Investigation on the Enzymatic Profile of Mulberry Alkaloids by Enzymatic Study and Molecular Docking

 , ,
, ,
Abstract
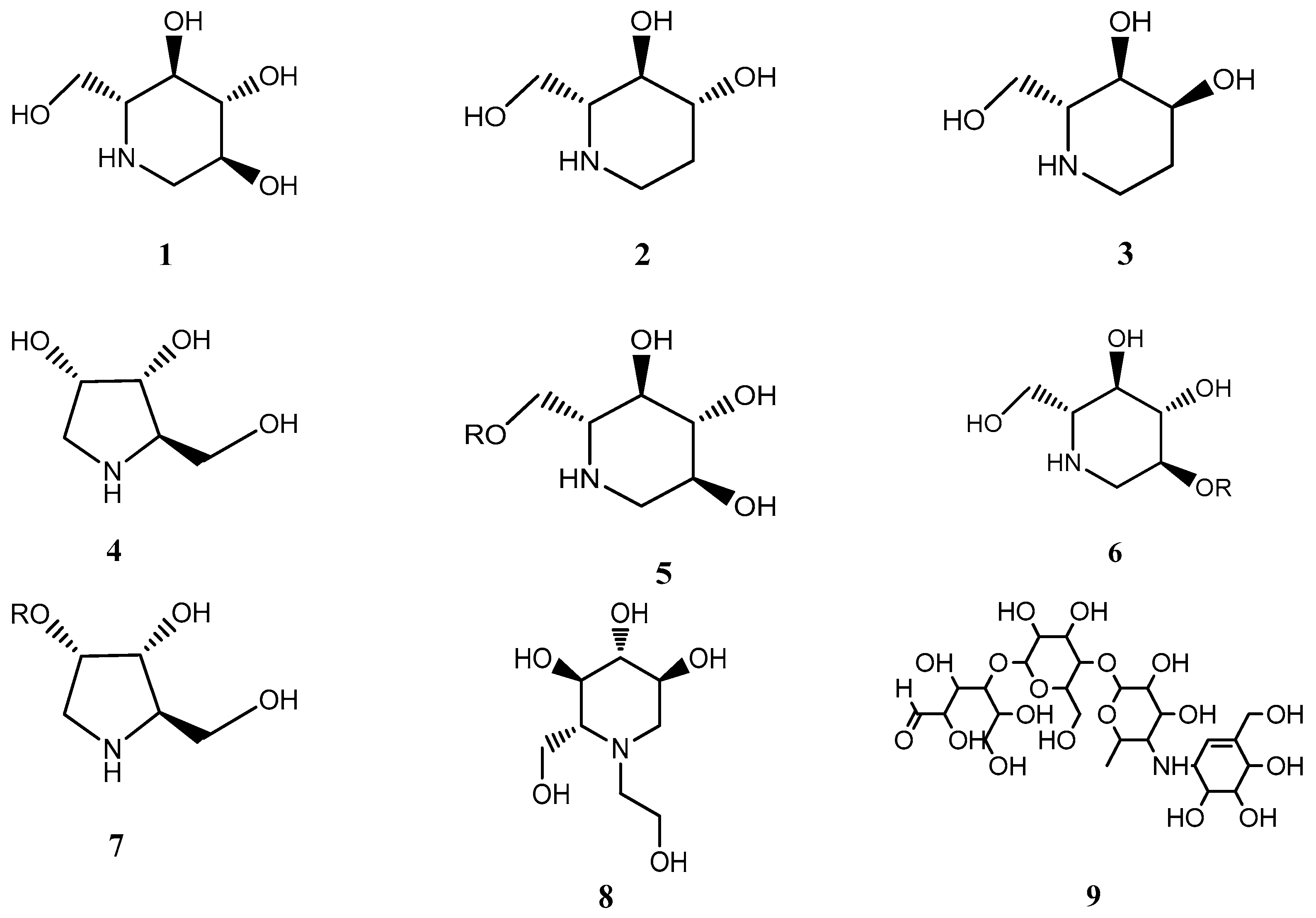
1. Introduction
2. Results and Discussion
2.1. Inhibition of α-Glucosidase by SZ-A
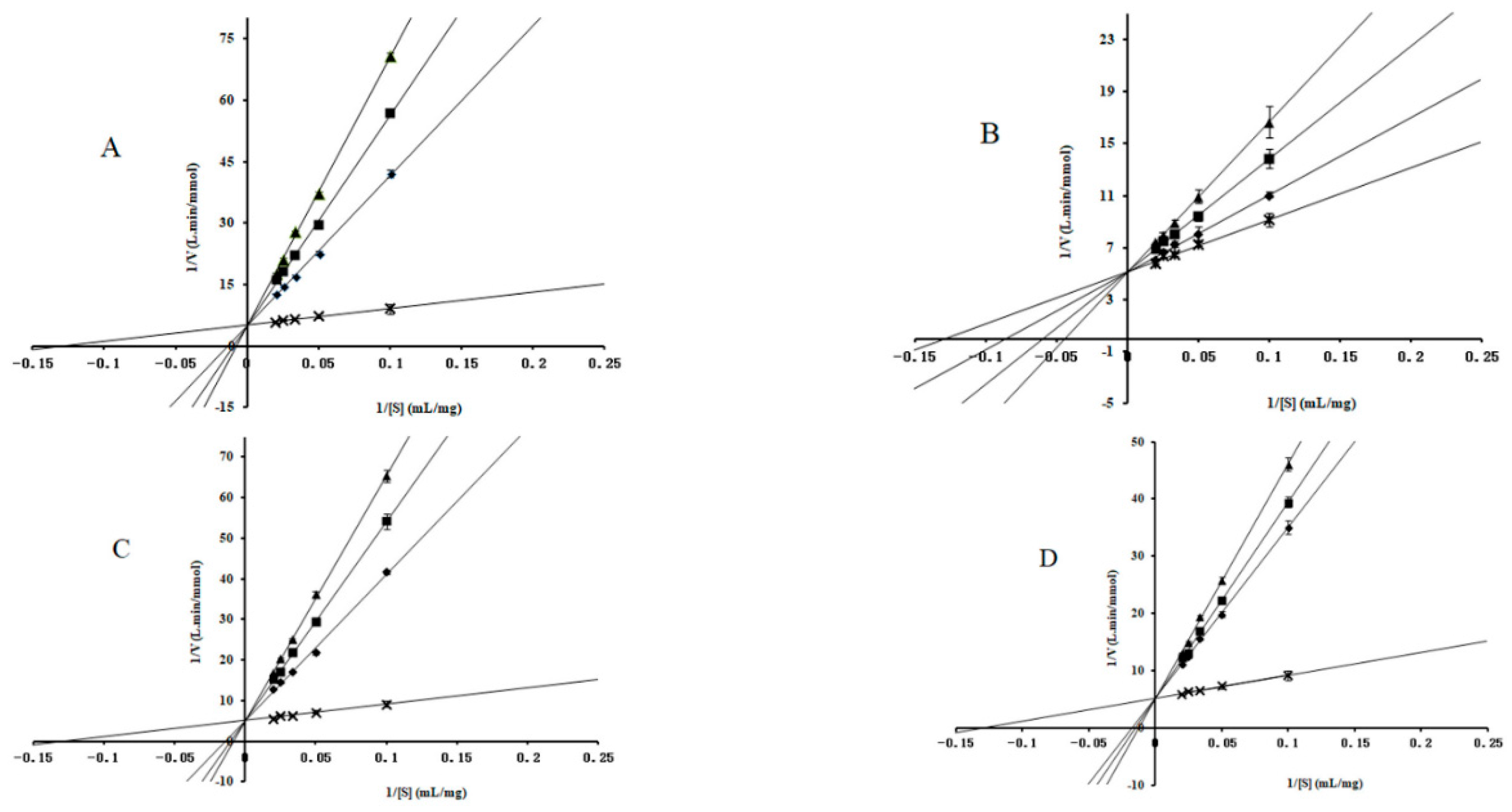
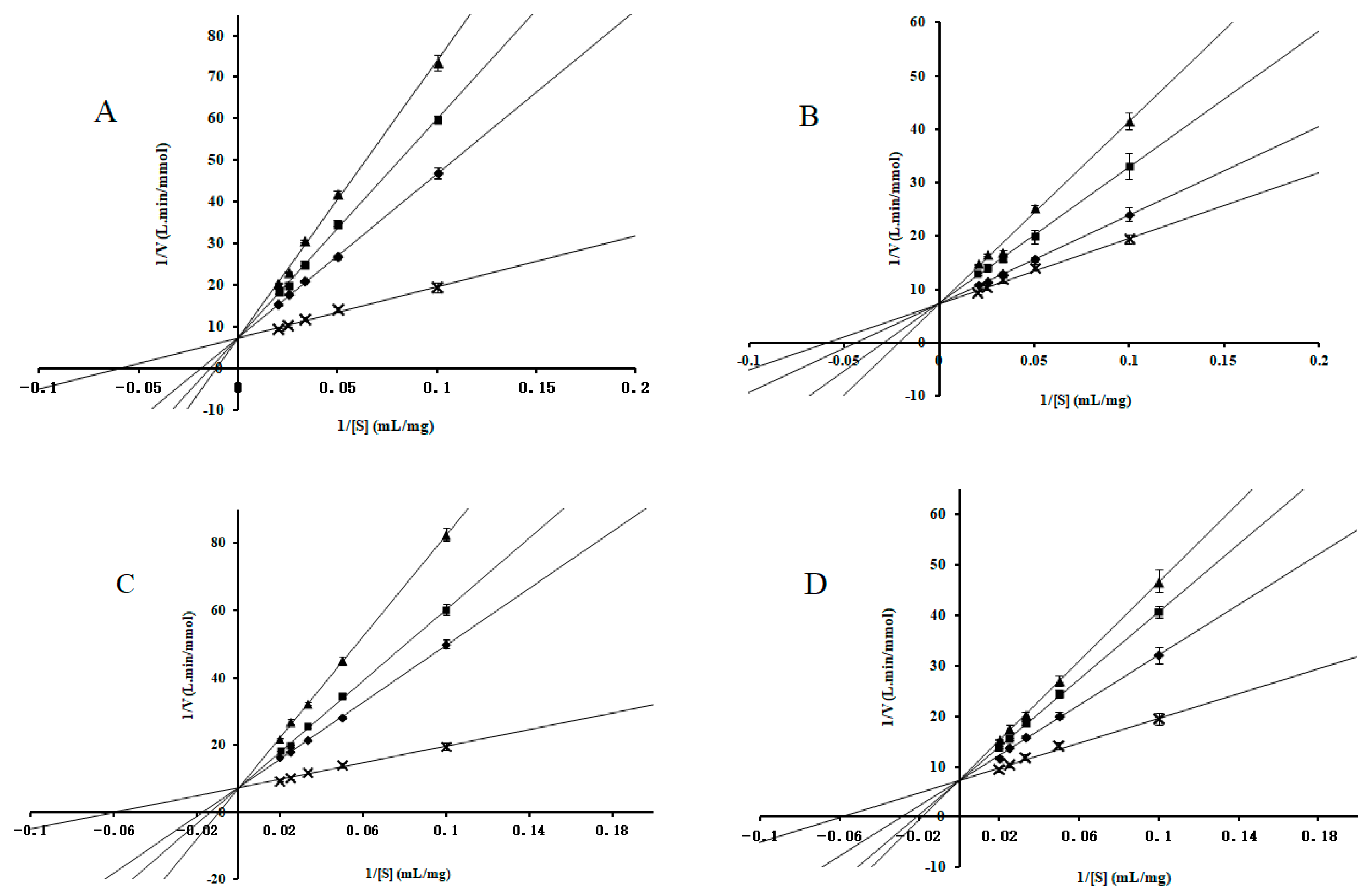
2.2. Enzyme-Kinetic Analysis
2.3. Molecular Docking Studies
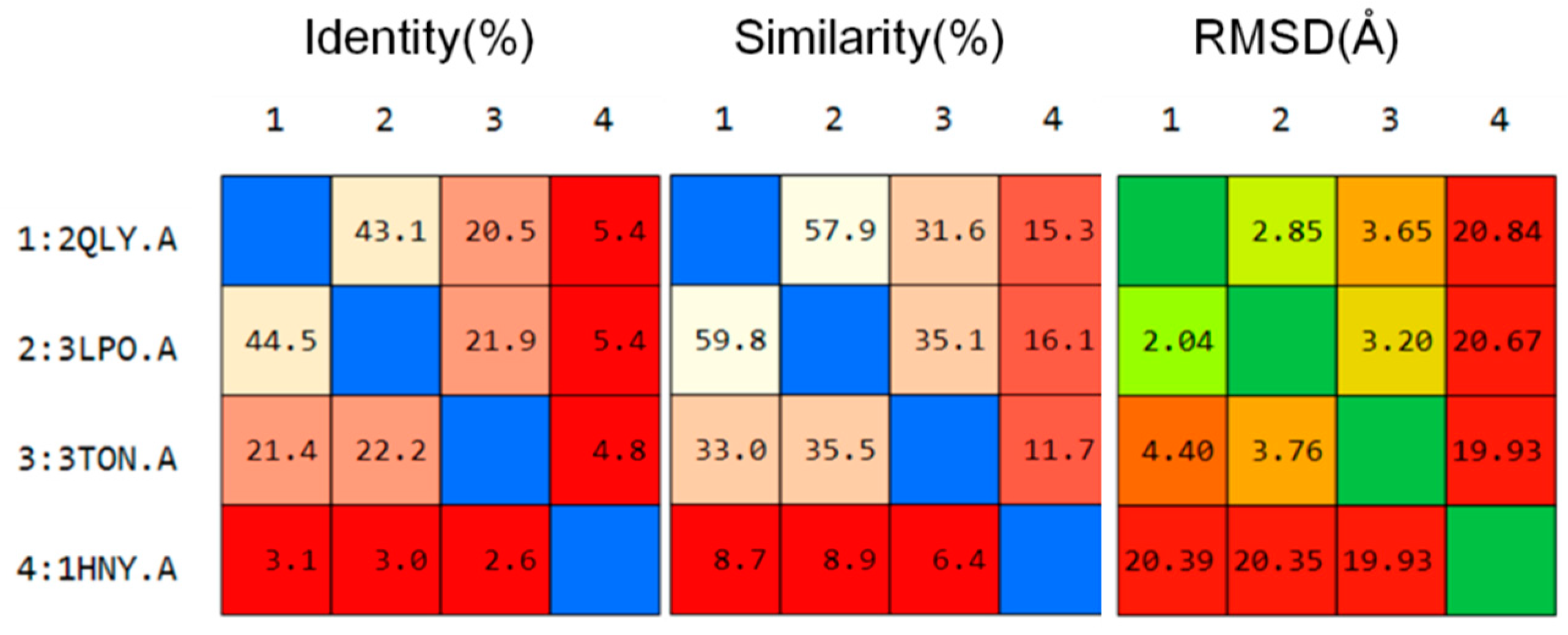
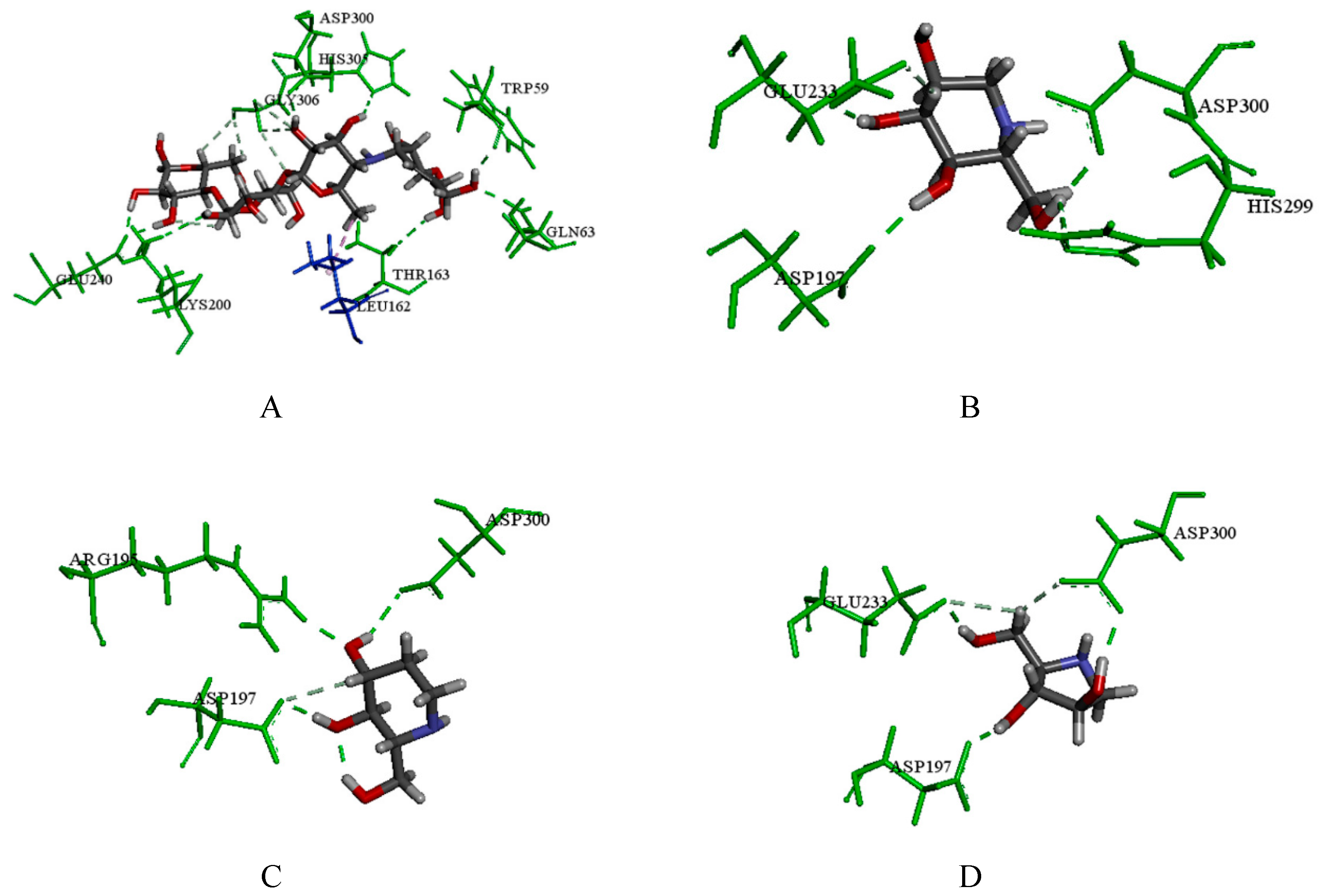
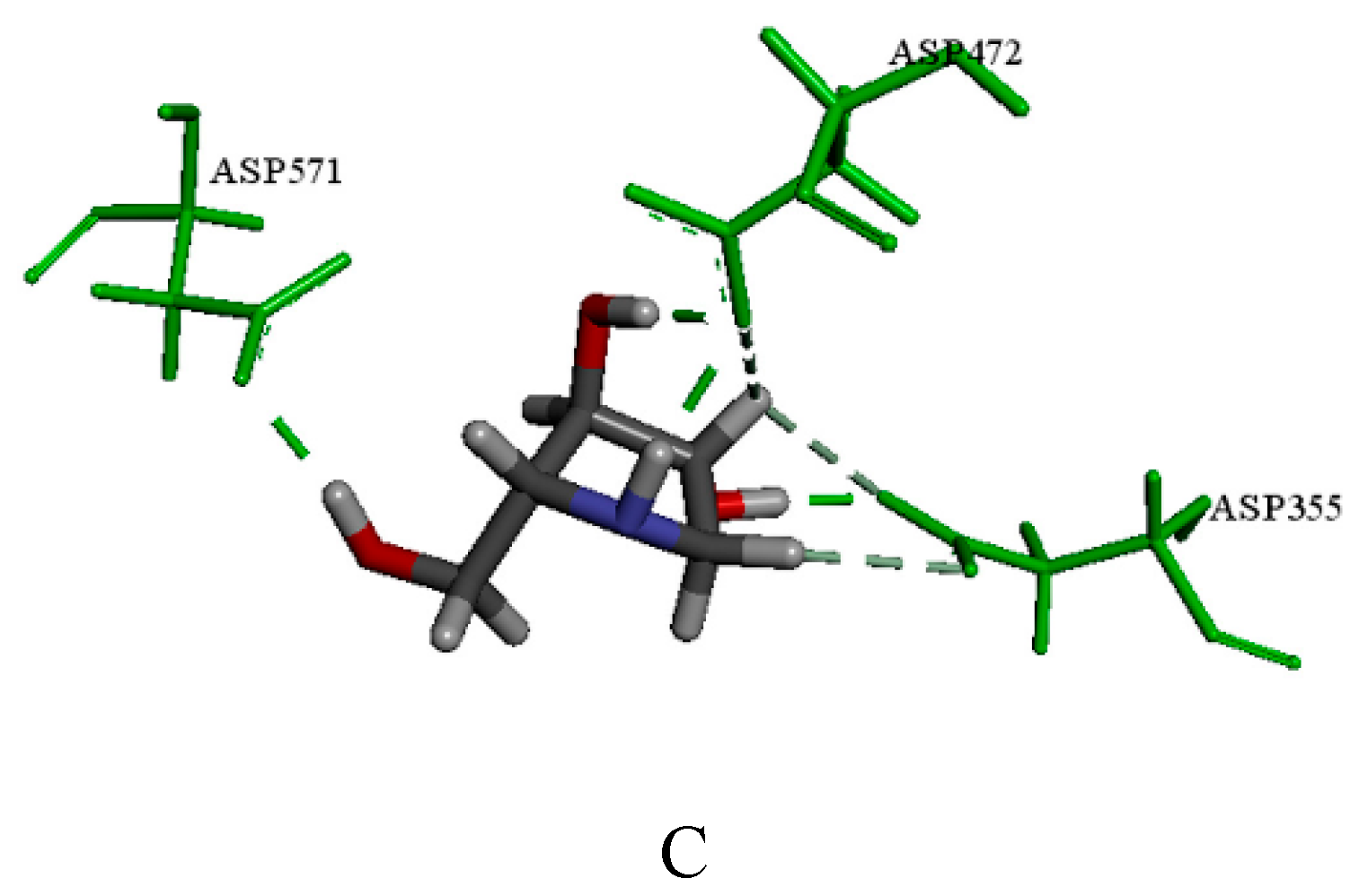
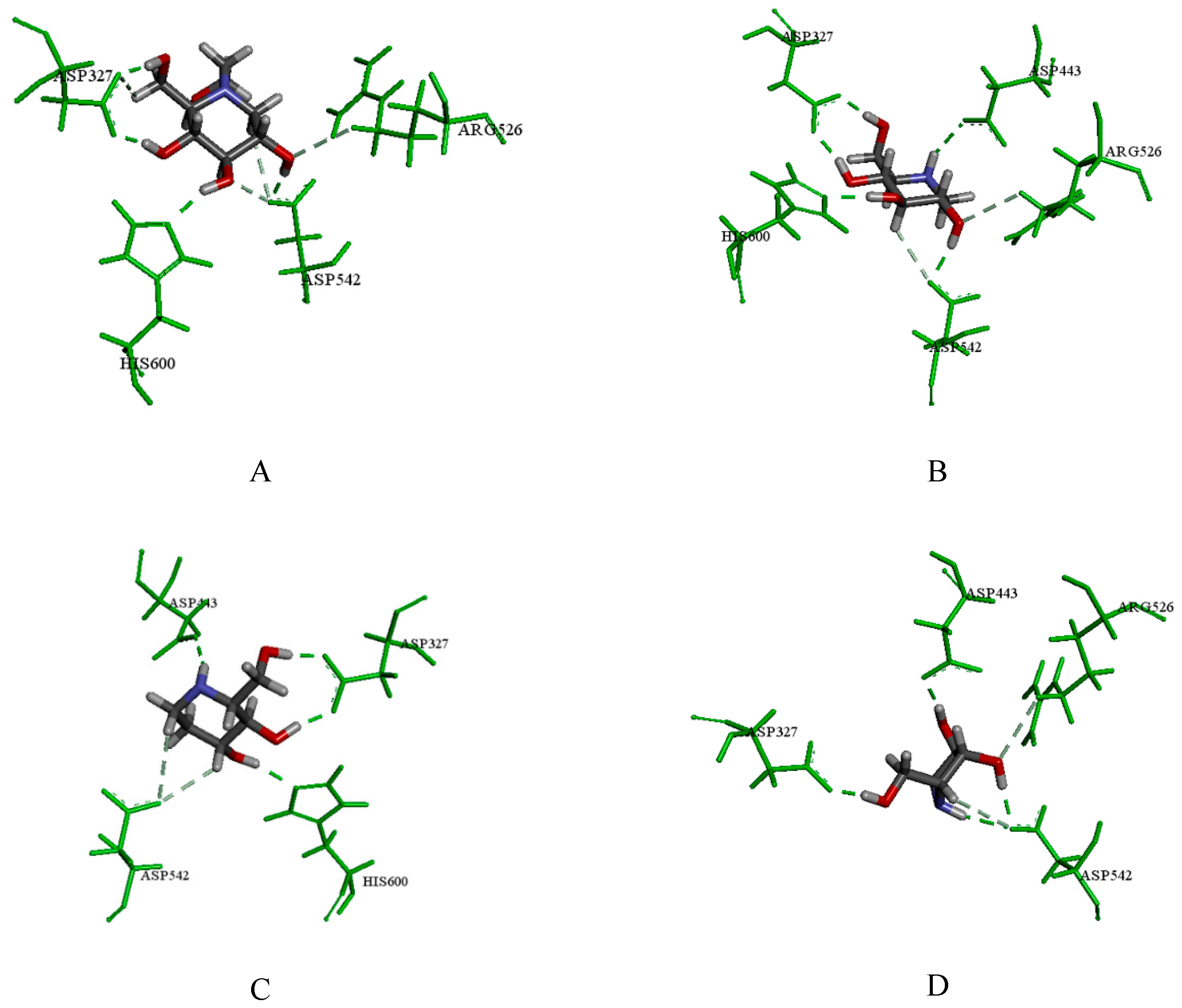
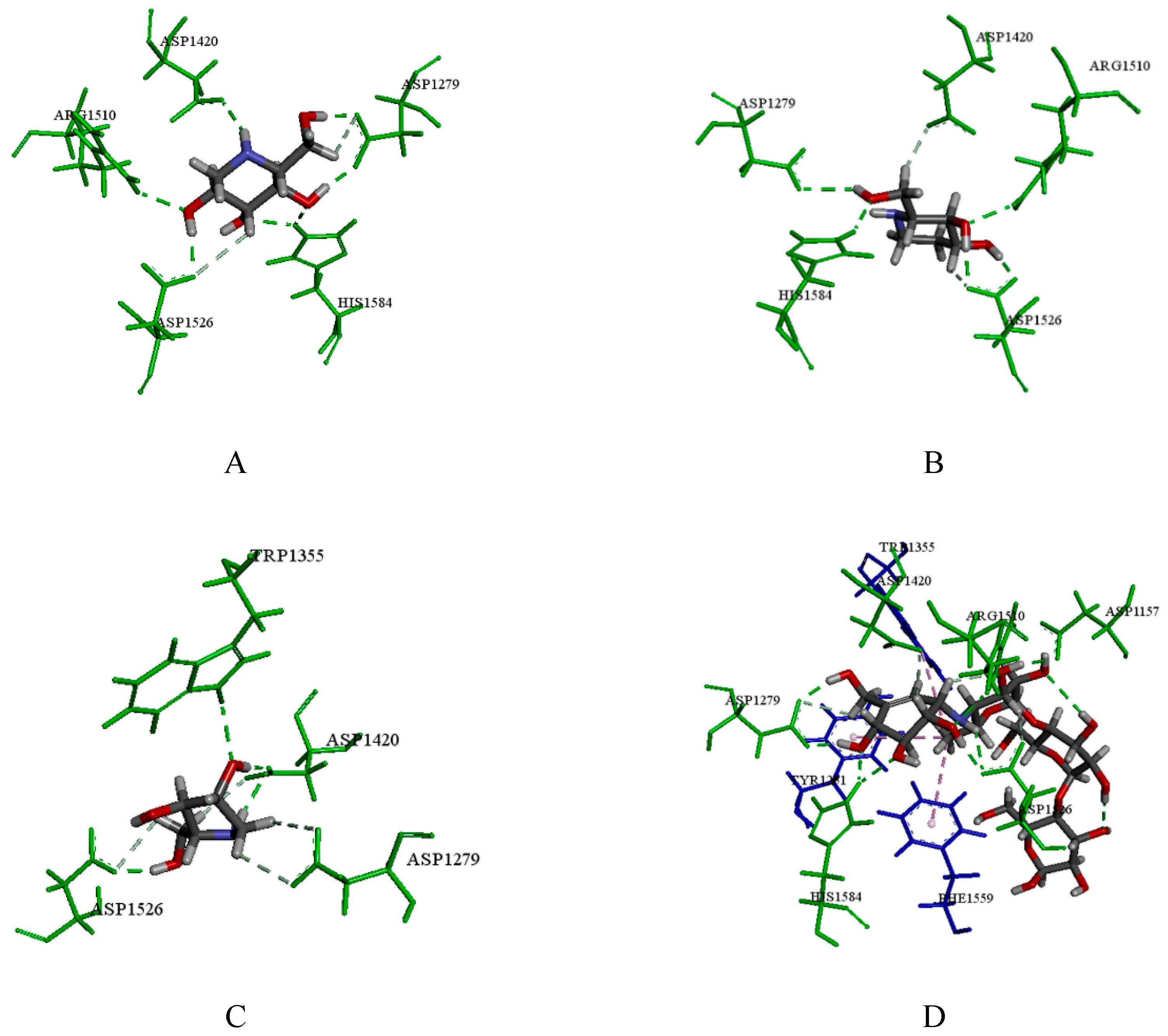
2.3.1. Catalytic Analysis of α-Glucosidases
2.3.2. Molecular Docking and Consensus Scoring
3. Materials and Methods
3.1. Materials
3.2. α-Glucosidase Inhibition Assay
3.3. Enzyme-Kinetic Analysis
3.4. Molecular Docking Studies
3.4.1. Sequence Alignment and Structural Superposition of Proteins
3.4.2. SAS Calculations
3.4.3. Molecular Docking Studies
3.4.4. Consensus Scoring
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Baron, A.D.; Baron, A.D. Postprandial hyperglycaemia and alphα-glucosidase inhibitors. Diabetes Res. Clin. Pract. 1998, 40, S51–S55. [Google Scholar] [CrossRef]
- O’Keefe, J.H.; Mohammad, A.; Lavie, C.J.; Bell, D.S.H. Strategies for optimizing glycemic control and cardiovascular prognosis in patients with type 2 diabetes mellitus. Mayo Clin. Proc. 2011, 86, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Yoshihiko, S.; Motoaki, S.; Kentaro, H.; Ikuroh, O.; Shigeo, O.; Keiichi, F. Are the effects of alphα-glucosidase inhibitors on cardiovascular events related to elevated levels of hydrogen gas in the gastrointestinal tract? Febs Lett. 2009, 583, 2157–2159. [Google Scholar]
