Interactions Controlling the Slow Dynamic Conformational Motions of Ubiquitin


Abstract
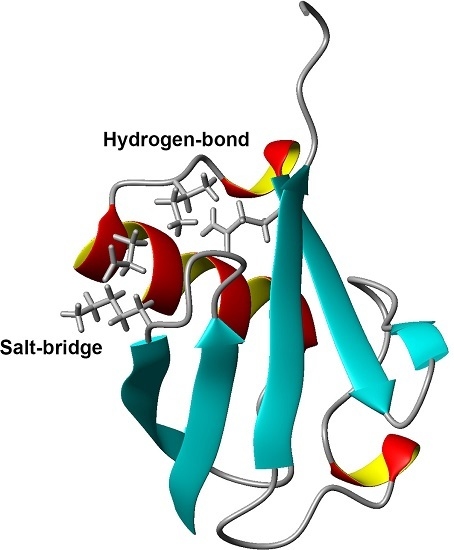
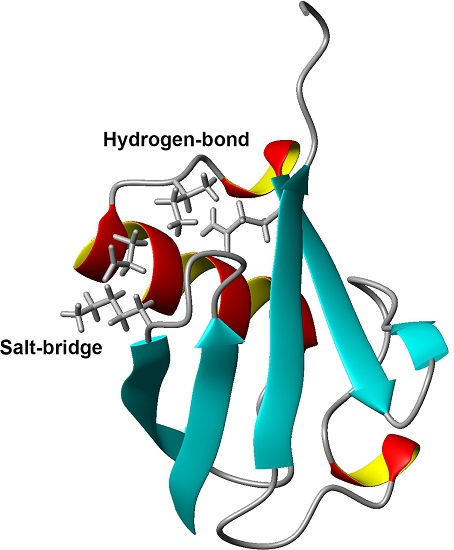
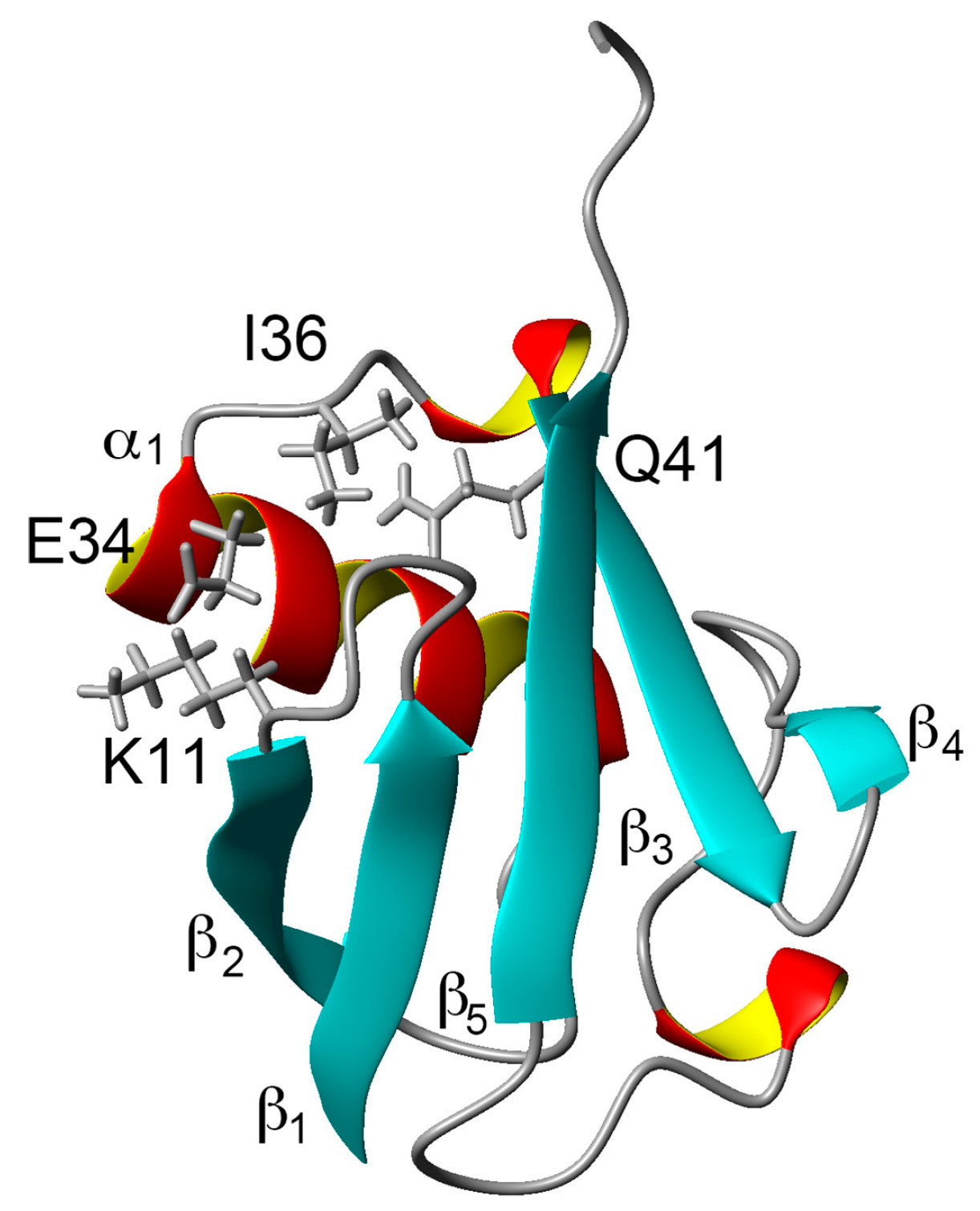
1. Introduction
2. Results and Discussion
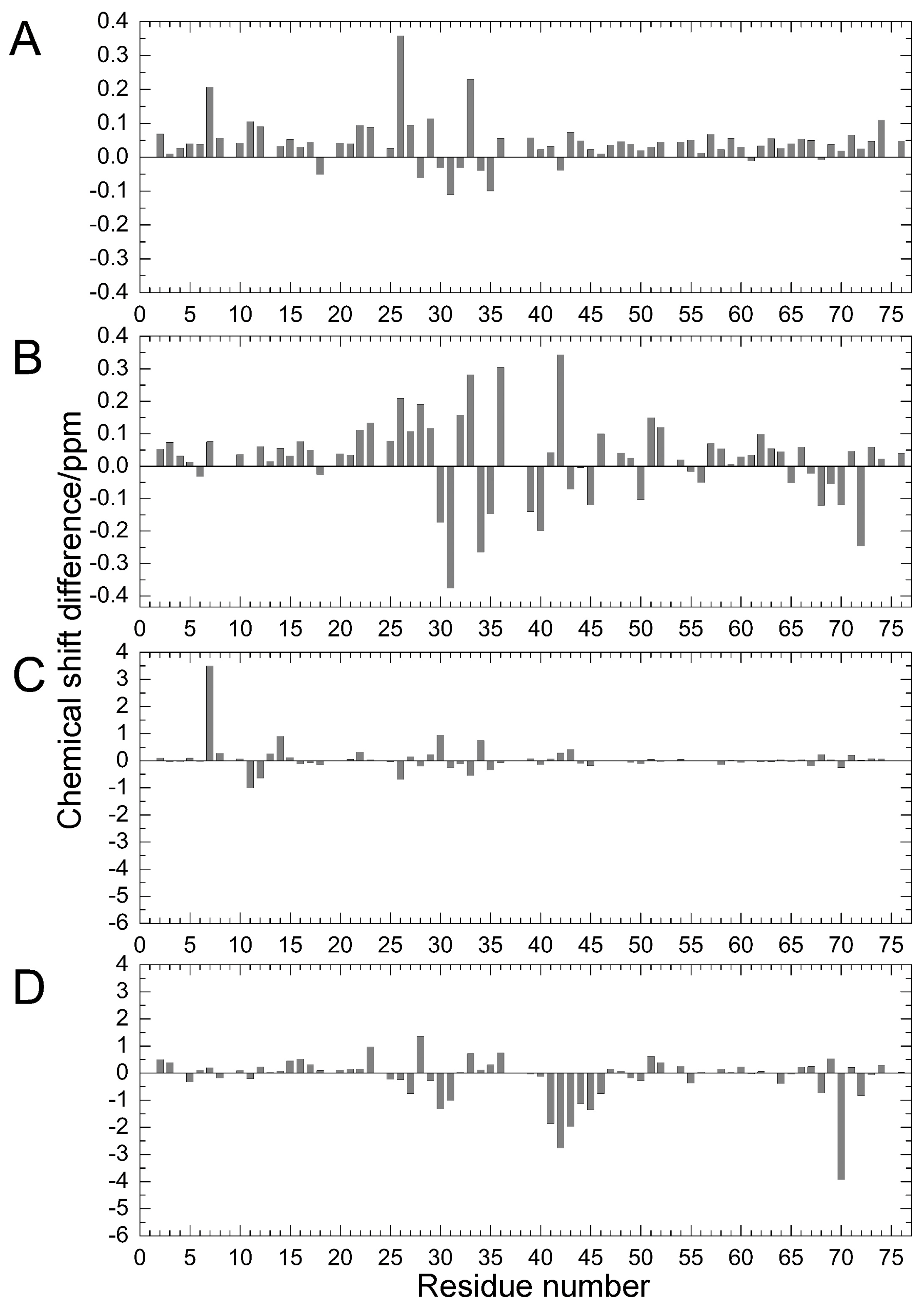
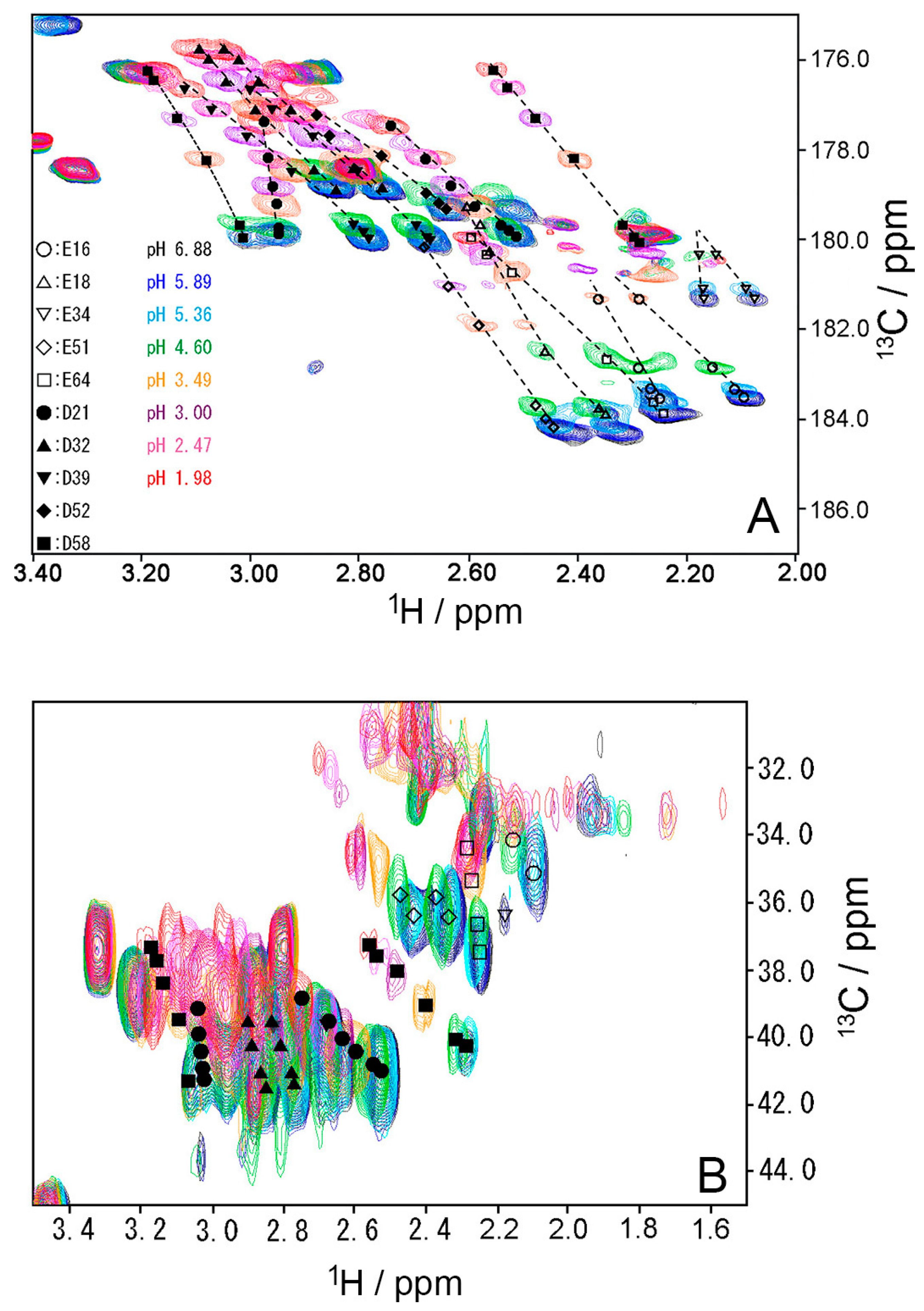
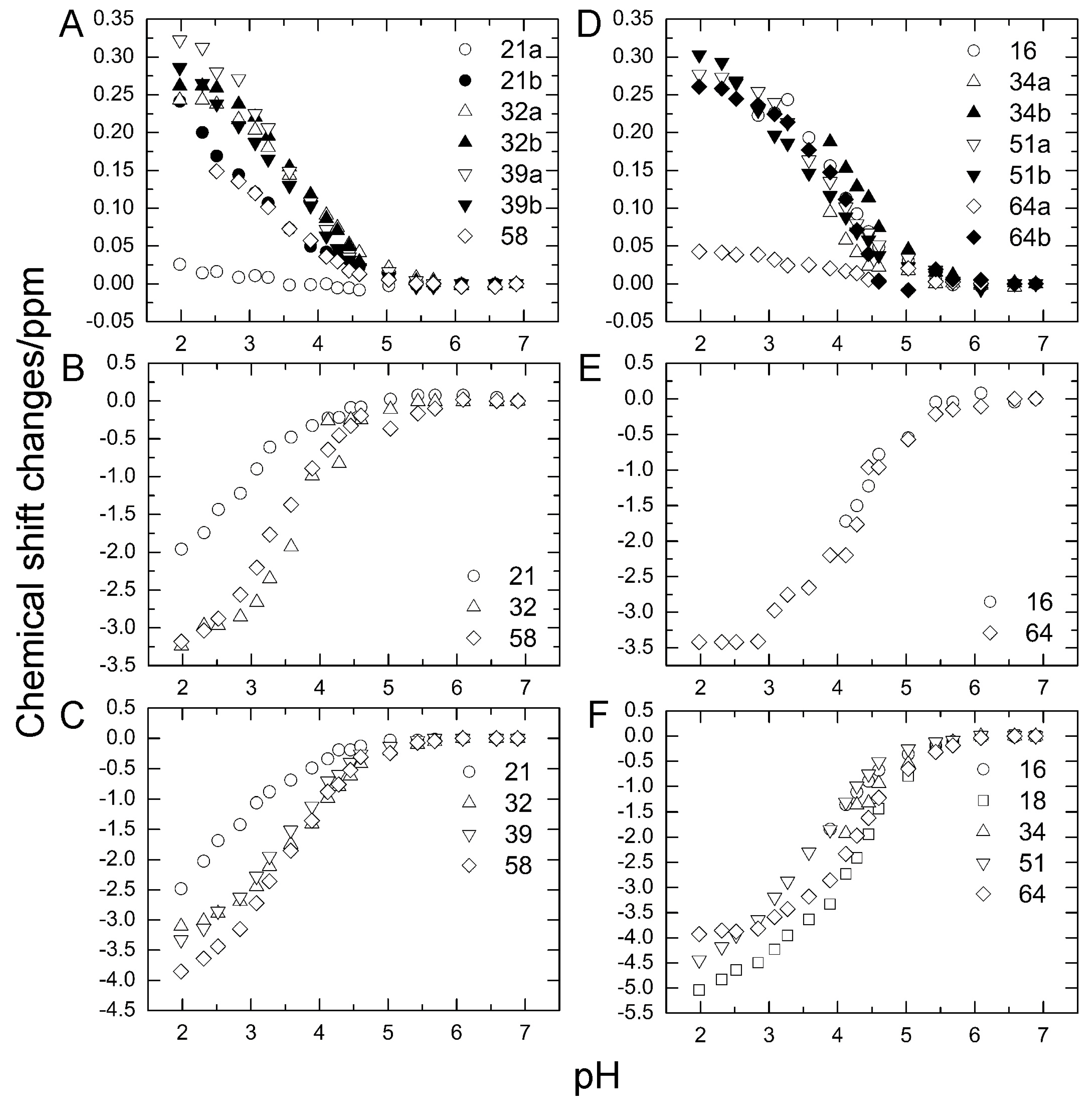
2.1. Chemical Shift Changes Induced by Mutations
2.2. Stability of WT and Mutant Proteins
2.3. pKa Values for Side Chains of Aspartate and Glutamate
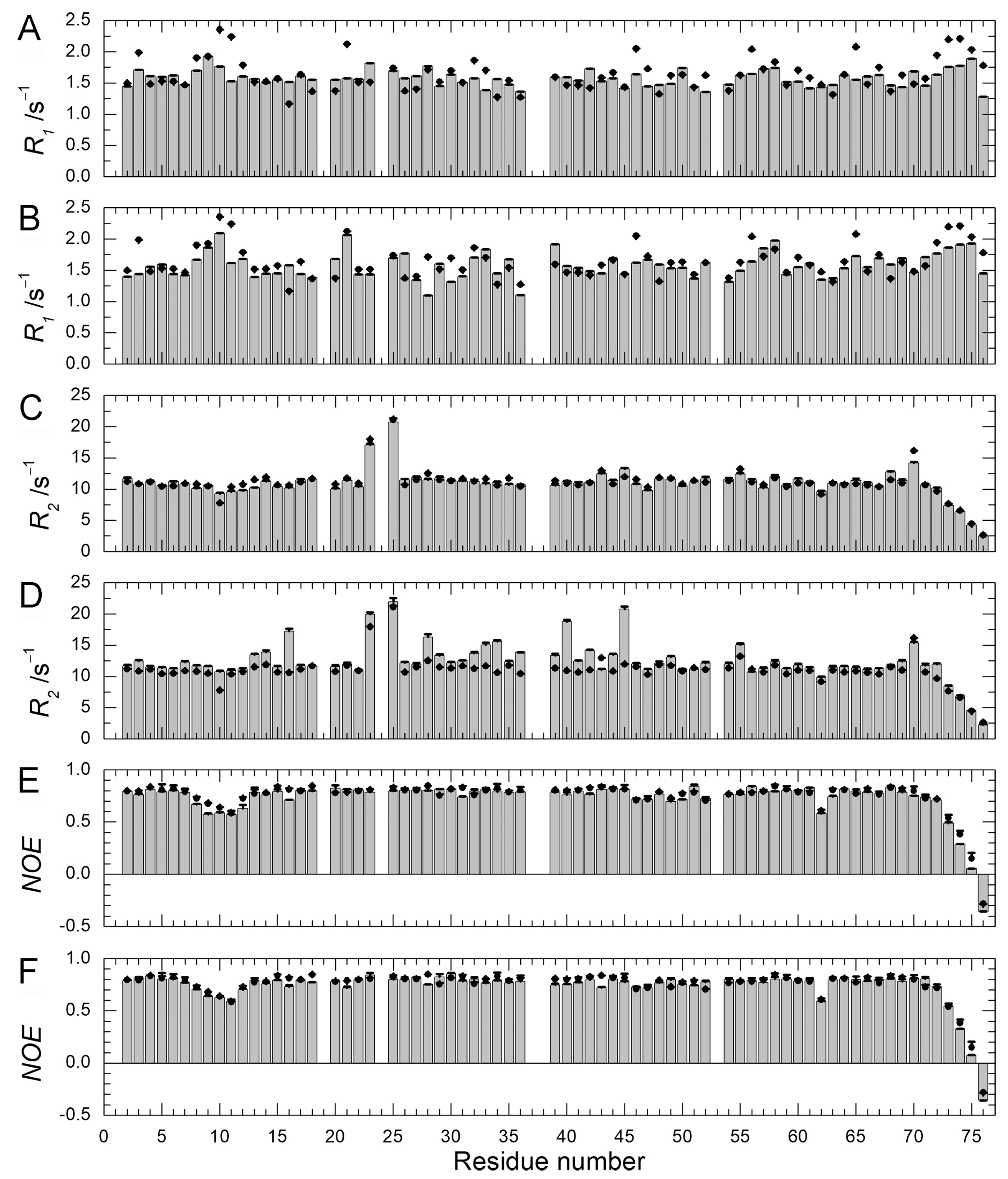
2.4. Backbone Dynamics and Hydration
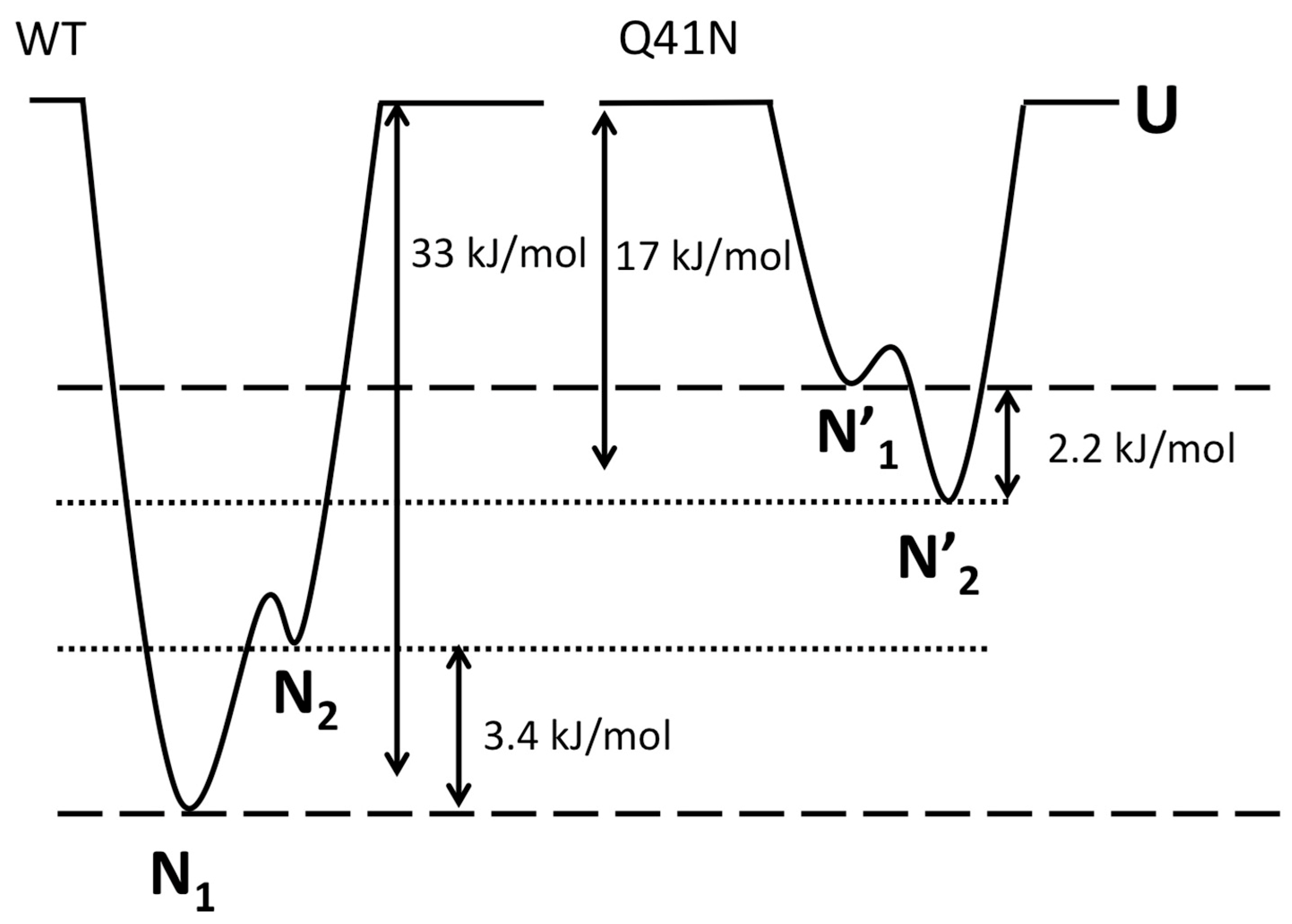
2.5. The Free-Energy Landscape
3. Materials and Methods
3.1. Sample Preparation
3.2. Circular Dichroism Measurements
3.3. NMR Measurements and Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boehr, D.D.; Dyson, H.J.; Wright, P.E. An NMR perspective on enzyme dynamics. Chem. Rev. 2006, 106, 3055–3079. [Google Scholar] [CrossRef] [PubMed]
- Eisenmesser, E.Z.; Millet, O.; Labeikovsky, W.; Korzhnev, D.M.; Wolf-Watz, M.; Bosco, D.A.; Skalicky, J.J.; Kay, L.E.; Kern, D. Intrinsic dynamics of an enzyme underlies