Synthesis of New Hydrazone Derivatives for MAO Enzymes Inhibitory Activity

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
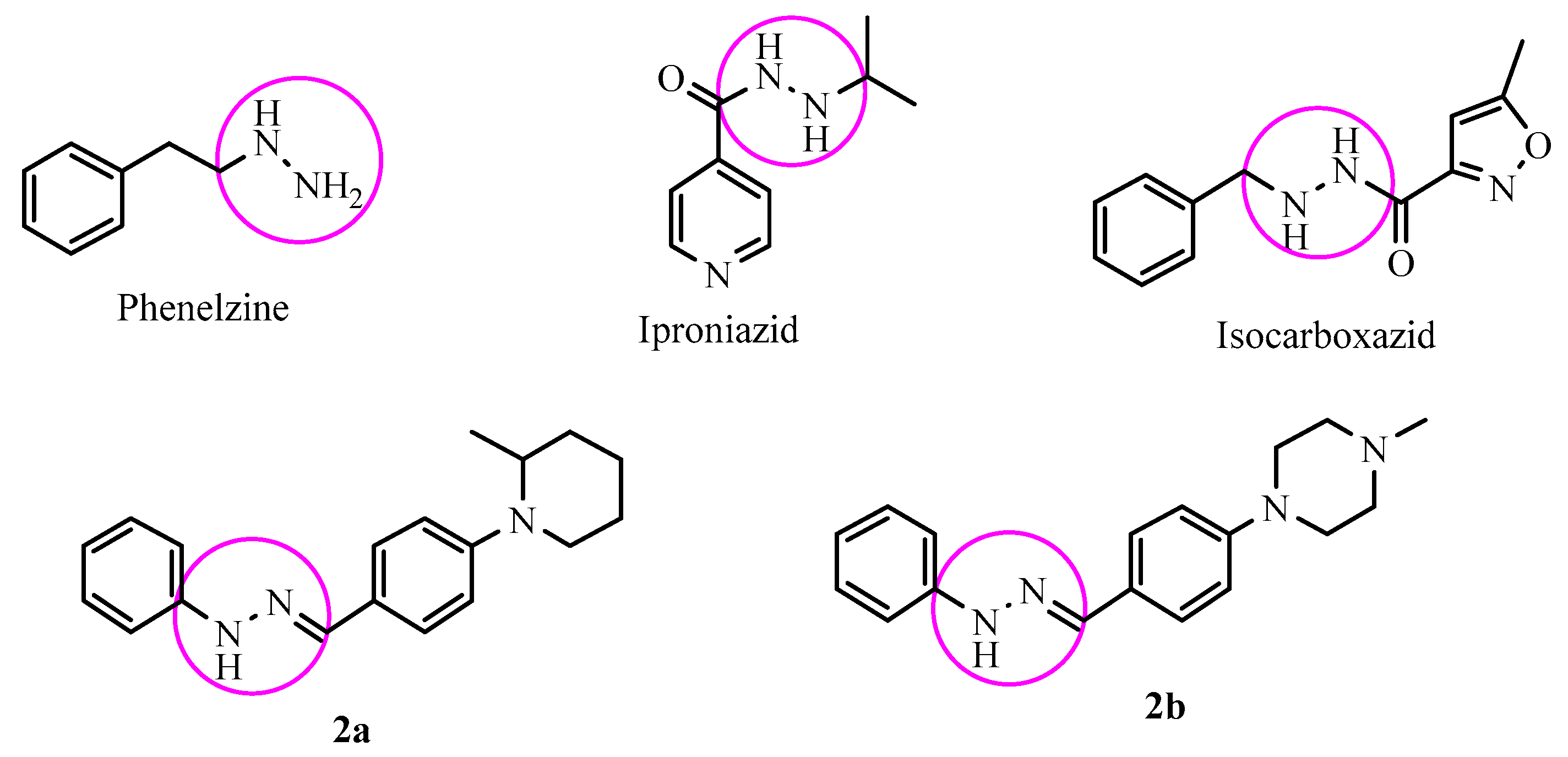
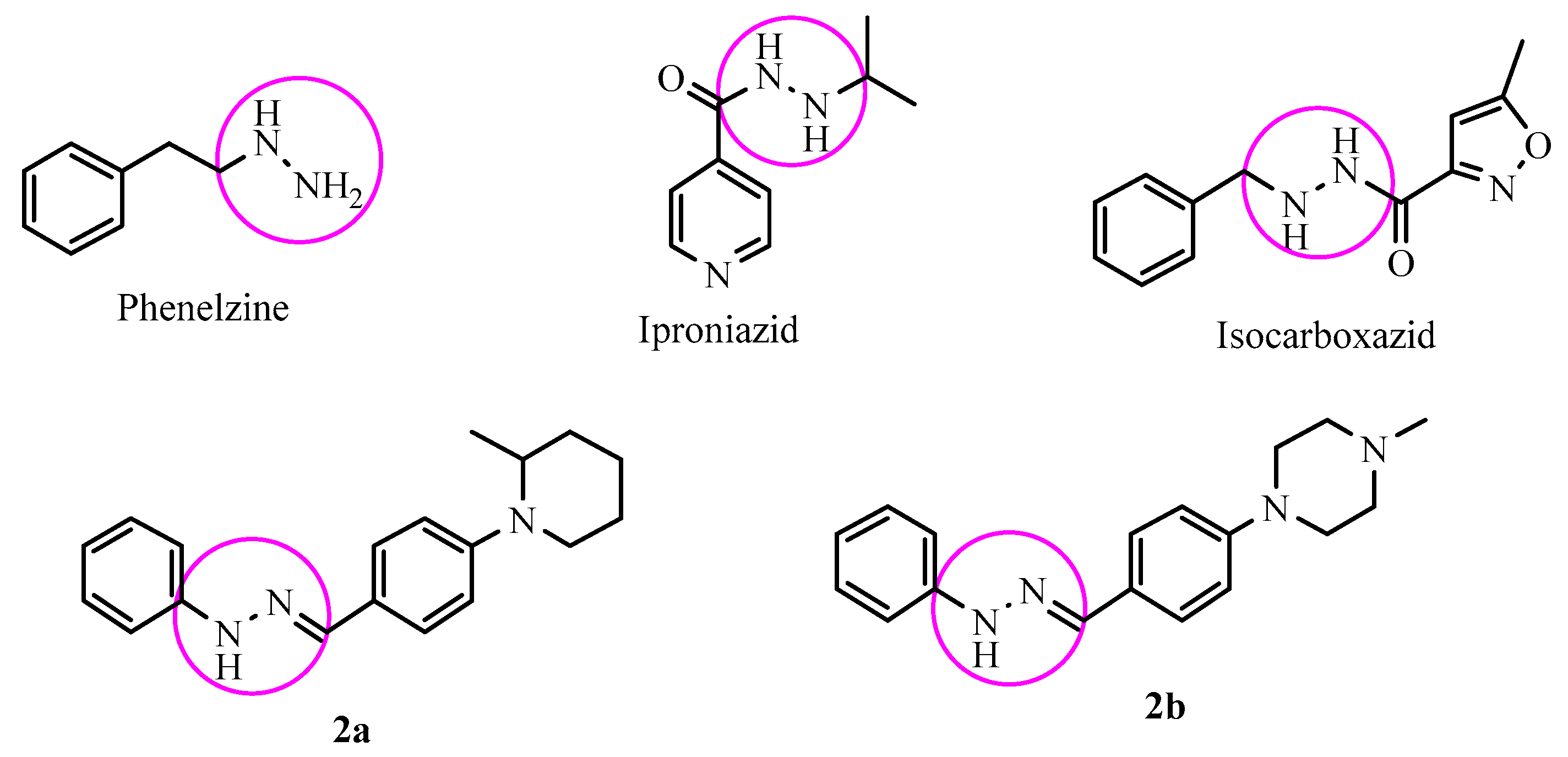
2.1. Chemistry
| Compounds | R |
| 2a | 2-Methylpiperidinyl |
| 2b | 4-Methylpiperazinyl |
| 2c | 4-Phenylpiperazinyl |
| 2d | 1-(4-Methoxyphenyl)piperazinyl |
| 2e | 4-Methoxyphenoxy |
| 2f | 4-Methoxyphenylthio |
| 2g | 4-Fluorophenoxy |
| 2h | 4-Fluorophenylthio |
| 2i | imidazolyl |
| 2j | triazolyl |
| 2k | 4-Chlorophenylthio |
| 2l | 4-Benzylpiperidinyl |
| 2m | 4-(2-Dimethylaminoethyl)piperazinyl |
| 2n | 4-(3-Dimethylaminopropyl)piperazinyl |
2.2. Enzymatic Studies
2.2.1. MAO-A and MAO-B Inhibition Assay
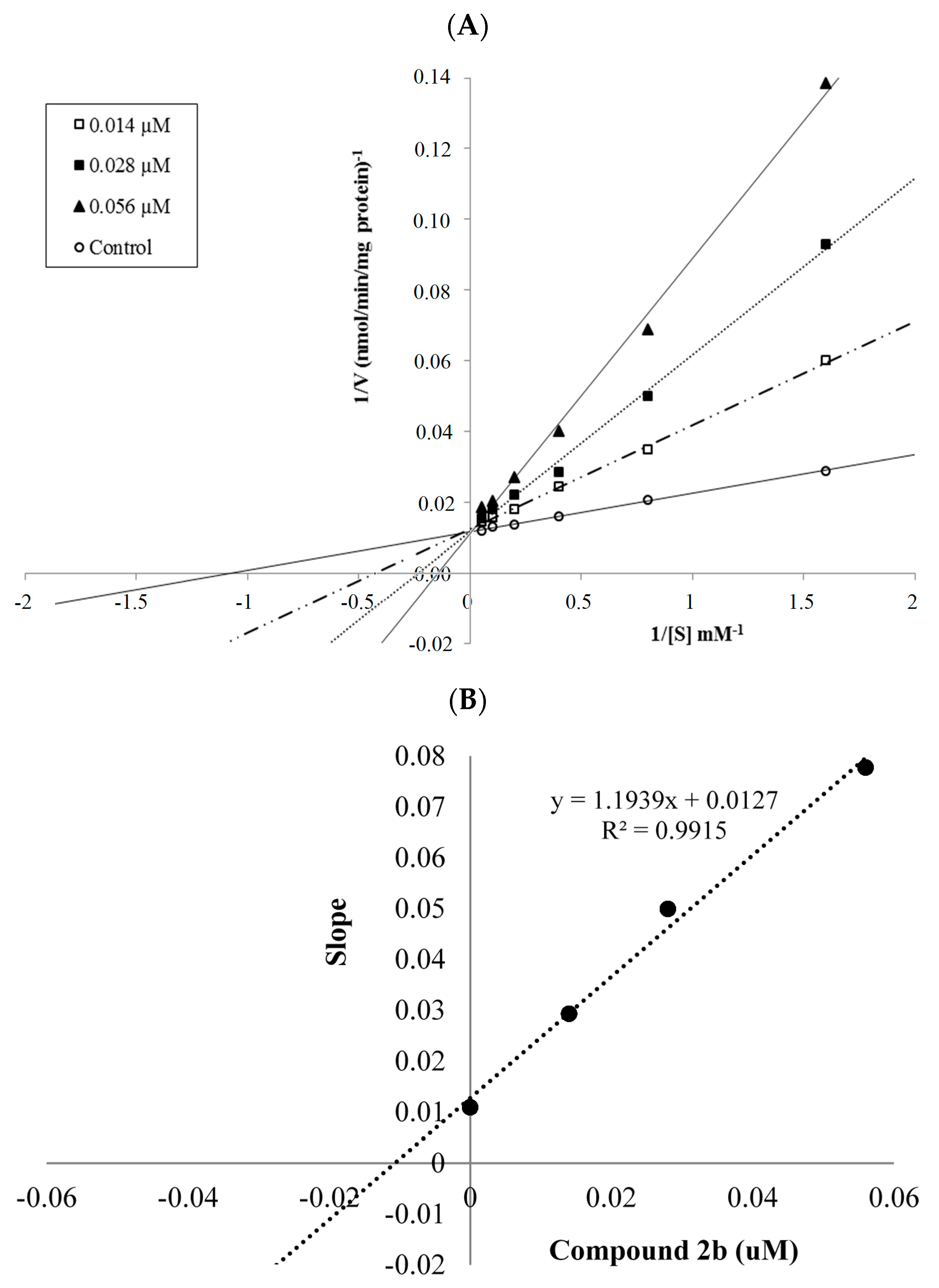
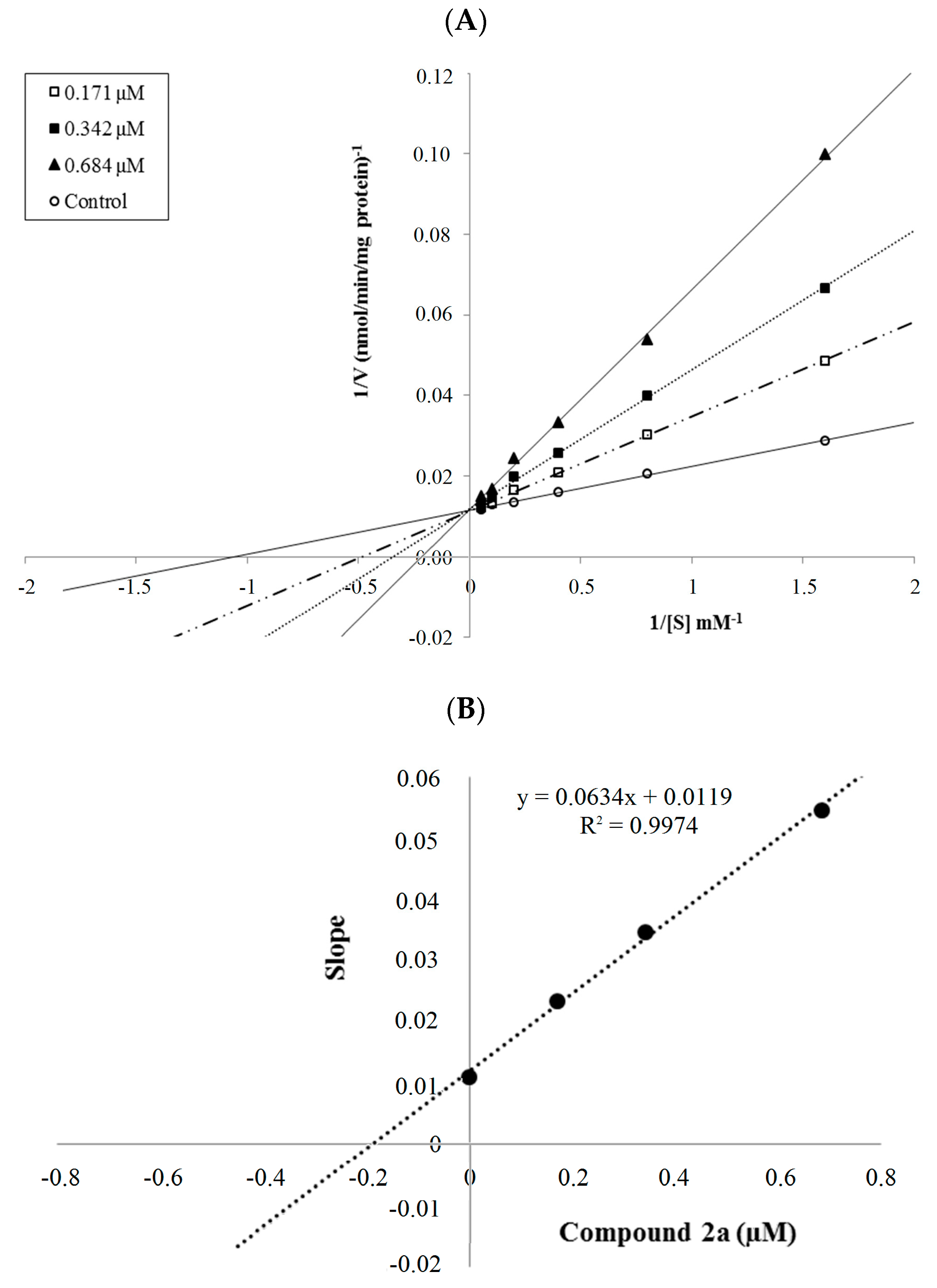
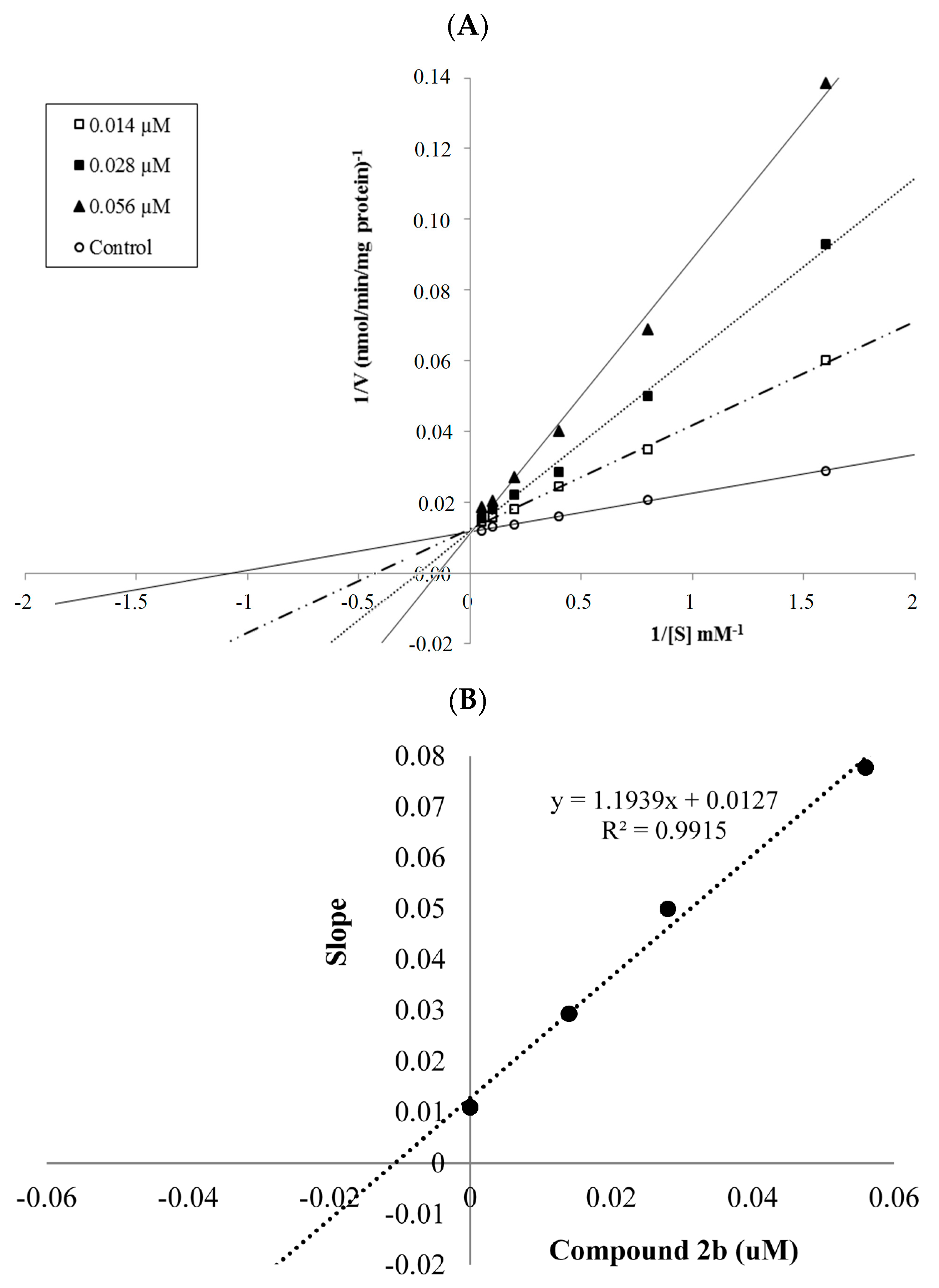
2.2.2. Enzyme Kinetics
2.3. Toxicological Studies
2.3.1. Cytotoxicity Test
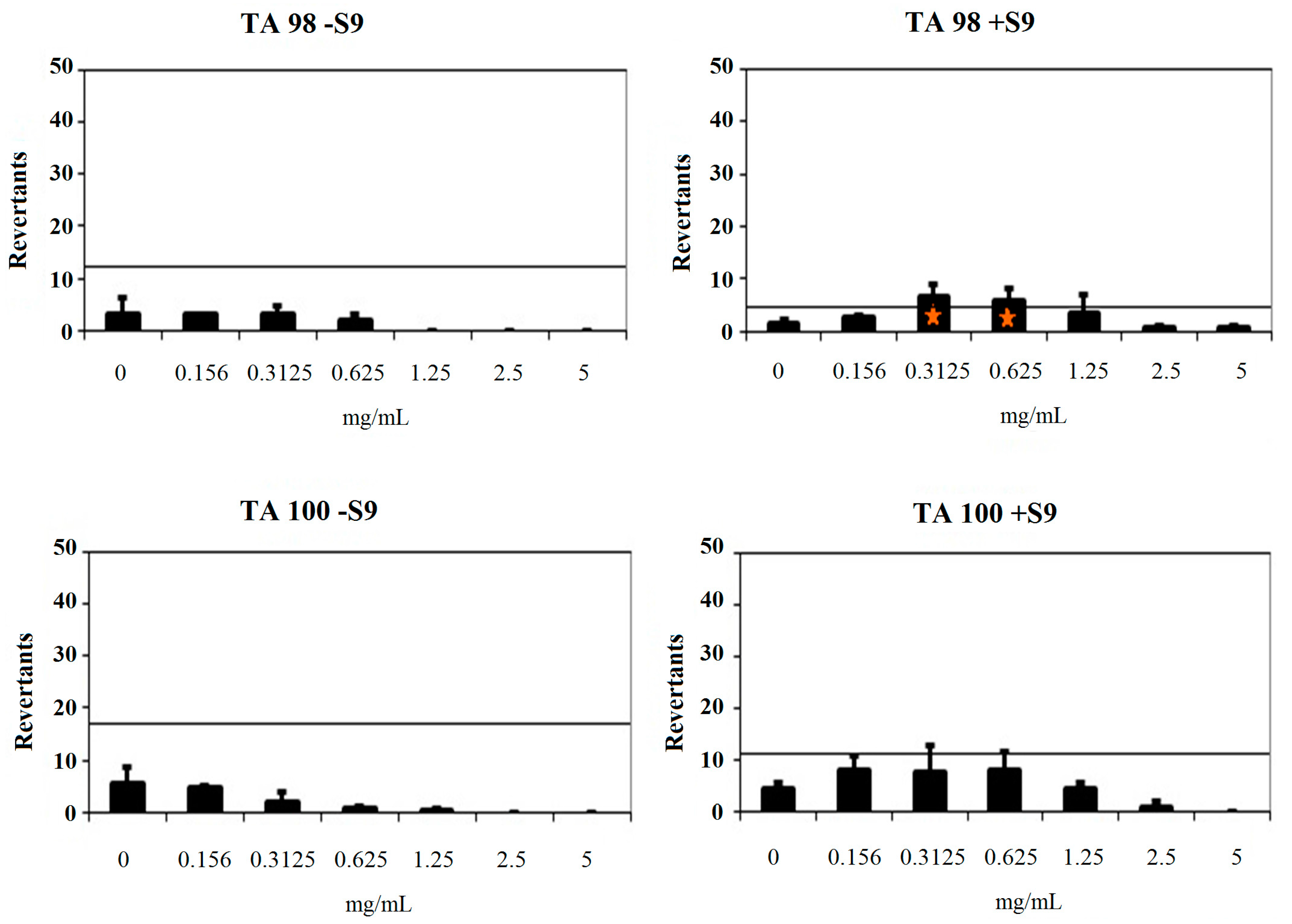
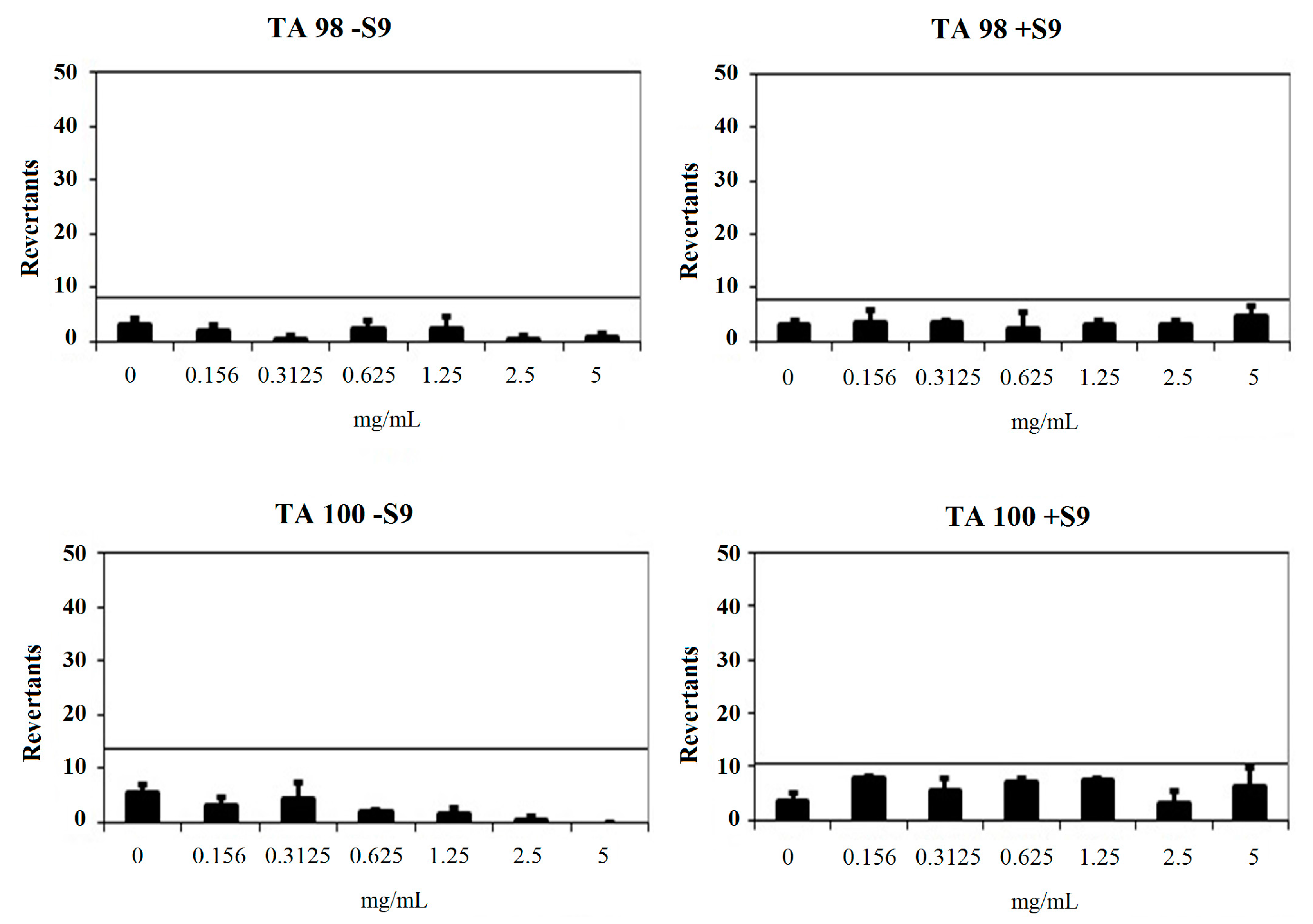
2.3.2. Genotoxicity Test
2.4. Prediction of ADME Parameters and BBB Permeability
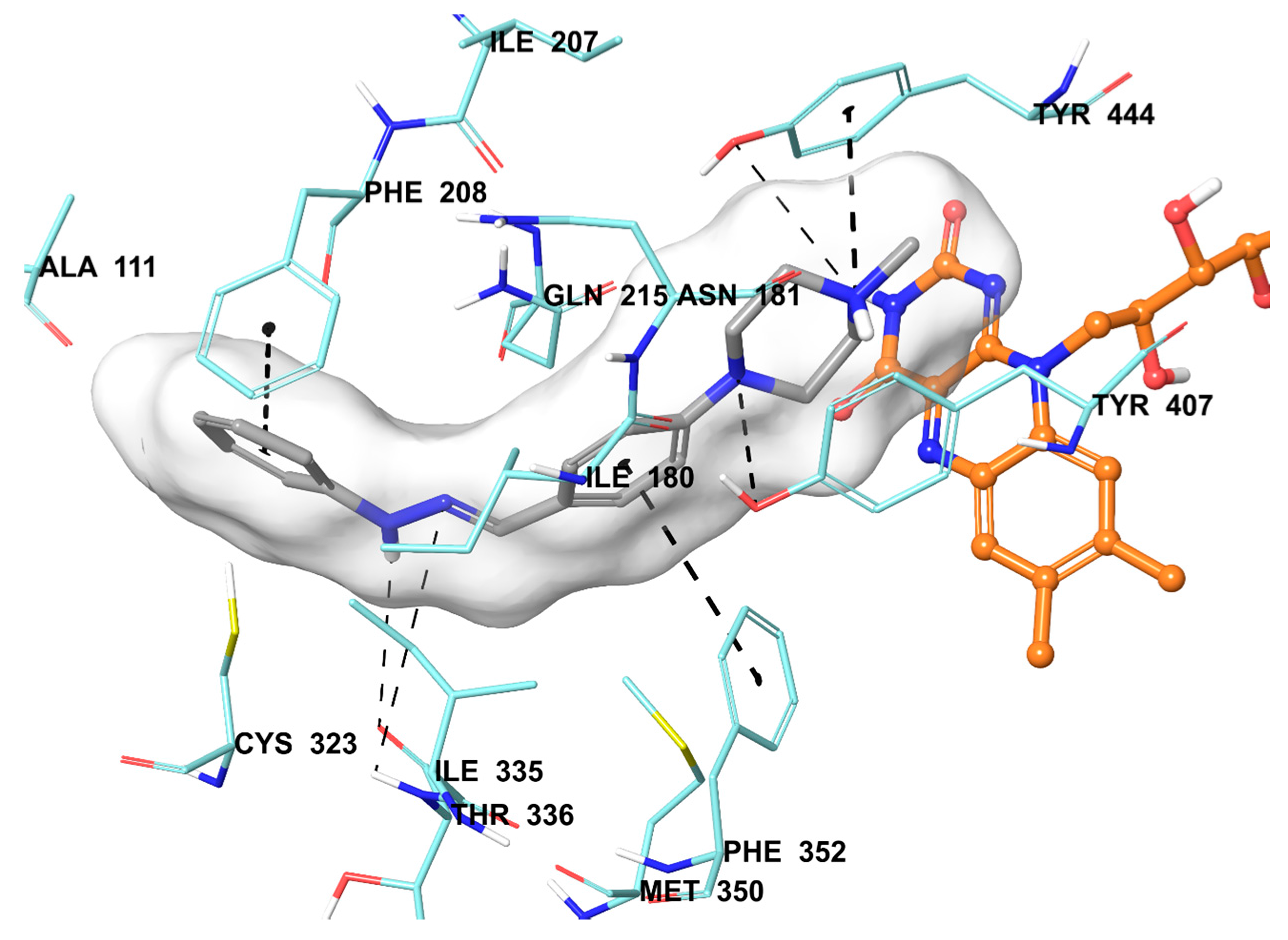
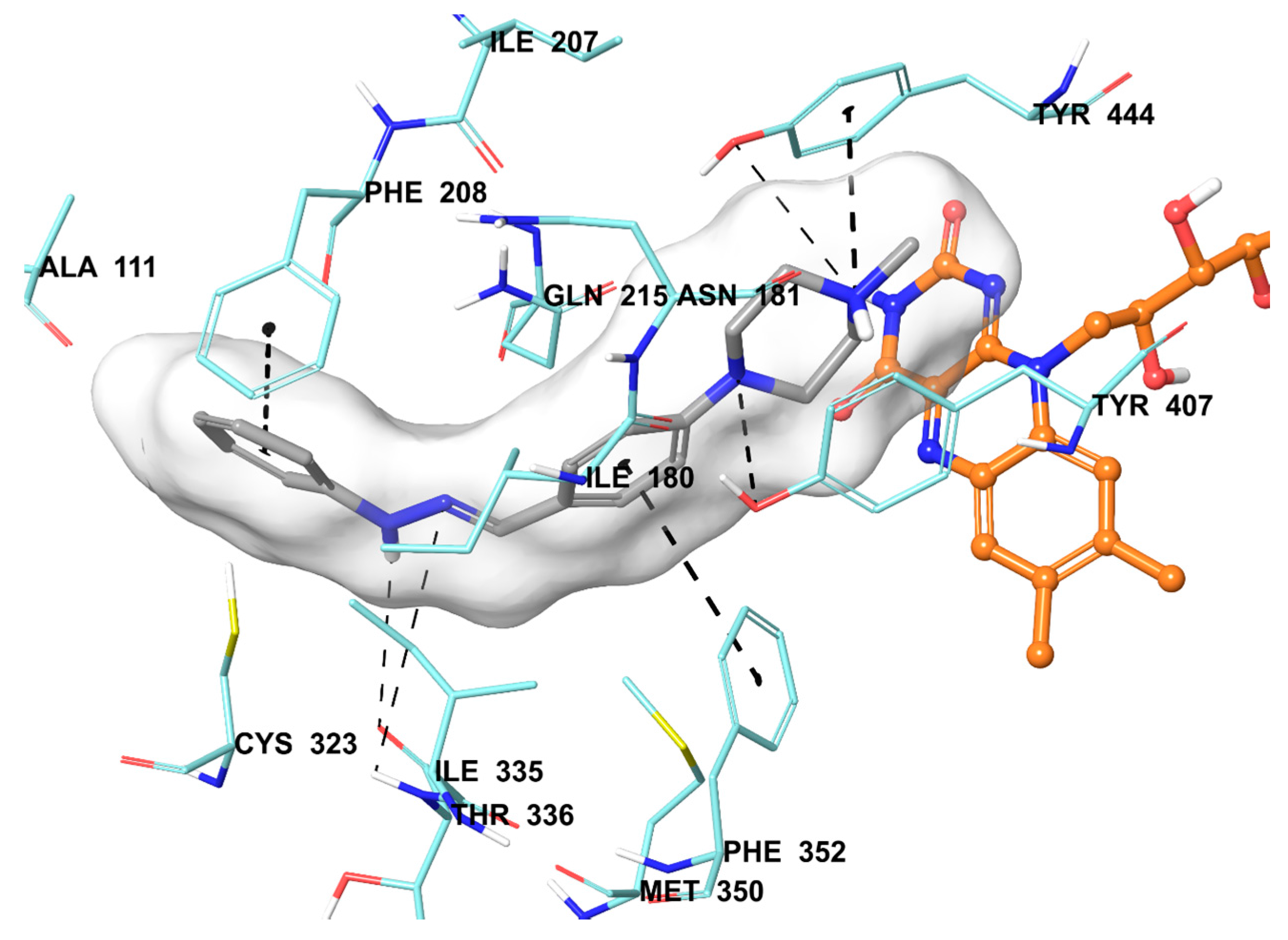
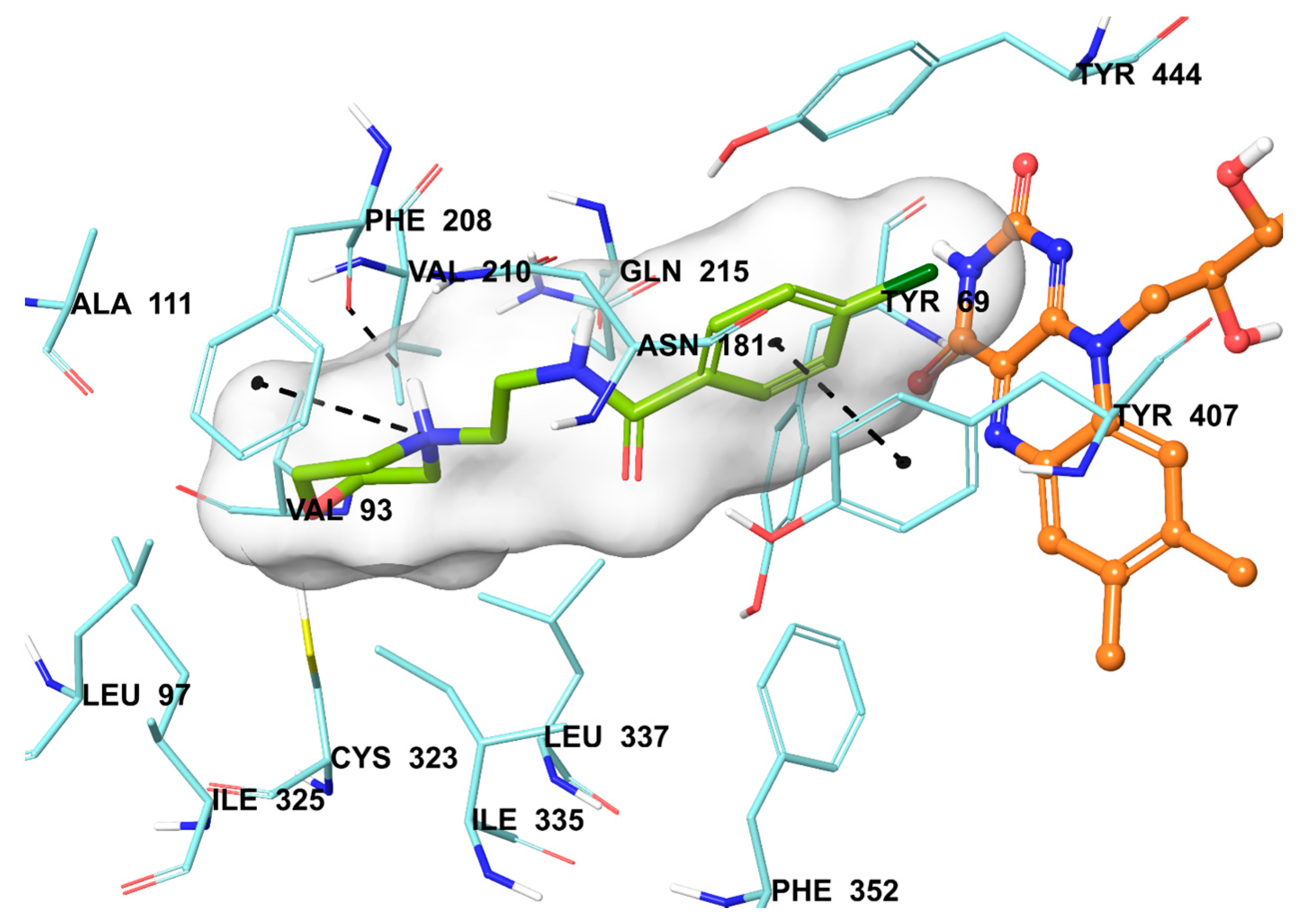
2.5. Molecular Docking Studies
3. Materials and Methods
3.1. General Information
3.2. Chemistry
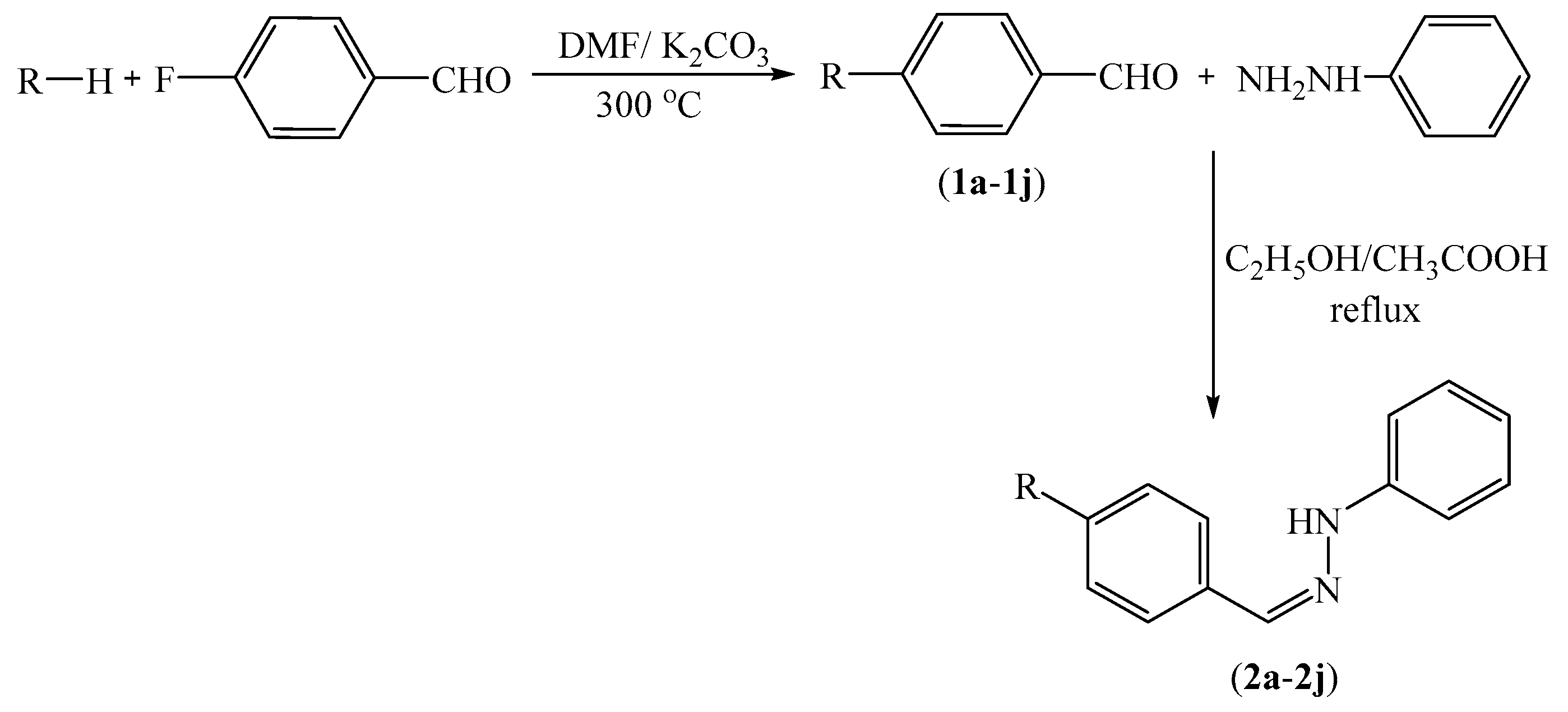
3.2.1. Synthesis of 4-substituted Benzaldehydes 1a–1n
3.2.2. General Procedure for the Synthesis of Target Compounds 2a–2n
3.3. Activity Studies