- Schmidt, D.D.; Frommer, W.; Junge, B.; Müller, L.; Wingender, W.; Truscheit, E.; Sch? Fer, D. alphα-glucosidase inhibitors. New complex oligosaccharides of microbial origin. Die Nat. 1977, 64, 535. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.; Anne, B. Gastrointestinal side effects of drugs. Exp. Opin. Drug Saf. 2003, 2, 421–429. [Google Scholar]
- Chiasson, J.L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M.; STOP-NIDDM Trial Research Group. Acarbose for prevention of type 2 diabetes mellitus: The STOP-NIDDM randomised trial. Lancet 2002, 359, 2072–2077. [Google Scholar] [CrossRef]
- Kim, G.N.; Kwon, Y.I.; Jang, H.D. Mulberry leaf extract reduces postprandial hyperglycemia with few side effects by inhibiting α-glucosidase in normal rats. J. Med. Food 2011, 14, 712–717. [Google Scholar] [CrossRef]
- Hao, H.; Yan-Hua, L. Comparison of inhibitory activities and mechanisms of five mulberry plant bioactive components against α-glucosidase. J. Agric. Food Chem. 2013, 61, 8110–8119. [Google Scholar]
- Jimin, P.; Hayoon, B.; Hyein, J.; Yeonkyoung, K.; Jiyeon, K.; Oran, K. Postprandial hypoglycemic effect of mulberry leaf in Goto-Kakizaki rats and counterpart control Wistar rats. Nutr. Res. Pract. 2009, 3, 272–278. [Google Scholar]
- Joubert, P.H.; Bam, W.J.; Manyane, N. Effect of an alphα-glucosidase inhibitor (BAY m 1099) on post-prandial blood glucose and insulin in type II diabetics. Eur. J. Clin. Pharmacol. 1986, 30, 253–255. [Google Scholar] [CrossRef]
