Investigation into Improving the Aqueous Solubility of the Thieno[2,3-b]pyridine Anti-Proliferative Agents


 ,
,  , ,
, ,  and
and 
Abstract
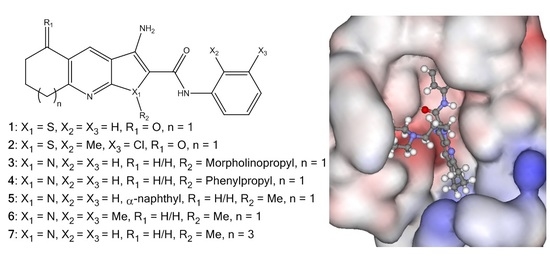
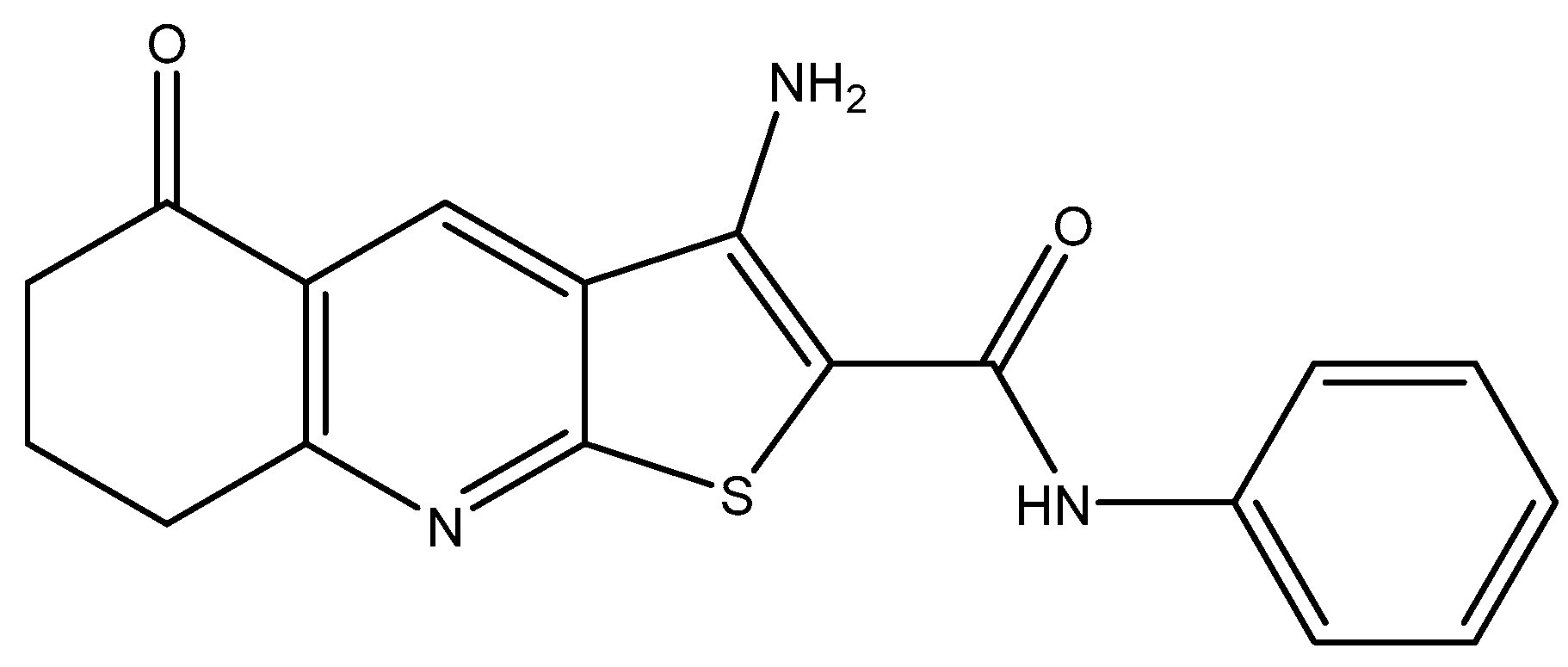
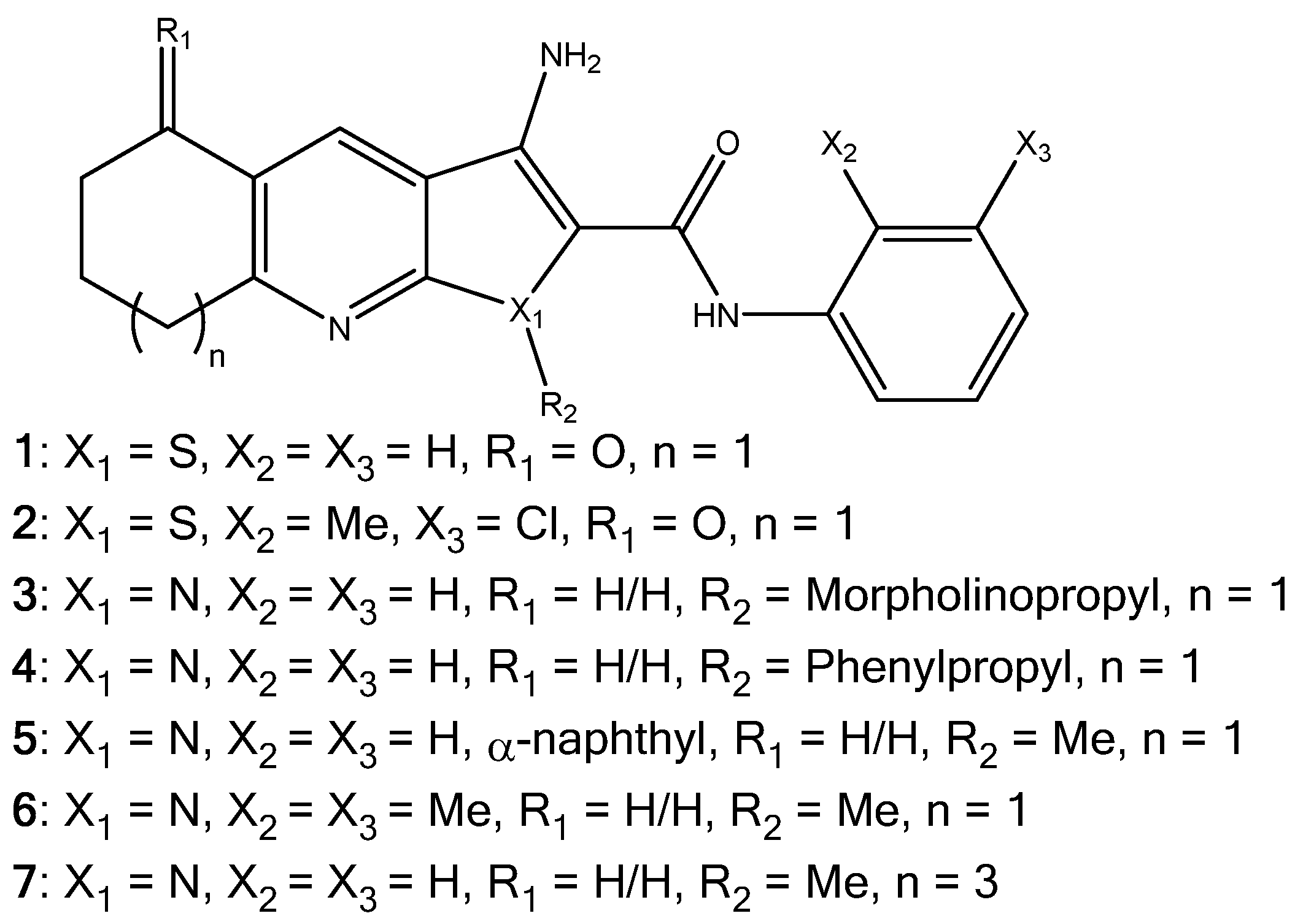
1. Introduction
2. Results and Discussion
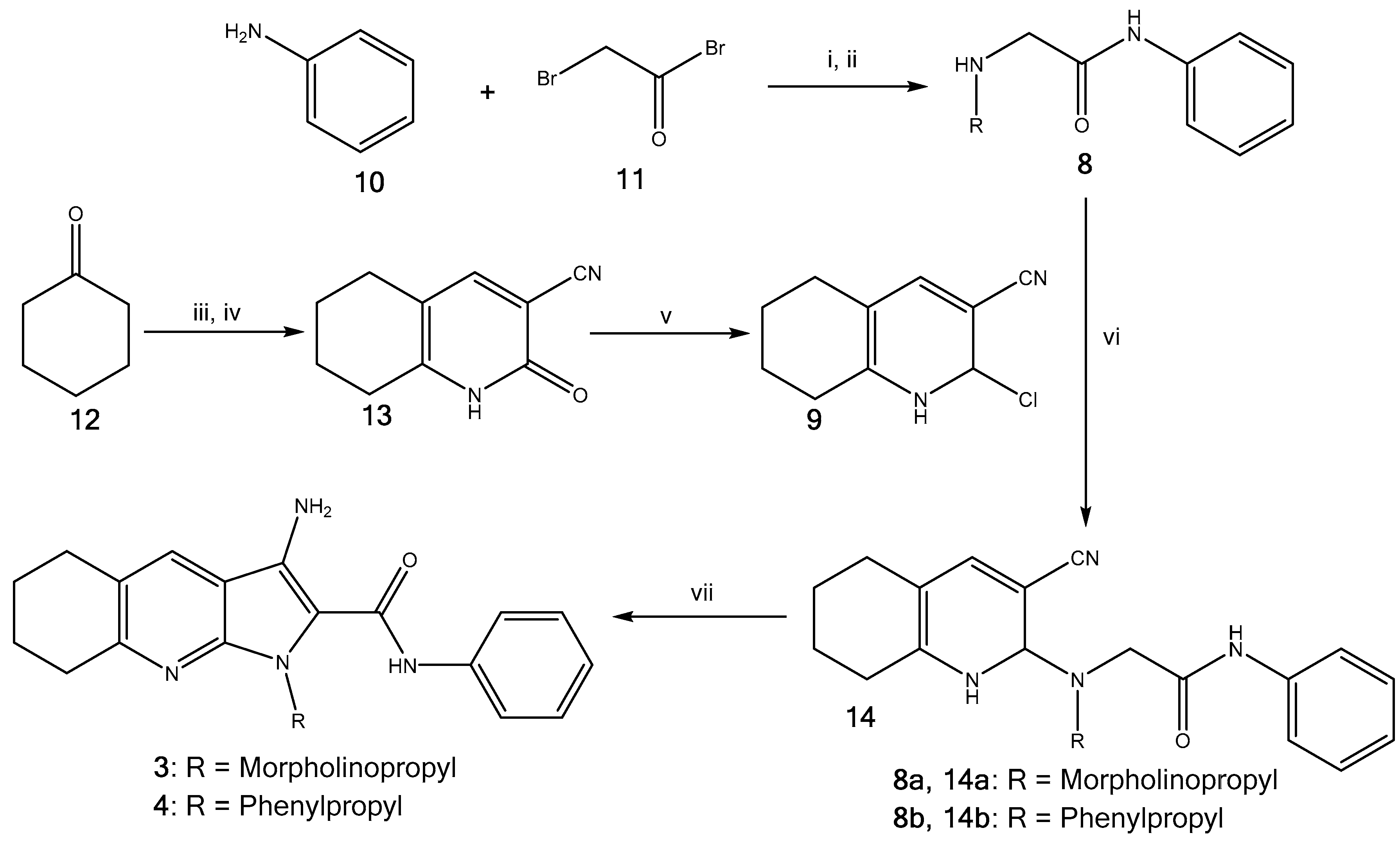
2.1. Synthesis of Pyrrolo[2,3-b]pyridine Derivatives
2.2. Water Solubility Testing
2.3. Cell Proliferation
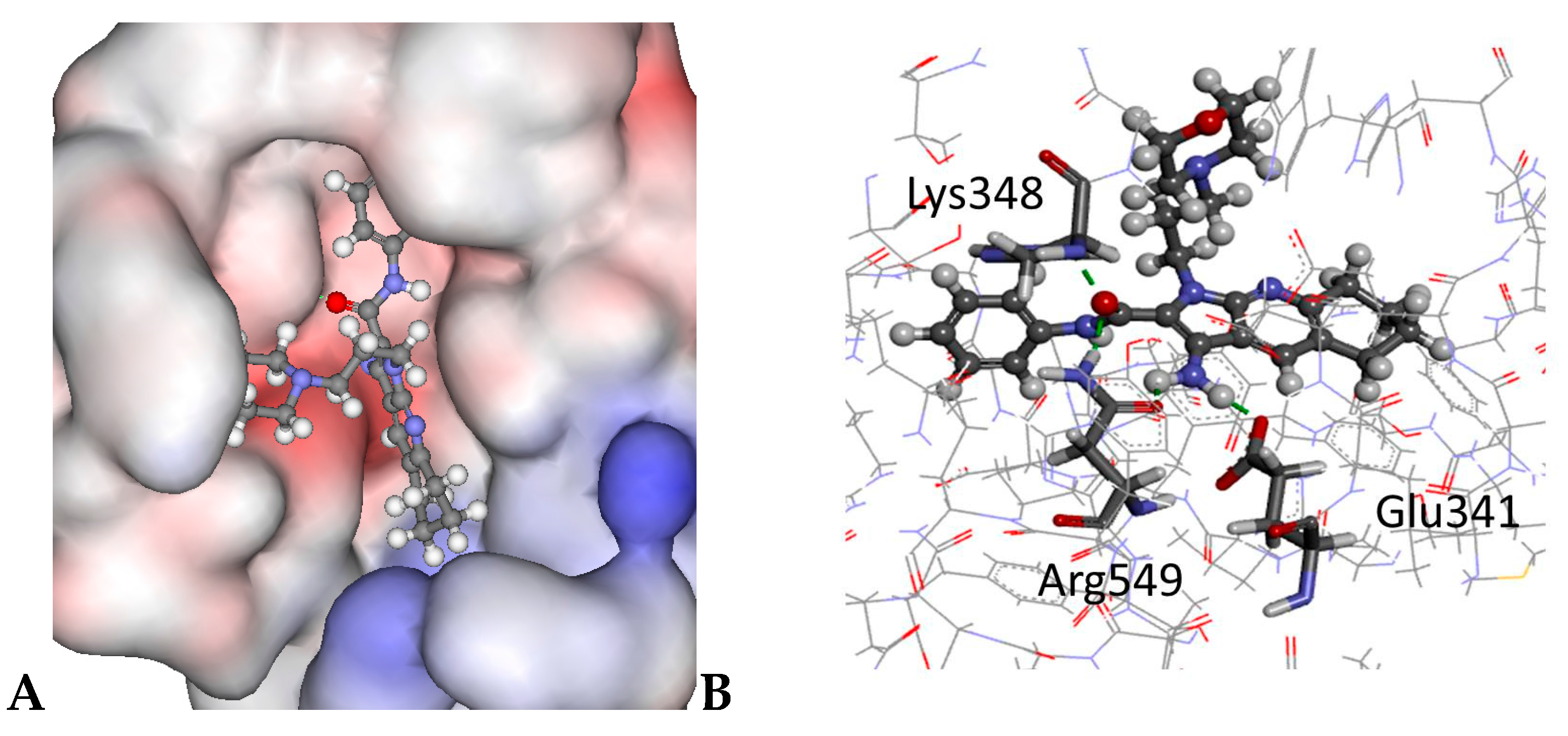
2.4. Molecular Modelling
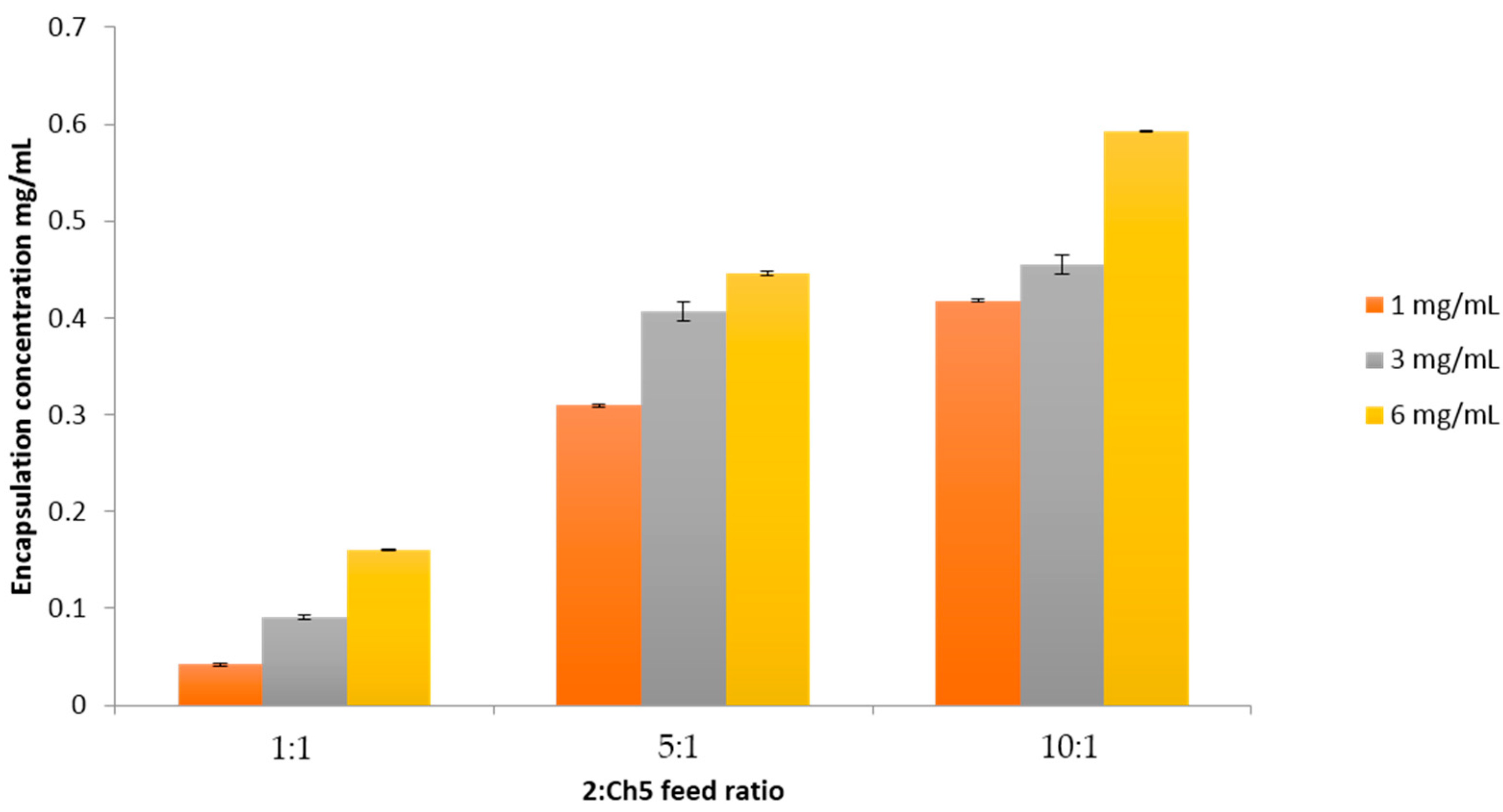
2.5. Polymer Encapsulation
3. Materials and Methods
3.1. Drug Solubility
3.2. Cell Proliferation Assay
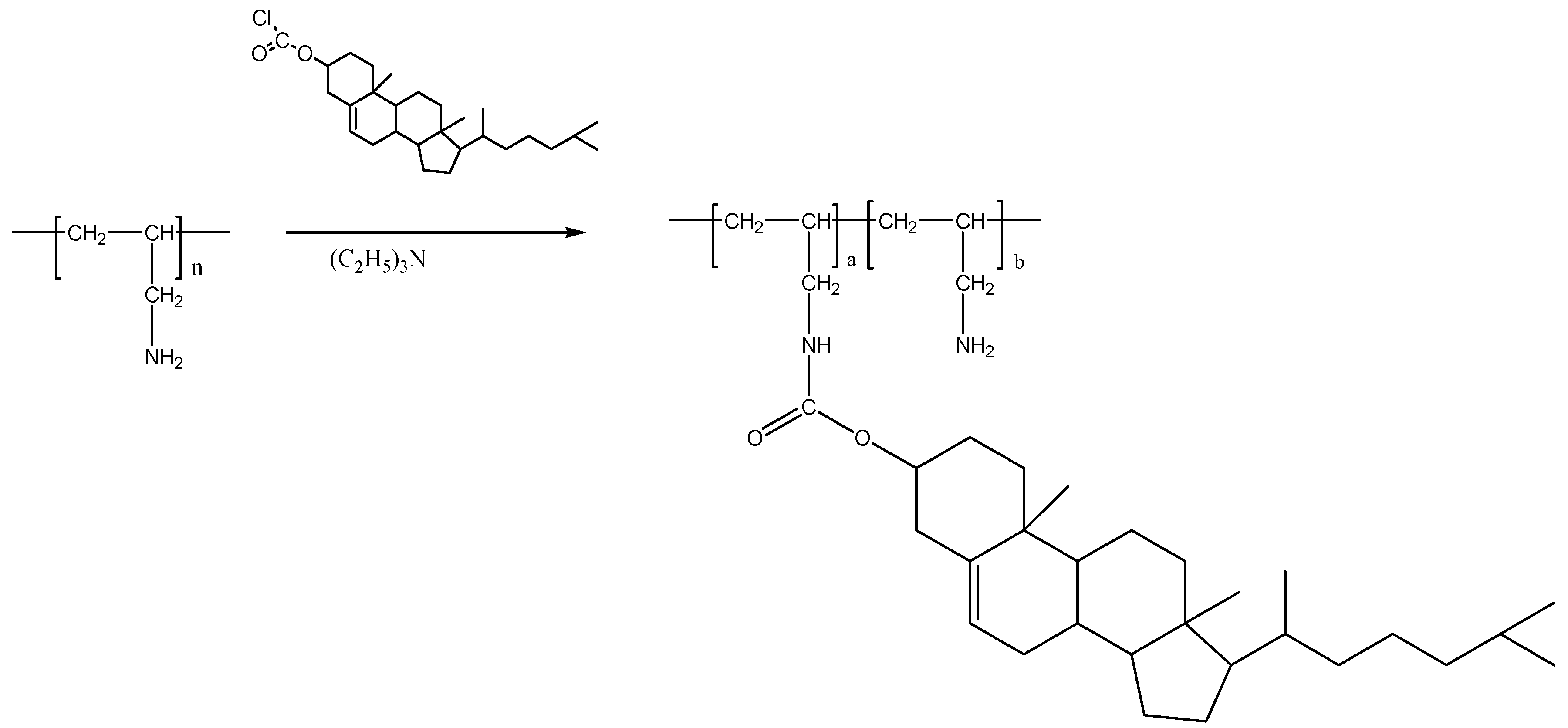
3.3. Preparation of Cholesteryl-Poly(allylamine) (Ch5)
3.3.1. 1H NMR Spectra of Ch5
3.3.2. Elemental Analysis of Ch5 Polymer
3.3.3. FTIR of Ch5 Polymer
3.4. Drug Loading
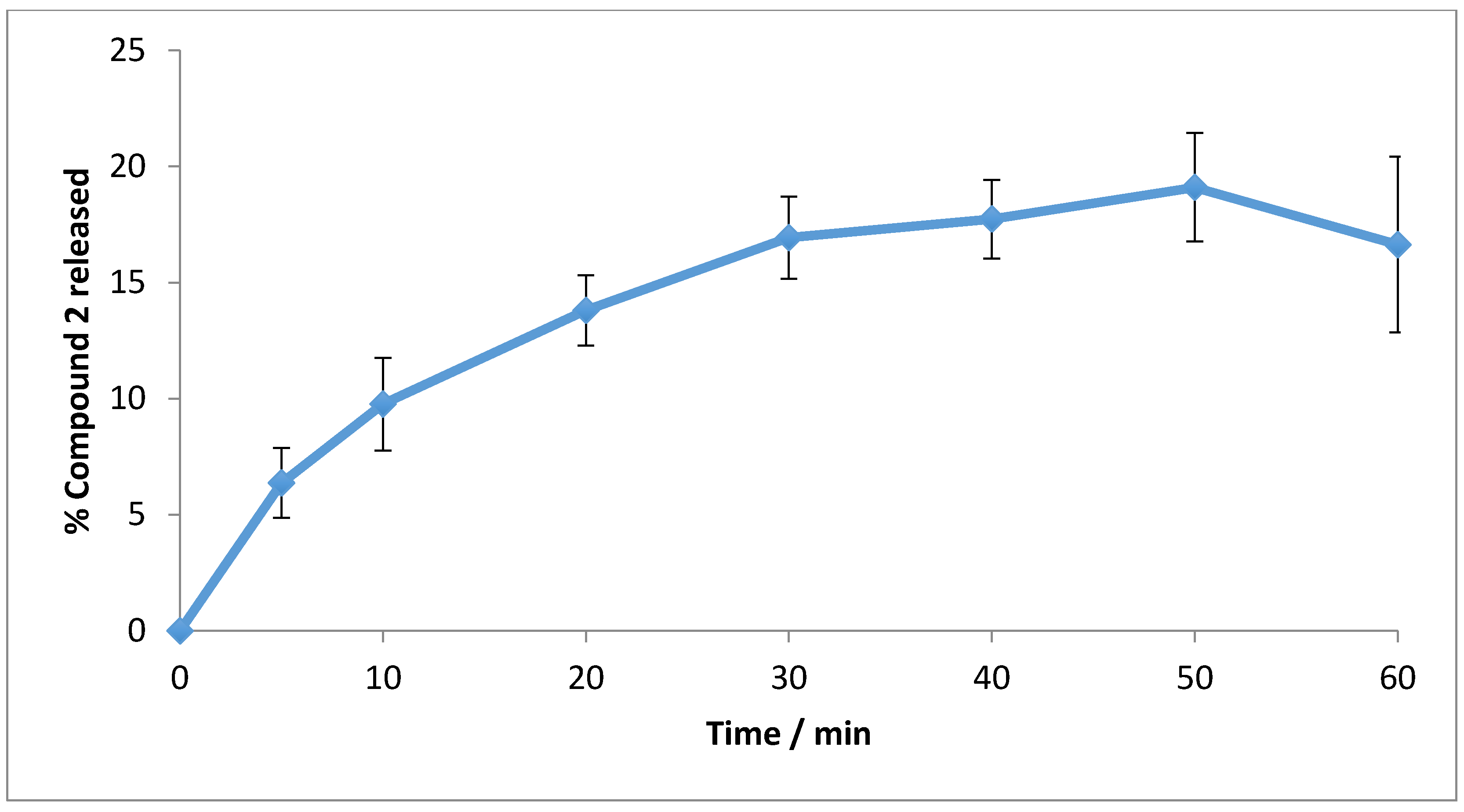
3.5. In Vitro Drug Release Studies
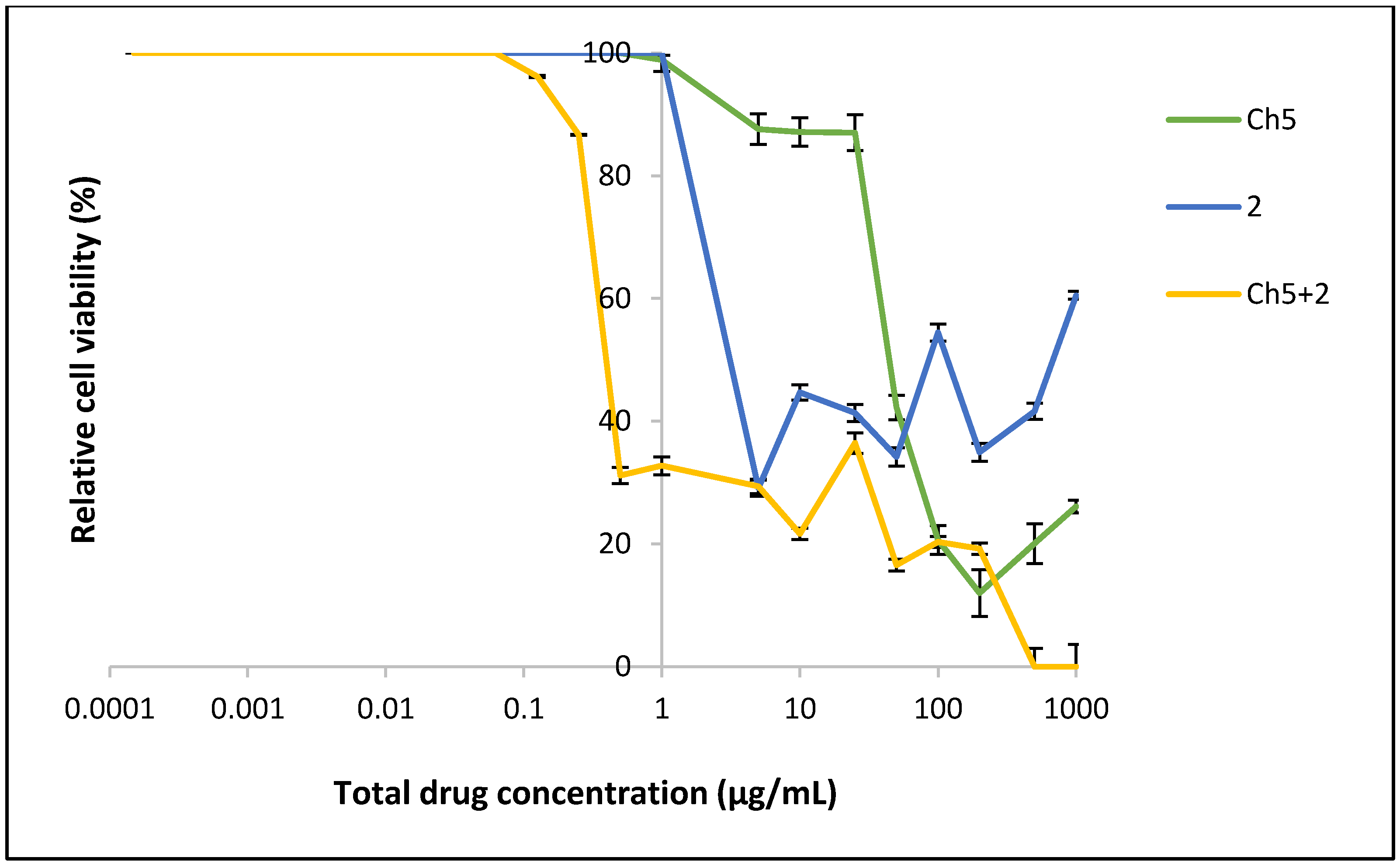
3.6. Cytotoxicity of Formulation Using MTT Assay
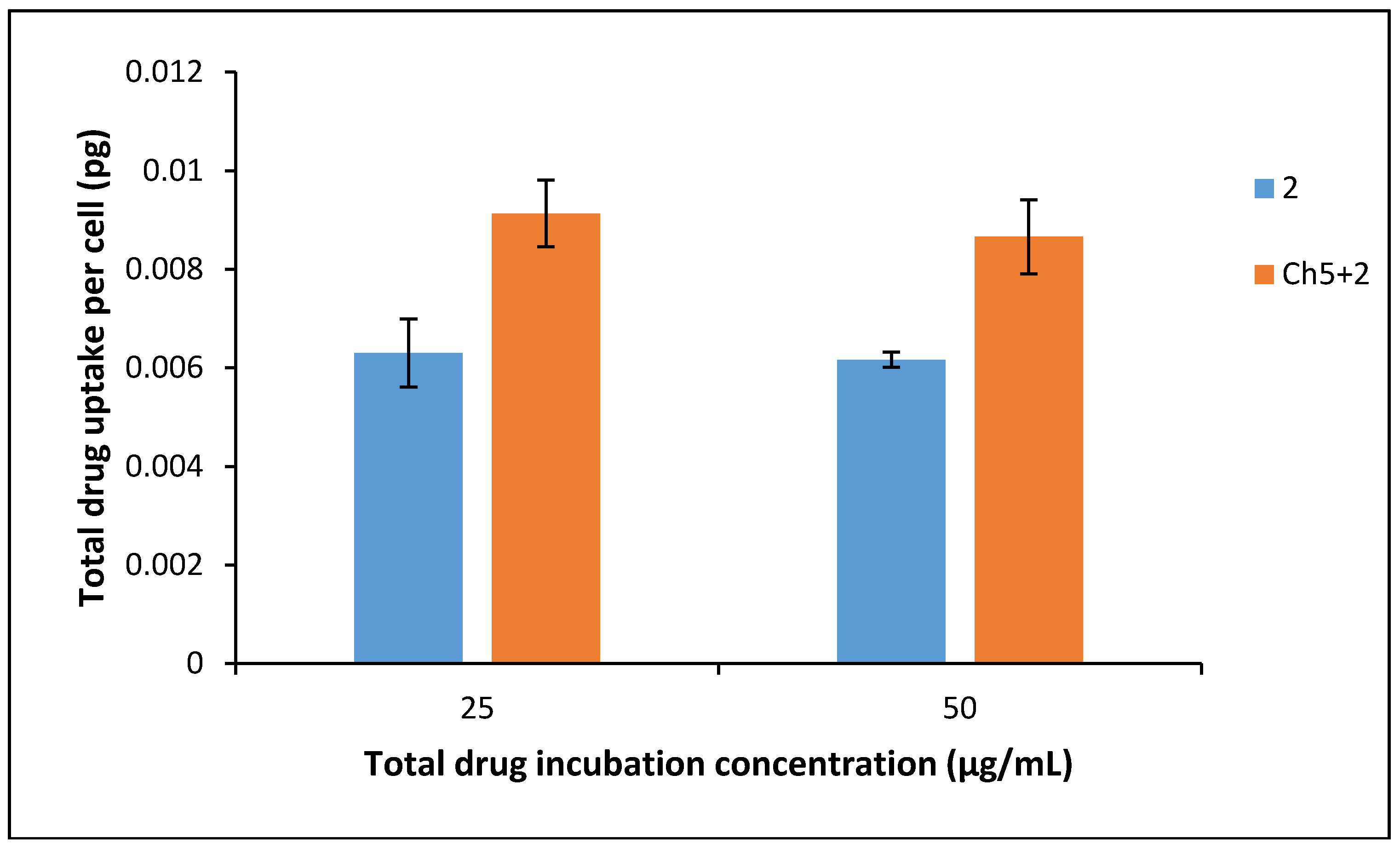
3.7. Drug Uptake into BxPC-3 Cells
3.8. Molecular Modelling
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Feng, L.; Reynisdóttir, I.; Reynisson, J. The effect of PLC-gamma2 inhibitors on the growth of human tumour cells. Eur. J. Med. Chem. 2012, 54, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Arabshahi, H.J.; Leung, E.; Barker, D.; Reynisson, J. The development of thieno[2,3-b]pyridine analogues as anticancer agents applying in silico methods. Med. Chem. Commun. 2014, 5, 186–191. [Google Scholar] [CrossRef]
