An Unusual Dimeric Inhibitor of Acetylcholinesterase: Cooperative Binding of Crystal Violet

Abstract
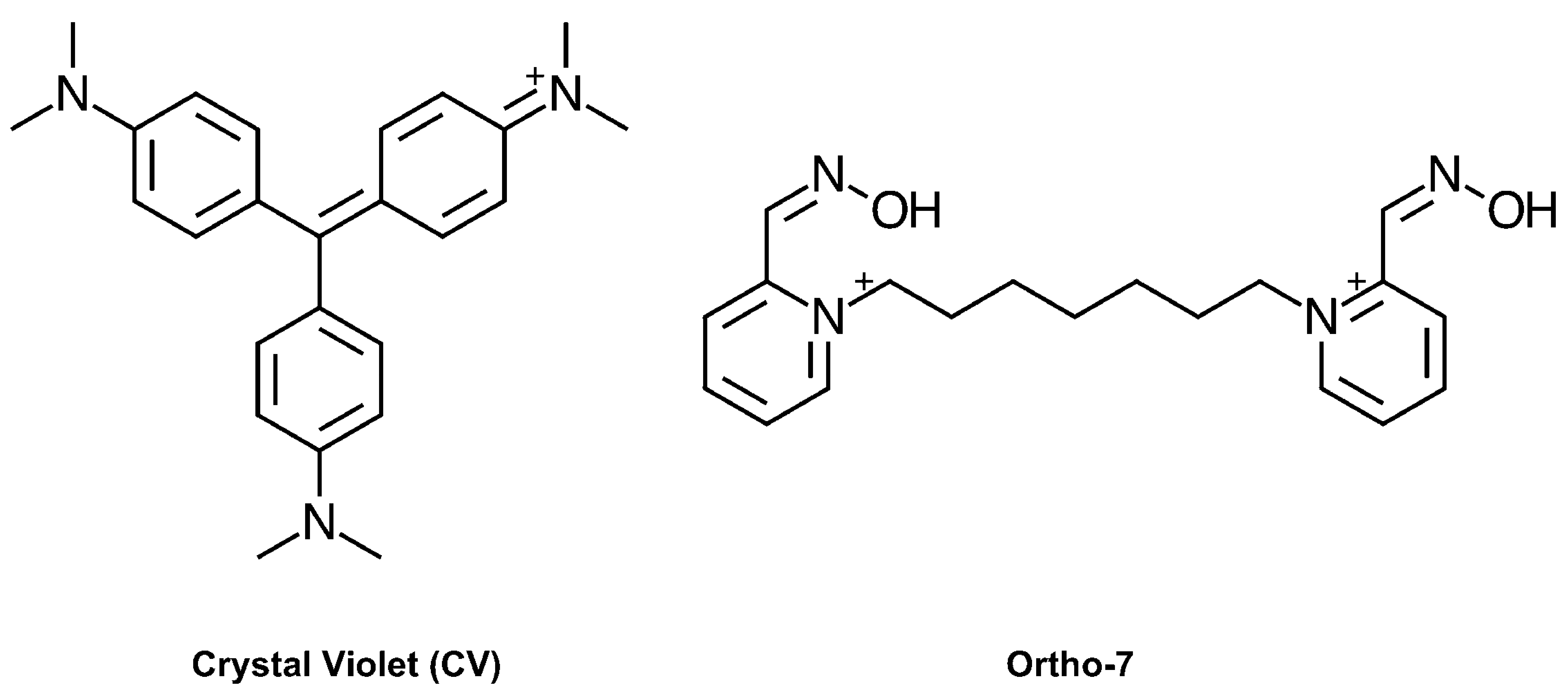
:1. Introduction
2. Results
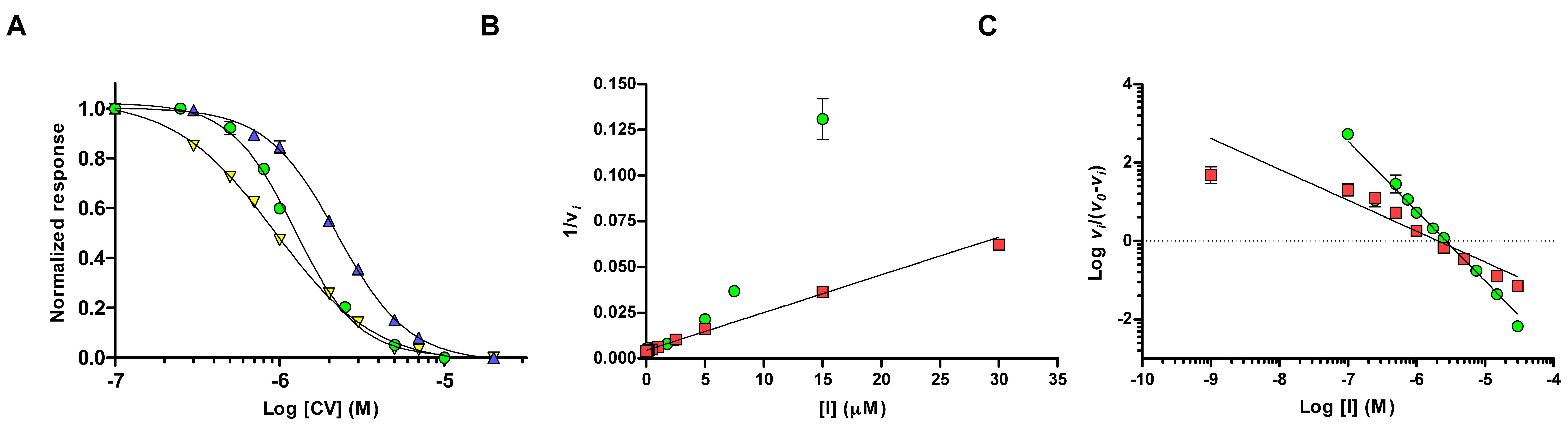
2.1. Kinetic Support of Positive Cooperative Inhibition
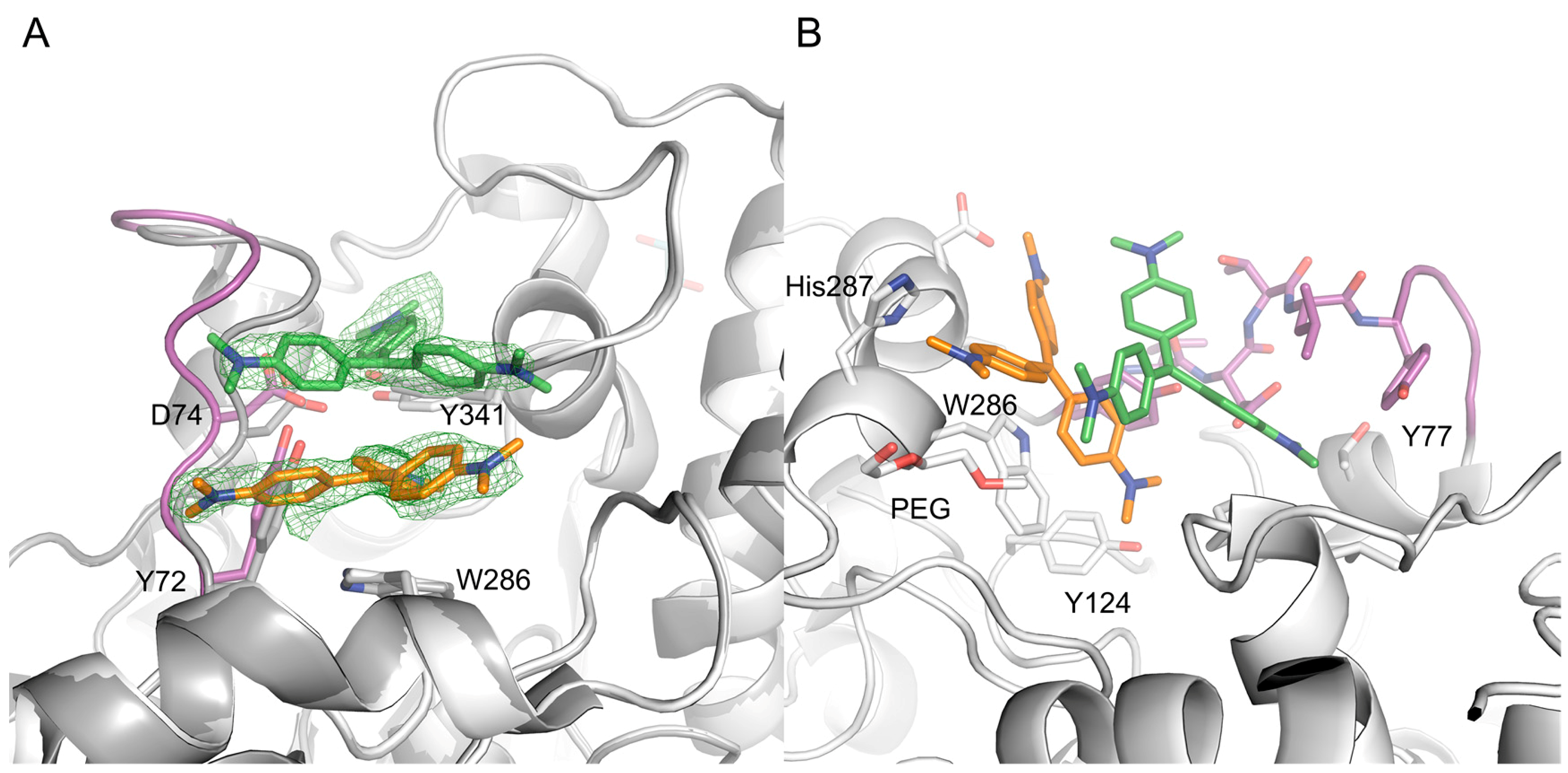
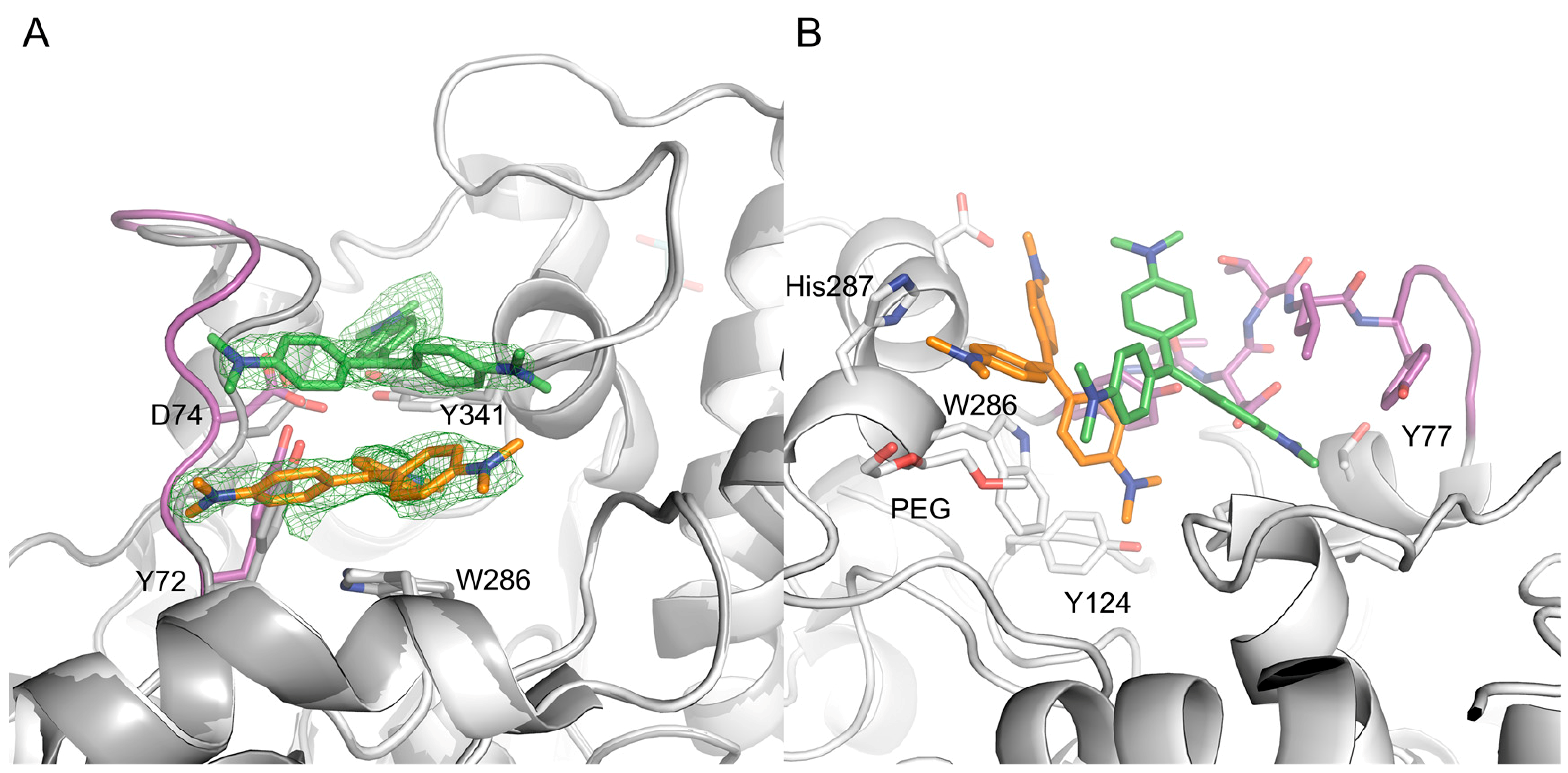
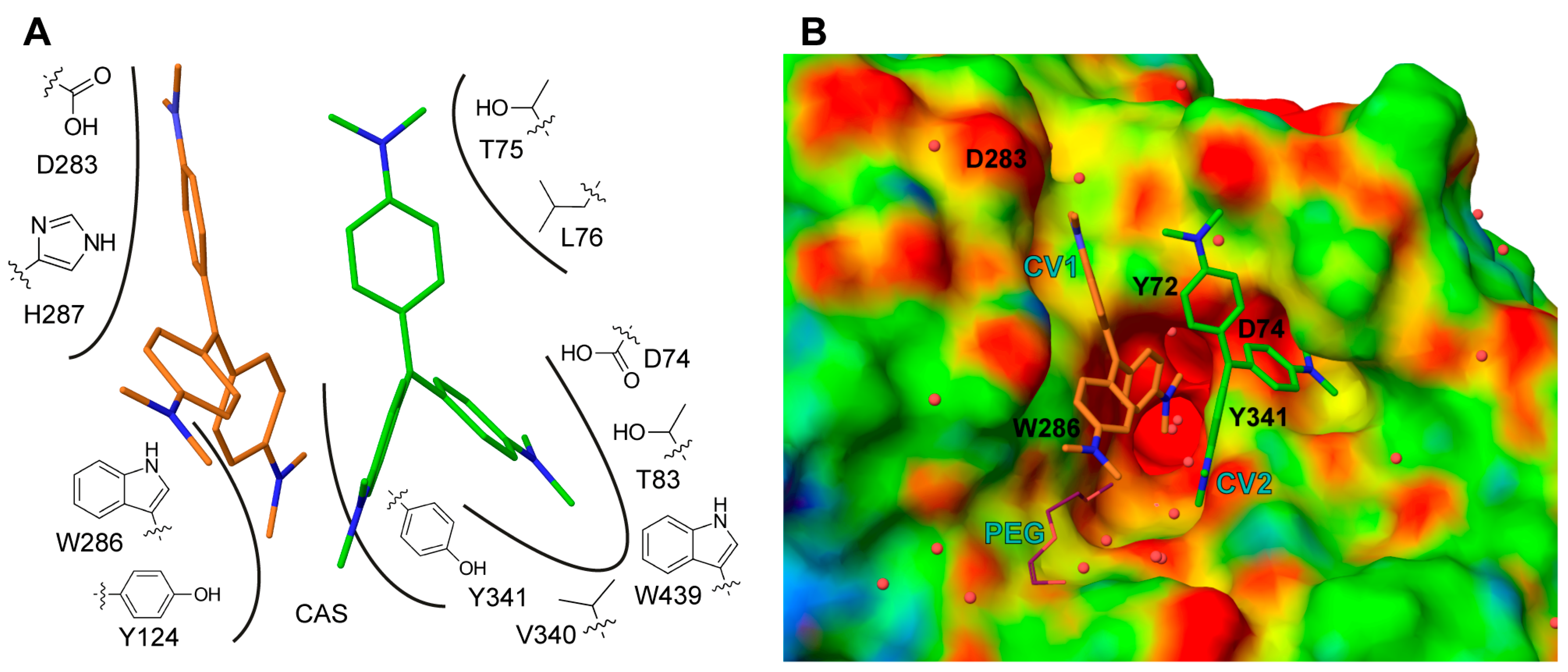
2.2. X-ray Crystal Structure of Crystal Violet in Complex with Mus musculus AChE Establish Structural Basis of Cooperativity
2.3. Site Directed Mutants Probing the Binding Site of CV
3. Discussion and Conclusions
4. Methods
4.1. Expression, Site Directed Mutagenesis and Determination of Kinetic Parameters
4.2. Purification, Crystallization and Structure Determination
4.3. Density Functional Theory Calculations
4.4. Dipole Moment and Electrostatic Potential Calculations
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AChE | Acetylcholinesterase |
| AChI | Acetylthiocholine iodide |
| AaAChE | Aedis aegypti Acetylcholinesterase |
| AgAChE | Anopheles gambiae Acetylcholinesterase |
| CV | Crystal Violet, Hexamethylpararosaniline chloride |
| DFT | Density Functional Theory |
| hAChE | Homo sapiens Acetylcholinesterase |
| hBuChE | Homo sapiens Butyrylcholinesterase |
| mAChE | Mus musculus Acetylcholinesterase |
| Ortho-7 | 1,7-heptylene-bis-N,N′-2-pyridiniumaldoxime dichloride |
| TcAChE | Torpedo californica Acetylcholinesterase |
| 4PL | Four Parameters Logistic |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Data Bank Accession Code | 5OV9 |
|---|---|
| Data Collection Statistics | |
| Resolution range (Å) | 29.1–2.4 (2.486–2.4) |
| Space group | P 212121 |
