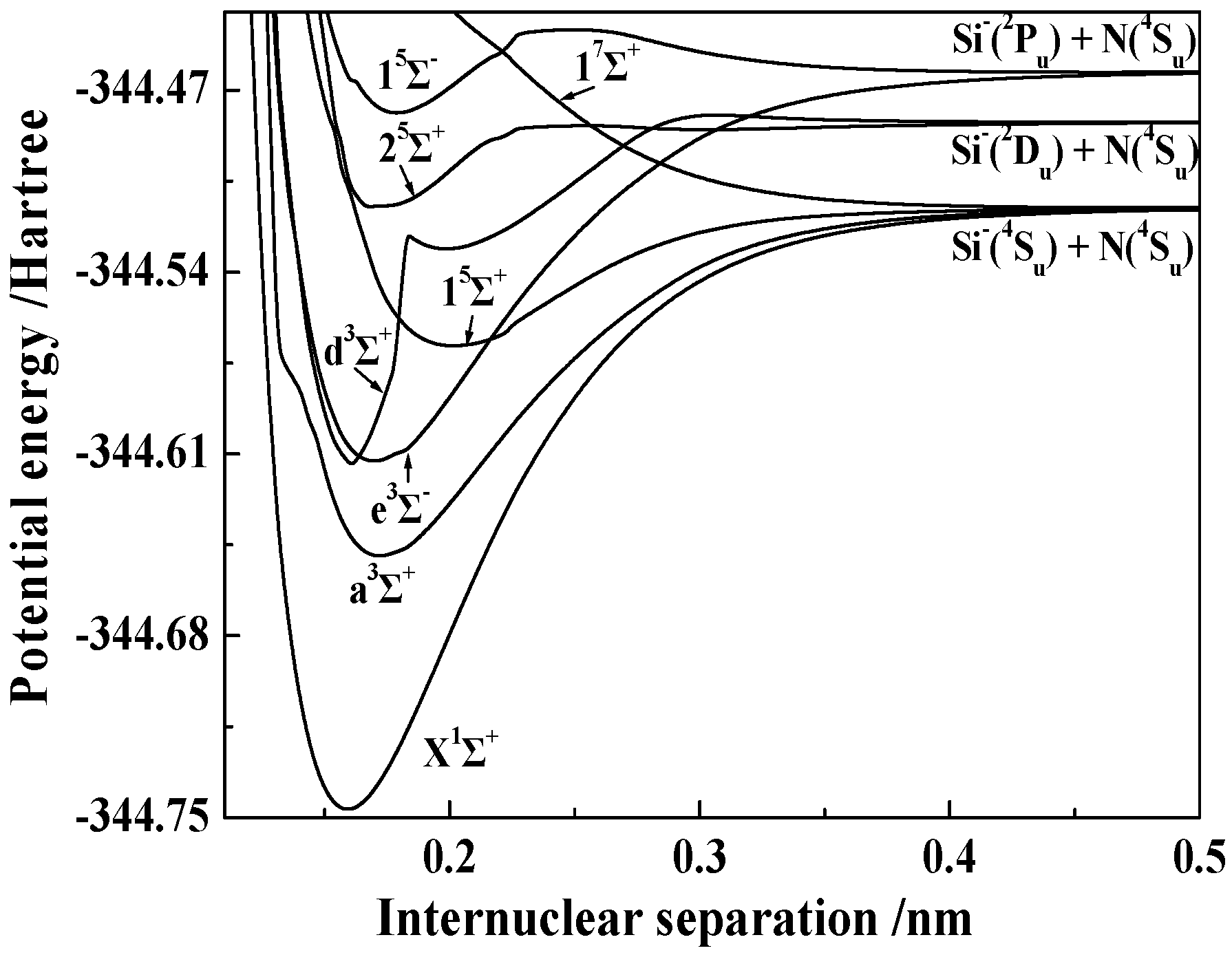
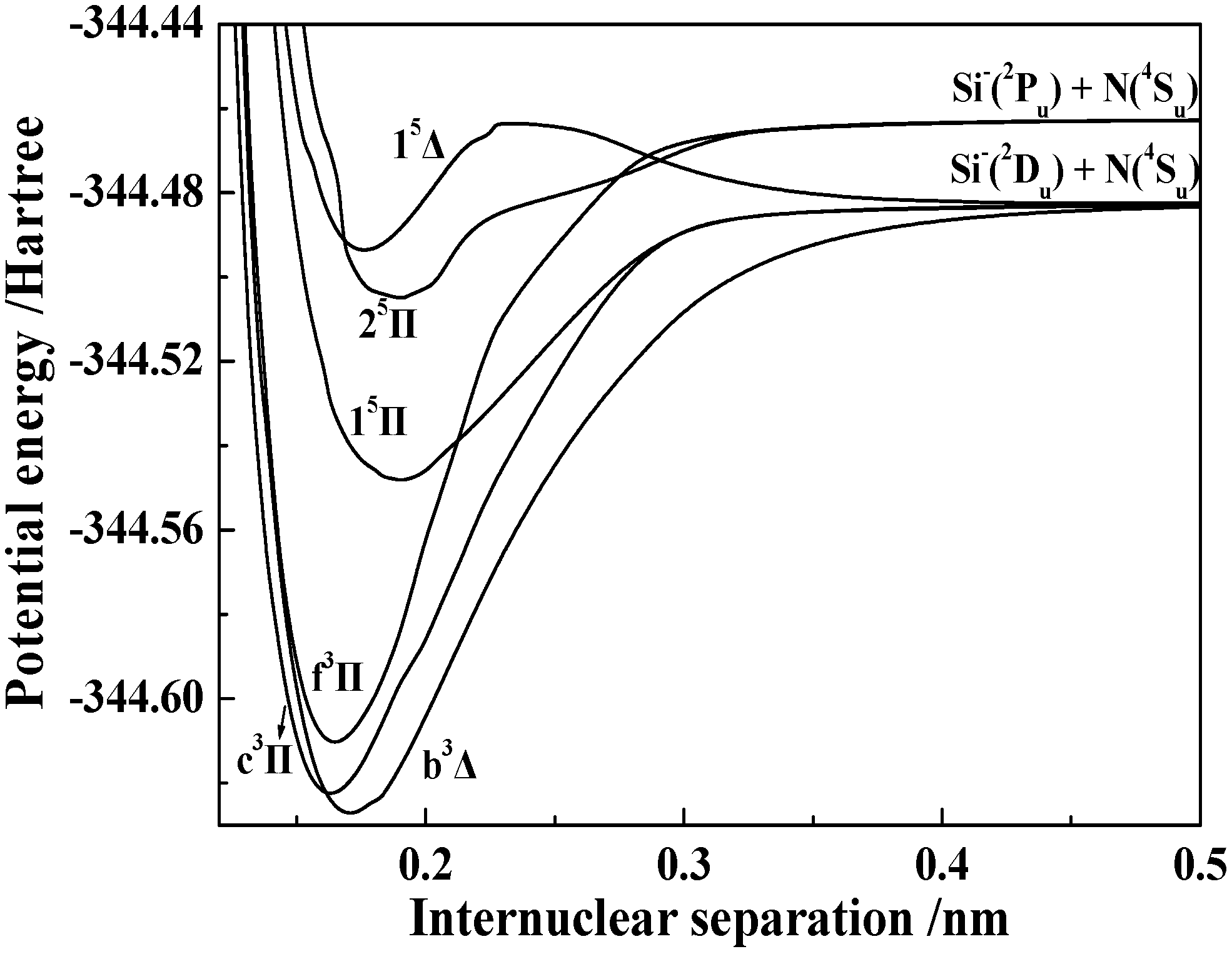
3.1.2. Spectroscopic Parameters of the 13 Stable and Metastable Λ-S States of SiN−
3.2. Spectroscopic Parameters and Vibrational Levels of the 45 Ω Bound States
3.2.1. Spectroscopic and Vibrational Properties of 16 Ω States with the Σ Symmetry
3.2.2. Spectroscopic and Vibrational Properties of 20 Ω States with the Π Symmetry
3.2.3. Spectroscopic and Vibrational Properties of Nine Ω States with the Δ Symmetry
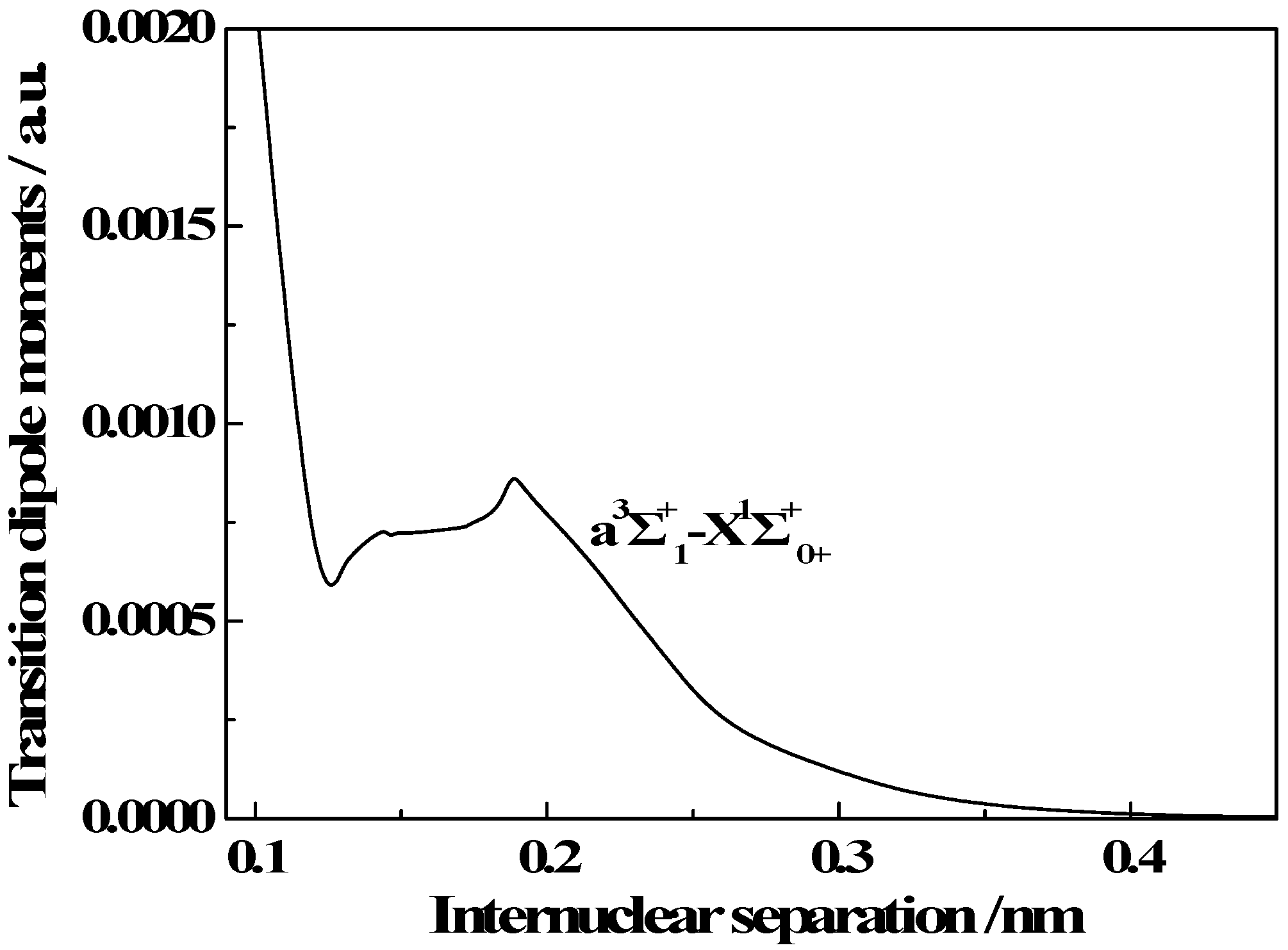
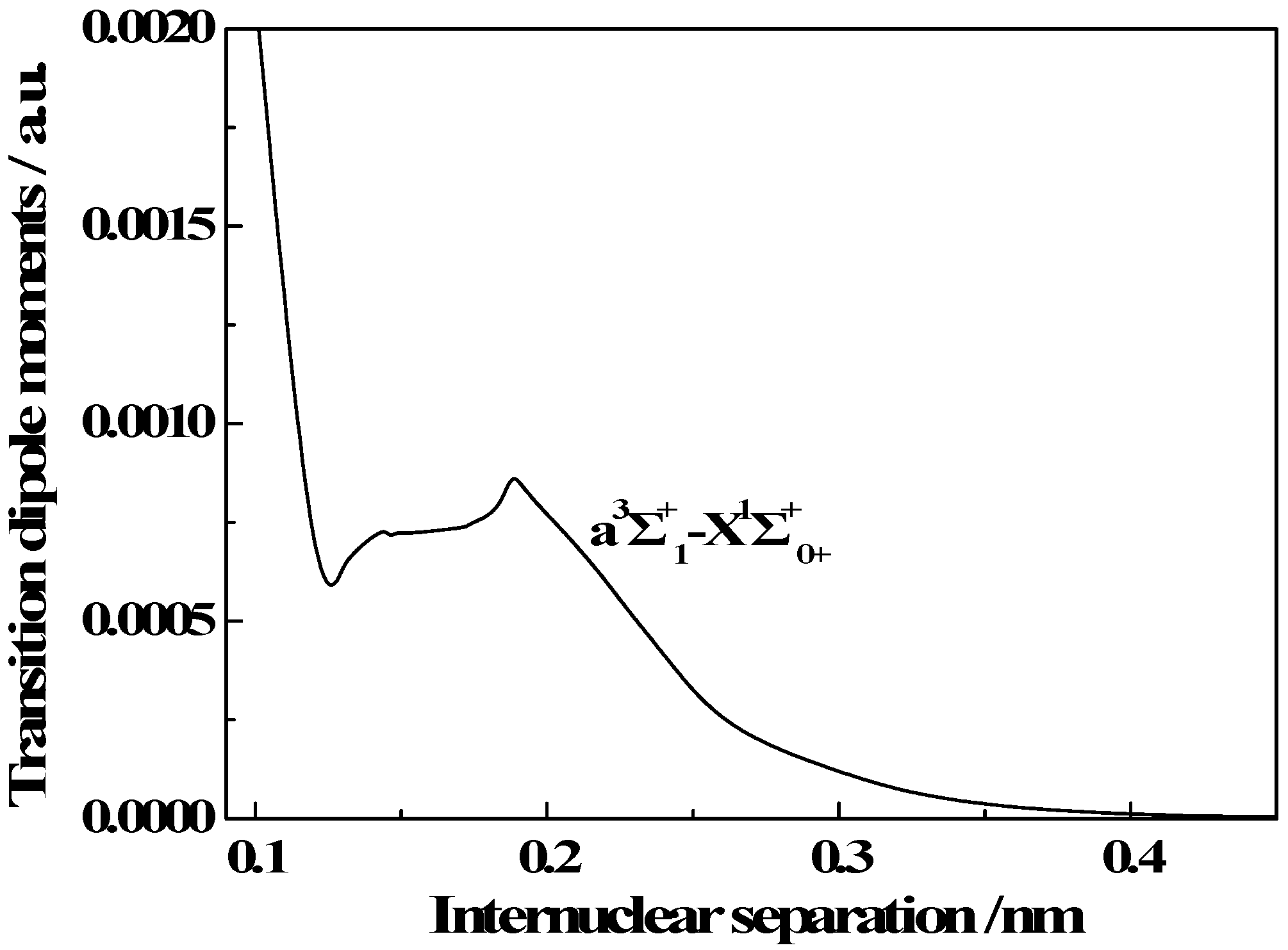
3.3. Transition Properties
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Agúndez, M.; Cernicharo, J.; Guélin, M.; Kahane, C.; Roueff, E.; Kłos, J.; Aoiz, F.J.; Lique, F.; Marcelino, N.; Goicoechea, J.R.; et al. Astronomical identification of CN−, the smallest observed molecular anion. Astron. Astrophys. 2010, 517, L2. [Google Scholar] [CrossRef]
- Turner, B.E. Detection of SiN in IRC + 10216. Astrophys. J. 1992, 388, L35–L38. [Google Scholar] [CrossRef]
- Guèlin, M.; Muller, S.; Cernicharo, J.; Apponi, A.J.; McCarthy, M.C.; Gottlieb, C.A.; Thaddeus, P. Astronomical detection of the free radical SiCN. Astrophys. J. 2000, 363, L9–L12. [Google Scholar]
- Guèlin, M.; Muller, S.; Cernicharo, J.; McCarthy, M.C.; Thaddeus, P. Detection of the SiNC Radical in IRC + 10216. Astron. Astrophys. 2004, 426, L49–L52. [Google Scholar] [CrossRef]