- Hung, J.M.; Arabshahi, H.J.; Leung, E.; Reynisson, J.; Barker, D. Synthesis and cytotoxicity of thieno[2,3-b]pyridine and furo[2,3-b]pyridine derivatives. Eur. J. Med. Chem. 2014, 86, 420–437. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, J.; Court, W.; O’Neill, C.; Day, J.; Patterson, L.; McDonald, E.; Workman, P.; Katan, M.; Eccles, S.A. The identification of novel PLC-c inhibitors using virtual high throughput screening. Bioorg. Med. Chem. 2009, 17, 3169–3176. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Hung, J.M.; Barker, D.; Reynisson, J. The effect of a thieno[2,3-b]pyridine PLC-γ inhibitor on DNA synthesis, morphology, migration and cell cycle of breast cancer cells. Med. Chem. Commun. 2014, 5, 99–106. [Google Scholar] [CrossRef]
- Reynisson, J.; Jaiswal, J.K.; Barker, D.; D’mello, S.A.N.; Denny, W.A.; Baguley, B.; Leung, E. Evidence that phospholipase C is involved in the antitumour action of NSC768313, a new thieno[2,3-b]pyridine derivative. Cancer Cell Int. 2016, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Arabshahi, H.J.; van Rensburg, M.; Pilkington, L.I.; Jeon, C.Y.; Song, M.; Gridel, L.-M.; Leung, E.; Barker, D.; Vuica-Ross, M.; Volcho, K.P.; et al. A synthesis, in silico, in vitro and in vivo study of thieno[2,3-b]pyridine anticancer analogues. Med. Chem. Commun. 2015, 6, 1987–1997. [Google Scholar] [CrossRef]
