Energetic Di- and Trinitromethylpyridines: Synthesis and Characterization


Abstract
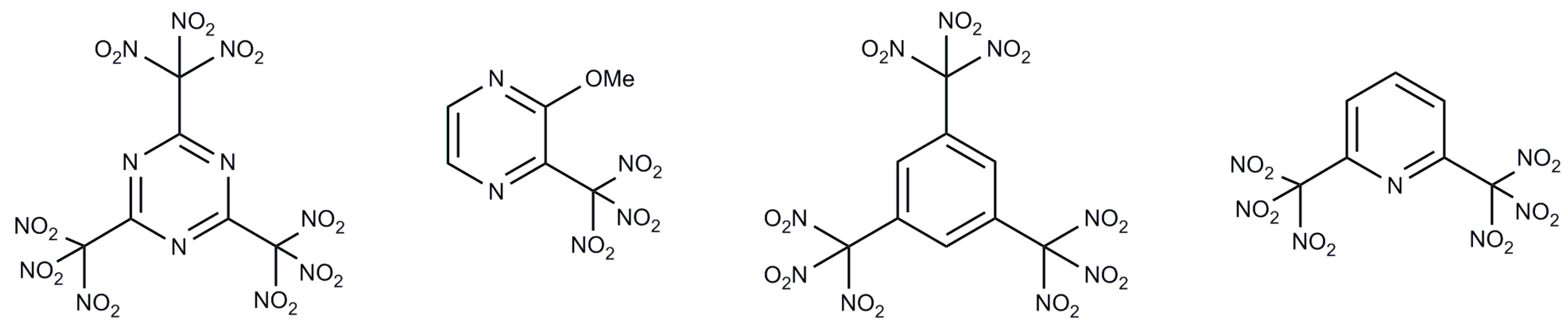
:1. Introduction
2. Results and Discussion
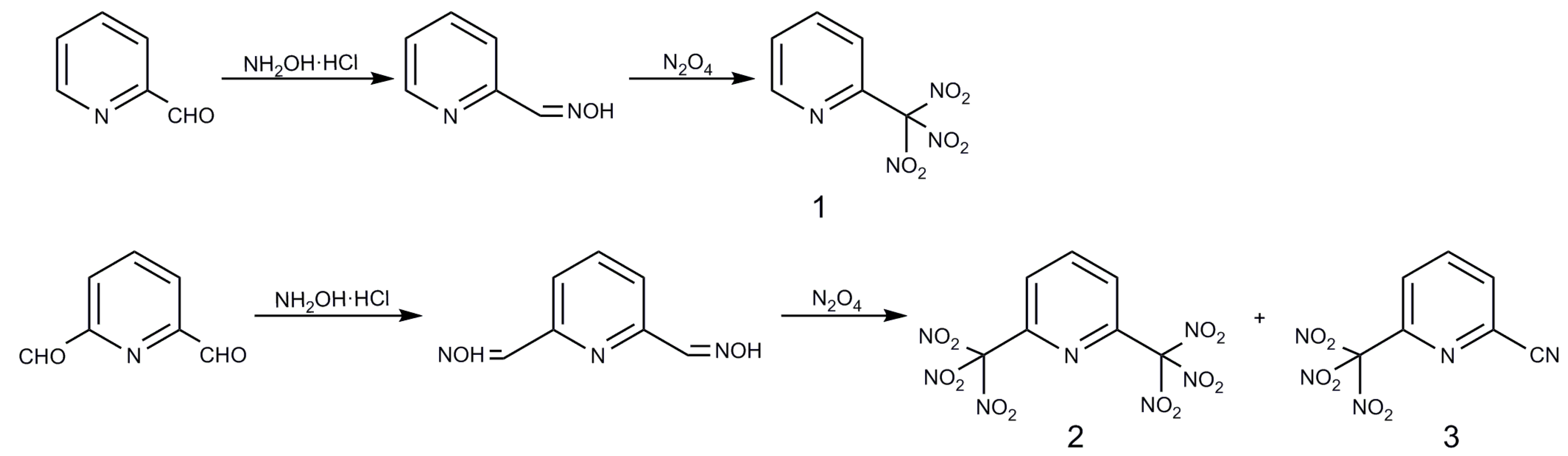
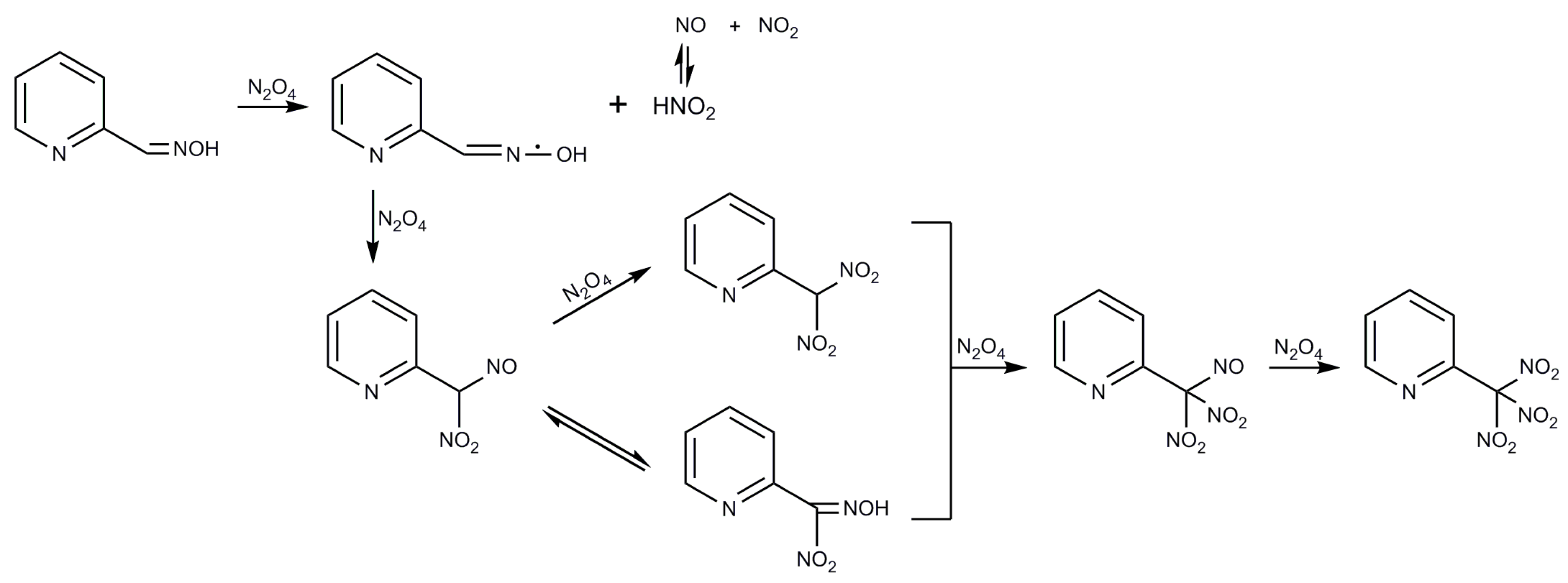
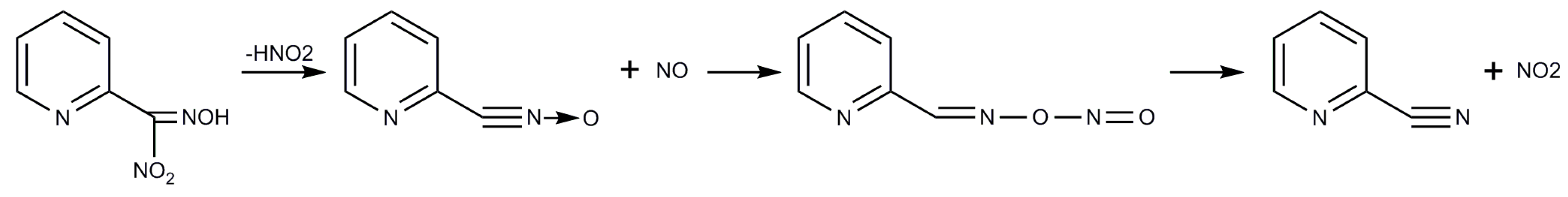
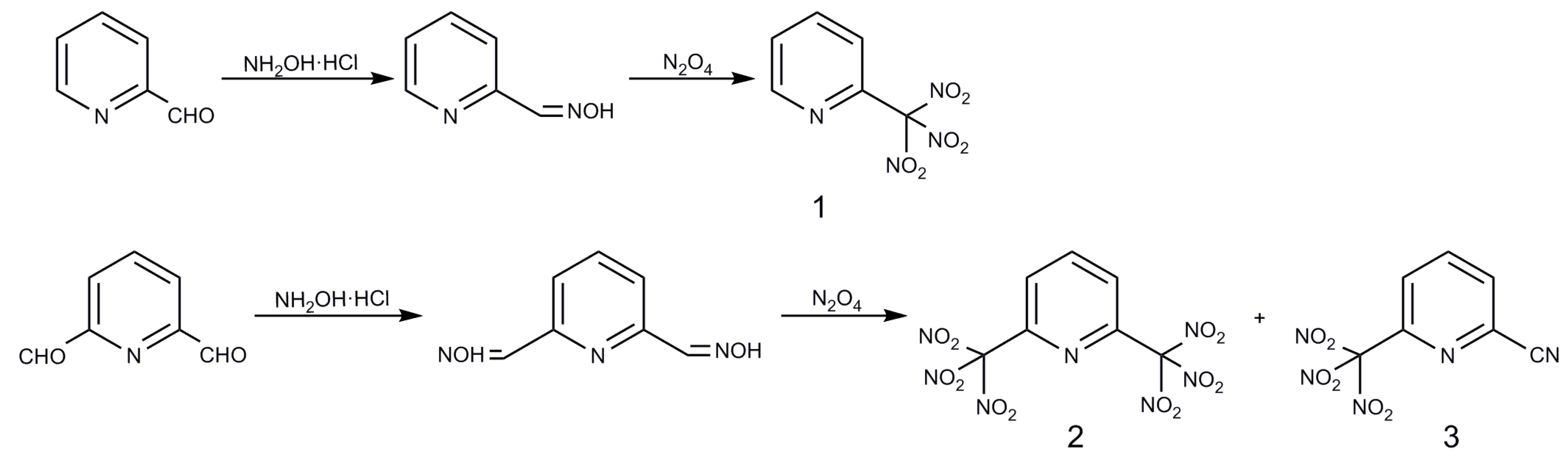
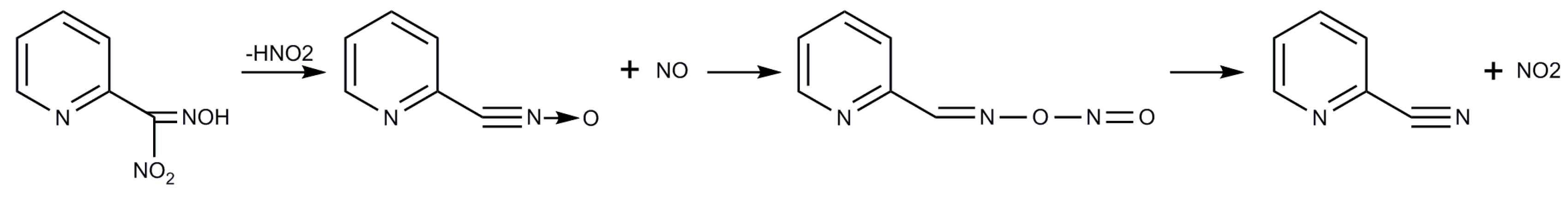
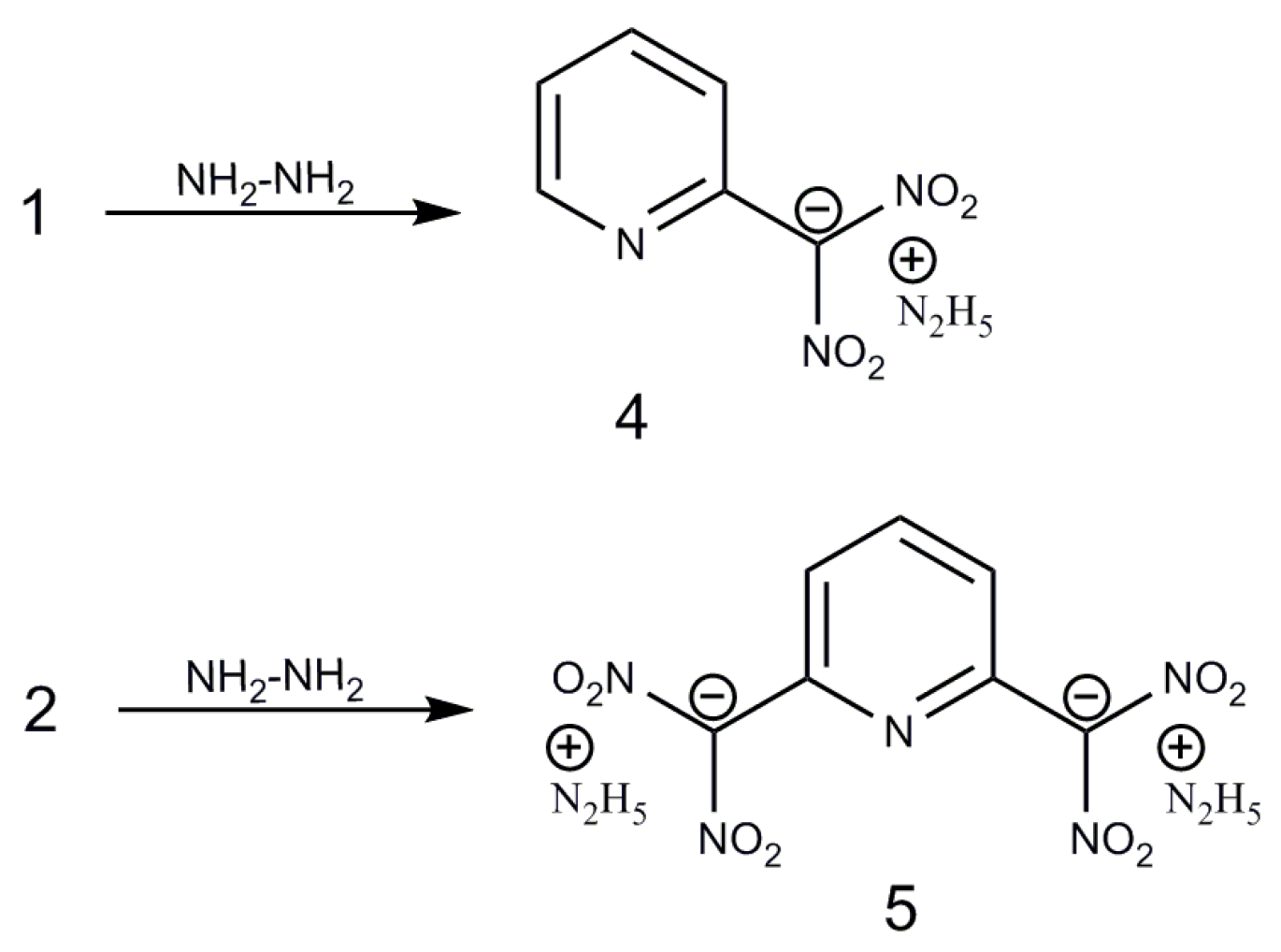
2.1. Syntheses
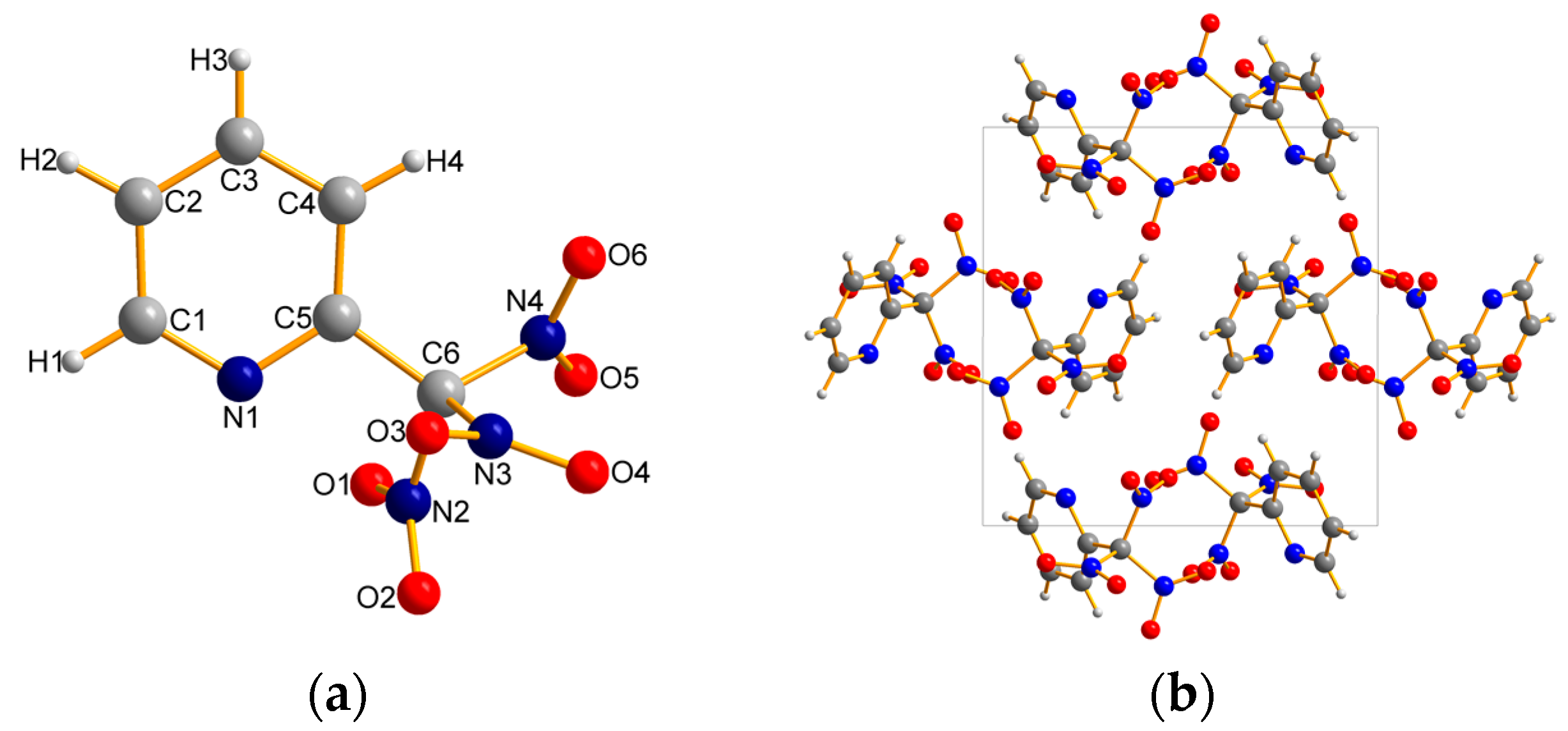
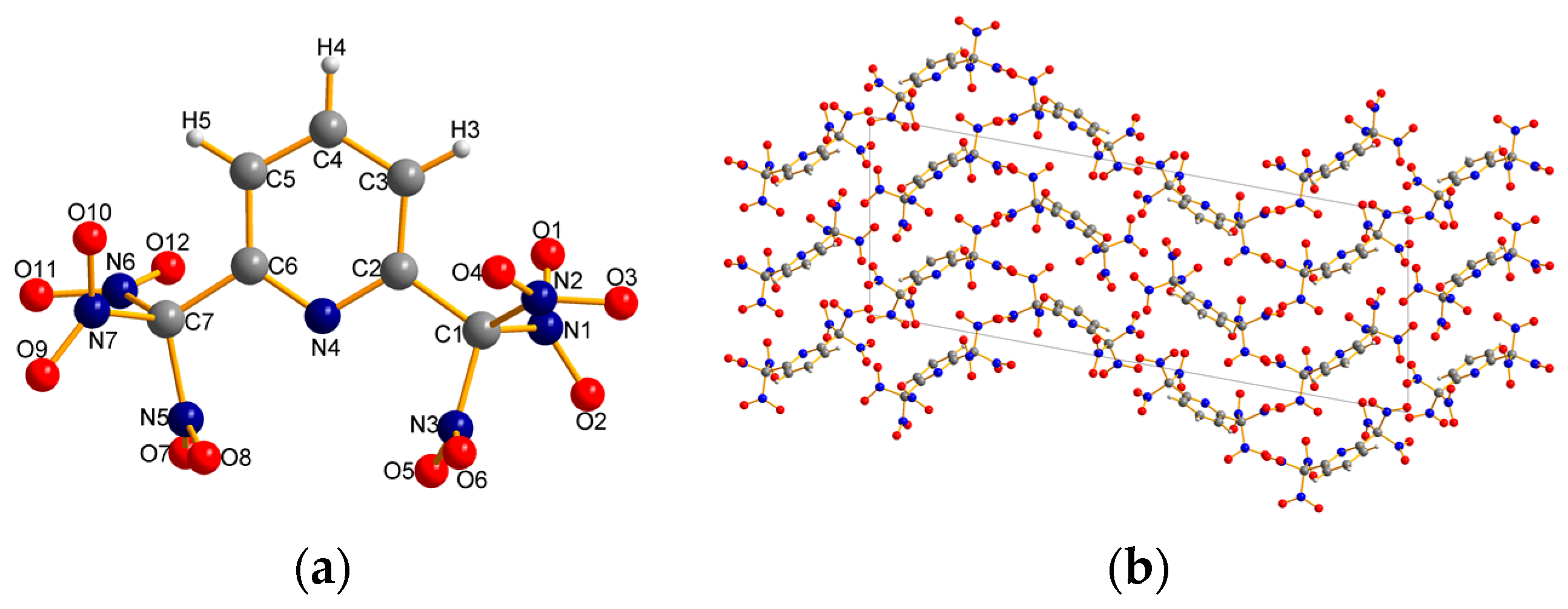
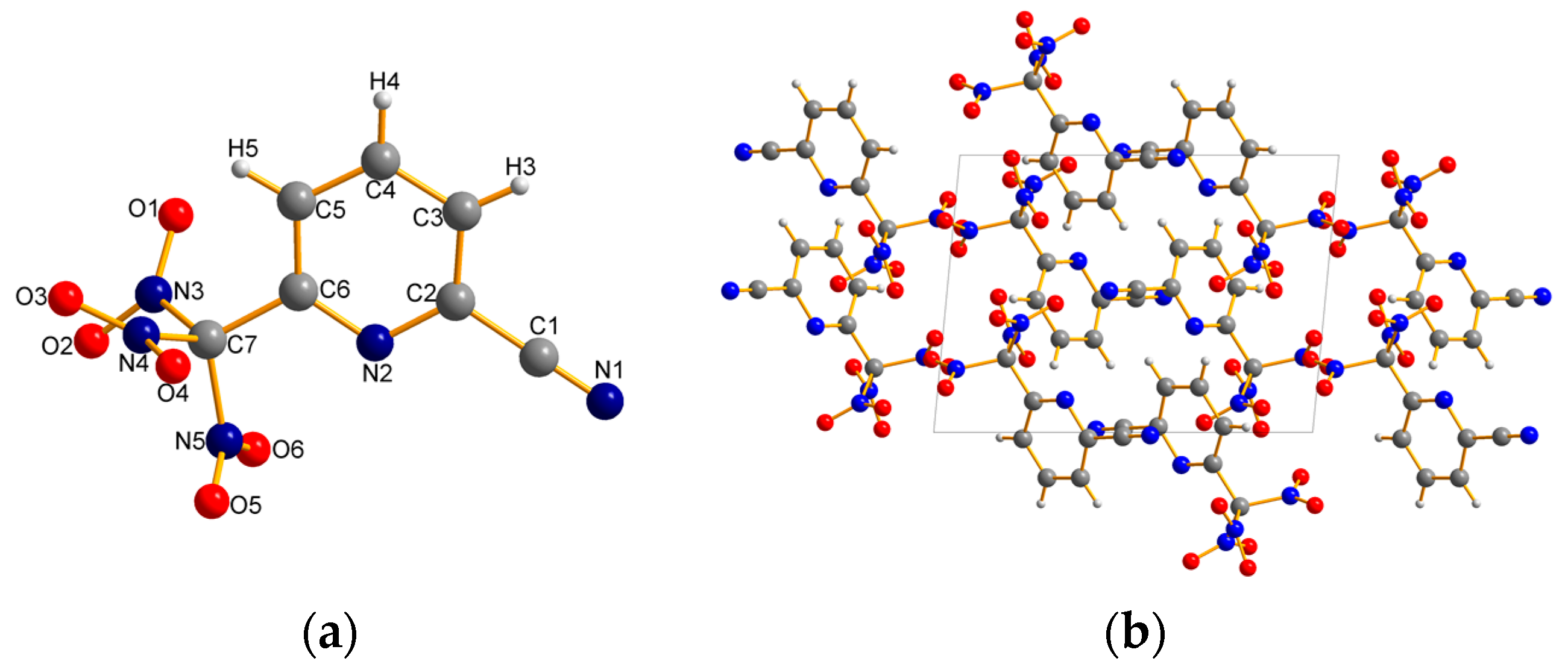
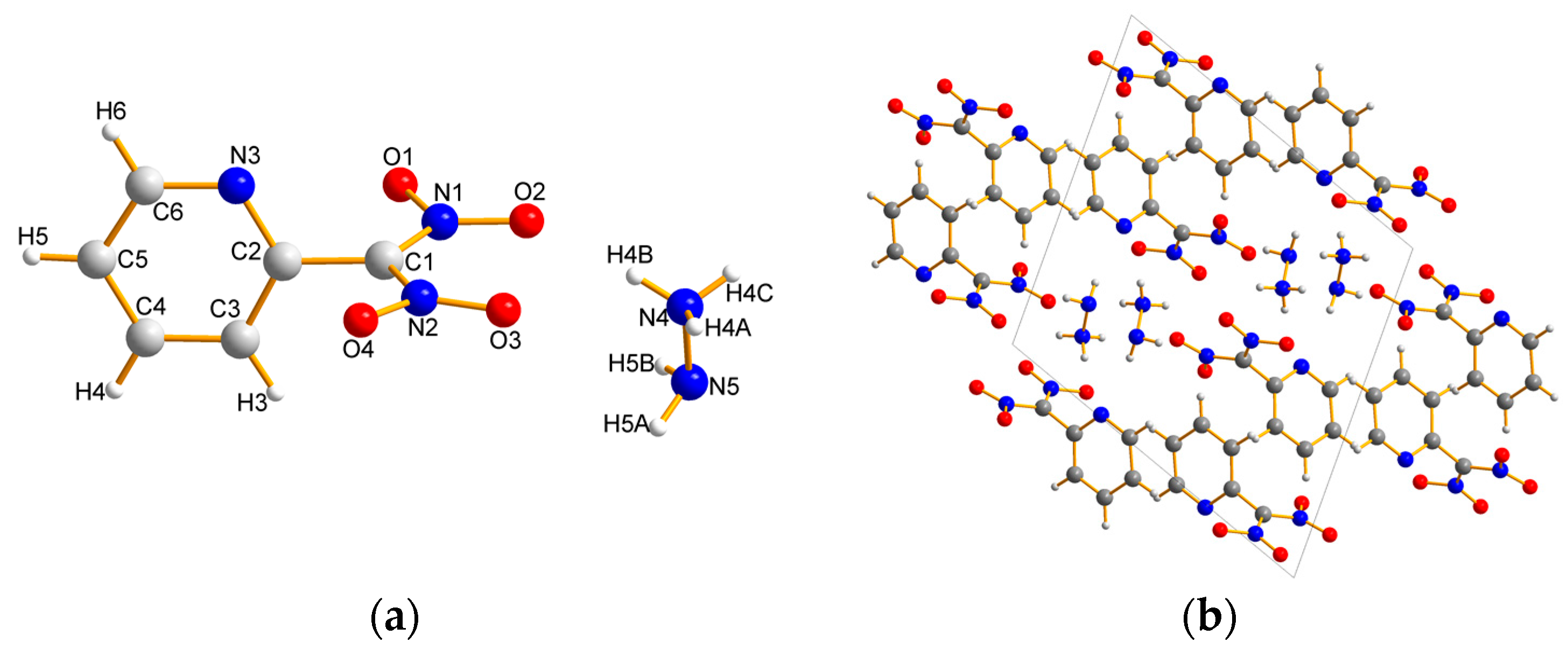
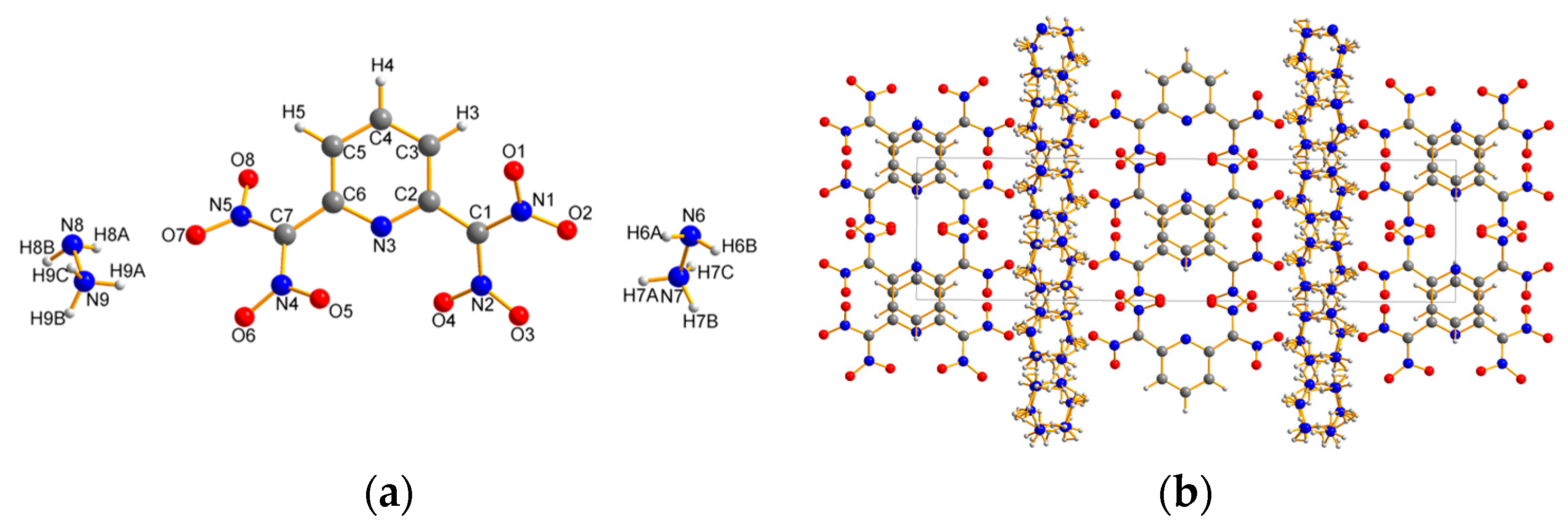
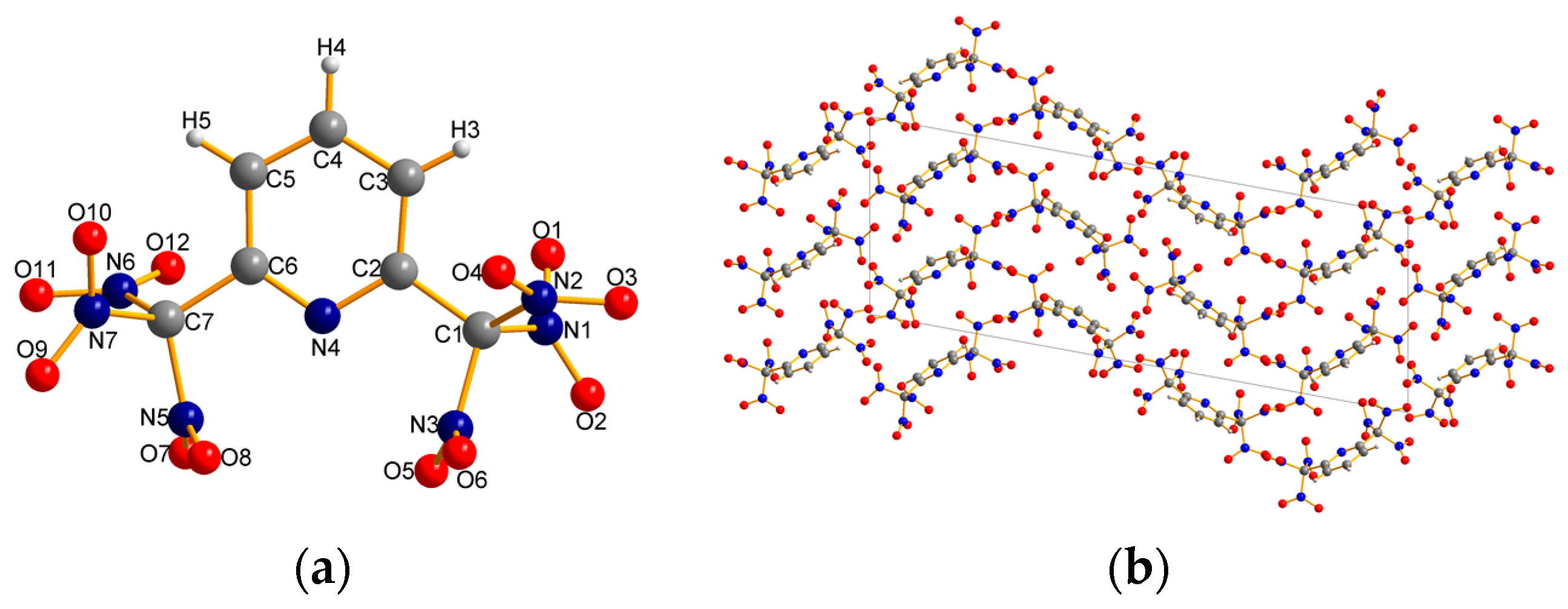
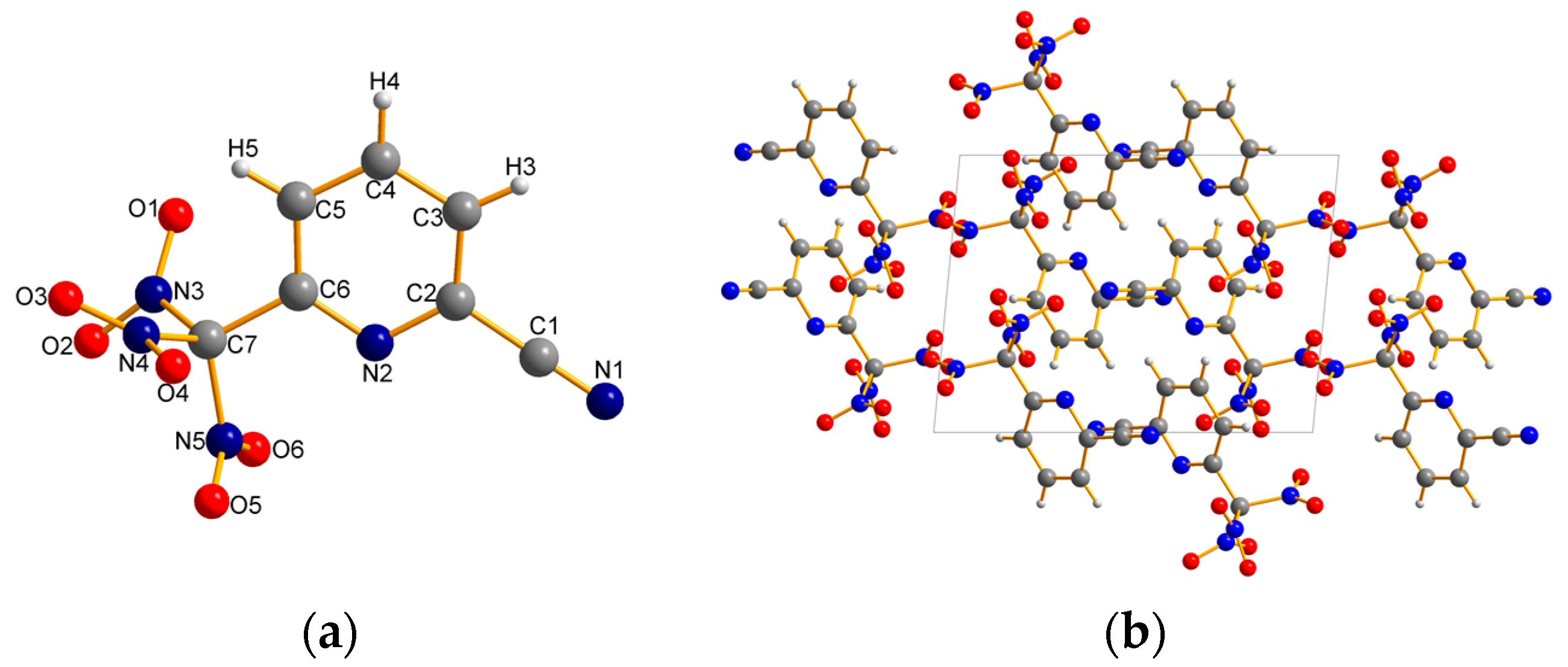
2.2. X-ray Crystallography
2.3. Physicochemical and Energetic Properties
3. Materials and Methods
3.1. General Information
3.2. Theoretical Studies
3.3. Experiment Procedures
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Note
- Ye, C.F.; Gao, H.X.; Boatz, J.A.; Drake, G.W.; Twamley, B.; Shreeve, J.M. Polyazidopyrimidines: High-energy compounds and precursors to carbon nanotubes. Angew. Chem. Int. Ed. 2006, 45, 7262–7265. [Google Scholar] [CrossRef] [PubMed]
- Göbel, M.; Klapötke, T.M. Development and testing of energetic materials: The concept of high densities based on the trinitroethyl functionality. Adv. Funct. Mater. 2009, 19, 347–365. [Google Scholar] [CrossRef]