- Fortenberry, R.C.; Daniel Crawford, T. Theoretical prediction of new dipole-bound singlet states for anions of interstellar interest. J. Chem. Phys. 2011, 134, 154304. [Google Scholar] [CrossRef] [PubMed]
- Drzaic, P.S.; Marks, J.; Brauman, J.I. Gas-Phase Ion Chemistry; Academic Press: New York, NY, USA, 1984; pp. 1–63. [Google Scholar]
- Damrauer, R.; Hankin, J.A. Chemistry and thermochemistry of silicon-containing anions in the gas phase. Chem. Rev. 1995, 95, 1137–1160. [Google Scholar] [CrossRef]
- Zhu, W.; Kochanski, G.P.; Jin, S. Low-field electron emission from undoped nanostructured diamond. Science 1998, 282, 1471–1473. [Google Scholar] [CrossRef] [PubMed]
- Shabanov, S.V.; Gornushkin, I.B. Anions in laser-induced plasmas. Appl. Phys. A 2016, 122, 676. [Google Scholar] [CrossRef]
- Meloni, G.; Sheehan, S.M.; Ferguson, M.J.; Neumark, D.M. Negative ion photoelectron spectroscopy of SiN−. J. Phys. Chem. A 2004, 108, 9750–9754. [Google Scholar] [CrossRef]
- Peterson, K.A.; Woods, R.C. Ground state spectroscopic and thermodynamic properties of AlO−, SiN−, CP−, BS−, BO−, and CN− from Møller-Plesset perturbation theory. J. Chem. Phys. 1989, 90, 7239–7250. [Google Scholar] [CrossRef]