| Unit cell (Å, °) | 79.6 113.9 226.7 90 90 90 |
| Total reflections | 332,637 (32,579) |
| Unique reflections | 80,635 (7919) |
| Multiplicity | 4.1 (4.1) |
| Completeness (%) | 98.22 (98.00) |
| Mean I/sigma (I) | 16.39 (4.94) |
| Wilson B-factor | 42.99 |
| Rmerge | 0.04764 (0.4941) |
| Rmeas | 0.0545 (0.5644) |
| Refinement Statistics | |
| Reflections used in refinement | 79975 (7902) |
| Reflections used for R-free | 1585 (143) |
| R-work | 0.1614 (0.2108) |
| R-free | 0.1989 (0.2587) |
| CC (work) | 0.967 (0.950) |
| CC (free) | 0.944 (0.905) |
| Number of non-hydrogen atoms | 9224 |
| Macromolecules | 8334 |
| Ligands | 259 |
| Solvent | 631 |
| Protein residues | 1067 |
| Rmsd from ideal values | |
| Bond length | 0.012 |
| Bond angles | 1.35 |
| Ramachandran plot (%) | |
| Favoured regions | 96.40 |
| Allowed regions | 3.31 |
| Disallowed regions | 0.28 |
References
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Radic, Z.; Sulzenbacher, G.; Kim, E.; Taylor, P.; Marchot, P. Substrate and product trafficking through the active center gorge of acetylcholinesterase analyzed by crystallography and equilibrium binding. J. Biol. Chem. 2006, 281, 29256–29267. [Google Scholar] [CrossRef] [PubMed]
- Colletier, J.P.; Fournier, D.; Greenblatt, H.M.; Stojan, J.; Sussman, J.L.; Zaccai, G.; Silman, I.; Weik, M. Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J. 2006, 25, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Berg, L.; Mishra, B.K.; Andersson, C.D.; Ekstrom, F.; Linusson, A. The nature of activated non-classical hydrogen bonds: A case study on acetylcholinesterase-ligand complexes. Chemistry 2016, 22, 2672–2681. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.D.; Forsgren, N.; Akfur, C.; Allgardsson, A.; Berg, L.; Engdahl, C.; Qian, W.; Ekstrom, F.; Linusson, A. Divergent structure-activity relationships of structurally similar acetylcholinesterase inhibitors. J. Med. Chem. 2013, 56, 7615–7624. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, H.M.; Kryger, G.; Lewis, T.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with (−)-galanthamine at 2.3 A resolution. FEBS Lett. 1999, 463, 321–326. [Google Scholar] [CrossRef]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef]
- Wilson, I.B.; Ginsburg, S. Reactivation of acetylcholinesterase inhibited by alkylphosphates. Arch. Biochem. Biophys. 1955, 54, 569–571. [Google Scholar] [CrossRef]
- Casida, J.E.; Quistad, G.B. Golden age of insecticide research: Past, present, or future? Annu. Rev. Entomol. 1998, 43, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Berg, L.; Andersson, C.D.; Artursson, E.; Hornberg, A.; Tunemalm, A.K.; Linusson, A.; Ekstrom, F. Targeting acetylcholinesterase: Identification of chemical leads by high throughput screening, structure determination and molecular modeling. PLoS ONE 2011, 6, e26039. [Google Scholar] [CrossRef] [PubMed]
- Engdahl, C.; Knutsson, S.; Ekstrom, F.; Linusson, A. Discovery of Selective Inhibitors Targeting Acetylcholinesterase 1 from Disease-Transmitting Mosquitoes. J. Med. Chem. 2016, 59, 9409–9421. [Google Scholar] [CrossRef] [PubMed]
- Stefan, M.I.; Le Novere, N. Cooperative binding. PLoS Comput. Biol. 2013, 9, e1003106. [Google Scholar] [CrossRef] [PubMed]
- Bohr, C.; Hasselbalch, K.; Krogh, A. Über einen in biologischer Beziehung wichtigen Einfluss, den die Kohlensäurespannung des Blutes auf dessen Sauerstoffbindung übt. Skand. Arch. Physiol. 1904, 16, 401–412. (In German) [Google Scholar] [CrossRef]