3.3.1. MAO-A and MAO-B Inhibition Assay
3.3.2. Enzyme Kinetics Studies
3.4. Toxicology Studies
3.4.1. Cytotoxicity Test
3.4.2. Genotoxicity Test
3.5. Prediction of ADME Parameters and BBB Permeability
3.6. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Oscar, S.; Gershanik, M.D. Improving l-dopa therapy: The development of enzyme inhibitors. Mov. Disord. 2015, 30, 103–113. [Google Scholar]
- Mıshra, V.; Kashaw, S.K.; Kumar, A. Quantitative structure-activity analysis of 1,3,5-trisubstituted pyrazoline derivatives as monoamine oxidase inhibitors. Asian J. Chem. 2011, 23, 4377–4379. [Google Scholar]
- Da Prad, M.; Kettler, R.; Keller, H.H.; Cesura, A.M.; Richards, J.G.; Saura Marti, J.; Muggli-Maniglio, D.; Wyss, P.C.; Kyburz, E. From moclobemide to the development of a new class of reversible, selective MAO-A and MAO-B inhibitors. J. Neural Transm. 1990, 29, 279–292. [Google Scholar]
- Ellen Billett, E. Monoamine oxidase (MAO) in human peripheral tissues. NeuroToxicology 2004, 25, 139–148. [Google Scholar] [CrossRef]
- Yang, D.; Wang, R.; Zhu, J.J. Synthesis, crystal structures, molecular docking, in vitro monoamine oxidase-B inhibitory activity of transition metal complexes with 2-{4-[bis (4-fluorophenyl)methyl]piperazin-1-yl} acetic acid. J. Mol. Struct. 2017, 1128, 493–498. [Google Scholar] [CrossRef]