- Xue, H.; Gao, H.X.; Twamley, B.; Shreeve, J.M. Energetic salts of 3-nitro-1,2,4-triazole-5-one, 5-nitroaminotetrazole, and other nitro-substituted azoles. Chem. Mater. 2007, 19, 1731–1739. [Google Scholar] [CrossRef]
- Thottempudi, V.; Shreeve, J.M. Synthesis and promising properties of a new family of high-density energetic salts of 5-nitro-3-trinitromethyl-1H-1,2,4-triazole and 5,5′-bis(trinitromethyl)-3,3′-azo-1H-1,2,4-triazole. J. Am. Chem. Soc. 2011, 133, 19982–19992. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.T.; Shreeve, J.M. 1,1-Diamino-2,2-dinitroethene (FOX-7) and 1-amino-1-hydrazino-2,2-dinitroethene (HFOX) as amphotères: Bases with strong acids. J. Mater. Chem. A 2015, 3, 8756–8763. [Google Scholar] [CrossRef]
- Tang, Y.X.; Zhang, J.H.; Mitchell, L.A.; Parrish, D.A.; Shreeve, J.M. Taming of 3,4-di(nitramino)furazan. J. Am. Chem. Soc. 2015, 137, 15984–15987. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H.; Xu, H.Y.; Guo, Y.; Sa, R.J.; Shreeve, J.M. Bis[3-(5-nitroimino-1,2,4-triazolate)]-based energetic salts: Synthesis and promising properties of a new family of high-density insensitive materials. J. Am. Chem. Soc. 2010, 132, 11904–11905. [Google Scholar] [CrossRef] [PubMed]
- Dippold, A.A.; Klapötke, T.M. A study of dinitro-bis-1,2,4-triazole-1,1′-diol and derivatives: Design of high-performance insensitive energetic materials by the introduction of N-oxides. J. Am. Chem. Soc. 2013, 135, 9931–9938. [Google Scholar] [CrossRef] [PubMed]