- Umbarger, H.E. Evidence for a negative-feedback mechanism in the biosynthesis of isoleucine. Science 1956, 123, 848. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.P.; Podleski, T.R. On the excitability and cooperativity of the electroplax membrane. Proc. Natl. Acad. Sci. USA 1968, 59, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.P.; Thiery, J.; Tung, Y.; Kittel, C. On the cooperativity of biological membranes. Proc. Natl. Acad. Sci. USA 1967, 57, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Ptashne, M.; Jeffrey, A.; Johnson, A.D.; Maurer, R.; Meyer, B.J.; Pabo, C.O.; Roberts, T.M.; Sauer, R.T. How the lambda repressor and cro work. Cell 1980, 19, 1–11. [Google Scholar] [CrossRef]
- Adams, E. The antibacterial action of crystal violet. J. Pharm. Pharmacol. 1967, 19, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Kucukkilinc, T.T.; Ozer, I. Inhibition of electric eel acetylcholinesterase by triarylmethane dyes. Chem. Biol. Interact. 2008, 175, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Arias, H.R.; Bhumireddy, P.; Spitzmaul, G.; Trudell, J.R.; Bouzat, C. Molecular mechanisms and binding site location for the noncompetitive antagonist crystal violet on nicotinic acetylcholine receptors. Biochemistry 2006, 45, 2014–2026. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, F.; Pang, Y.P.; Boman, M.; Artursson, E.; Akfur, C.; Borjegren, S. Crystal structures of acetylcholinesterase in complex with HI-6, Ortho-7 and obidoxime: Structural basis for differences in the ability to reactivate tabun conjugates. Biochem. Pharmacol. 2006, 72, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, L.; Smulevich, G.; Marzocchi, M.P. Resonance Raman spectrum of crystal violet. J. Raman Spectrosc. 1979, 8, 305–310. [Google Scholar] [CrossRef]