- Katritch, V.; Jaakola, V.; Lane, J.R.; Lin, J.; IJzerman, A.P.; Yeager, M.; Kufareva, I.; Stevens, R.C.; Abagyan, R. Structure-Based Discovery of Novel Chemotypes for Adenosine A2A Receptor Antagonists. J. Med. Chem. 2010, 53, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, C.; Shan, C.; You, Q.; Lu, J.; Elf, S.; Zhou, Y.; Wen, Y.; Vinkenborg, J.L.; Fan, J.; et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem. 2015, 7, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Eurtivong, C.; Semenov, V.; Semenova, M.; Konyushkin, L.; Atamanenko, O.; Reynisson, J.; Kiselyov, A. 3-Amino-thieno[2,3-b]pyridines as microtubule-destabilising agents: Molecular modelling and biological evaluation in the sea urchin embryo and human cancer cells. Bioorg. Med. Chem. 2017, 25, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Bassetto, M.; Brancale, A.; Hamel, E.; Castagliuolo, I.; Bortolozzi, R.; et al. Synthesis and Biological Evaluation of 2-(Alkoxycarbonyl)-3-Anilinobenzo[b]thiophenes and Thieno[2,3-b]pyridines as New Potent Anticancer Agents. J. Med. Chem. 2013, 56, 2606–2618. [Google Scholar] [CrossRef] [PubMed]
- Huuskonen, J.; Salo, M.; Taskinen, J. Aqueous Solubility Prediction of Drugs Based on Molecular Topology and Neural Network Modeling. J. Chem. Inf. Comput. Sci. 1998, 38, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Llinàs, A.; Glen, R.C.; Goodman, J.M. Solubility Challenge: Can You Predict Solubilities of 32 Molecules Using a Database of 100 Reliable Measurements? J. Chem. Inf. Model. 