Insights into the Effect of the G245S Single Point Mutation on the Structure of p53 and the Binding of the Protein to DNA

 ,
, 

Abstract
:1. Introduction
2. Results
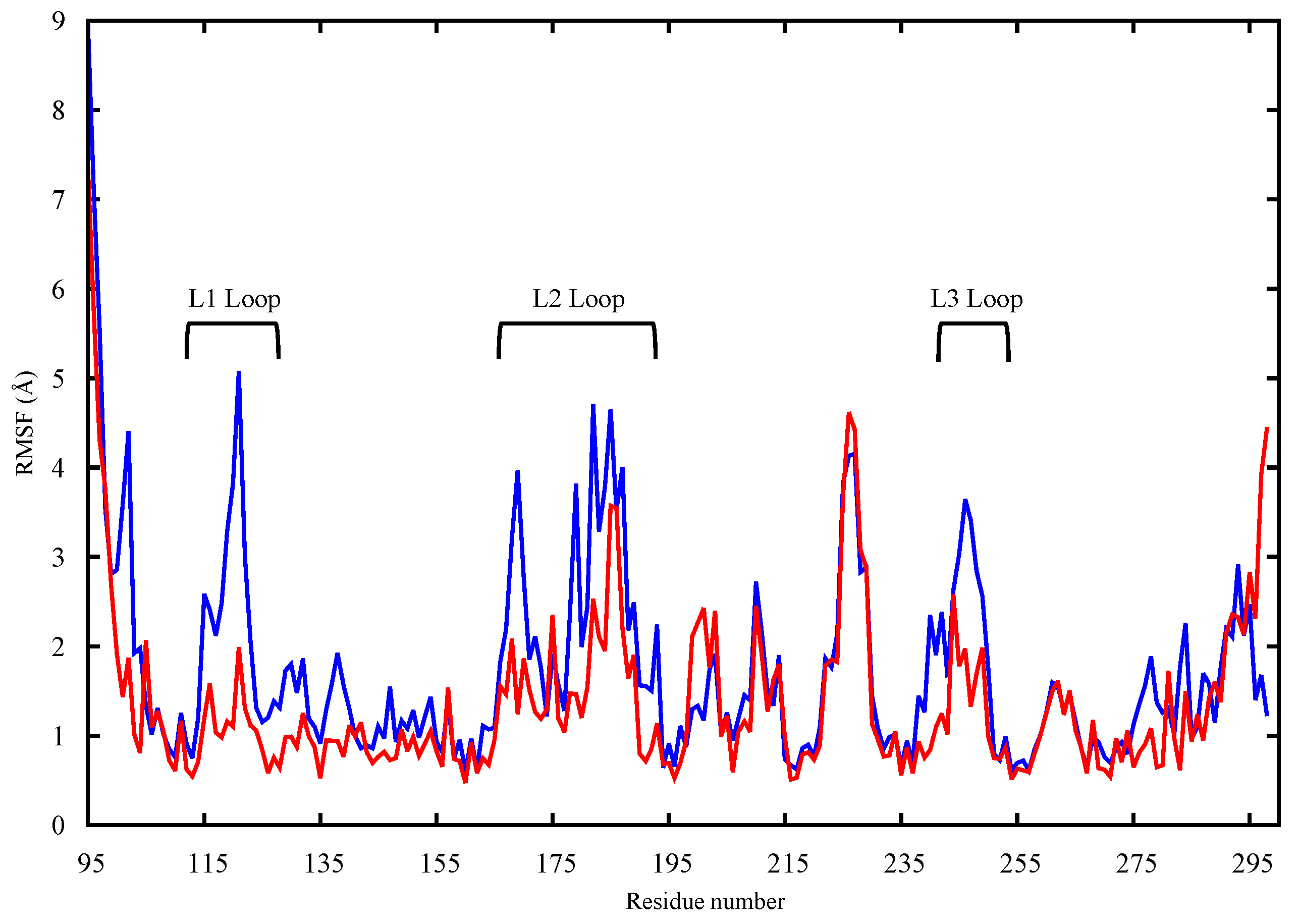
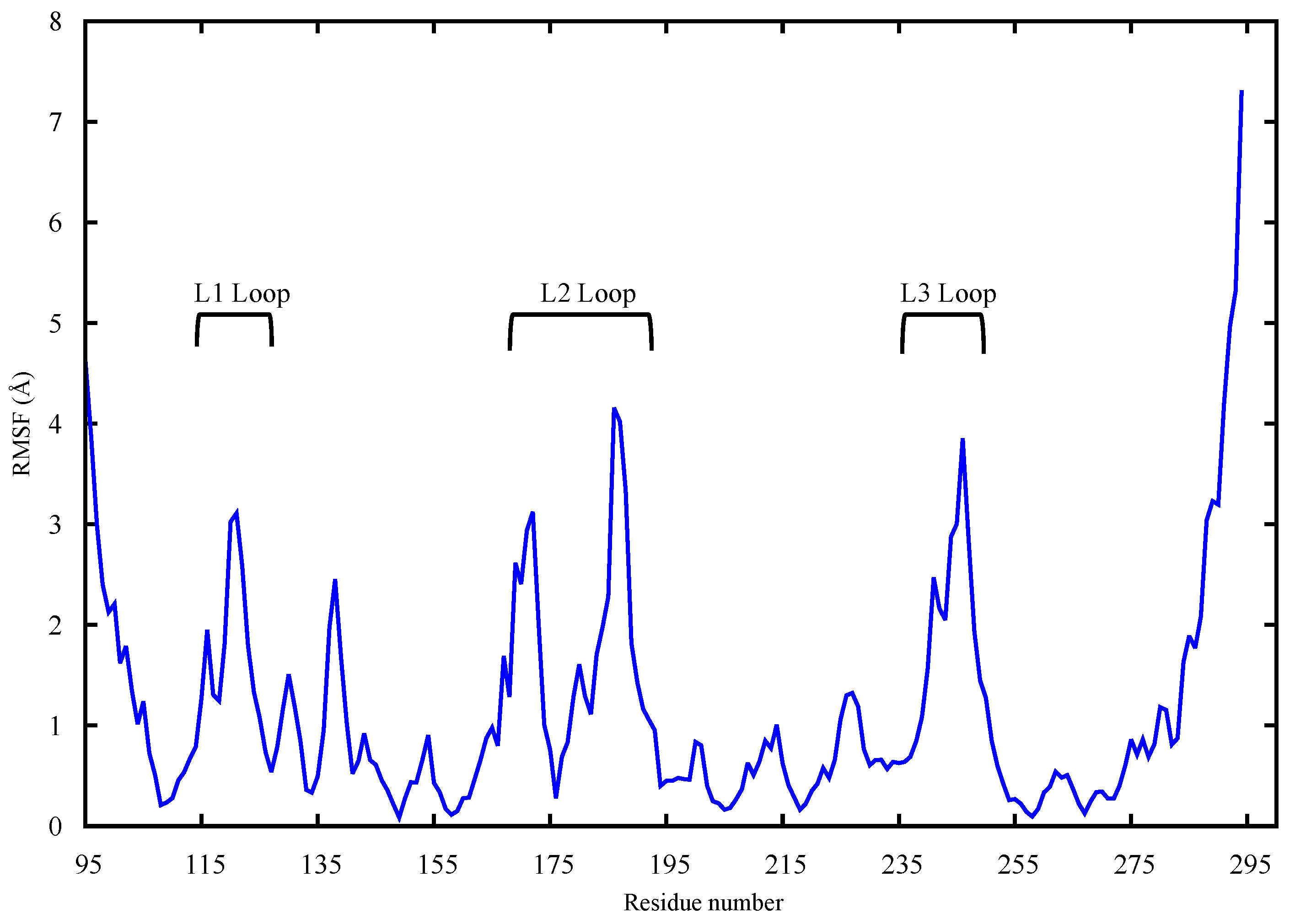
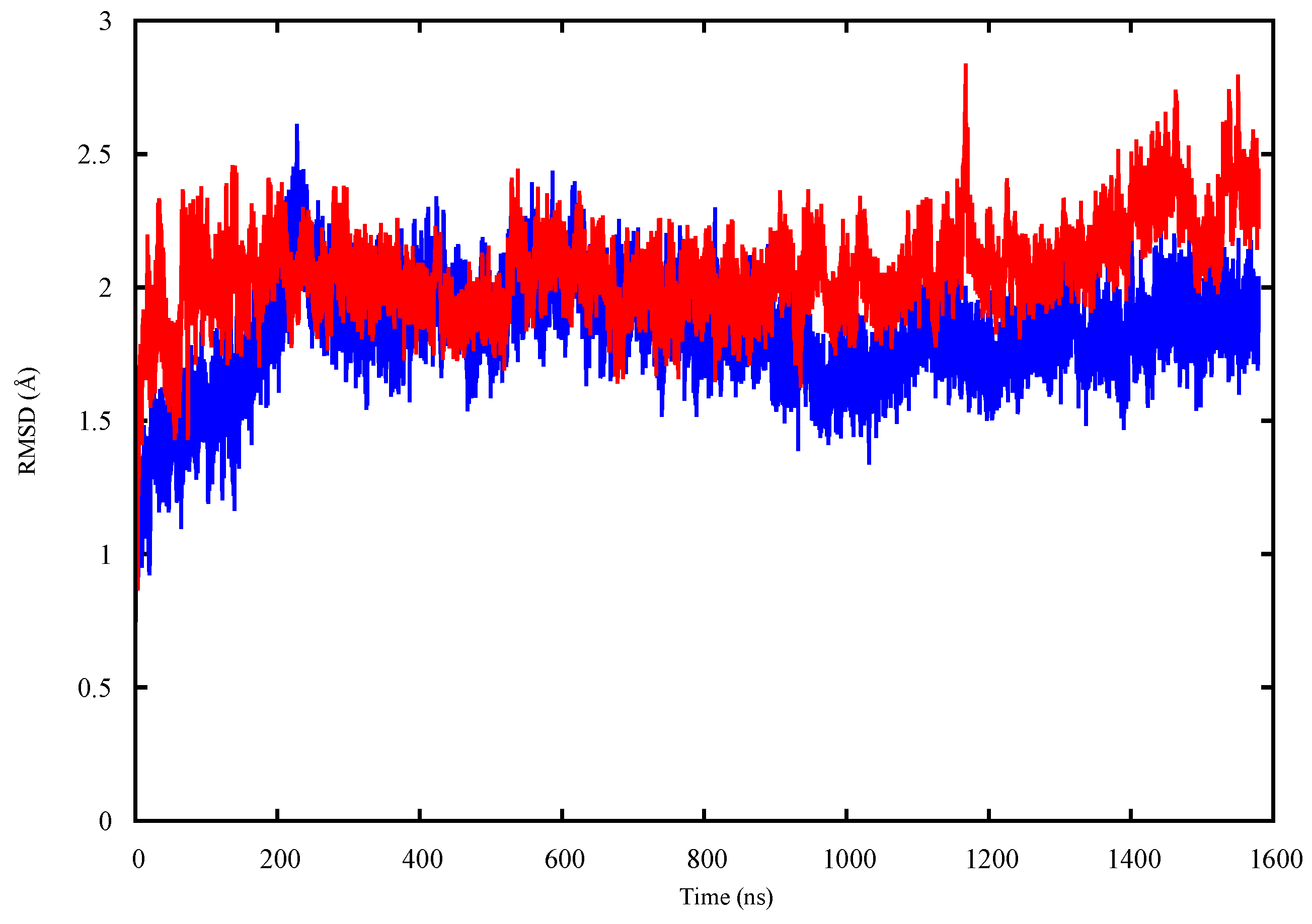
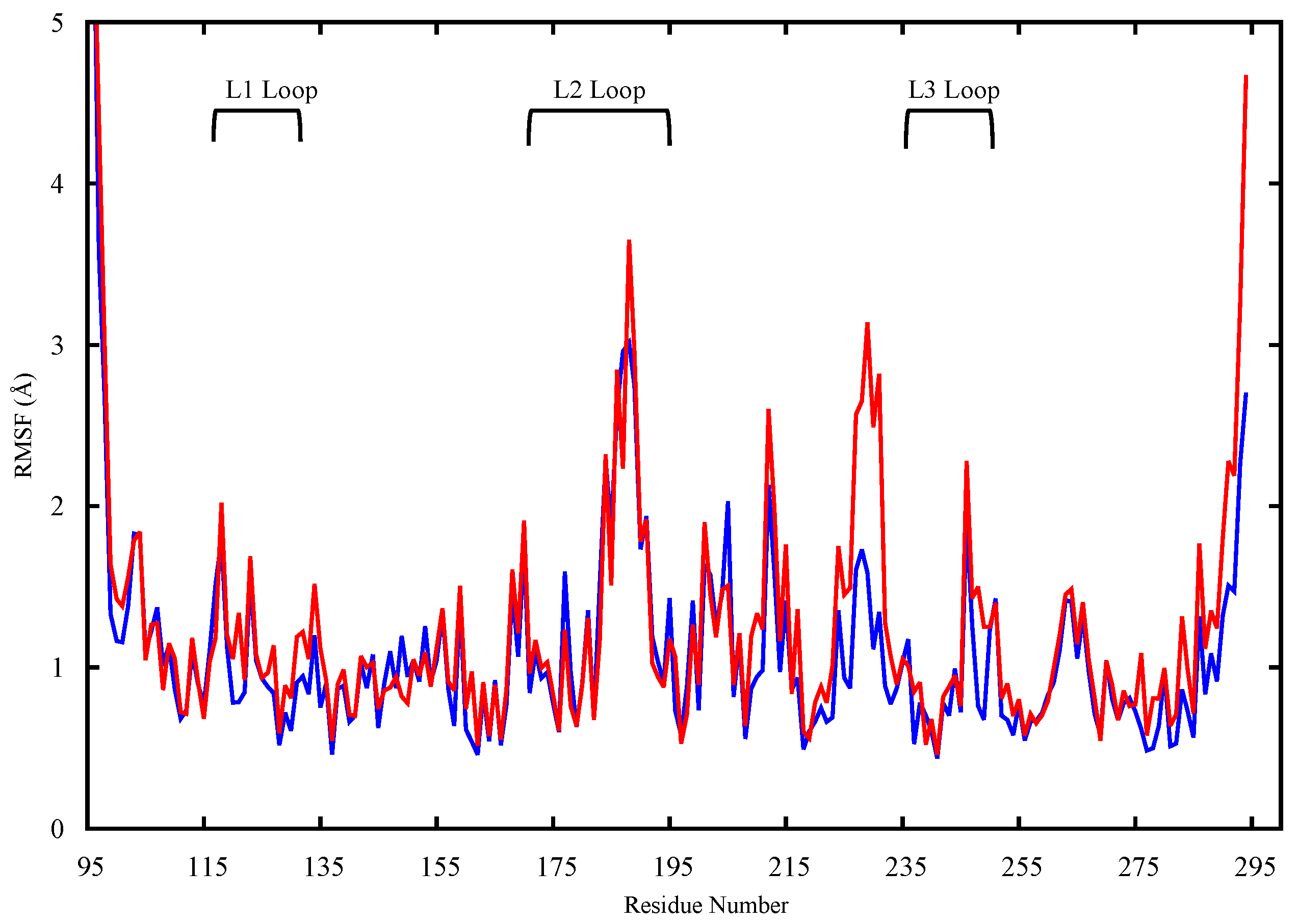
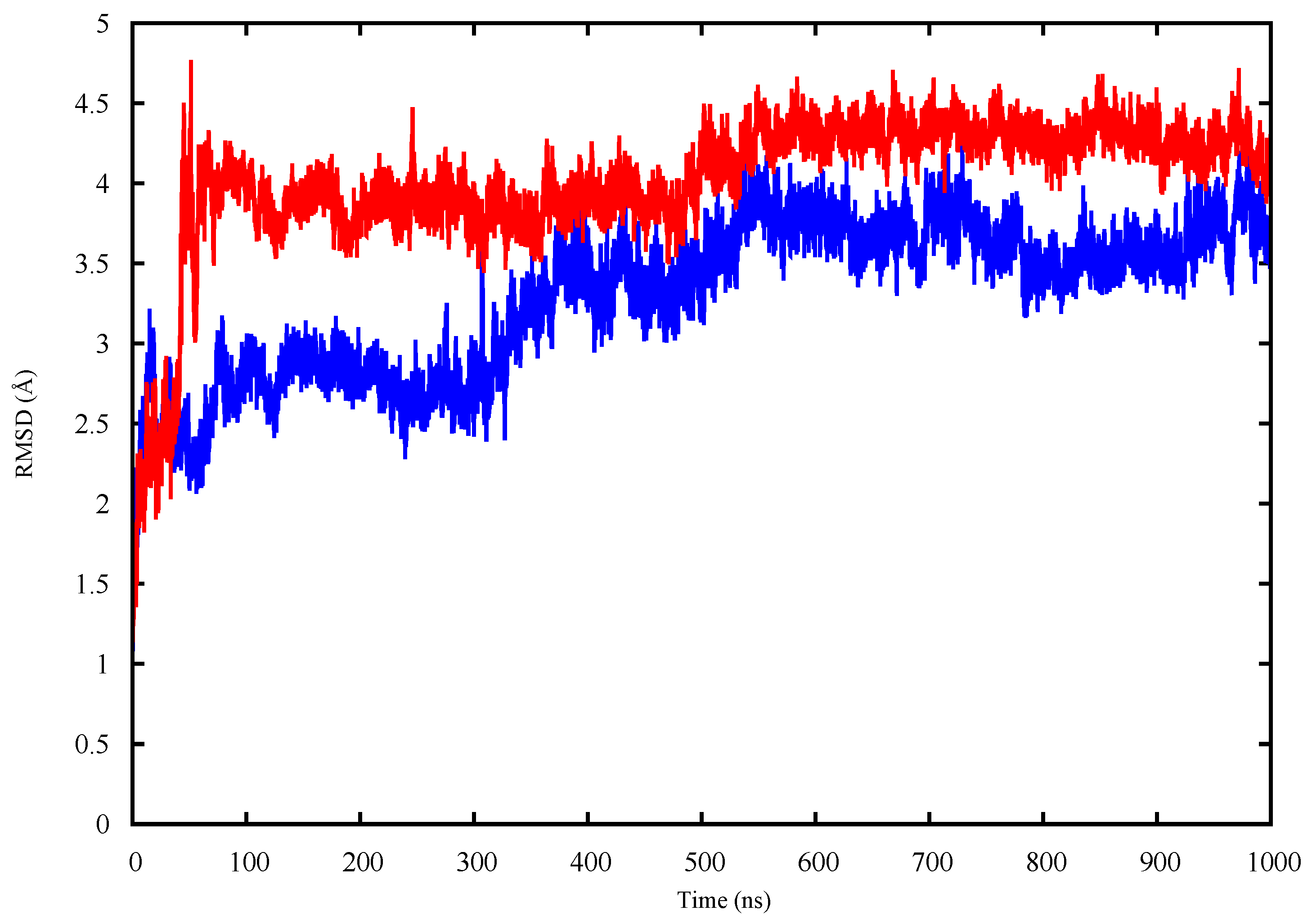
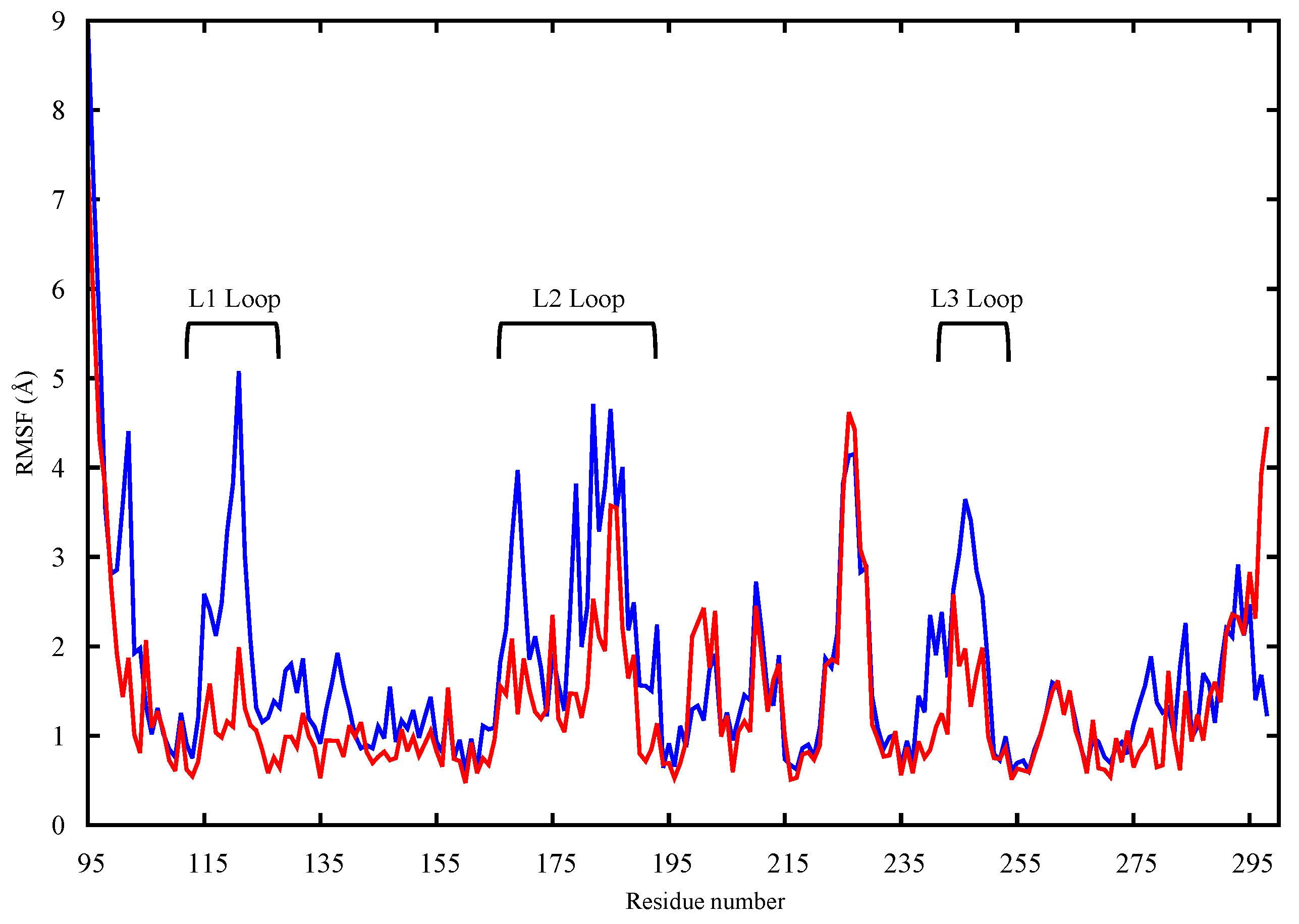
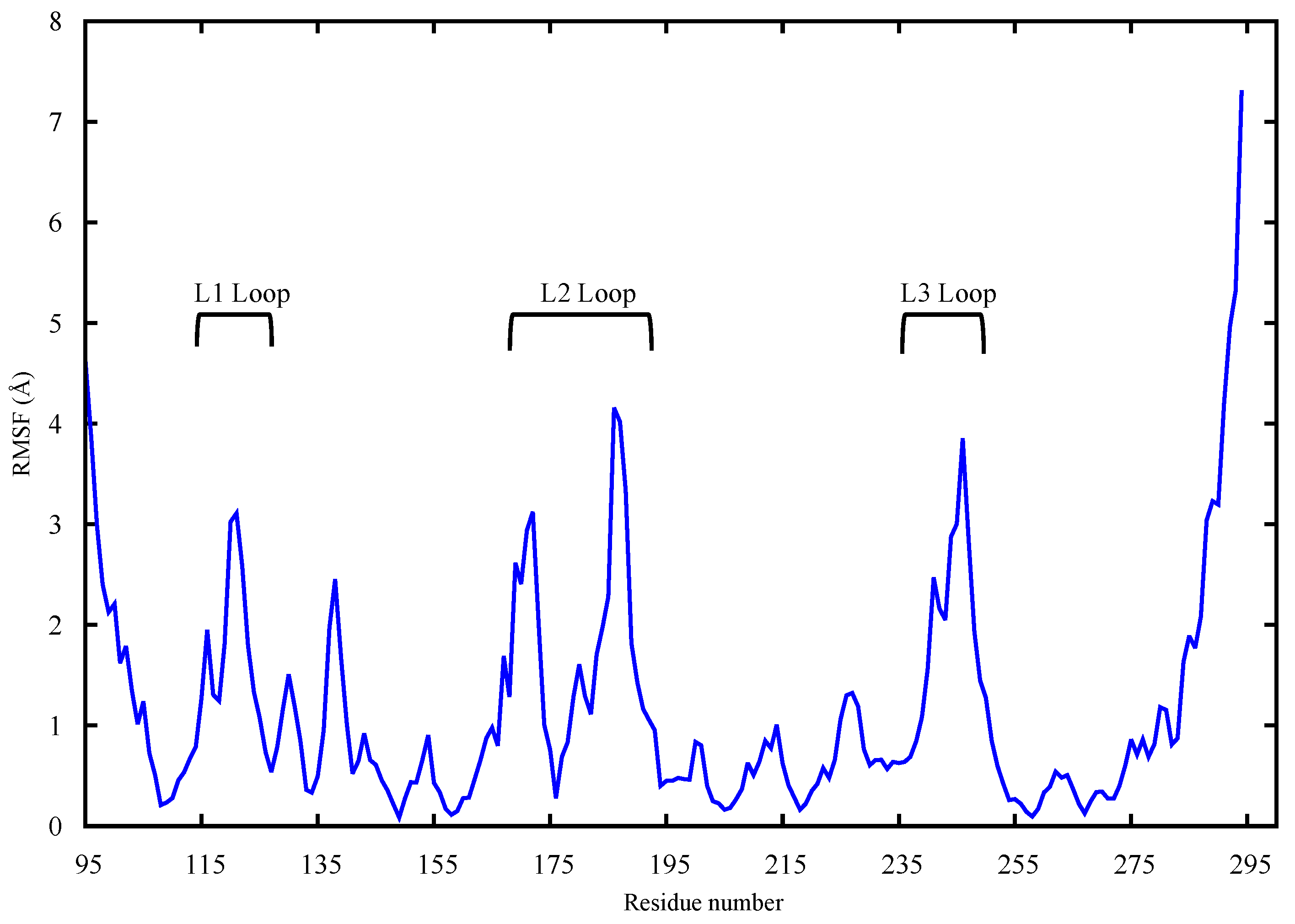
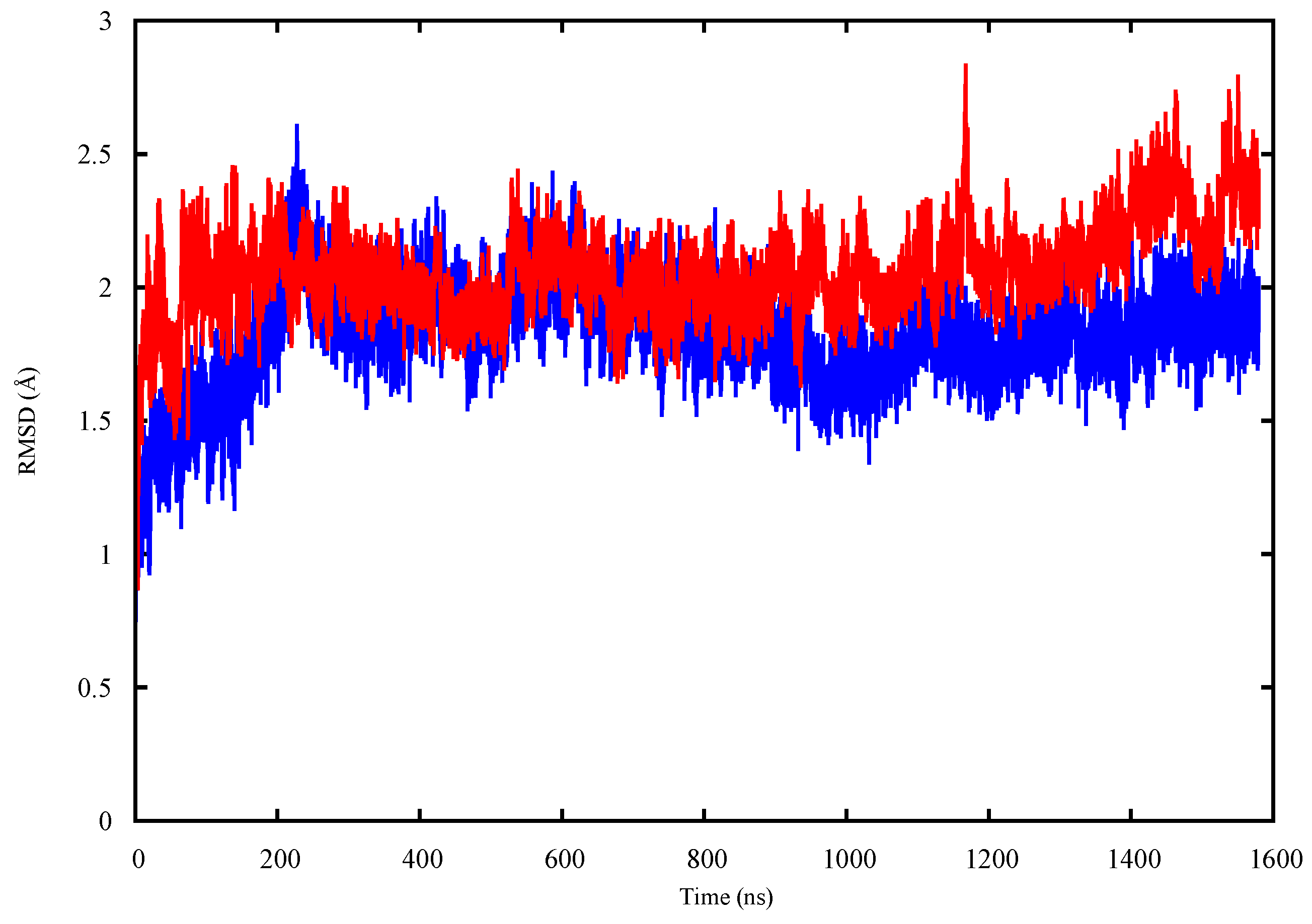
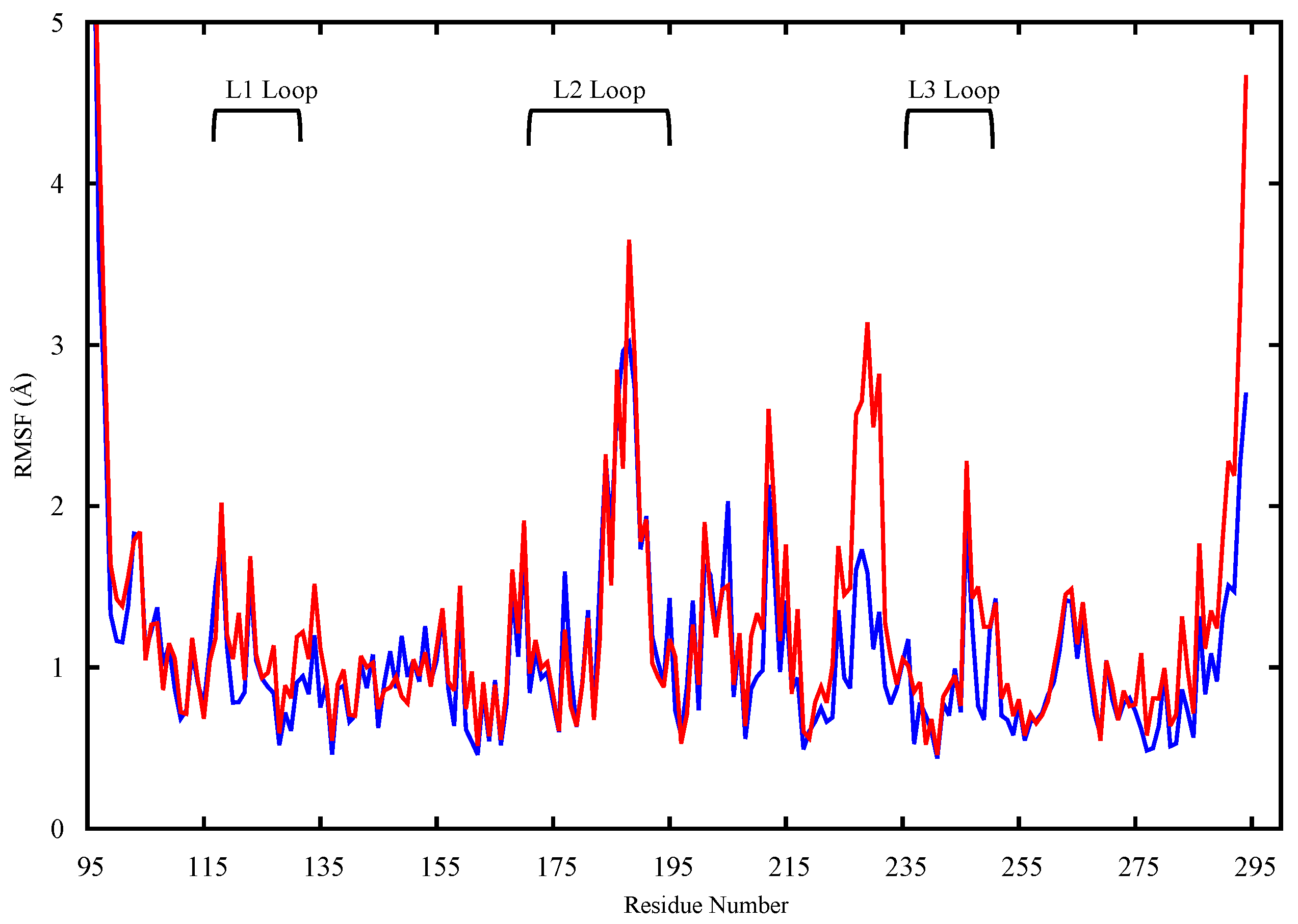
2.1. Conformational Analysis
2.1.1. Apo p53 Proteins
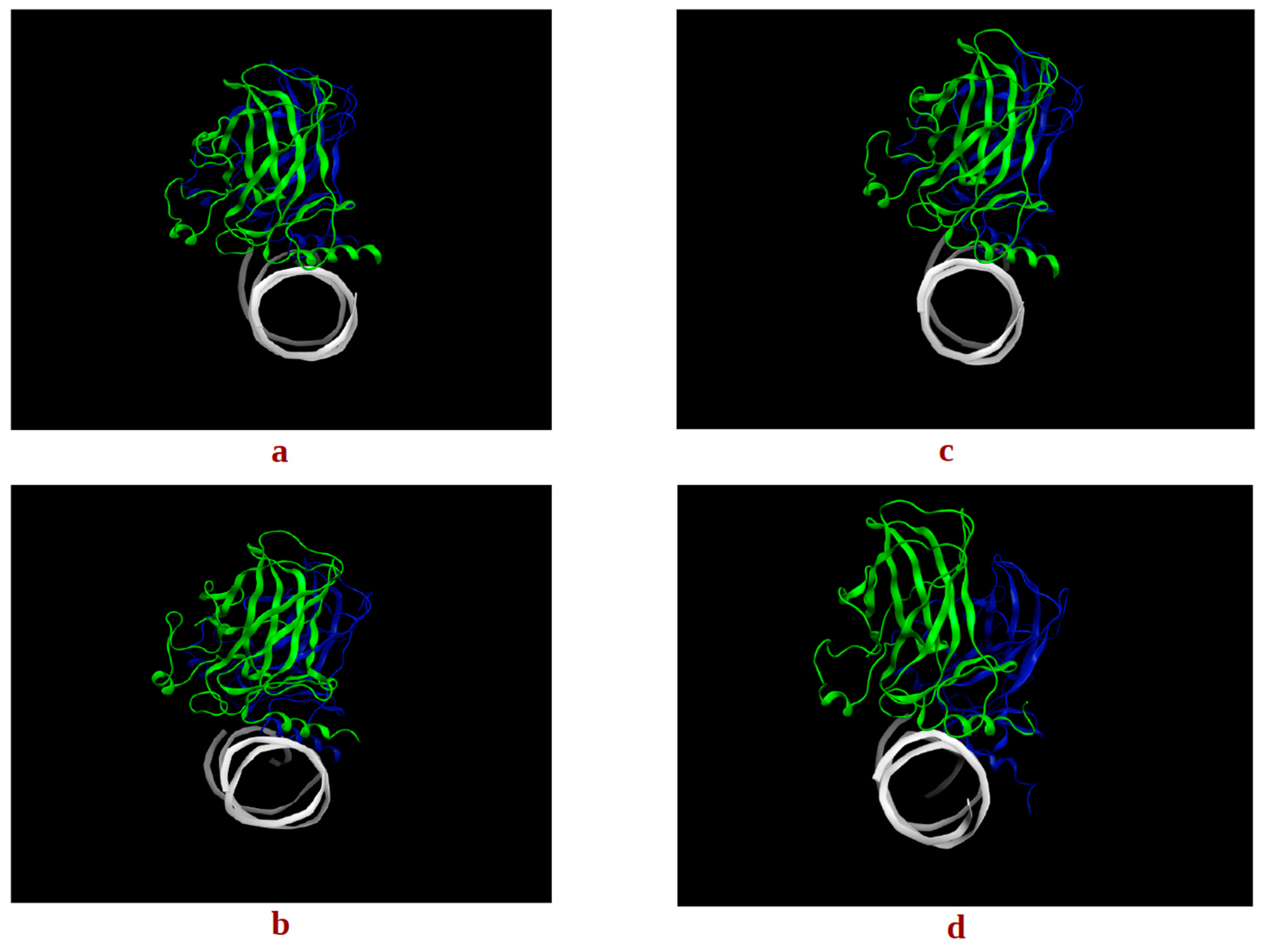