- Knutsson, S.; Kindahl, T.; Engdahl, C.; Nikjoo, D.; Forsgren, N.; Kitur, S.; Ekstrom, F.; Kamau, L.; Linusson, A. N-Aryl-N′-ethyleneaminothioureas effectively inhibit acetylcholinesterase 1 from disease-transmitting mosquitoes. Eur. J. Med. Chem 2017, 134, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.; Marquardt, B.J.; Kahr, B. Crystal violet´s shoulder. J. Chem. Soc. Perkin Trans. 2 1999, 11, 2241–2247. [Google Scholar] [CrossRef]
- Radic, Z.; Duran, R.; Vellom, D.C.; Li, Y.; Cervenansky, C.; Taylor, P. Site of fasciculin interaction with acetylcholinesterase. J. Biol. Chem. 1994, 269, 11233–11239. [Google Scholar] [PubMed]
- Harel, M.; Kleywegt, G.J.; Ravelli, R.B.; Silman, I.; Sussman, J.L. Crystal structure of an acetylcholinesterase-fasciculin complex: Interaction of a three-fingered toxin from snake venom with its target. Structure 1995, 3, 1355–1366. [Google Scholar] [CrossRef]
- Weiss, J.N. The Hill equation revisited: Uses and misuses. FASEB J. 1997, 11, 835–841. [Google Scholar] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Rosenberry, T.L.; Martin, P.K.; Nix, A.J.; Wildman, S.A.; Cheung, J.; Snyder, S.A.; Tan, R.X. Hopeahainol A binds reversibly at the acetylcholinesterase (AChE) peripheral site and inhibits enzyme activity with a novel higher order concentration dependence. Chem. Biol. Interact. 2016, 259, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Miller, M.C.; Grkovic, S.; Brown, M.H.; Skurray, R.A.; Brennan, R.G. Structural mechanisms of QacR induction and multidrug recognition. Science 2001, 294, 2158–2163. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Nikaido, E.; Nakashima, R.; Sakurai, K.; Fujiwara, D.; Fujii, I.; Nishino, K. The crystal structure of multidrug-resistance regulator RamR with multiple drugs. Nat. Commun. 2013, 4, 2078. [Google Scholar] [CrossRef] [PubMed]
- Kielkopf, C.L.; White, S.; Szewczyk, J.W.; Turner, J.M.; Baird, E.E.; Dervan, P.B.; Rees, D.C. A structural basis for recognition of A.T and T.A base pairs in the minor groove of B-DNA. Science 1998, 282, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.P. Nonbonded bivalence approach to cell-permeable molecules that target DNA sequences. Bioorg. Med. Chem. 2004, 12, 3063–3068. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, F.; Hornberg, A.; Artursson, E.; Hammarstrom, L.G.; Schneider, G.; Pang, Y.P. Structure of HI-6*sarin-acetylcholinesterase determined by X-ray crystallography and molecular dynamics simulation: Reactivator mechanism and design. PLoS ONE 2009, 4, e5957. [Google Scholar] [CrossRef] [PubMed]
- Artursson, E.; Akfur, C.; Hornberg, A.; Worek, F.; Ekstrom, F. Reactivation of tabun-hAChE investigated by structurally analogous oximes and mutagenesis. Toxicology 2009, 265, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Ekstrom, F.; Akfur, C.; Tunemalm, A.K.; Lundberg, S. Structural changes of phenylalanine 338 and histidine 447 revealed by the crystal structures of tabun-inhibited murine acetylcholinesterase. Biochemistry 2006, 45, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Berg, L.; Niemiec, M.S.; Qian, W.; Andersson, C.D.; Wittung-Stafshede, P.; Ekstrom, F.; Linusson, A. Similar but different: Thermodynamic and structural characterization of a pair of enantiomers binding to acetylcholinesterase. Angew. Chem. Int. Ed. Engl. 2012, 51, 12716–12720. [Google Scholar] [CrossRef] [PubMed]