2008, 48, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Pilkington, L.I.; Haverkate, N.A.; van Rensburg, M.; Reynisson, J.; Leung, E.; Barker, D. Synthesis of 3-Amino-2-carboxamide Tetrahydropyrrolo[2,3-b]quinolines. Synlett 2016, 27, 2811–2814. [Google Scholar] [CrossRef]
- Apley, M.; Crist, G.B.; Fellner, V.; Gonzalez, M.A.; Hunter, R.P.; Martinez, M.N.; Messenheimer, J.R.; Modric, S.; Papich, M.G.; Parr, A.F.; et al. Determination of Thermodynamic Solubility of Active Pharmaceutical Ingredients for Veterinary Species: A New USP General Chapter. Pharm. Forum 2015, 41. [Google Scholar] [CrossRef]
- Loftsson, T.; Vogensen, S.B.; Desbos, C.; Jansook, P. Carvedilol: Solubilization and cyclodextrin complexation. A technical note. AAPS PharmSciTech 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Reynisson, J. Hydration Free Energy as a Molecular Descriptor in Drug Design: A Feasibility Study. Mol. Inf. 2016, 35, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The NCI60 Human Tumour Cell line Anticancer Drug Screen. Nat. Rev. Drug Dis. 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.; Pilkington, L.I.; van Rensburg, M.; Jeon, C.Y.; Song, M.; Arabshahi, H.J.; De Zoysa, G.H.; Sarojini, V.; Denny, W.A.; Reynisson, J.; et al. Synthesis and cytotoxicity of thieno[2,3-b]quinoline-2-carboxamide and cycloalkyl[b]thieno [3,2-e]pyridine-2-carboxamide