- McLean, A.D.; Liu, B.; Chandler, G.S. Computed self-consistent field and singles and doubles configuration interaction spectroscopic data and dissociation energies for the diatomics B2, C2, N2, O2, F2, CN, CP, CS, PN, SiC, SiN, SiO, SiP, and their ions. J. Chem. Phys. 1992, 97, 8459–8464. [Google Scholar] [CrossRef]
- Kalcher, J. Trends in ground and excited state electron affinities of group 14, 15, and 16 mixed diatomic anions: A computational study. Phys. Chem. Chem. Phys. 2002, 4, 3311–3317. [Google Scholar] [CrossRef]
- Midda, S.; Das, A.K. Theoretical study of spectroscopic constants and molecular properties of diatomic anions using B3LYP method. J. Mol. Struct. 2003, 640, 183–189. [Google Scholar] [CrossRef]
- Kerkines, I.S.K.; Mavridis, A. On the electron affinity of SiN and spectroscopic constants of SiN−. J. Chem. Phys. 2005, 123, 124301. [Google Scholar] [CrossRef] [PubMed]
- Mogren, M.M.A.; El-Azhary, A.A.; Alkiali, W.Z.; Hochlaf, M. Electronic structure and properties of neutral, anionic and cationic silicon-nitrogen nanoclusters. J. Mol. Model. 2013, 19, 2657–2668. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Li, R.; Gai, Z.Q.; Ai, R.B.; Zhang, H.M.; Zhang, X.M.; Yan, B. Configuration interaction studies on the spectroscopic properties of PbO including spin-Orbit coupling. Chin. Phys. B 2016, 25, 073101. [Google Scholar] [CrossRef]
- Zhao, S.T.; Yan, B.; Li, R.; Wu, S.; Wang, Q.L. MRCI + Q study of the low-lying electronic states of CdF including spin-orbit coupling. Chin. Phys. B 2017, 26, 023105. [Google Scholar] [CrossRef]