- Ramsay, R.R. Molecular aspects of monoamine oxidase B. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 69, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Stockmeier, C.A.; Meyer, H.J.; Austin, M.C.; Albert, P.R.; Wang, J.; May, W.; Rajkowska, G.; Overholser, J.C.; Jurjus, G.; et al. The reduction of R1, a novel repressor protein for monoamine oxidase A, in major depressive disorder. Neuropsychopharmacology 2011, 36, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, M.; Chen, K.; Shih, J.C. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Matveychuk, D.; Nunes, E.; Ullah, N.; Aldawsari, F.S.; Velazquez-Martinez, C.A.; Baker, G.B. Elevation of rat brain tyrosine levels by phenelzine is mediated by its active metabolite β-phenylethylidenehydrazine. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 53, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Li, M.; Hubalek, F.; Matteyi, A.; Edmondson, D.E. Structural and mechanistic studies of arylalkylhydrazine inhibition of human monoamine oxidases A and B. Biochemistry 2008, 47, 5616–5625. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Nguyen, P.V.; Baker, G.B. Phenylethylıdenehydrazıne, a novel GABA-transamınase inhıbıtor, reduces epıleptıform activity in rat hippocampal slices. Neuroscience 2004, 126, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, E.M.; Fassihi, A.; Davood, A.; Chen, Q.; Rauw, G.; Rauw, G.; Knaus, E.E.; Baker, G.B. N-Propynyl analogs of β-phenylethylidenehydrazines: Synthesis and evaluation of effects on glycine, GABA, and monoamine oxidase. Bioorg. Med. Chem. 2008, 16, 8254–8263. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Yasuhara, H. Clinical pharmacology of MAO inhibitors: Safety and future. NeuroToxicology 2004, 25, 215–221. [Google Scholar] [CrossRef]