derivatives. Bioorg. Med. Chem. 2016, 24, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Van Rensburg, M.; Leung, E.; Haverkate, N.A.; Eurtivong, C.; Pilkington, L.I.; Reynisson, J.; Barker, D. Synthesis and antiproliferative activity of 2-chlorophenyl carboxamide thienopyridines. Bioorg. Med. Chem. Lett. 2017, 27, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.N.; Pommier, Y.; Marchand, C. Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) inhibitors. Expert Opin. Ther. Pat. 2011, 21, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Gillies, E.R.; Fréchet, J.M.J. Dendrimers and dendritic polymers in drug delivery. Drug Discov. Today 2005, 10, 35–43. [Google Scholar] [CrossRef]
- Aliabadi, H.M.; Lavasanifar, A. Polymeric micelles for drug delivery. Expert Opin. Drug Deliv. 2006, 3, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, C.; Ouaissi, M.; Lima, S.C.; Cheng, W.P.; Loureirio, I.; Mas, E.; Lombardo, D.; Cordeiro-da-Silva, A.; Ouaissi, A.; Kong Thoo Lin, P. In vitro and in vivo anticancer activity of a novel nano-sized formulation based on self-assembling polymers against pancreatic cancer. Pharm. Res. 2010, 27, 2694–2703. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Barnett, C.M.; Gueorguieva, M.; Lees, M.R.; McGarvey, D.J.; Hoskins, C. Physical stability, biocompatibility and potential use of hybrid iron oxide-gold nanoparticles as drug carriers. J. Nanopart. Res. 2013, 15, 1706. [Google Scholar] [CrossRef]