- Chung, H.I.; Kim, J.; Ji, Y.K.; Kwon, O. Acute intake of mulberry leaf aqueous extract affects postprandial glucose response after maltose loading: Randomized double-blind placebo-controlled pilot study. J. Funct. Foods 2013, 5, 1502–1506. [Google Scholar] [CrossRef]
- Toshiyuki, K.; Kiyotaka, N.; Hiroyuki, K.; Yoshihiro, K.; Yuko, G.; Kenji, Y.; Shigeru, O.; Shinichi, O.; Teruo, M. Food-grade mulberry powder enriched with 1-deoxynojirimycin suppresses the elevation of postprandial blood glucose in humans. J. Agric. Food Chem. 2007, 55, 5869–5874. [Google Scholar]
- Shuang, Y.; Wang, B.; Xia, X.; Xue, L.; Wang, R.; Li, S.; Dan, L.; Liu, Y.; Yan, L. Simultaneous quantification of three active alkaloids from a traditional Chinese medicine Ramulus Mori (Sangzhi) in rat plasma using liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. Anal. 2015, 109, 177–183. [Google Scholar]
- Li, W.; Tan, G.; Zhao, L.; Chen, X.; Zhang, X.; Zhu, Z.; Chai, Y. Computer-aided molecular modeling study of enantioseparation of iodiconazole and structurally related triadimenol analogues by capillary electrophoresis: Chiral recognition mechanism and mathematical model for predicting chiral separation. Anal. Chim. Acta 2012, 718, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Wei, L.; Higai, K.; Koike, K. Canthinone alkaloids are novel protein tyrosine phosphatase 1B inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1979–1981. [Google Scholar] [CrossRef] [PubMed]