catalysis. Nature 2005, 438, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Yamada, H.; Akasaka, K.; Herrmann, C.; Kremer, W.; Maurer, T.; Doker, R.; Kalbitzer, H.R. Pressure-induced local unfolding of the ras binding domain of RalGDS. Nat. Struct. Biol. 2000, 7, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A. The ubiquitin system for protein degradation and some of its roles in the control of the cell-division cycle (Nobel lecture). Angew. Chem. Int. Ed. 2005, 44, 5932–5943. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Ann. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Vijay-Kumar, S.; Bugg, C.E.; Cook, W.J. Structure of ubiquitin refined at 1.8 A resolution. J. Mol. Biol. 1987, 194, 531–544. [Google Scholar] [CrossRef]
- Cornilescu, G.; Marquardt, J.L.; Ottiger, M.; Bax, A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J. Am. Chem. Soc. 1998, 120, 6836–6837. [Google Scholar] [CrossRef]
- Sakata, E.; Satoh, T.; Yamamoto, S.; Yamaguchi, Y.; Yagi-Utsumi, M.; Kurimoto, E.; Tanaka, K.; Wakatsuki, S.; Kato, K. Crystal structure of UbcH5b~ubiquitin intermediate: Insight into the formation of the self-assembled E2~Ub conjugates. Structure 