- Cartiera, M.S.; Johnson, K.M.; Rajendran, V.; Caplan, M.J.; Saltzman, W.M. The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials 2009, 30, 2790–2798. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.Y.; Kim, J.E.; Askarian-Amiri, M.; Rewcastle, G.W.; Finlay, G.J.; Baguley, B.C. Relationships between Signaling Pathway Usage and Sensitivity to a Pathway Inhibitor: Examination of Trametinib Responses in Cultured Breast Cancer Lines. PLoS ONE 2014, 9, e105792. [Google Scholar] [CrossRef] [PubMed]
- Alley, M.C.; Scudiero, D.A.; Monks, P.A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrazolium Assay. Cancer Res. 1988, 48, 589–601. [Google Scholar] [PubMed]
- Boyd, M.R.; Paull, K.D. Some Practical Considerations and Applications of the National Cancer Institute In Vitro Anticancer Drug Discovery Screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Essen, L.O.; Perisic, O.; Katan, M.; Wu, Y.; Roberts, M.F.; Williams, R.L. Structural mapping of the catalytic mechanism for a mammalian phosphoinositide-specific phospholipase C. Biochemistry 1997, 36, 1704–1718. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R.; Interthal, H.; Champoux, J.J.; Hol, W.G.J. Crystal Structure of a Human Tyrosyl-DNA Phosphodiesterase (Tdp1)-Tungstate Complex. J. Mol. Biol. 2003, 324, 917–932. [Google Scholar] [CrossRef]