- Andersen, T.; Haugen, H.K.; Hotop, H. Binding energies in atomic negative ions: III. J. Phys. Chem. Ref. Data 1999, 28, 1511–1533. [Google Scholar] [CrossRef]
- Moore, C.E. CRC Series in Evaluated Data in Atomic Physics; CRC Press: Boca Raton, FL, USA, 1993; p. 339. [Google Scholar]
- Langhoff, S.R.; Davidson, E.R. Configuration interaction calculations on the nitrogen molecule. Int. J. Quantum. Chem. 1974, 8, 61–72. [Google Scholar] [CrossRef]
- Richartz, A.; Buenker, R.J.; Peyerimhoff, S.D. Ab initio MRD-CI study of ethane: The 14–25 eV PES region and Rydberg states of positive ions. Chem. Phys. 1978, 28, 305–312. [Google Scholar] [CrossRef]
- Van Mourik, T.; Wilson, A.K.; Dunning, T.H. Benchmark calculations with correlated molecular wavefunctions. XIII. Potential energy curves for He2, Ne2 and Ar2 using correlation consistent basis sets through augmented sextuple zeta. Mol. Phys. 1999, 99, 529–547. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Lindh, R.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; et al. MOLPRO 2010.1 is a Package of Ab Initio Programs. Available online: http://www.molpro.net (accessed on 17 September 2010).