- Borhani, D.W.; Shaw, D.E. The future of molecular dynamics simulations in drug discovery. J. Comput.-Aided Mol. Des. 2012, 26, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Taekyung, H.; Seunghee, E.; Jusung, K. Molecular docking studies for discovery of plant-derived α-glucosidase inhibitors. Plant Omics 2014, 7, 166–170. [Google Scholar]
- Azam, S.S.; Uddin, R.; Syed, A.A.S.; Ul-Haq, Z. Molecular docking studies of potent inhibitors of tyrosinase and α-glucosidase. Med. Chem. Res. 2012, 21, 1677–1683. [Google Scholar] [CrossRef]
- Ghosh, S.; Rangan, L. Molecular Docking and Inhibition Kinetics of α-glucosidase Activity by Labdane Diterpenes Isolated from Tora Seeds (Alpinia nigra B.L. Burtt.). Appl. Biochem. Biotechnol. 2014, 175, 1–13. [Google Scholar] [CrossRef]
- Sultan, S.; Choudhary, M.I.; Khan, S.N.; Fatima, U.; Atif, M.; Ali, R.A.; Rahman, A.U.; Fatmi, M.Q. Fungal transformation of cedryl acetate and α-glucosidase inhibition assay, quantum mechanical calculations and molecular docking studies of its metabolites. Eur. J. Med. Chem. 2013, 62, 764–770. [Google Scholar] [CrossRef]
- Lin, H.M.; Lee, B.H.; Chang, W.J. Small intestine mucosal α-glucosidase: A missing feature of invitro starch digestibility. Food Hydrocoll. 2016, 53, 163–171. [Google Scholar] [CrossRef]
- Ao, Z.; Quezada-Calvillo, R.; Sim, L.; Nichols, B.L.; Rose, D.R.; Sterchi, E.E.; Hamaker, B.R. Evidence of native starch degradation with human small intestinal maltase-glucoamylase (recombinant). FEBS Lett. 2007, 581, 2381–2388. [Google Scholar] [CrossRef]
- Asano, N. Glycosidase inhibitors: update and perspectives on practical use. Glycobiology 2003, 13, 93R–104R. [Google Scholar] [CrossRef]