- Wernimont, A.K.; Huffman, D.L.; Lamb, A.L.; O'Halloran, T.V.; Rosenzweig, A.C. Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins. Nat. Struct. Biol. 2000, 7, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The Novel Microtubule-Destabilizing Drug BAL27862 Binds to the Colchicine Site of Tubulin with Distinct Effects on Microtubule Organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willet, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.; Murray, C.; Auton, T.R.; Paolini, G.V.; Mee, P.M. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput.-Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Mooij, W.T.M.; Verdonk, M.L. General and targeted statistical potentials for protein–ligand interactions. Proteins 2005, 61, 272–287. [Google Scholar] [CrossRef] [PubMed]
- Scigress Ultra v. F.J 2.6 FujitsuLimited; Fujistu: Tokyo, Japan, 2000.
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- QikProp; v3.2; Schrödinger: New York, NY, USA, 2009.
- Ioakimidis, L.; Thoukydidis, L.; Naeem, S.; Mirza, A.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental S | Calculated S | ||||
|---|---|---|---|---|---|
| μg/mL | log S a | μg/mL | log S a | log P | |
| 1 | 1.21 | −5.4 | 13.4 | −4.4 | 2.9 |
| 2 | 3.53 | −5.0 | 96.9 | −3.6 | 2.2 |
| 3 | 1339.8/1336.7 b | −2.5 c | 1089.1 | −2.6 | 2.9 |
| 4 | 0.20 | −6.3 | 1.1 | −5.6 | 5.3 |
| 5 | 0.18 | −6.3 | 5.9 | −4.8 | 4.0 |
| 6 | 0.76 | −5.7 | 27.7 | −4.1 | 3.3 |
| 7 | 0.61 | −5.8 | 13.9 | −4.4 | 3.5 |
| Hydrodynamic Diameter (nm) | Polydispersity Index | Zeta Potential, mV | |
|---|---|---|---|
| Ch5 | 189 ± 1.800 | 0.246 ± 0.010 | +39.6 (2.242) |