- Anderson, M.C.; Hasan, F.; McCrodden, J.M.; Tipton, K.H. Monoamine oxidase inhibitors and the cheese effect. Neurochem. Res. 1993, 18, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.G.; Livingston, H.M. Monoamine oxidase inhibitors. Drug Exp. 1996, 14, 219–227. [Google Scholar] [CrossRef]
- Ali, M.R.; Marella, A.; Alam, M.T.; Naz, R.; Akhter, M.; Shaquiquzzaman, M.; Saha, R.; Tanwar, O.; Alam, M. M.; Hooda, J. Revıew of biological activities of hydrazones. Indones. J. Pharm. 2012, 23, 193–202. [Google Scholar]
- Kamal, R.; Kumar, V.; Bhardwaj, V.; Kumar, V.; Aneja, K.R. Synthesis, characterization and in vitro antimicrobial evaluation of some novel hydrazone derivatives bearing pyrimidinyl and pyrazolyl moieties as a promising heterocycles. Med. Chem. Res. 2015, 24, 2551–2560. [Google Scholar] [CrossRef]
- Belskaya, N.P.; Dehaen, W.; Bakuleva, V.A. Synthesis and properties of hydrazones bearing amide, thioamide and amidine functions. Arkıvoc 2010, 275–332. [Google Scholar]
- Evranos-Aksöz, B.; Yabanoğlu Çiftçi, S.; Uçar, G.; Yelekçi, K.; Ertan, R. Synthesis of some novel hydrazone and 2-pyrazoline derivatives: Monoamine oxidase inhibitory activities and docking studies. Bioorg. Med. Chem. Lett. 2014, 24, 3278–3284. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Granese, A.; Carradori, S.; D’Ascenzio, M.; Yanez, M.; Orallo, F. Synthesis and selective inhibition of human Monoamine Oxidases of a large scaffold of (4,5-substituted-thiazol-2-yl)hydrazones. Med. Chem. Commun. 2010, 1, 61–72. [Google Scholar] [CrossRef]