- Robayo–Torres, C.C.; Quezada–Calvillo, R.; Nichols, B.L. Disaccharide Digestion: Clinical and Molecular Aspects. Clin. Gastroenterol. Hepatol. 2006, 4, 276–287. [Google Scholar] [CrossRef]
- Sim, L.; Willemsma, C.S.; Naim, H.Y.; Pinto, B.M.; Rose, D.R. Structural basis for substrate selectivity in human maltase-glucoamylase and sucrase-isomaltase N-terminal domains. J. Biol. Chem. 2010, 285, 17763. [Google Scholar] [CrossRef]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human Intestinal Maltase–Glucoamylase: Crystal Structure of the N-Terminal Catalytic Subunit and Basis of Inhibition and Substrate Specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef]
- Lyann, S.; Kumarasamy, J.; Sankar, M.; Ravindranath, N.; Johnston, B.D.; Pinto, B.M.; Rose, D.R. New glucosidase inhibitors from an ayurvedic herbal treatment for type 2 diabetes: Structures and inhibition of human intestinal maltase-glucoamylase with compounds from Salacia reticulata. Biochemistry 2010, 49, 443–451. [Google Scholar]
- Ren, L.; Qin, X.; Cao, X.; Wang, L.; Bai, F.; Bai, G.; Shen, Y. Structural insight into substrate specificity of human intestinal maltase-glucoamylase. Protein Cell 2011, 2, 827. [Google Scholar] [CrossRef]
- Cao, X.; Chen, Z.; Dong, Y.; Peng, G.; Fang, B.; Gang, B. Modeling of cooked starch digestion process using recombinant human pancreatic α-amylase and maltase-glucoamylase for invitro evaluation of α-glucosidase inhibitors. Carbohyd. Res. 2015, 414, 15–21. [Google Scholar] [CrossRef]
- Ren, L.; Cao, X.; Geng, P.; Bai, F.; Bai, G. Study of the inhibition of two human maltase-glucoamylases catalytic domains by different α-glucosidase inhibitors. Carbohyd. Res. 2011, 346, 2688–2692. [Google Scholar] [CrossRef]