- Hermann, T.S.; Klapötke, T.M.; Krumm, B.; Stierstorfer, J. The energetic 3-trinitromethyl-5-nitramino-1H-1,2,4-triazole and nitrogen-rich salts. New J. Chem. 2017, 41, 3068–3072. [Google Scholar] [CrossRef]
- Yin, P.; He, C.L.; Shreeve, J.M. Fully C/N-polynitro-functionalized 2,2′-biimidazole derivatives as nitrogen- and oxygen-rich energetic salts. Chem. Eur. J. 2016, 22, 2108–2113. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Zhang, J.H.; He, C.L.; Parrish, D.A.; Shreeve, J.M. Polynitro-substituted pyrazoles and triazoles as potential energetic materials and oxidizers. J. Mater. Chem. A 2014, 2, 3200–3208. [Google Scholar] [CrossRef]
- Pagoria, P.F.; Lee, G.S.; Mitchell, A.R.; Schmidt, R.D. A review of energetic materials synthesis. Thermochim. Acta 2002, 384, 187–204. [Google Scholar] [CrossRef]
- Gao, H.X.; Ye, C.F.; Gupta, O.D.; Xiao, J.C.; Hiskey, M.A.; Twamley, B.; Shreeve, J.M. 2,4,5-Trinitroimidazole-based energetic salts. Chem. Eur. J. 2007, 13, 3853–3860. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.X.; Li, S.H.; Wang, Y.; Li, Y.C.; Zhao, F.Q.; Pang, S.P. Design and synthesis of energetic materials towards high density and positive oxygen balance by N-dinitromethyl functionalization of nitroazoles. J. Mater. Chem. A 2016, 4, 5495–5504. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Steemann, F.X. Dinitromethyltetrazole and its salts—A comprehensive study. Propellants Explos. Pyrotech. 2010, 35, 114–129. [Google Scholar] [CrossRef]