- Chimenti, F.; Secci, D.; Bolasco, A.; Chimenti, P.; Granese, A.; Carradori, S.; D’Ascenzio, M.; Yanez, M.; Orallo, F.; Sanna, M.L.; et al. Synthesis, stereochemical separation, and biological evaluation of selective inhibitors of human MAO-B: 1-(4-Arylthiazol-2-yl)-2-(3-methylcyclohexylidene)hydrazines. J. Med. Chem. 2010, 53, 6516–6520. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.K.; Ayyannan, S.R. Design, synthesis, and evaluation of 2-amino-6-nitrobenzothiazole-derived hydrazones as MAO inhibitors: Role of the methylene spacer group. Chem. Med. Chem. 2016, 11, 1551–1567. [Google Scholar] [CrossRef] [PubMed]
- Bhagavan, N.V. Essentials of Medical Biochemistry: With Clinical Cases, 1st ed.; Elsevier: Burlington, MA, USA, 2011; pp. 47–58. [Google Scholar]
- International Organization for Standardization. Biological Evaluation of Medical Devices. In Tests for In Vitro Cytotoxicity, 3rd ed.; International Organization for Standardization: Geneva, Switzerland, 2009; Volume 5, pp. 1–34. [Google Scholar]
- Flückiger-Isler, S.; Kamber, M. Direct comparison of the Ames microplate format (MPF) test in liquid medium with the standard Ames pre-incubation assay on agar plates by use of equivocal to weakly positive test compounds. Mutat. Res. 2012, 747, 36–45. [Google Scholar] [CrossRef] [PubMed]