- De Jong, W.A.; Harrison, R.J.; Dixon, D.A. Parallel Douglas-Kroll energy and gradients in NWChem: Estimating scalar relativistic effects using Douglas-Kroll contracted basis sets. J. Chem. Phys. 2001, 114, 48–53. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Berning, A.; Schweizer, M.; Werner, H.-J.; Knowles, P.J.; Palmieri, P. Spin-orbit matrix elements for internally contracted multireference configuration interaction wavefunctions. Mol. Phys. 2000, 98, 1823–1833. [Google Scholar]
- Oyeyemi, V.B.; Krisiloff, D.B.; Keith, J.A.; Libisch, F.; Pavone, M.; Carter, E.A. Size-extensivity-corrected multireference configuration interaction schemes to accurately predict bond dissociation energies of oxygenated hydrocarbons. J. Chem. Phys. 2014, 140, 044317. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Shi, D.H.; Sun, J.F.; Zhu, Z.L. Calculations of 21 Λ-S and 42 Ω states of BC molecule: Potential energy curves, spectroscopic parameters and spin-orbit coupling effect. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 153, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Shi, D.H.; Sun, J.F.; Zhu, Z.L. Accurate spectroscopic calculations of the 19 Λ-S states and 36 Ω states of the BC+ cation. Mol. Phys. 2017, 115, 387–402. [Google Scholar] [CrossRef]
- Le Roy, R.J. LEVEL 8.0: A Computer Program for Solving the Radial Schrödinger Equation for Bound and Quasi-Bound Levels; University of Waterloo Chemical Physics Research Report CP-663; University of Waterloo: Waterloo, ON, Canada, 2007. [Google Scholar]
