SistematX, an Online Web-Based Cheminformatics Tool for Data Management of Secondary Metabolites

 ,
,  , ,
, ,  and
and
Abstract
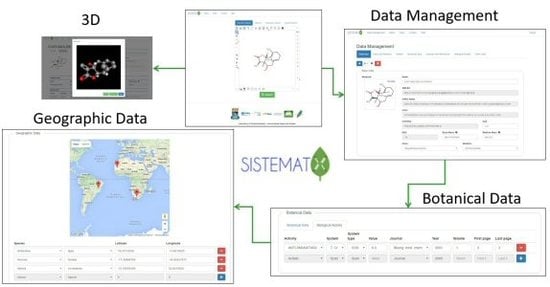
1. Introduction
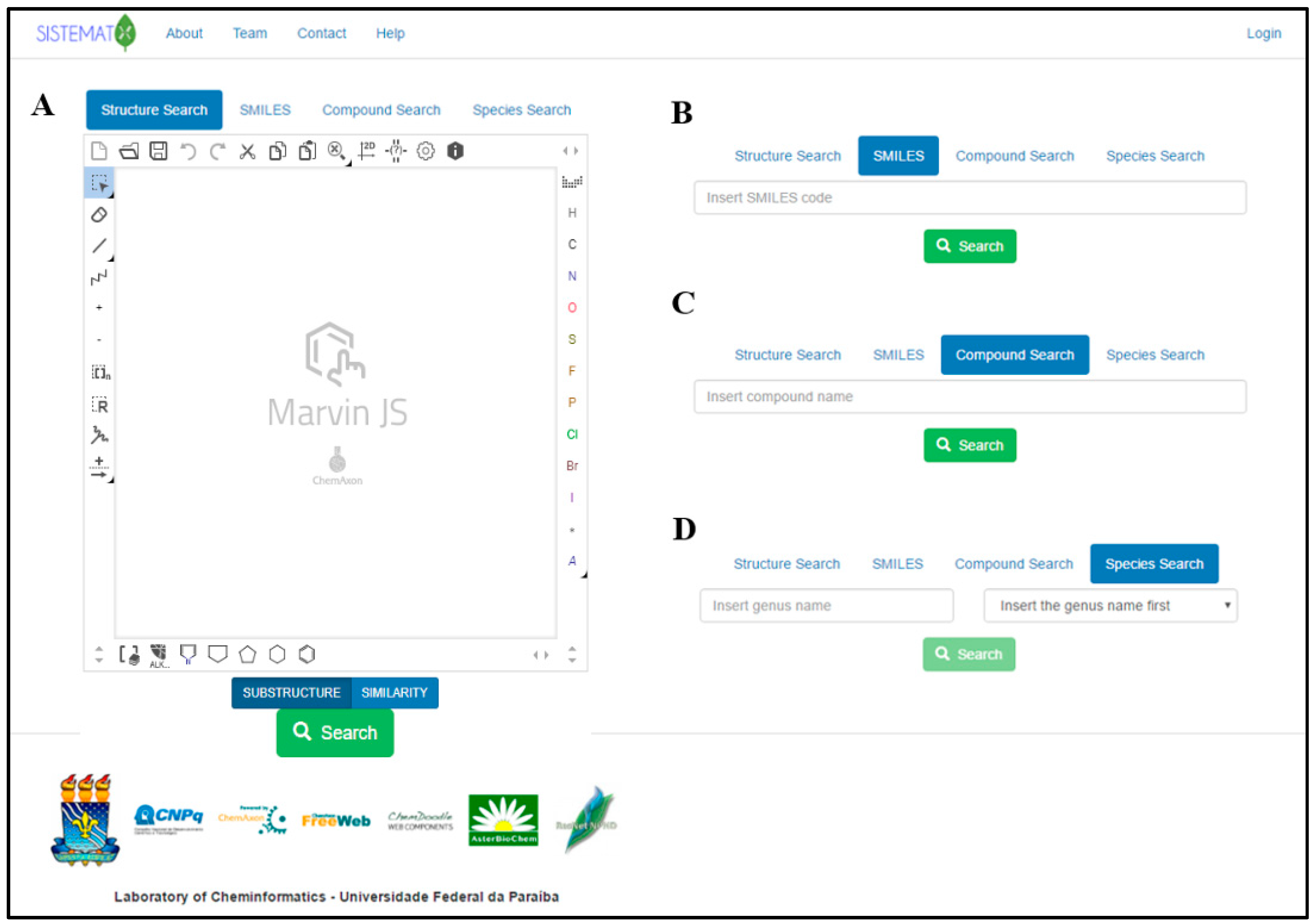
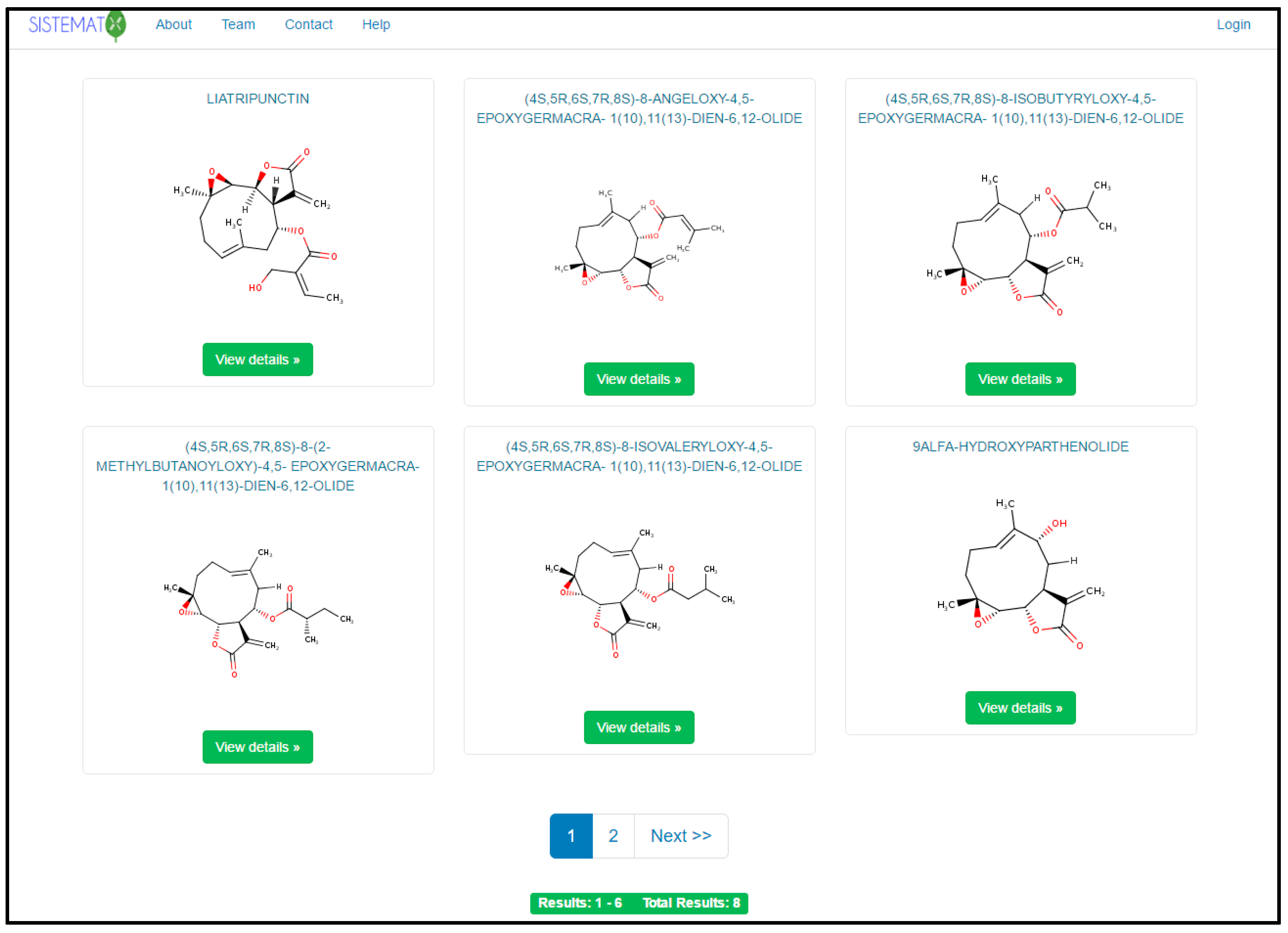
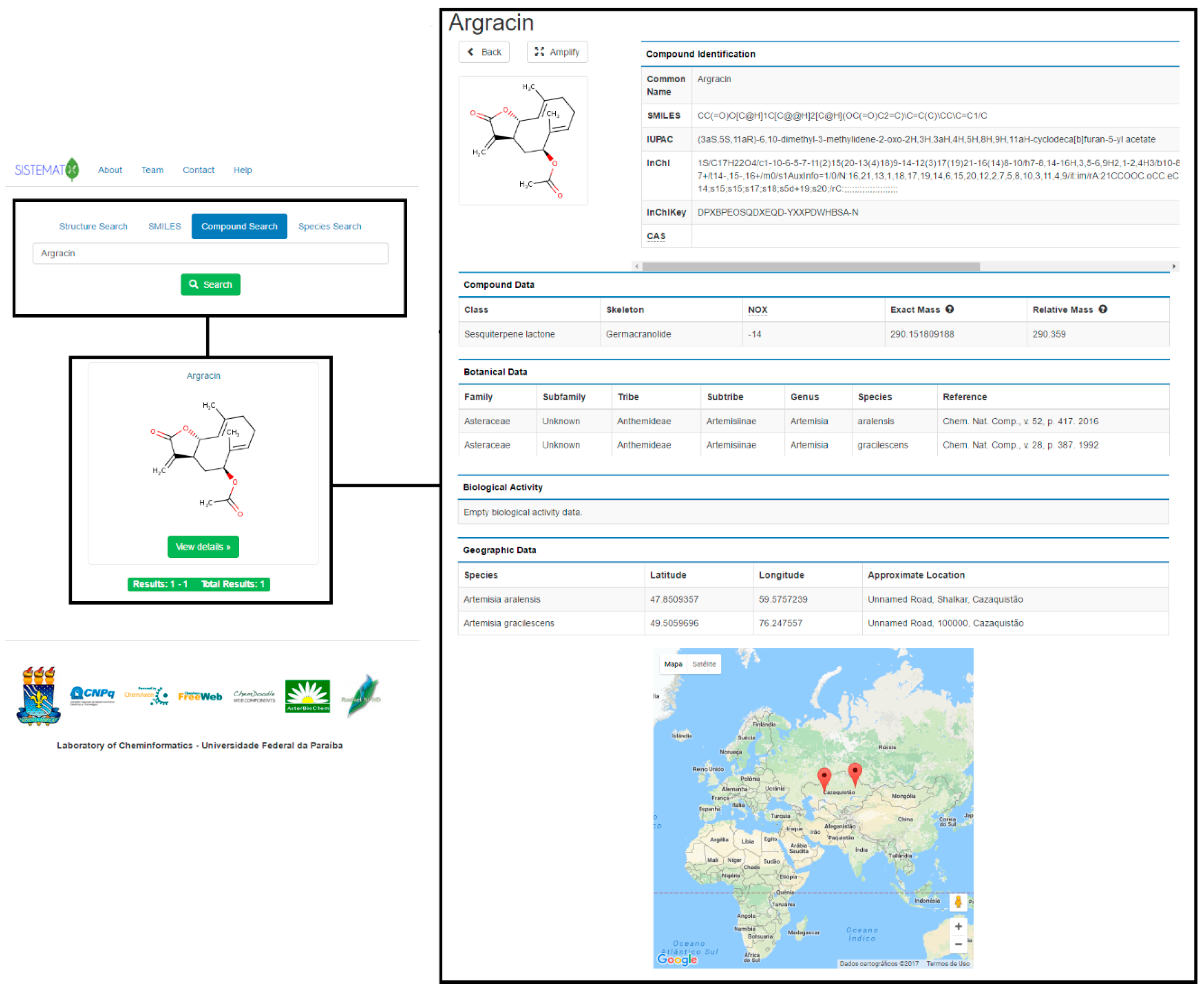
2. Results and Discussion
2.1. Utility and Discussion
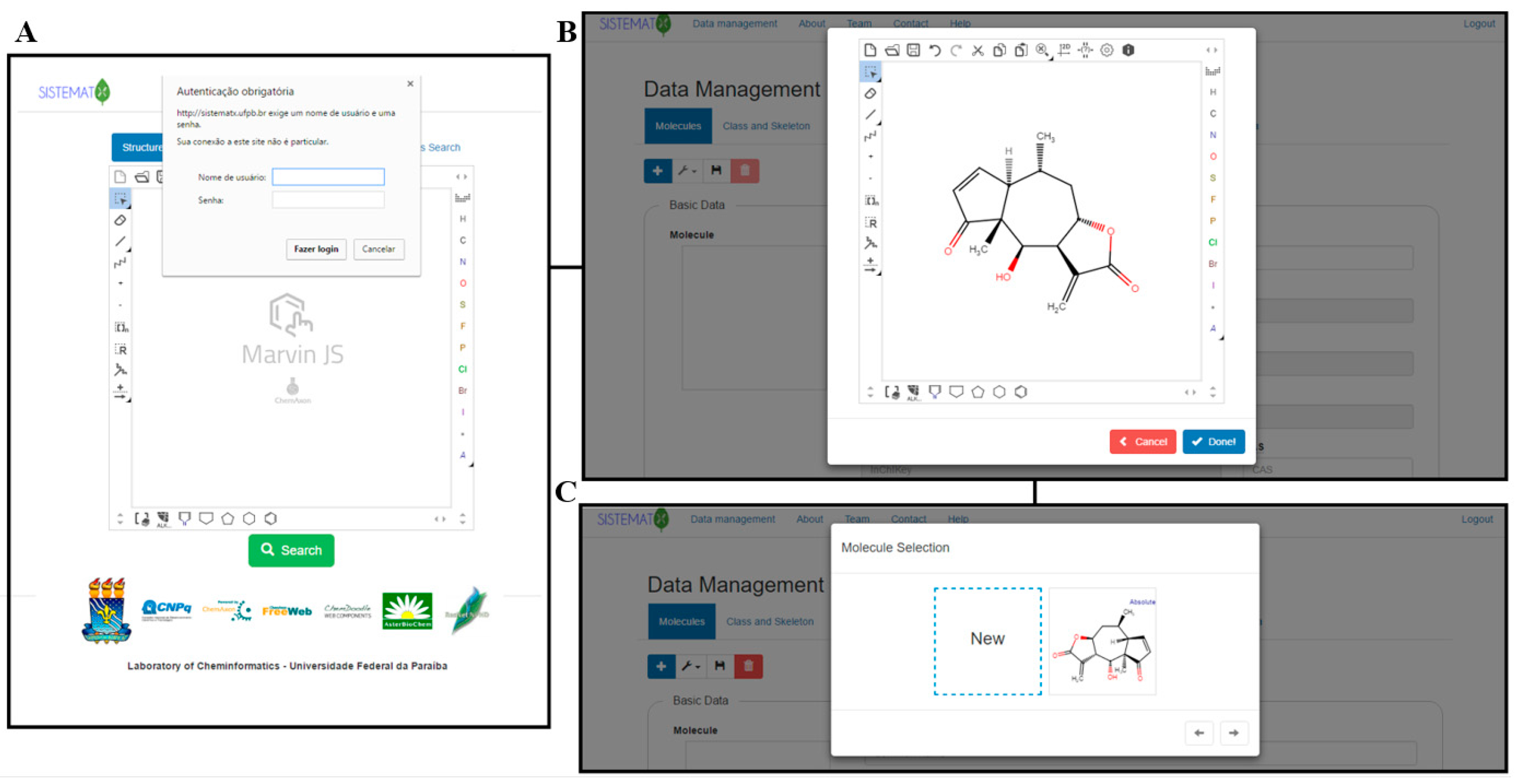
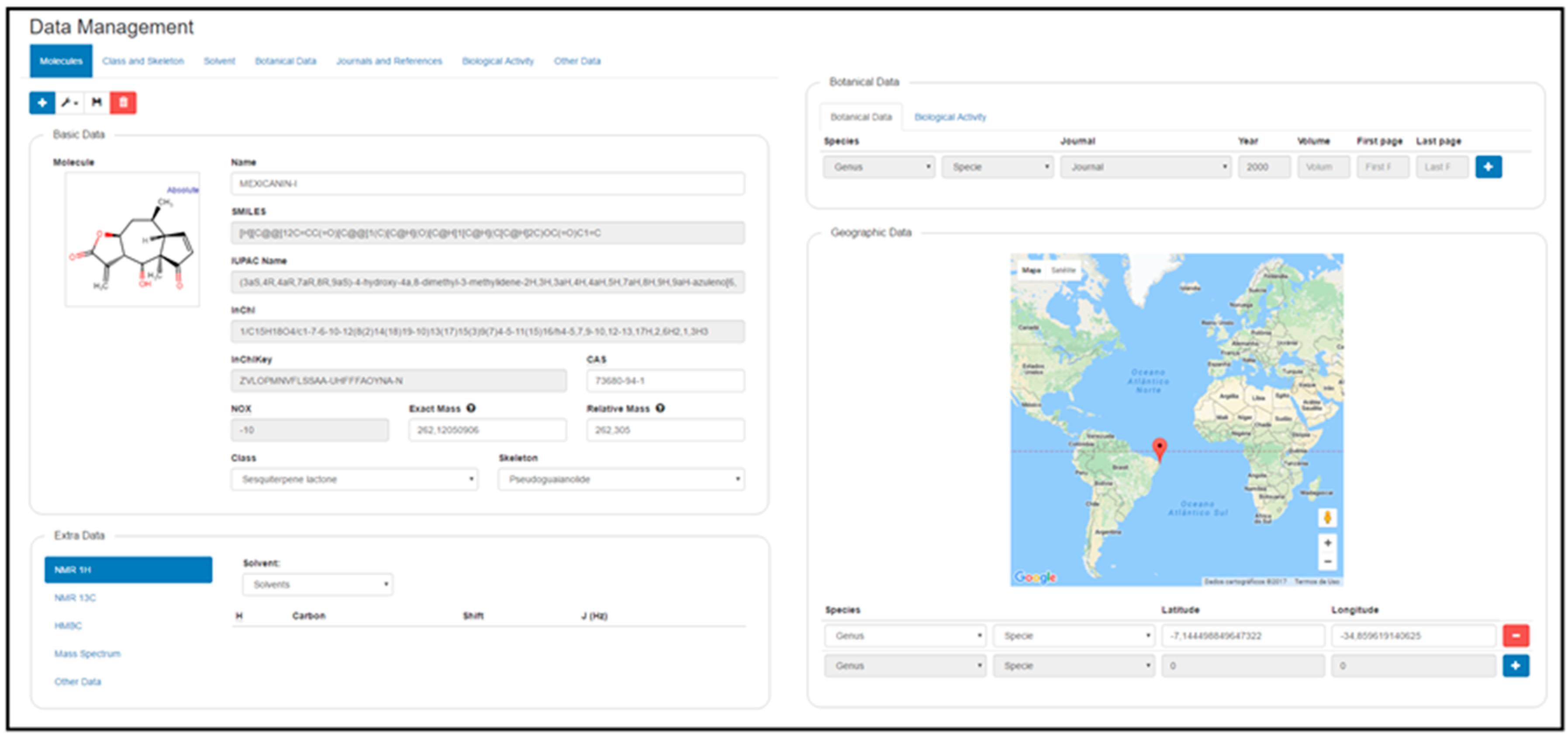
2.2. Data Management