2010, 18, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Page, R.C.; Pruneda, J.N.; Amick, J.; Klevit, R.E.; Misra, S. Structural insights into the conformation and oligomerization of E2~ubiquitin conjugates. Biochemistry 2012, 51, 4175–4187. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, R.B.; Esteban-Martin, S.; Richter, B.; Lee, D.; Walter, K.F.; Milovanovic, D.; Becker, S.; Lakomek, N.A.; Griesinger, C.; Salvatella, X. Weak long-range correlated motions in a surface patch of ubiquitin involved in molecular recognition. J. Am. Chem. Soc. 2011, 133, 10336–10339. [Google Scholar] [CrossRef] [PubMed]
- Kiel, C.; Serrano, L. The ubiquitin domain superfold: Structure-based sequence alignments and characterization of binding epitopes. J. Mol. Biol. 2006, 355, 821–844. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Kasinath, V.; Moorman, V.R.; Nucci, N.V.; Hilser, V.J.; Wand, A.J. Coupled motion in proteins revealed by pressure perturbation. J. Am. Chem. Soc. 2012, 134, 8543–8550. [Google Scholar] [CrossRef] [PubMed]
- Babu, C.R.; Hilser, V.J.; Wand, A.J. Direct access to the cooperative substructure of proteins and the protein ensemble via cold denaturation. Nat. Struct. Mol. Biol. 2004, 11, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Lange, O.F.; Lakomek, N.A.; Fares, C.; Schroder, G.F.; Walter, K.F.; Becker, S.; Meiler, J.; Grubmuller, H.; Griesinger, C.; de Groot, B.L. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 2008, 320, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Salvi, N.; Ulzega, S.; Ferrage, F.; Bodenhausen, G. Time scales of slow motions in ubiquitin explored by heteronuclear double resonance. J. Am. Chem. Soc. 2012, 134, 2481–2484. [Google Scholar] [CrossRef] [PubMed]
- Massi, F.; Grey, M.J.; Palmer, A.G., 3rd. Microsecond timescale backbone conformational dynamics in ubiquitin studied with NMR R1rho relaxation experiments. Protein Sci. 2005, 14, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, R.; Yamada, H.; Akasaka, K. Two folded conformers of ubiquitin revealed by high-pressure NMR. Biochemistry 2001, 40, 13556–13563. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, R.; Yokoyama, S.; Akasaka, K. NMR snapshots of a fluctuating protein structure: Ubiquitin at 30 bar-3 kbar. J. Mol. Biol. 2005, 347, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, R.; Akasaka, K. Close identity of a pressure-stabilized intermediate with a kinetic intermediate in protein folding. Proc. Natl. Acad. Sci. USA 2003, 100, 3167–3172. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wand, A.J. Partial alignment and measurement of residual dipolar couplings of proteins under high hydrostatic pressure. J. Biomol. NMR 2013, 56, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Sugita, Y. Dynamic Correlation between Pressure-Induced Protein Structural Transition and Water Penetration. J. Phys. Chem. B 2010, 114, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, S.; Kameda, T.; Yagi-Utsumi, M.; Sugase, K.; Baxter, N.J.; Kato, K.; Williamson, M.P.; Kitahara, R. Solution structure of the Q41N variant of ubiquitin as a model for the alternatively folded N2 state of ubiquitin. Biochemistry 2013, 52, 1874–1885. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, S.; Kameda, T.; Kumo, A.; Yagi-Utsumi, M.; Baxter, N.J.; Kato, K.; Williamson, M.P.; Kitahara, R. Close Identity between Alternatively Folded State N2 of Ubiquitin and the Conformation of the Protein Bound to the Ubiquitin-Activating Enzyme. Biochemistry 2014, 53, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, R.; Yamaguchi, Y.; Sakata, E.; Kasuya, T.; Tanaka, K.; Kato, K.; Yokoyama, S.; Akasaka, K. Evolutionally conserved intermediates between ubiquitin and NEDD8. J. Mol. Biol. 2006, 363, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, R.; Zhao, C.; Saito, K.; Koshiba, S.; Inoue, M.; Kigawa, T.; Yokoyama, S.; Akasaka, K. Basic folded and low-populated locally disordered conformers of SUMO-2 characterized by NMR spectroscopy at varying pressures. Biochemistry 2008, 47, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.H.; Ullah, S.; Hansen, P.E.; Williamson, M.P. Characterization of Salt Bridges to Lysines in the Protein G B1 Domain. J. Am. Chem. Soc. 2009, 131, 4674–4684. [Google Scholar] [CrossRef] [PubMed]
- Sundd, M.; Iverson, N.; Ibarra-Molero, B.; Sanchez-Ruiz, J.M.; Robertson, A.D. Electrostatic interactions in ubiquitin: Stabilization of carboxylates by lysine amino groups. Biochemistry 2002, 41, 7586–7596. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, W.R.; Antosiewicz, J.M.; Robertson, A.D. Empirical relationships between protein structure and carboxyl pK(a) values in proteins. Proteins 2002, 48, 388–403. [Google Scholar] [CrossRef] [PubMed]