- De Waterbeemd, H.V.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2013, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P. Molinspiration Cheminformatics. Available online: http://www.molinspiration.com/cgi-bin/properties.html (accessed 30 July 2017).
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef] [PubMed]
- About the Blood-brain Barrier (BBB) Prediction Server. Available online: http://www.cbligand.org/BBB/index.php (accessed 30 July 2017).
- Regis, N.; Ivan, H. Optimizing the reversibility of hydrazone formation for dynamic combinatorial chemistry. Chem. Commun. 2003, 8, 942–943. [Google Scholar]
- Son, S.Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of human monoamine oxidase A at 2.2-Å resolution: The control of opening the entry for substrates/inhibitors. PNAS 2008, 105, 5739–5744. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.A.; Binda, C.; Mattevi, A. The FAD binding sites of human monoamine oxidases A and B. Neurotoxicology 2004, 25, 63–72. [Google Scholar] [CrossRef]
- Hubalek, F.; Binda, C.; Khalil, A.; Li, M.; Mattevi, A.; Castagnoli, N.; Edmondson, D.E. Demonstration of isoleucine 199 as a structural determinant for the selective inhibition of human monoamine oxidase B by specific reversible inhibitors. J. Biol. Chem. 2005, 280, 15761–15766. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yoshimura, M.; Yamashita, E.; Nakagawa, A.; Ito, A.; Tsukihara, T. Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J. Mol. Biol. 2004, 338, 103–104. [Google Scholar] [CrossRef] [PubMed]