3. Materials and Methods
Implementation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Munro, M.H.G. Is There an Ideal Database for Natural Products Research? In Natural Products: Discourse, Diversity, and Design; Osbourn, A., Goss, R.J., Carter, G.T., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 413–431. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.G.; Durley, R.C. Strategies for Database Dereplication of Natural Products. J. Nat. Prod. 1994, 57, 1484–1490. [Google Scholar] [CrossRef]
- Oliveira, T.; Chagas-Paula, D.; Rosa, A.; Gobbo-Neto, L.; Schmidt, T.J.; Da Costa, F.B. Temporal characteristics of a natural products in-house database. Planta Med. 2013, 79, 1113–1114. [Google Scholar] [CrossRef]
- Pence, H.; Williams, A. ChemSpider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Bolton, E.E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. Chapter 12 PubChem: Integrated Platform of Small Molecules and Biological Activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. [Google Scholar] [CrossRef]
- Degtyarenko, K.; de Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcántara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008, 36, D344–D350. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.B.; O’Neil-Johnson, M.; Williams, A.J.; Wheeler, P.; Pol, R.; Moser, A. Dereplication of natural products using minimal NMR data inputs. Org. Biomol. Chem. 2015, 13, 9957–9962. [Google Scholar] [CrossRef] [PubMed]
- Eugster, P.J.; Boccard, J.; Debrus, B.; Bréant, L.; Wolfender, J.L.; Martel, S.; Carrupt, P.A. Retention time prediction for dereplication of natural products (CxHyOz) in LC–MS metabolite profiling. Phytochemistry 2014, 108, 196–207. [Google Scholar] [CrossRef] [PubMed]
- DNP Database, Dictionary of Natural Products. CRC Press. Available online: http://dnp.chemnetbase.com/ (accessed on 4 March 2017).