- Haririnia, A.; Verma, R.; Purohit, N.; Twarog, M.Z.; Deshaies, R.J.; Bolon, D.; Fushman, D. Mutations in the hydrophobic core of ubiquitin differentially affect its recognition by receptor proteins. J. Mol. Biol. 2008, 375, 979–996. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Blevins, R.A. NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 1994, 4, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Iwahara, J.; Koshiba, S.; Tomizawa, T.; Tochio, N.; Guntert, P.; Kigawa, T.; Yokoyama, S. KUJIRA, a package of integrated modules for systematic and interactive analysis of NMR data directed to high-throughput NMR structure studies. J. Biomol. NMR 2007, 39, 31–52. [Google Scholar] [CrossRef] [PubMed]
- Farrow, N.A.; Muhandiram, R.; Singer, A.U.; Pascal, S.M.; Kay, C.M.; Gish, G.; Shoelson, S.E.; Pawson, T.; Formankay, J.D.; Kay, L.E. Backbone Dynamics of a Free and a Phosphopeptide-Complexed Src Homology-2 Domain Studied by N-15 Nmr Relaxation. Biochemistry 1994, 33, 5984–6003. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | pKa a | pKa b |
|---|---|---|
| D21 | 2.98 ± 0.03 | 3.1 |
| D32 | 3.68 ± 0.03 | 3.8 |
| D39 | 3.52 ± 0.02 | 3.6 |
| D58 | 3.47 ± 0.02 | 3.7 |
| E34 | 4.16 ± 0.03 | 4.5 |
| E16 | 4.07 ± 0.02 | 3.9 |
| E18 | 4.26 ± 0.03 | 4.3 |
| E51 | 3.69 ± 0.02 | 3.8 |
| E64 | 4.26 ± 0.02 | 4.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitazawa, S.; Yagi-Utsumi, M.; Kato, K.; Kitahara, R. Interactions Controlling the Slow Dynamic Conformational Motions of Ubiquitin. Molecules 2017, 22, 1414. https://doi.org/10.3390/molecules22091414
Kitazawa S, Yagi-Utsumi M, Kato K, Kitahara R. Interactions Controlling the Slow Dynamic Conformational Motions of Ubiquitin. Molecules. 2017; 22(9):1414. https://doi.org/10.3390/molecules22091414
Chicago/Turabian StyleKitazawa, Soichiro, Maho Yagi-Utsumi, Koichi Kato, and Ryo Kitahara. 2017. "Interactions Controlling the Slow Dynamic Conformational Motions of Ubiquitin" Molecules 22, no. 9: 1414. https://doi.org/10.3390/molecules22091414
APA StyleKitazawa, S., Yagi-Utsumi, M., Kato, K., & Kitahara, R. (2017). Interactions Controlling the Slow Dynamic Conformational Motions of Ubiquitin. Molecules, 22(9), 1414. https://doi.org/10.3390/molecules22091414