- Dharavath, S.; Zhang, J.H.; Imler, G.H.; Parrishc, D.A.; Shreeve, J.M. 5-(Dinitromethyl)-3-(trinitromethyl)-1,2,4-triazole and its derivatives: A new application of oxidative nitration towards gem-trinitro-based energetic materials. J. Mater. Chem. A 2017, 5, 4785–4790. [Google Scholar] [CrossRef]
- Zhang, J.H.; Dharavath, S.; Mitchell, L.A.; Parrish, D.A.; Shreeve, J.M. Energetic salts based on 3,5-bis(dinitromethyl)-1,2,4-triazole monoanion and dianion: Controllable preparation, characterization, and high performance. J. Am. Chem. Soc. 2016, 138, 7500–7503. [Google Scholar] [CrossRef] [PubMed]
- Shastin, A.V.; Godovikova, T.I.; Golova, S.P.; Kuz’min, V.S.; Khmel’nitskii, L.I.; Korsunskii, B.L. Synthesis of 2,4,6-tris(trinitromethyl)-1,3,5-triazine. Mendeleev Commun. 1995, 5, 17–18. [Google Scholar] [CrossRef]
- Yudin, I.L.; Palysaeva, N.V.; Averkiev, B.B.; Sheremetev, A.B. Unexpected formation of (trinitromethyl)pyrazines. Mendeleev Commun. 2015, 25, 193–195. [Google Scholar] [CrossRef]
- Gakh, A.A.; Khutoretskii, V.M. Formation of α-trinitromethylpyridines in the reaction of trinitromethane with pyridinium salts. Bull. Acad. Sci. USSR Div. Chem. Sci. 1983, 32, 2386. [Google Scholar] [CrossRef]