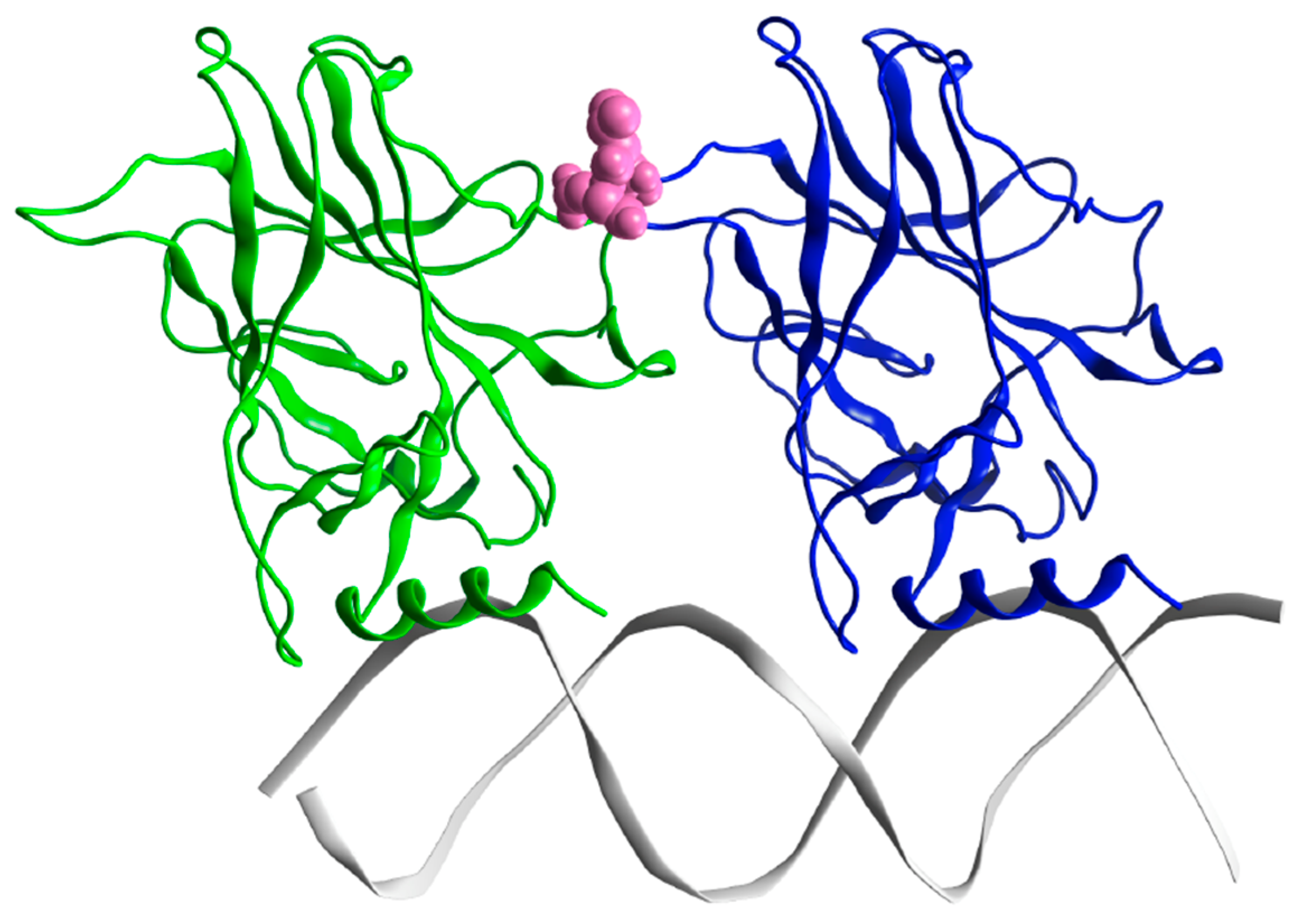
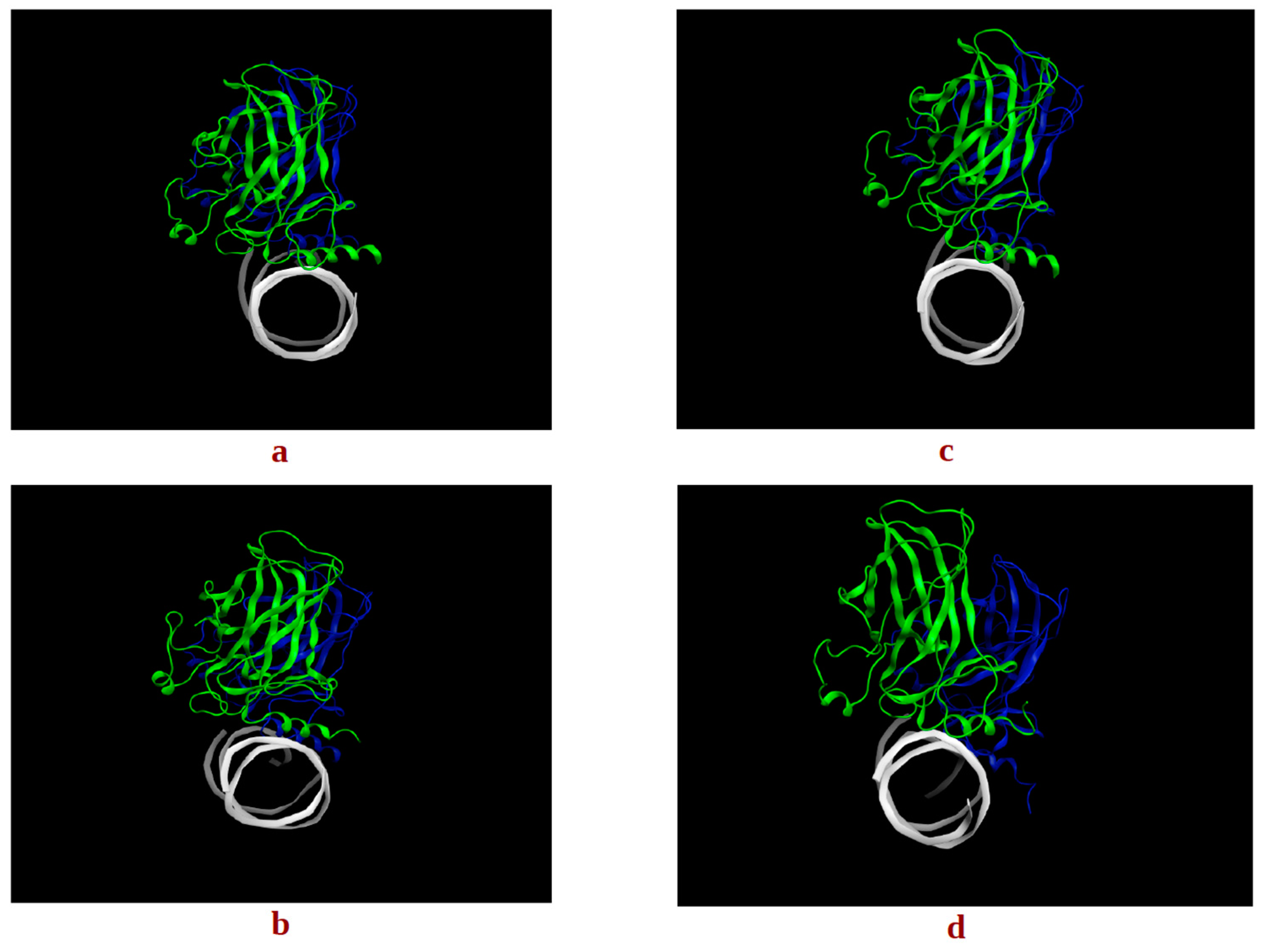
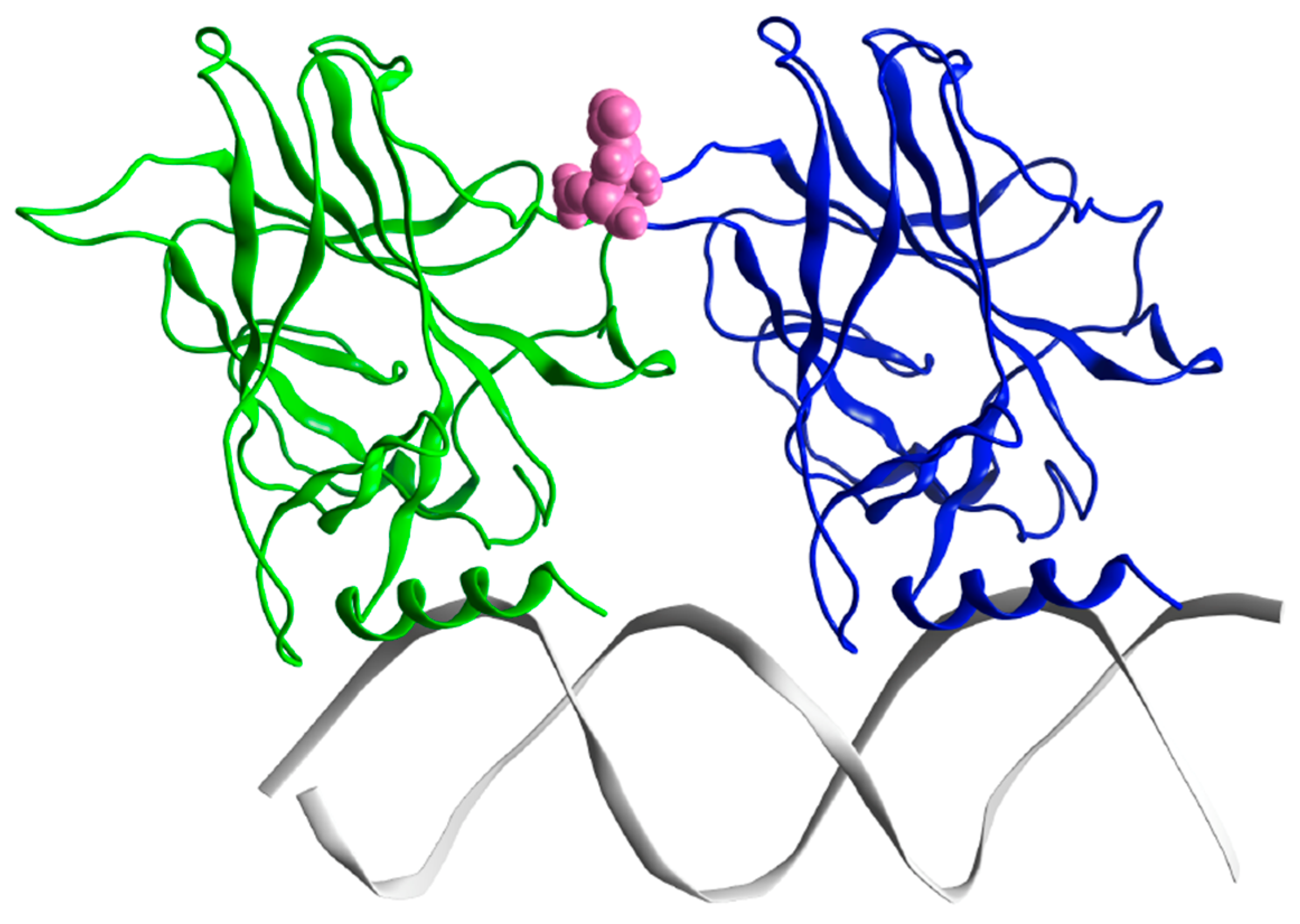
2.1.2. p53 Dimers in Complex with the DNA
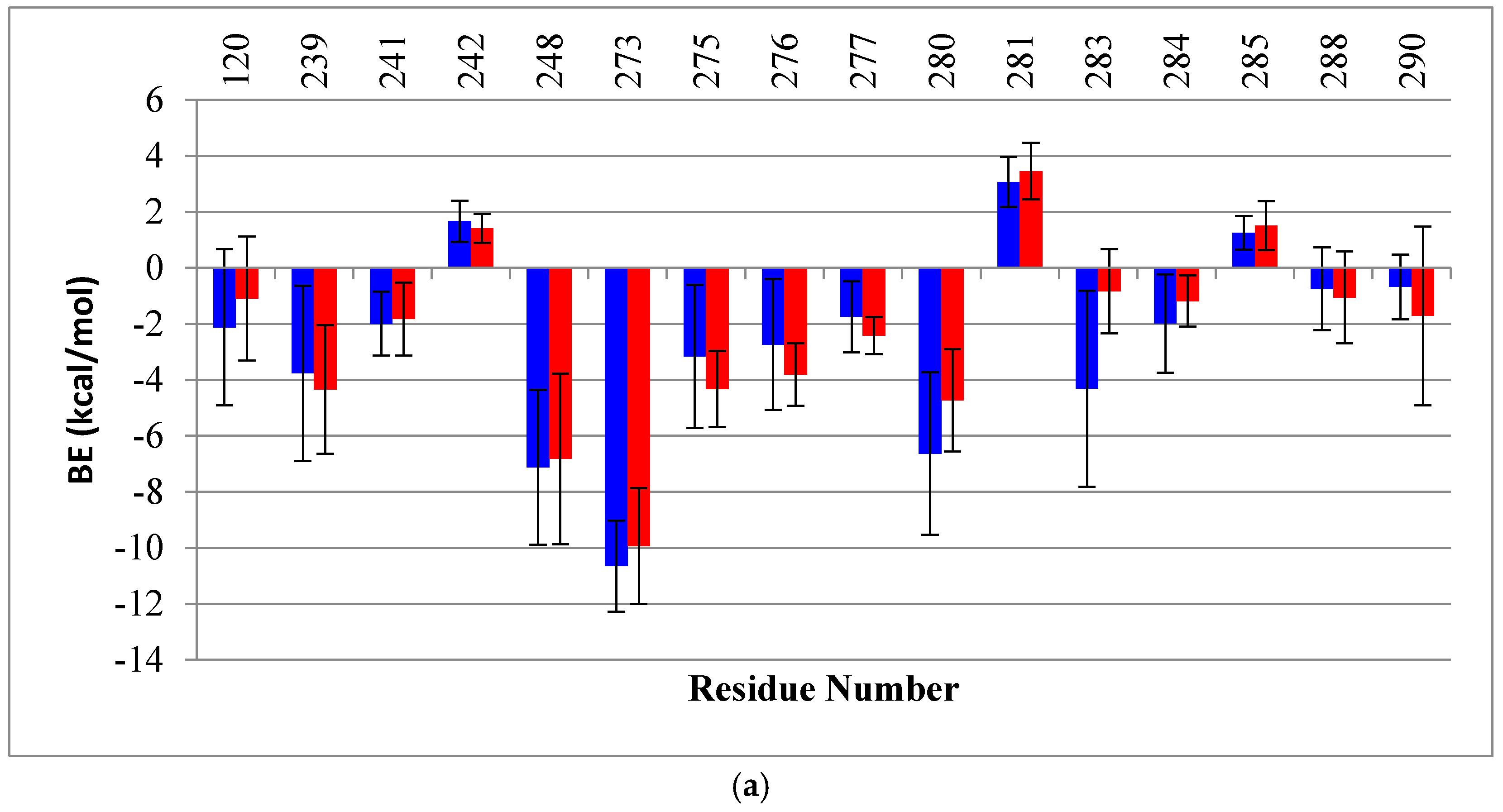
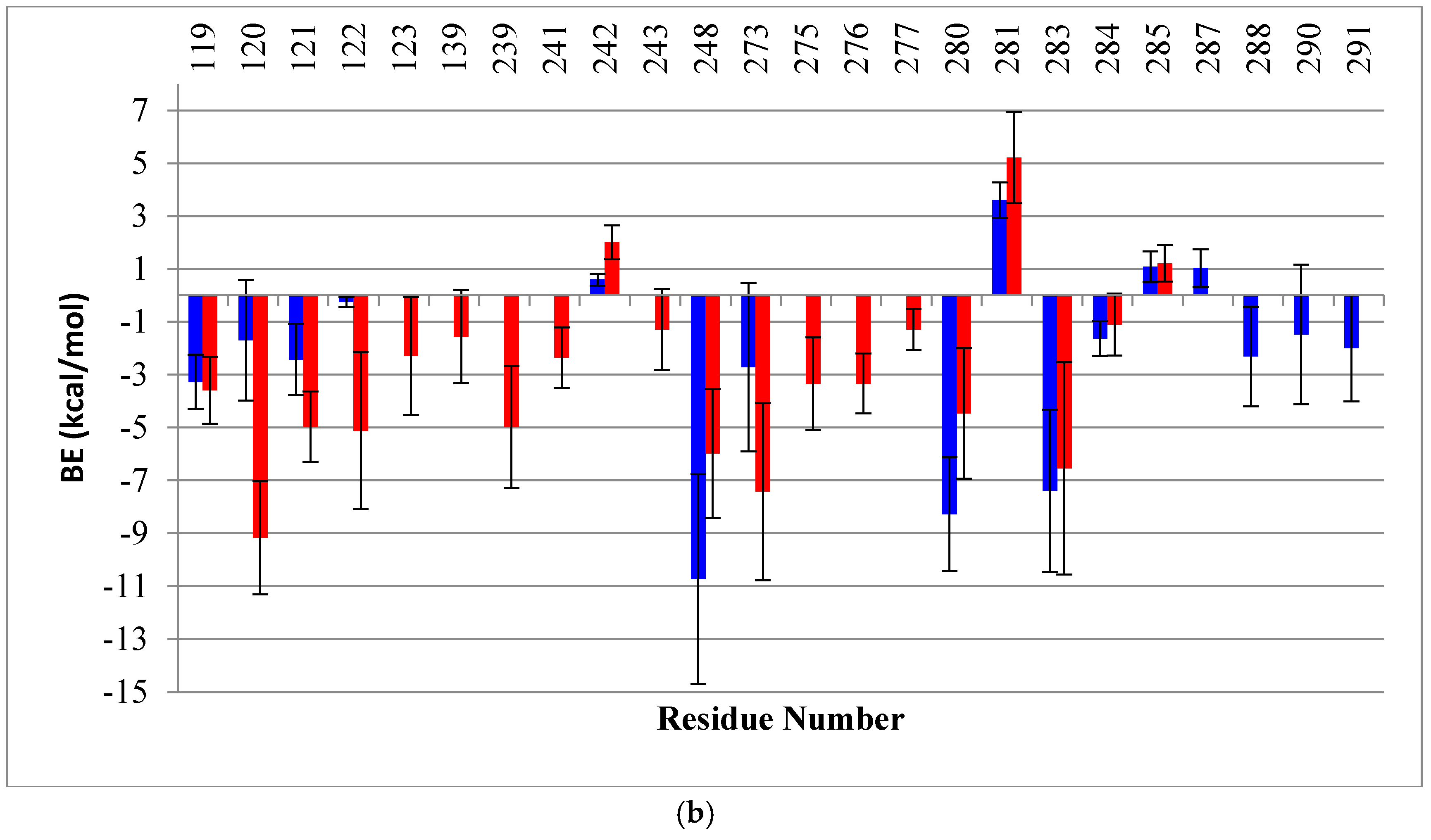
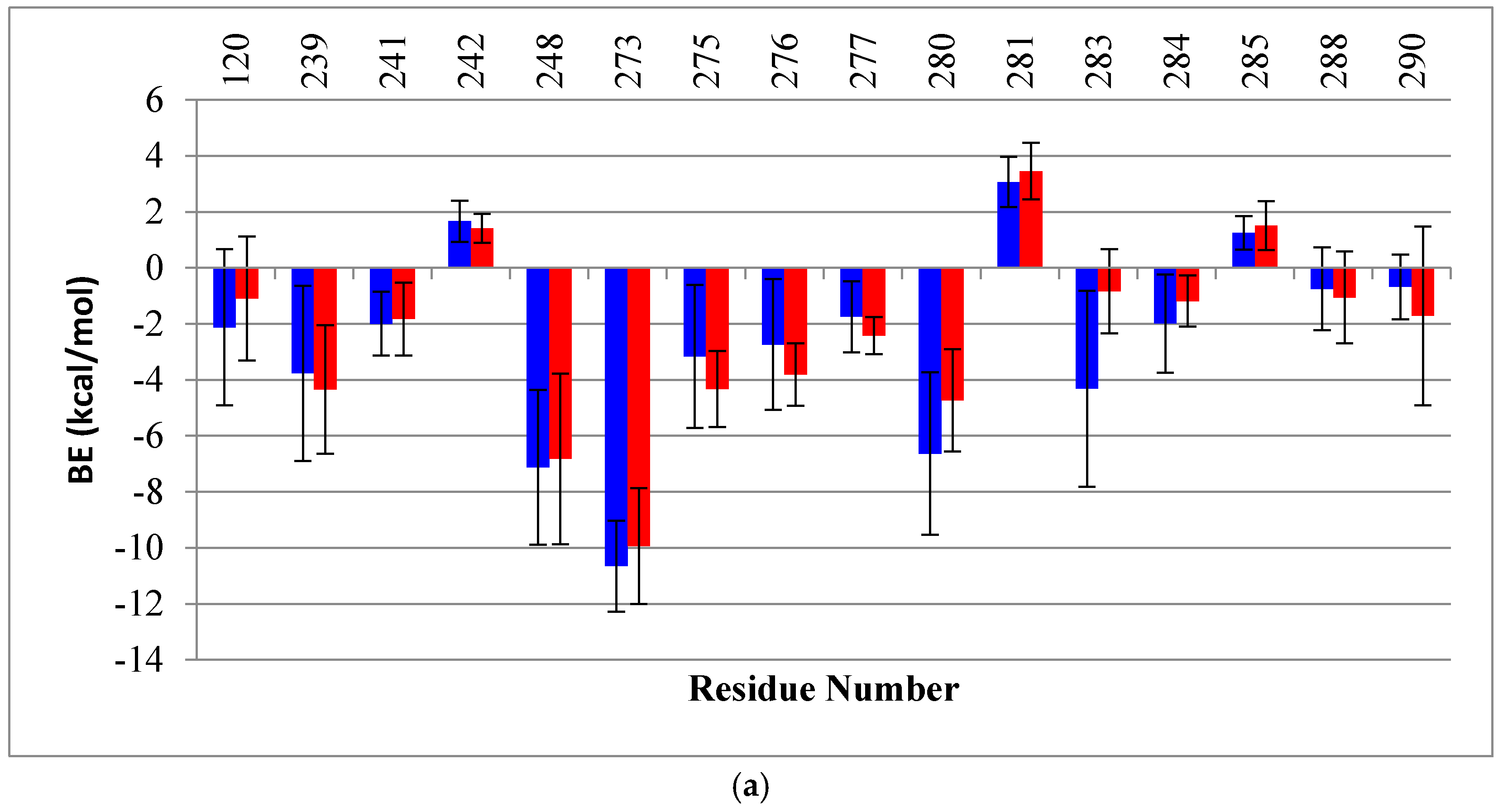
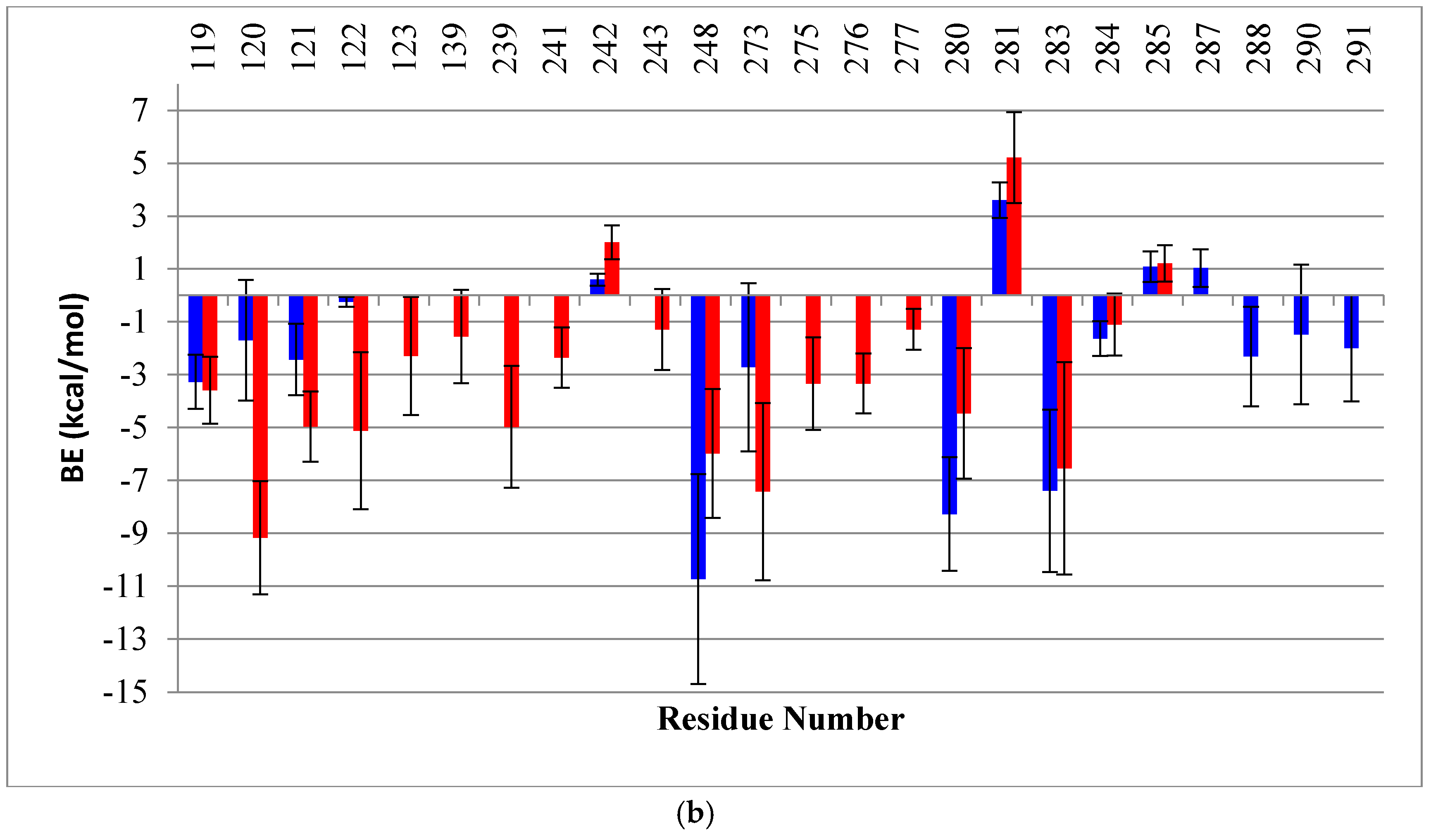
2.2. Binding Free Energy (BE) Analysis
3. Discussion
4. Materials and Methods
4.1. 3D Structure Preparation
4.2. Molecular Dynamics Simulation
4.3. Conformational Change Analysis
4.3.1. RMSD and RMSF
4.3.2. FMA
4.3.3. Clustering
4.4. Binding Energy Calculation