- Lachin, J.M.; Saul, G.; Nathan, D.M.; Bernard, Z.; Rutledge, B.N. Effect of glycemic exposure on the risk of microvascular complications in the diabetes control and complications trial--revisited. Diabetes 2008, 57, 995–1001. [Google Scholar] [CrossRef]
- Yoshida, K.; Hishida, A.; Iida, O.; Hosokawa, K.; Kawabata, J. Flavonol caffeoylglycosides as alphα-glucosidase inhibitors from Spiraea cantoniensis flower. J. Agric. Food Chem. 2008, 56, 4367–4371. [Google Scholar] [CrossRef]
- Syed Ausaf, A.; Md Imtaiyaz, H.; Asimul, I.; Faizan, A. A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci. 2014, 15, 5. [Google Scholar]
- Marie, E.K.; Batra, T.; Karthikeyan, C.; Deora, G.S.; Rathore, V.; Mulakayala, C.; Mulakayala, N.; Nusbaum, A.C.; Chen, J.; Amawi, H. 2,3-Diaryl-3H-imidazo[4,5-b]pyridine derivatives as potential anticancer and anti-inflammatory agents. Acta Pharm. Sin. B 2017, 7, 73–79. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Wang, R.; Wang, S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J. Chem. Inf. Comput. Sci. 2001, 41, 1422–1426. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds in the paper are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Sucrase Inhibition | Maltase Inhibition | α-Amylase Inhibition | |||
|---|---|---|---|---|---|---|
| Inhibition | Ic50 | Inhibition | IC50 | Inhibition | IC50 | |
| Ratio (%) | (μmol/L) | Ratio (%) | (μmol/L) | Ratio (%) | (μmol/L) | |
| 1-DNJ | 100 | 0.078 | 100 | 0.113 | 0 | >100 |
| FA | 84.6 | 13.53 | 65.7 | 25.97 | 0 | >100 |
| DAB | 79.7 | 21.27 | 77.9 | 12.33 | 0 | >100 |