- Andrés, A.M.; Soldevila, M.; Navarro, A.; Kidd, K.K.; Oliva, B.; Bertranpetit, J. Positive selection in MAO A gene is human exclusive: determination of the putative amino acid change selected in the human lineage. J. Hum. Genet. 2004, 115, 377–386. [Google Scholar]
- Karaca Gençer, H.; Acar Çevik, U.; Kaya Çavuşoğlu, B.; Sağlık, B.N.; Levent, S.; Atlı, Ö.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Design, synthesis, and evaluation of novel 2-phenylpropionic acid derivatives as dual COX inhibitory-antibacterial agents. J. Enzyme Inhib. Med. Chem. 2017, 32, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Can, Ö.D.; Osmaniye, D.; Demir Özkay, Ü.; Sağlık, B.N.; Levent, S.; Ilgın, S.; Baysal, M.; Özkay, Y.; Kaplancıklı, Z.A. MAO enzymes inhibitory activity of new benzimidazole derivatives including hydrazone and propargyl side chains. Eur. J. Med. Chem. 2017, 131, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Demir Özkay, Ü.; Can, Ö.D.; Sağlık, B.N.; Acar Çevik, U.; Levent, S.; Özkay, Y.; Ilgın, S.; Atlı, Ö. Design, synthesis, and AChE inhibitory activity of new benzothiazole-piperazines. Bioorg. Med. Chem. Lett. 2016, 26, 5387–5394. [Google Scholar] [CrossRef] [PubMed]
- Maestro. version 10.6. Schrödinger, LLC: New York, NY, USA, 2016.
- Protein Preparation Wizard. version 2016-2. Schrödinger, LLC: New York, NY, USA, 2016.
- LigPrep. version 3.8. Schrödinger, LLC: New York, NY, USA, 2016.
- Glide. version 7.1. Schrödinger, LLC: New York, NY, USA, 2016.