| Ch5+2 | 286 ± 5.839 | 0.267 ± 0.011 | +36.5 (1.635) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zafar, A.; Pilkington, L.I.; Haverkate, N.A.; Van Rensburg, M.; Leung, E.; Kumara, S.; Denny, W.A.; Barker, D.; Alsuraifi, A.; Hoskins, C.; et al. Investigation into Improving the Aqueous Solubility of the Thieno[2,3-b]pyridine Anti-Proliferative Agents. Molecules 2018, 23, 145. https://doi.org/10.3390/molecules23010145
Zafar A, Pilkington LI, Haverkate NA, Van Rensburg M, Leung E, Kumara S, Denny WA, Barker D, Alsuraifi A, Hoskins C, et al. Investigation into Improving the Aqueous Solubility of the Thieno[2,3-b]pyridine Anti-Proliferative Agents. Molecules. 2018; 23(1):145. https://doi.org/10.3390/molecules23010145
Chicago/Turabian StyleZafar, Ayesha, Lisa I. Pilkington, Natalie A. Haverkate, Michelle Van Rensburg, Euphemia Leung, Sisira Kumara, William A. Denny, David Barker, Ali Alsuraifi, Clare Hoskins, and et al. 2018. "Investigation into Improving the Aqueous Solubility of the Thieno[2,3-b]pyridine Anti-Proliferative Agents" Molecules 23, no. 1: 145. https://doi.org/10.3390/molecules23010145
APA StyleZafar, A., Pilkington, L. I., Haverkate, N. A., Van Rensburg, M., Leung, E., Kumara, S., Denny, W. A., Barker, D., Alsuraifi, A., Hoskins, C., & Reynisson, J. (2018). Investigation into Improving the Aqueous Solubility of the Thieno[2,3-b]pyridine Anti-Proliferative Agents. Molecules, 23(1), 145. https://doi.org/10.3390/molecules23010145