Sample Availability: Not Available. |

{kind=link}
{kind=link}
{kind=link}
| Dissociation Channel | Electronic State | Relative Energy/cm−1 | |
|---|---|---|---|
| This Work a | Exp. [19] | ||
| Si− (4Su) + N(4Su) | X1Σ+, a3Σ+, 15Σ+, 17Σ+ | 0.0 | 0.0 |
| Si− (2Du) + N(4Su) | b3Δ, c3Π, d3Σ+, 25Σ+, 15Π, 15Δ | 6973.48 | 6961.85 b |
| Si− (2Pu) + N(4Su) | e3Σ−, 15Σ−, f3Π, 25Π | 10,974.90 | (10,977.24 + x c)/2 |
| Atomic State | Possible Ω States | Relative Energy/cm−1 | |
|---|---|---|---|
| This Work a | Exp. [19] | ||
| Si− (4S3/2) + N(4S3/2) | 0− (2), 0+(2), 1(3), 2(2), 3 | 0.00 | 0.00 |
| Si− (2D3/2) + N(4S3/2) | −1, 0−(2), 0+(2), 1(2), 2(2), 3 | 6968.32 | 6954.81 |
| Si− (2D5/2) + N(4S3/2) | 0−(2), 0+(2), 1(4), 2(3), 3(2), 4 | 6978.63 | 6968.89 |
| Si− (2P1/2) + N(4S3/2) | −1, 0−, 0+, 1, 2 | 10,970.08 | 10,977.24 |
| Si− (2P3/2) + N(4S3/2) | 0−(2), 0+(2), 1(3), 2(2), 3 | 10,979.76 | x b |
| State to State | υ′′ = 0 | υ′′ = 1 | υ′′ = 2 | υ′′ = 3 | υ′′ = 4 | υ′′ = 5 | υ′′ = 6 | υ′′ = 7 | υ′′ = 8 | υ′′ = 9 |
|---|---|---|---|---|---|---|---|---|---|---|
| a3Σ+1 to X1Σ+0+ | ||||||||||
| υ′ = 0 | 0.0970 | 0.2393 | 0.2796 | 0.2068 | 0.1099 | 0.0457 | 0.0159 | 0.0047 | 0.0011 | 0.0002 |
| 0.3370 | 0.7136 | 0.7129 | 0.4506 | 0.2054 | 0.0736 | 0.0220 | 0.0056 | 0.0012 | 0.0002 | |
| υ′ = 1 | 0.1886 | 0.1666 | 0.0131 | 0.0455 | 0.1663 | 0.1929 | 0.1328 | 0.0644 | 0.0230 | 0.0057 |