| Acarbose | 100 | 1.046 | 98.0 | 0.104 | 93.6 | 2.69 |
| SZ-A | 100 | 0.03 μg/mL | 100 | 0.06 μg/mL | 0 | >100 μg/mL |
| Enzyme | Inhibitor | Inhibition Type | Km (M) | Ki (M) |
|---|---|---|---|---|
| 1-DNJ | maltase | Competitive inhibition | 2.14 × 10−2 | 1.12 × 10−6 |
| FA | maltase | Competitive inhibition | 2.14 × 10−2 | 3.24 × 10−4 |
| DAB | maltase | Competitive inhibition | 2.14 × 10−2 | 5.21 × 10−5 |
| Acarbose | maltase | Competitive inhibition | 2.14 × 10−2 | 1.75 × 10−6 |
| 1−DNJ | sucrose | Competitive inhibition | 4.85 × 10−2 | 1.37 × 10−8 |
| FA | maltase | Competitive inhibition | 4.85 × 10−2 | 3.97 × 10−6 |
| DAB | maltase | Competitive inhibition | 4.85 × 10−2 | 1.91 × 10−5 |
| acarbose | maltase | Competitive inhibition | 4.85 × 10−2 | 5.32 × 10−8 |
| Protein | PDB Code | Ligand | Solvent Accessibility Surface (Å2) | Resolution (Å) | |
|---|---|---|---|---|---|
| HPA | Apo | 1HNY | - | 17,891.6 | 1.8 |
| Complex | 3OLD | Acarviostatin I03 | 17,413.6 | 2.0 | |
| NtSI | Apo | 3LPO | - | 30,056.4 | 3.2 |
| Complex | 3LPP | Kotalanol | 30,331.4 | 3.2 | |
| NtMGAM | Apo | 2QLY | - | 30,738 | 2.0 |
| Complex | 2QMJ | acarbose | 29,942.5 | 1.9 | |
| 3L4W | Miglitol | 31,044.5 | 2.0 | ||
| CtMGAM | Apo | 3TON | - | 31,142.5 | 2.95 |
| Complex | 3TOP | acarbose | 30,812.4 | 2.88 | |
| Name | PDB | Ligand | Radius (Å) | RMSD (Å) |
|---|---|---|---|---|
| HPA | 3OLD | Acarviostatin I03 | 13.70 | 2.70 |
| NtSI | 3LPP | Kotalanol | 8.21 | 1.06 |
| NtMGAM | 3L4W | Miglitol | 5.66 | 0.92 |
| CtMGAM | 3TOP | acarbose | 9.13 | 1.55 |
| Name | Consensus (Number of Votes) | Consensus (Normalized Average) | ||||||