- Pal’mbakh, G.G.; Gakh, A.A.; Khutoretskii, V.M.; Chizhov, O.S. Electron-impact mass spectra of nitromethylpyridines. Bull. Acad. Sci. USSR Div. Chem. Sci. 1985, 34, 2582–2584. [Google Scholar] [CrossRef]
- Suzuki, H.; Kozai, I.; Murashima, T. Ozone-mediated C-nitration of pyridine and methylpyridines with nitrogen dioxide. J. Chem. Res. Synop. 1993, 4, 156–157. [Google Scholar]
- Katritzky, A.R.; Scriven, E.F.V.; Majumder, S.; Akhmedova, R.G.; Vakulenko, A.V.; Akhmedov, N.G.; Murugan, R.; Abboud, K.A. Preparation of nitropyridines by nitration of pyridines with nitric acid. Org. Biomol. Chem. 2005, 3, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Novikov, S.S.; Khmel’nitskii, L.I.; Novikova, T.S.; Lebedev, O.V. Reactions of N2O4 with organic compounds. VIII. Trinitromethyl derivatives of pyridine and thiophene. Chem. Heterocycl. Comp. 1970, 6, 543–545. [Google Scholar] [CrossRef]
- Lebedev, O.V.; Epishina, L.V.; Novikova, T.S.; Makhova, N.N.; Godovikova, T.I.; Golova, S.P.; Shastin, A.V.; Arnautova, E.A.; Pivana, T.S.; Khmel’nitskii, L.I. Trinitromethylarenes and trinitromethylhetarenes-synthetic ways and characterization. In Proceedings of the International Annual Conference of ICT on Energetic Materials, Karlsruhe, Germany, 30 June–3 July 1998. [Google Scholar]
- Novikov, S.S.; Khmel’nitskii, L.I.; Novikova, T.S.; Lebedev, O.V.; Epishina, L.V. The reaction of N2O4 with organic compounds IX. Nitration of the imidazole ring in the 2-position. Chem. Heterocycl. Compd. 1970, 6, 619–623. [Google Scholar] [CrossRef]