- Graham, J.G.; Farnsworth, N.R. The NAPRALERT database as an aid for discovery of novel bioactive compounds. In Comprehensive Natural Products II: Chemistry and Biology; Elsevier Ltd.: Amsterdam, The Netherlands, 2010; Volume 3, pp. 81–94. ISBN 9780080453828. [Google Scholar]
- Dabb, S.; Blunt, J.; Munro, M. MarinLit: Database and essential tools for the marine natural products community. In Proceedings of the 248th National Meeting of the American-Chemical-Society (ACS), San Francisco, CA, USA, 10–14 August 2014. [Google Scholar]
- Banerjee, P.; Erehman, J.; Gohlke, B.-O.; Wilheim, T.; Preissner, R.; Dunkel, M. Super Natural II—A database of natural products. Nucleic Acids Res. 2015, 43, D935–D939. [Google Scholar] [CrossRef] [PubMed]
- Drewry, D.H.; Macarron, R. Enhancements of screening collections to address areas of unmet medical need: an industry perspective. Curr. Opin. Chem. Biol. 2010, 14, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Valli, M.; dos Santos, R.N.; Figueira, L.D.; Nakajima, C.H.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. Development of a natural products database from the biodiversity of Brazil. J. Nat. Prod. 2013, 76, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Hatherley, R.; Brown, D.K.; Musyoka, T.M.; Penkler, D.L.; Faya, N.; Lobb, K.A.; Tastan Bishop, Ö. SANCDB: A South African natural compound database. J. Cheminform. 2015, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Nam, S.; Jang, H.; Kim, A.; Lee, J.J. TM-MC: A database of medicinal materials and chemical compounds in Northeast Asian traditional medicine. BMC Complement. Altern. Med. 2015, 15, 218. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.-C. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Telukunta, K.K.; Döring, K.; Simoben, C.V.; Moumbock, A.F.A.; Malange, Y.I.; Njume, L.E.; Yong, J.N.; Sippl, W.; Günther, S. NANPDB: A Resource for Natural Products from Northern African Sources. J. Nat. Prod. 2017, 80, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Fang, Z.; Zhang, M.; Yi, Z.; Wen, C.; Shi, T. TCMID: Traditional Chinese medicine integrative database for herb molecular mechanism analysis. Nucleic Acids Res. 2013, 41, D1089–D1095. [Google Scholar] [CrossRef] [PubMed]
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. KNApSAcK Family Databases: Integrated Metabolite-Plant Species Databases for Multifaceted Plant Research. Plant Cell Physiol. 2012, 53, e1. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.W.; Lin, Y.C.; Chang, H.S.; Wang, C.C.; Chen, I.S.; Jheng, J.L.; Li, J.H. TIPdb-3D: The three-dimensional structure database of phytochemicals from Taiwan indigenous plants. Database (Oxford) 2014. [Google Scholar] [CrossRef] [PubMed]
- AsterDB, AsterBioChem in House Database. Available online: http://www.asterbiochem.org/asterdb (accessed on 4 March 2017).
- Sampaio, B.L.; Edrada-Ebel, R.; Da Costa, F.B. Effect of the environment on the secondary metabolic profile of Tithonia diversifolia: A model for environmental metabolomics of plants. Sci. Rep. 2016, 6, 29265. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Rzeppa, S.; Kaiser, M.; Brun, R. Larrea tridentata—Absolute configuration of its epoxylignans and investigations on its antiprotozoal activity. Phytochem. Lett. 2012, 5, 632–638. [Google Scholar] [CrossRef]
- Gaquerel, E.; Kuhl, C.; Neumann, S. Computational annotation of plant metabolomics profiles via a novel network-assisted approach. Metabolomics 2013, 9, 904–918. [Google Scholar] [CrossRef]
- Hendrickson, J.B.; Cram, D.J.; Hammond, G.S. Organic Chemistry, 3rd ed.; McGraw-Hill: New York, NY, USA, 1970; ISBN 07-028150-5. [Google Scholar]
- Gottlieb, O. The role of oxygen in phytochemical evolution towards diversity. Phytochemistry 1989, 28, 2545–2558. [Google Scholar] [CrossRef]
- Züst, T.; Heichinger, C.; Grossniklaus, U.; Harrington, R.; Kliebenstein, D.J.; Turnbull, L.A. Natural enemies drive geographic variation in plant defenses. Science 2012, 338, 116–119. [Google Scholar] [CrossRef] [PubMed]
- MySQL. Available online: http://dev.mysql.com/downloads/mysql/ (accessed on 31 January 2017).