|---|---|---|---|---|---|---|---|---|
| HPA | NtMGAM | CtMGAM | NtSI | HPA | NtMGAM | CtMGAM | NtSI | |
| 1-DNJ | 2 | 10 | 11 | 8 | 0.27 | 0.93 | 0.70 | 0.89 |
| DAB | 3 | 1 | 2 | 5 | 0.40 | 0.63 | 0.54 | 0.73 |
| Fagomine | 2 | 5 | 1 | 3 | 0.38 | 0.74 | 0.55 | 0.62 |
| acarbose | 11 | 7 | 10 | 7 | 0.93 | 0.62 | 0.84 | 0.75 |
| Miglitol | 5 | 11 | 10 | 6 | 0.35 | 0.81 | 0.67 | 0.76 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Yang, Y.; Dong, W.; Liu, Q.; Wang, R.; Pang, J.; Xia, X.; Zhu, X.; Liu, S.; Shen, Z.; et al. Investigation on the Enzymatic Profile of Mulberry Alkaloids by Enzymatic Study and Molecular Docking. Molecules 2019, 24, 1776. https://doi.org/10.3390/molecules24091776
Liu Z, Yang Y, Dong W, Liu Q, Wang R, Pang J, Xia X, Zhu X, Liu S, Shen Z, et al. Investigation on the Enzymatic Profile of Mulberry Alkaloids by Enzymatic Study and Molecular Docking. Molecules. 2019; 24(9):1776. https://doi.org/10.3390/molecules24091776
Chicago/Turabian StyleLiu, Zhihua, Ying Yang, Wujun Dong, Quan Liu, Renyun Wang, Jianmei Pang, Xuejun Xia, Xiangyang Zhu, Shuainan Liu, Zhufang Shen, and et al. 2019. "Investigation on the Enzymatic Profile of Mulberry Alkaloids by Enzymatic Study and Molecular Docking" Molecules 24, no. 9: 1776. https://doi.org/10.3390/molecules24091776
APA StyleLiu, Z., Yang, Y., Dong, W., Liu, Q., Wang, R., Pang, J., Xia, X., Zhu, X., Liu, S., Shen, Z., Xiao, Z., & Liu, Y. (2019). Investigation on the Enzymatic Profile of Mulberry Alkaloids by Enzymatic Study and Molecular Docking. Molecules, 24(9), 1776. https://doi.org/10.3390/molecules24091776