- Liu, W.; Li, S.H.; Li, Y.C.; Yang, Y.Z.; Yu, Y.; Pang, S.P. Nitrogen-rich salts based on polyamino substituted N,N′-azo-1,2,4-triazole: A new family of high performance energetic materials. J. Mater. Chem. A 2014, 2, 15978–15986. [Google Scholar] [CrossRef]
- Bakharev, V.V.; Gidaspov, A.A.; Parfenov, V.E. Chemistry of (dinitromethyl)azines. Chem. Heterocycl. Comp. 2017, 53, 659–669. [Google Scholar] [CrossRef]
- Thottempudi, V.; Kim, T.K.; Chung, K.H.; Kim, J.S. Synthesis and characterization of some polynitro imidazoles. Bull. Korean Chem. Soc. 2009, 30, 2152–2154. [Google Scholar]
- Haiges, R.; Christe, K.O. Energetic high-nitrogen compounds: 5-(trinitromethyl)-2H-tetrazole and -tetrazolates, preparation, characterization, and conversion into 5-(dinitromethyl)tetrazole. Inorg. Chem. 2013, 52, 7249–7260. [Google Scholar] [CrossRef] [PubMed]
- Frojmovic, M.M.; Just, G. On the lead tetraacetate and related oxidations of aromatic ketoximes. Can. J. Chem. 1968, 46, 3719–3726. [Google Scholar] [CrossRef]
- Rakitin, O.A.; Ogurtsov, V.A.; Godovikova, T.I.; Khmel’nitskii, L.I. Reactions of nitrile oxides with nitrogen oxides. 2. Reactions with nitrogen monoxide and nitrogen sesquioxide. Bull. Acad. Sci. USSR Div. Chem. Sci. 1990, 39, 1472–1474. [Google Scholar] [CrossRef]