| 0.7245 | 0.5493 | 0.0359 | 0.1140 | 0.3519 | 0.3484 | 0.2064 | 0.0870 | 0.0274 | 0.0062 | |
| υ′ = 2 | 0.2373 | 0.0262 | 0.0709 | 0.1362 | 0.0207 | 0.0289 | 0.1378 | 0.1661 | 0.1101 | 0.0479 |
| 1.0072 | 0.0954 | 0.2258 | 0.3692 | 0.0462 | 0.0613 | 0.2448 | 0.2544 | 0.1468 | 0.0556 |
| Radiative Lifetimes | |||
|---|---|---|---|
| Transitions | υ′ = 0 | υ′ = 1 | υ′ = 2 |
| a3Σ+1 to X1Σ+0+ (ms) | 396.5 | 407.9 | 396.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, W.; Sun, J.; Shi, D.; Zhu, Z. icMRCI+Q Study of the Spectroscopic Properties of the 14 Λ-S and 49 Ω States of the SiN− Anion in the Gas Phase. Molecules 2018, 23, 210. https://doi.org/10.3390/molecules23010210
Xing W, Sun J, Shi D, Zhu Z. icMRCI+Q Study of the Spectroscopic Properties of the 14 Λ-S and 49 Ω States of the SiN− Anion in the Gas Phase. Molecules. 2018; 23(1):210. https://doi.org/10.3390/molecules23010210
Chicago/Turabian StyleXing, Wei, Jinfeng Sun, Deheng Shi, and Zunlue Zhu. 2018. "icMRCI+Q Study of the Spectroscopic Properties of the 14 Λ-S and 49 Ω States of the SiN− Anion in the Gas Phase" Molecules 23, no. 1: 210. https://doi.org/10.3390/molecules23010210
APA StyleXing, W., Sun, J., Shi, D., & Zhu, Z. (2018). icMRCI+Q Study of the Spectroscopic Properties of the 14 Λ-S and 49 Ω States of the SiN− Anion in the Gas Phase. Molecules, 23(1), 210. https://doi.org/10.3390/molecules23010210