- Allgardsson, A.; Berg, L.; Akfur, C.; Hornberg, A.; Worek, F.; Linusson, A.; Ekstrom, F.J. Structure of a prereaction complex between the nerve agent sarin, its biological target acetylcholinesterase, and the antidote HI-6. Proc. Natl. Acad. Sci. USA 2016, 113, 5514–5519. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-Functional Thermochemistry 3. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin-density calculations—A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum. Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.C.; W. Zhang, W.; et al. AMBER 10; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Felder, C.E.; Prilusky, J.; Silman, I.; Sussman, J.L. A server and database for dipole moments of proteins. Nucleic Acids Res. 2007, 35, W512–W521. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |

| Protein | IC50 1 (µM) | Hill Coefficient |
|---|---|---|
| mAChE | 2.2 (2.13–2.36) | −2.0 (−2.18–−1.81) |
| hAChE | 1.2 (1.16–1.32) | −2.2 (−2.53–−1.91) |
| hBuChE | 0.9 (0.87–1.00) | −1.4 (−1.56–−1.29) |
| Protein | Substitution | IC50 1 (µM) | Factor 2 | Hill Coefficient |
|---|---|---|---|---|
| mAChE | Wild-type | 2.2 (2.13–2.36) | −2.0 (−2.18–−1.81) | |
| D283A | 7.4 (6.85–7.89) | 3.4 | −1.5 (−1.62–−1.31) | |
| D283N | 6.5 (6.25–6.80) | 3.0 | −1.8 (−1.92–−1.64) | |
| hAChE | Wild-type | 1.2 (1.16–1.32) | −2.2 (−2.53–−1.91) | |
| Y72A | 1.4 (1.37–1.49) | 1.2 | −1.8 (−1.96–−1.73) | |
| D74A | 5.0 (4.70–5.36) | 4.2 | −1.7 (−1.83–−1.49) | |
| D74N | 5.0 (4.66–5.31) | 4.2 | −2.1 (−2.44–−1.86) | |