- Klapöke, T.M.; Stierstorfer, J.; Weyrauther, M.; Witkowski, T.G. Synthesis and investigation of 2,6-bis(picrylamino)-3,5-dinitropyridine (PYX) and its salts. Chem. Eur. J. 2016, 22, 8619–8626. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wang, Z.X.; Wu, B.; Yang, H.W.; Ju, X.H.; Lu, C.X.; Cheng, G.B. A study of N-trinitroethyl-substituted aminofurazans: High detonation performance energetic compounds with good oxygen balance. J. Mater. Chem. A 2015, 3, 8156–8164. [Google Scholar] [CrossRef]
- Bakharev, V.V.; Gidaspov, A.A. Synthesis and molecular structure of 2-methoxy-4-amino-6-dinitromethyl-1,3,5-triazines zwitterionic salts. Russ. J. Org. Chem. 2007, 43, 1238–1242. [Google Scholar] [CrossRef]
- Bakharev, V.V.; Gidaspov, A.A.; Kosareva, E.A. Molecular structure of a zwitter-ionic 2-piperidinyl-4-dinitromethyl-1,3,5-triazin×(1H,5H)-6-one salt. J. Struct. Chem. 2007, 48, 181–185. [Google Scholar] [CrossRef]
- Ma, C.M.; Liu, Z.L.; Xu, X.J.; Yao, Q.Z. Research progress on the synthesis of energetic pyridines. Chin. J. Org. Chem. 2014, 34, 1288–1299. [Google Scholar] [CrossRef]
- Xu, Y.G.; Shen, C.; Lin, Q.H.; Wang, P.C.; Jiang, C.; Lu, M. 1-Nitro-2-trinitromethyl substituted imidazoles: A new family of high performance energetic materials. J. Mater. Chem. A 2016, 4, 17791–17800. [Google Scholar] [CrossRef]
- Tests were conducted according to the UN Recommendations on the Transport of Dangerous Goods, Manual of Tests and Criteria, 5th Rev. Ed., United Nations Publication, New York, 2009.