- Scotti, M.; da Silva, R.O., Jr.; Yudi, S.; Brayner, R.; Scotti, L. SISTEMAT X—A web tool to manage databases of secondary metabolites. In Proceedings of the MOL2NET, 5–15 December 2015. [Google Scholar]
- Bootstrap. Available online: http://getbootstrap.com/ (accessed on 10 January 2017).
- jQuery. Available online: http://jquery.com/ (accessed on 17 January 2017).
- jQuery Autocomplete. Available online: http://jqueryui.com/autocomplete/ (accessed on 20 January 2017).
- ChemAxon. Marvin JS. Available online: http://chemaxon.com/products/marvin/marvin-js/ (accessed on 24 January 2017).
- ChemAxon. Jchem Web Services. Available online: http://chemaxon.com/products/jchem-web-services/ (accessed on 24 January 2017).
- ChemDoodle by iChemLabs. Available online: http://web.chemdoodle.com/ (accessed on 24 January 2017).
- Google Maps by Google, Inc. Available online: http://developers.google.com/maps/ (accessed on 13 January 2017).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| API | Description | Engine |
|---|---|---|
| 1. Structure | ||
| 2D drawing | Allows drawing and visualization of chemical structures | ChemAxon |
| 3D generator | Uses 2D drawing to generate a 3D representation of the molecule | ChemAxon |
| 3D | Graphical visualization of 3D molecules with JavaScript | ChemDoodl |
| 2. Compound Identification | ||
| SMILES | Simplified Molecular Input Line Entry System | ChemAxon |
| IUPAC | IUPAC Nomenclature | ChemAxon |
| InChI | IUPAC International Chemical Identifier | ChemAxon |
| InChIKey | InChIKey is a compact format of the InChI code | ChemAxon |
| CAS | Chemical Abstracts Service Registry Number | ChemAxon |
| 3. Compound Data | ||
| NOX | Oxidation number (NOX) of an organic compound | ChemAxon |
| Exact Mass | Uses the mass of the most abundant isotope of each element | ChemAxon |
| Relative Mass | Uses the average atomic mass of each element | ChemAxon |
| 4. Geographic data | ||
| Latitude | Can be inserted by the administrator or appears by clicking in the world map | Google Inc. |
| Longitude | Can be inserted by the administrator or appears by clicking in the world map | Google Inc. |
| Approximate | Using the latitude and longitude, appears an an approximate location of the specie | Google Inc. |
| Visualization | Uses the world map to possible to visualize the localization of the species | Google Inc. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scotti, M.T.; Herrera-Acevedo, C.; Oliveira, T.B.; Costa, R.P.O.; Santos, S.Y.K.d.O.; Rodrigues, R.P.; Scotti, L.; Da-Costa, F.B. SistematX, an Online Web-Based Cheminformatics Tool for Data Management of Secondary Metabolites. Molecules 2018, 23, 103. https://doi.org/10.3390/molecules23010103
Scotti MT, Herrera-Acevedo C, Oliveira TB, Costa RPO, Santos SYKdO, Rodrigues RP, Scotti L, Da-Costa FB. SistematX, an Online Web-Based Cheminformatics Tool for Data Management of Secondary Metabolites. Molecules. 2018; 23(1):103. https://doi.org/10.3390/molecules23010103
Chicago/Turabian StyleScotti, Marcus Tullius, Chonny Herrera-Acevedo, Tiago Branquinho Oliveira, Renan Paiva Oliveira Costa, Silas Yudi Konno de Oliveira Santos, Ricardo Pereira Rodrigues, Luciana Scotti, and Fernando Batista Da-Costa. 2018. "SistematX, an Online Web-Based Cheminformatics Tool for Data Management of Secondary Metabolites" Molecules 23, no. 1: 103. https://doi.org/10.3390/molecules23010103
APA StyleScotti, M. T., Herrera-Acevedo, C., Oliveira, T. B., Costa, R. P. O., Santos, S. Y. K. d. O., Rodrigues, R. P., Scotti, L., & Da-Costa, F. B. (2018). SistematX, an Online Web-Based Cheminformatics Tool for Data Management of Secondary Metabolites. Molecules, 23(1), 103. https://doi.org/10.3390/molecules23010103