- Toprakçı, M.; Yelekçi, K. Docking studies on monoamine oxidase-B inhibitors: Estimation of inhibition constants (Ki) of a series of experimentally tested compounds. Bioorg. Med. Chem. Lett. 2005, 15, 4438–4446. [Google Scholar] [CrossRef] [PubMed]
- Gökhan-Kelekçi, N.; Özgün Şimşek, Ö.; Ercan, A.; Yelekçi, K.; Sibel Şahin, Z.; Işık, Ş.; Uçar, G.; Bilgin, A.A. Synthesis and molecular modeling of some novel hexahydroindazole derivatives as potent monoamine oxidase inhibitors. Bioorg. Med. Chem. 2009, 17, 6761–6772. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1a–1n and 2a–2n are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MAO-A Inhibition % | MAO-B Inhibition % | ||
|---|---|---|---|---|
| 10−3 M | 10−4 M | 10−3 M | 10−4 M | |
| 2a | 88.198 ± 0.821 | 84.288 ± 0.641 | 35.011 ± 0.766 | 22.150 ± 0.443 |
| 2b | 93.530 ± 0.412 | 88.862 ± 0.214 | 63.747 ± 1.028 | 42.951 ± 1.203 |
| 2c | 70.926 ± 1.064 | 43.245 ± 0.861 | 38.507 ± 0.732 | 34.740 ± 0.556 |
| 2d | 68.645 ± 1.501 | 40.212 ± 0.970 | 39.226 ± 0.863 | 30.630 ± 0.613 |
| 2e | 32.492 ± 0.844 | 25.018 ± 0.425 | 26.528 ± 0.478 | 19.401 ± 0.327 |
| 2f | 30.522 ± 0.817 | 26.705 ± 0.748 | 33.302 ± 0.833 | 21.880 ± 0.416 |
| 2g | 54.538 ± 1.003 | 20.380 ± 0.469 | 21.683 ± 0.520 | 17.080 ± 0.412 |
| 2h | 28.552 ± 0.570 | 21.045 ± 0.548 | 30.210 ± 0.574 | 24.126 ± 0.355 |
| 2i | 40.926 ± 0.900 | 18.326 ± 0.618 | 38.215 ± 1.095 | 30.718 ± 0.707 |
| 2j | 38.645 ± 0.743 | 33.013 ± 0.660 | 27.511 ± 0.633 | 22.055 ± 0.419 |
| 2k | 29.212 ± 0.672 | 21.526 ± 0.468 | 36.278 ± 0.774 | 18.978 ± 0.825 |
| 2l | 74.251 ± 1.614 | 44.828 ± 0.917 | 45.194 ± 0.692 | 28.560 ± 0.629 |
| 2m | 68.276 ± 1.048 | 39.471 ± 0.719 | 41.072 ± 0.732 | 27.607 ± 0.961 |
| 2n | 64.435 ± 1.104 | 36.154 ± 0.817 | 40.629 ± 0.933 | 29.068 ± 0.734 |
| Moclobemide | 94.121 ± 2.760 | 82.143 ± 2.691 | - | - |
| Selegiline | - | - | 98.910 ± 1.280 | 96.882 ± 1.312 |
| Compound | MAO-A Inhibition % | MAO-A IC50 (µM) | ||||||
|---|---|---|---|---|---|---|---|---|
| 10−3 M | 10−4 M | 10−5 M | 10−6 M | 10−7 M | 10−8 M | 10−9 M | ||
| 2a | 88.198 ± 0.821 | 84.288 ± 0.641 | 75.098 ± 0.693 | 50.828 ± 0.582 | 34.748 ± 0.378 | 30.548 ± 0.542 | 25.458 ± 0.517 | 0.342 ± 0.015 |
| 2b | 93.530 ± 0.412 | 88.862 ± 0.214 | 80.418 ± 0.470 | 66.218 ± 0.540 | 48.243 ± 0.631 | 45.273 ± 0.480 | 38.150 ± 0.367 | 0.028 ± 0.001 |
| Moclobemide | 94.121 ± 2.760 | 82.143 ± 2.691 | 60.458 ± 2.559 | 36.151 ± 1.984 | 22.135 ± 1.337 | 18.166 ± 0.812 | 14.128 ± 0.725 | 6.061 ± 0.262 |
| Compound | IC50 (µM) |
|---|---|
| 2a | 930 ± 15.43 |
| 2b | 20 ± 1.32 |
| Comp. | Concentration (mg/mL) | Revertants Fold Increase (Over Baseline) | |||
|---|---|---|---|---|---|
| TA 98 | TA 100 | ||||
| S9− | S9+ | S9− | S9+ | ||
| 2a | 0.156 | 0.42 | 0.92 | 0.44 | 0.99 |
| 0.3125 | 0.08 * | 0.83 | 0.63 | 0.69 | |
| 0.625 | 0.58 | 0.58 | 0.24 * | 0.91 | |
| 1.25 | 0.50 | 0.75 | 0.19 * | 0.95 | |
| 2.5 | 0.08 * | 0.75 | 0.05 * | 0.39 | |
| 5 | 0.17 * | 1.17 | 0.00 * | 0.82 | |
| 2b | 0.156 | 1.24 * | 0.85 | 0.40 | 1.46 * |
| 0.3125 | 0.36 | 2.44 * | 0.17 * | 1.40 | |
| 0.625 | 0.22 | 1.59 * | 0.06 * | 1.46 | |
| 1.25 | 0.00 | 0.37 | 0.03 * | 0.79 | |
| 2.5 | 0.00 | 0.37 | 0.00 * | 0.12 * | |
| 5 | 0.00 | 0.12 | 0.00 * | 0.00 * | |
| Compound | MW | logP | TPSA | HBA | HBD | MV | Vio | BBB |
|---|---|---|---|---|---|---|---|---|
| 2a | 293.41 | 6.10 | 27.63 | 1 | 1 | 293.65 | 1 | + |
| 2b | 294.40 | 4.76 | 105.11 | 2 | 1 | 289.61 | 0 | + |
| 2c | 356.47 | 6.46 | 30.87 | 1 | 1 | 344.46 | 1 | + |
| 2d | 386.50 | 6.51 | 40.10 | 2 | 1 | 370.00 | 1 | + |
| 2e | 318.38 | 6.57 | 42.86 | 3 | 1 | 297.05 | 1 | + |
| 2f | 334.44 | 6.79 | 33.62 | 3 | 1 | 306.20 | 1 | + |
| 2g | 306.34 | 6.68 | 33.62 | 2 | 1 | 276.44 | 1 | + |
| 2h | 322.41 | 6.89 | 24.39 | 1 | 1 | 285.58 | 1 | + |
| 2i | 262.32 | 4.09 | 42.22 | 2 | 1 | 243.73 | 0 | + |
| 2j | 263.30 | 3.56 | 55.11 | 3 | 1 | 239.58 | 0 | + |
| 2k | 338.86 | 7.41 | 24.39 | 2 | 1 | 294.19 | 1 | + |
| 2l | 369.51 | 7.44 | 27.63 | 3 | 1 | 365.30 | 1 | + |
| 2m | 351.50 | 4.79 | 34.10 | 5 | 1 | 352.56 | 0 | + |
| 2n | 365.52 | 5.06 | 34.10 | 5 | 1 | 369.36 | 1 | + |
| Moclobemide | 268.74 | 1.69 | 41.57 | 4 | 1 | 240.70 | 0 | + |
| Selegiline | 187.29 | 2.64 | 3.24 | 1 | 0 | 202.64 | 0 | + |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Can, N.Ö.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; İnci, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Hydrazone Derivatives for MAO Enzymes Inhibitory Activity. Molecules 2017, 22, 1381. https://doi.org/10.3390/molecules22081381
Can NÖ, Osmaniye D, Levent S, Sağlık BN, İnci B, Ilgın S, Özkay Y, Kaplancıklı ZA. Synthesis of New Hydrazone Derivatives for MAO Enzymes Inhibitory Activity. Molecules. 2017; 22(8):1381. https://doi.org/10.3390/molecules22081381
Chicago/Turabian StyleCan, Nafiz Öncü, Derya Osmaniye, Serkan Levent, Begüm Nurpelin Sağlık, Beril İnci, Sinem Ilgın, Yusuf Özkay, and Zafer Asım Kaplancıklı. 2017. "Synthesis of New Hydrazone Derivatives for MAO Enzymes Inhibitory Activity" Molecules 22, no. 8: 1381. https://doi.org/10.3390/molecules22081381
APA StyleCan, N. Ö., Osmaniye, D., Levent, S., Sağlık, B. N., İnci, B., Ilgın, S., Özkay, Y., & Kaplancıklı, Z. A. (2017). Synthesis of New Hydrazone Derivatives for MAO Enzymes Inhibitory Activity. Molecules, 22(8), 1381. https://doi.org/10.3390/molecules22081381