- Yin, P.; Parrish, D.A.; Shreeve, J.M. Energetic multifunctionalized nitraminopyrazoles and their ionic derivatives: Ternary hydrogen-bond induced high energy density materials. J. Am. Chem. Soc. 2015, 137, 4778–4786. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian Development Version; Revision F.02; Gaussian, Inc.: Wallingford, CT, USA, 2006. [Google Scholar]
- Suceska, M. EXPLO5 V6.01; Brodarski Institute: Zagreb, Croatia, 2012. [Google Scholar]
- Hehre, W.J.; Ditchfield, R.; Radom, L.; Pople, J.A. Molecular orbital theory of the electronic structure of organic compounds. V. molecular theory of bond separation. J. Am. Chem. Soc. 1970, 92, 4796–4801. [Google Scholar] [CrossRef]
- Guo, Y.; Tao, G.H.; Zeng, Z.; Gao, H.X.; Parrish, D.A.; Shreeve, J.M. Energetic salts based on monoanions of N,N-bis(1H-tetrazol-5-yl)amine and 5,5′-bis(tetrazole). Chem. Eur. J. 2010, 16, 3753–3762. [Google Scholar] [CrossRef] [PubMed]
- Kamlet, M.J.; Jacobs, S.J. Chemistry of detonations. I. A simple method for calculating detonation properties of C-H-N-O explosives. J. Chem. Phys. 1968, 48, 23–35. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Ablard, J.E. Chemistry of detonations. II. Buffered equilibria. J. Chem. Phys. 1968, 48, 36–42. [Google Scholar] [CrossRef]
- Kamlet, M.J.; Dicknison, C. Chemistry of detonations. III. Evaluation of the simplified calculational method for Chapman-Jouguet detonation pressures on the basis of available experimental information. J. Chem. Phys. 1968, 48, 43–49. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| CCDC | 1,582,435 | 1,583,118 | 1,582,533 | 1,582,458 | 1,583,040 |
| Empirical formula | C6H4N4O6 | C7H3N7O12 | C7H3N5O6 | C6H9N5O4 | C7H13N9O8 |
| Formula mass | 228.12 | 377.14 | 253.13 | 215.17 | 351.26 |
| Temperature/K | 153(2) | 105 | 126 | 153(2) | 153(2) |
| Crystal system | monoclinic | triclinic | monoclinic | monoclinic | orthorhombic |
| Space group | P21/c | P | P21/c | P21/n | Pbcn |
| a/Å | 8.0672(16) | 7.5655(3) | 11.2611(5) | 11.672(2) | 27.413(6) |
| b/Å | 10.496(2) | 11.7552(4) | 10.6182(4) | 6.5976(13) | 7.2092(14) |
| c/Å | 11.234(2) | 31.9329(11) | 8.2766(3) | 12.193(2) | 6.6259(13) |
| α/° | 90.00 | 79.435(3) | 90.00 | 90.00 | 90.00 |
| β/° | 109.66(3) | 87.936(3) | 95.406(3) | 109.73(3) | 90.00 |
| γ/° | 90.00 | 78.473(3) | 90.00 | 90.00 | 90.00 |
| Z | 4 | 8 | 4 | 4 | 4 |
| Volume/Å3 | 895.7(3) | 2735.47(17) | 985.26(7) | 883.8(3) | 1309.4(4) |
| ρcalc/g∙cm−3 | 1.692 | 1.832 | 1.707 | 1.617 | 1.782 |
| μ/mm−1 | 0.154 | 0.178 | 0.152 | 0.137 | 0.160 |
| F(000) | 464 | 1520 | 512 | 448 | 728 |
| Crystal size/mm3 | 0.13 × 0.12 × 0.08 | 0.50 × 0.15 × 0.14 | 0.40 × 0.35 × 0.30 | 0.15 × 0.13 × 0.10 | 0.16 × 0.14 × 0.03 |
| θ range/° | 3.31–27.49 | 3.04–26.00 | 3.13–26.00 | 2.97–27.50 | 2.92–27.48 |
| Index ranges | −10 ≤ h ≤ 10; −13 ≤ k ≤ 13; −13 ≤ l ≤ 14 | −9 ≤ h ≤ 9; −14 ≤ k ≤ 14; −32 ≤ l ≤ 39 | −13 ≤ h ≤ 9; −13 ≤ k ≤ 12; −10 ≤ l ≤ 10 | −14 ≤ h ≤ 15; −8 ≤ k ≤ 8; −15 ≤ l ≤ 15 | −35 ≤ h ≤ 35; −9 ≤ k ≤ 6; −7 ≤ l ≤ 8 |
| Reflections collected | 6328 | 24085 | 4005 | 5836 | 5938 |
| Independent reflections | 2028 | 10754 | 1927 | 2007 | 1498 |
| Goodness-of-fit on F2 | 1.203 | 1.077 | 1.073 | 1.122 | 1.157 |
| Final R indexes [I > 2σ(I) i.e., Fo > 4σ(Fo)] | R1 = 0.0556; wR2 = 0.1121 | R1 = 0.0552; wR2 = 0.1422 | R1 = 0.0339; wR2 = 0.0785 | R1 = 0.0456; wR2 = 0.1158 | R1 = 0.0865; wR2 = 0.2420 |
| Final R indexes [all data] | R1 = 0.0633; wR2 = 0.1171 | R1 = 0.0607; wR2 = 0.1481 | R1 = 0.0397; wR2 = 0.0820 | R1 = 0.0477; wR2 = 0.1175 | R1 = 0.0920; wR2 = 0.2500 |
| Compd | Tm a | Td b | ρ c | △fHM d | D e | P f | IS g | FS h | OB i |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 70 | 114 | 1.62/1.69 j | 160.65 | 7678 | 24.2 | 16 | >360 | −56.11 |
| 2 | 96 | 101 | 1.77/1.83 j | 258.96 | 8700 | 33.2 | 9 | 192 | −14.85 |
| 3 | 68 | 106 | 1.62/1.71 j | 314.22 | 7683 | 23.9 | 27 | >360 | −60.04 |
| 4 | - | 181 | 1.56/1.62 j | 151.32 | 7438 | 19.6 | 14 | >360 | −92.94 |
| 5 | - | 125 | 1.71/1.78 j | 150.80 | 8383 | 27.7 | 11.5 | >360 | −56.94 |
| TNPyO k | 170 | 170 | 1.86 | - | 8369 | - | 1.5–3.0 | 160 | −27.8 |
| RDX l | 204 | 230 | 1.80 | 92.6 | 8795 | 34.9 | 7.4 | 120 | −21.62 |
| TNT l | 81 | 295 | 1.65 | −59.4 | 7303 | 21.3 | 15 | >353 | −74.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Sun, X.; Yu, S.; Bao, L.; Sun, C.; Pang, S. Energetic Di- and Trinitromethylpyridines: Synthesis and Characterization. Molecules 2018, 23, 2. https://doi.org/10.3390/molecules23010002
Zhang Y, Sun X, Yu S, Bao L, Sun C, Pang S. Energetic Di- and Trinitromethylpyridines: Synthesis and Characterization. Molecules. 2018; 23(1):2. https://doi.org/10.3390/molecules23010002
Chicago/Turabian StyleZhang, Yiying, Xiaoyu Sun, Shannan Yu, Lingxiang Bao, Chenghui Sun, and Siping Pang. 2018. "Energetic Di- and Trinitromethylpyridines: Synthesis and Characterization" Molecules 23, no. 1: 2. https://doi.org/10.3390/molecules23010002
APA StyleZhang, Y., Sun, X., Yu, S., Bao, L., Sun, C., & Pang, S. (2018). Energetic Di- and Trinitromethylpyridines: Synthesis and Characterization. Molecules, 23(1), 2. https://doi.org/10.3390/molecules23010002