| N283A | 3.7 (3.54–3.78) | 3.1 | −1.8 (−1.86–−1.68) | |
| N283D | 3.5 (3.38–3.68) | 2.9 | −1.9 (−2.01–−1.75) | |
| W286A | 4.4 (4.06–4.73) | 3.7 | −2.1 (−2.49–−1.77) | |
| Y341A | 2.9 (2.76–3.09) | 2.4 | −2.0 (−2.26–−1.82) |
| Protein | Substitution | PDB Code | Dipole Moment (Debye) 1 | Net Charge |
|---|---|---|---|---|
| mAChE | Wild-type | 1J06 | 615 (778) | −8 (−8) |
| D283A | 605 | −7 | ||
| hAChE | Wild-type | 4EY4 | 737 (824) | −9 (−10) |
| D74A | 659 | −8 | ||
| Y72A | 739 | −9 | ||
| W286A | 738 | −9 | ||
| TcAChE 2 | Wild-type | 1ACE | 1744 (1592) | −5 (−9) |
| hBuChE | Wild-type | 1P0I | 1691 (1655) | 8 (5) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allgardsson, A.; David Andersson, C.; Akfur, C.; Worek, F.; Linusson, A.; Ekström, F. An Unusual Dimeric Inhibitor of Acetylcholinesterase: Cooperative Binding of Crystal Violet. Molecules 2017, 22, 1433. https://doi.org/10.3390/molecules22091433
Allgardsson A, David Andersson C, Akfur C, Worek F, Linusson A, Ekström F. An Unusual Dimeric Inhibitor of Acetylcholinesterase: Cooperative Binding of Crystal Violet. Molecules. 2017; 22(9):1433. https://doi.org/10.3390/molecules22091433
Chicago/Turabian StyleAllgardsson, Anders, C. David Andersson, Christine Akfur, Franz Worek, Anna Linusson, and Fredrik Ekström. 2017. "An Unusual Dimeric Inhibitor of Acetylcholinesterase: Cooperative Binding of Crystal Violet" Molecules 22, no. 9: 1433. https://doi.org/10.3390/molecules22091433
APA StyleAllgardsson, A., David Andersson, C., Akfur, C., Worek, F., Linusson, A., & Ekström, F. (2017). An Unusual Dimeric Inhibitor of Acetylcholinesterase: Cooperative Binding of Crystal Violet. Molecules, 22(9), 1433. https://doi.org/10.3390/molecules22091433