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, I.; Marcel, V.; Olivier, M.; Oren, M.; Rotter, V.; Hainaut, P. Understanding wild-type and mutant p53 activities in human cancer: New landmarks on the way to targeted therapies. Cancer Gene Ther. 2011, 18, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, Y. Targeting p53 for Novel Anticancer Therapy. Transl. Oncol. 2010, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Kogan, S.; Carpizo, D. Pharmacological targeting of mutant p53. Transl. Cancer Res. 2016, 5, 698–706. [Google Scholar] [CrossRef]
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614. [Google Scholar] [CrossRef] [PubMed]
- McLure, K.G.; Lee, P.W.K. How p53 binds DNA as a tetramer. EMBO J. 1998, 17, 3342–3350. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, X.; Dantas Machado, A.C.; Ding, Y.; Chen, Z.; Qin, P.Z.; Rohs, R.; Chen, L. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 2013, 41, 8368–8376. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dey, R.; Chen, L. Crystal Structure of the p53 Core Domain Bound to a Full Consensus Site as a Self-Assembled Tetramer. Structure 2010, 18, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Nilsson, L. Effect of Zn2+ on DNA recognition and stability of the p53 DNA-binding domain. Biochemistry 2006, 45, 7483–7492. [Google Scholar] [CrossRef] [PubMed]
- Hanel, W.; Marchenko, N.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Qian, J.; Hu, Y.; Wang, J.; Zhou, X.; Chen, H.; Fang, J.Y. Heterogeneity of Li-Fraumeni syndrome links to unequal gain-of-function effects of p53 mutations. Sci. Rep. 2014, 4, 4223. [Google Scholar] [CrossRef] [PubMed]
- Samowitz, W.S.; Curtin, K.; Ma, K.; Edwards, S.; Schaffer, D.; Leppert, M.F.; Slattery, M.L. Prognostic significance of p53 mutations in colon cancer at the population level. Int. J. Cancer 2002, 99, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Bykov, V.J.N.; Ali, D.; Andreń, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef] [PubMed]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, G.W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Chai, X.; Johnston, K.; Clements, A.; Marmorstein, R. Crystal Structure of the Mouse p53 Core DNA-binding Domain at 2.7 Å Resolution. J. Biol. Chem. 2001, 276, 12120–12127. [Google Scholar] [CrossRef] [PubMed]
- Cañadillas, J.M.P.; Tidow, H.; Freund, S.M.V.; Rutherford, T.J.; Ang, H.C.; Fersht, A.R. Solution structure of p53 core domain: Structural basis for its instability. Proc. Natl. Acad. Sci. USA 2006, 103, 2109–2114. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Wiman, K.G. 25 Years of p53 Research; Springer: Berlin, Germany, 2005; ISBN 9781402029226. [Google Scholar]
- Wong, K.B.; DeDecker, B.S.; Freund, S.M.; Proctor, M.R.; Bycroft, M.; Fersht, A.R. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. USA 1999, 96, 8438–8442. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061. [Google Scholar] [CrossRef] [PubMed]
- Demir, Ö.; Baronio, R.; Salehi, F.; Wassman, C.D.; Hall, L.; Hatfield, G.W.; Chamberlin, R.; Kaiser, P.; Lathrop, R.H.; Amaro, R.E. Ensemble-based computational approach discriminates functional activity of p53 cancer and rescue mutants. PLoS Comput. Biol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Koulgi, S.; Achalere, A.; Sharma, N.; Sonavane, U.; Joshi, R. QM-MM simulations on p53-DNA complex: A study of hot spot and rescue mutants. J. Mol. Model. 2013, 19, 5545–5559. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.U.N.; Schwedes, J.F.; Parks, D.; Mann, K.; Tegtmeyer, P. Interaction of p53 with its consensus DNA-binding site. Mol. Cell. Biol. 1995, 15, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Soussi, T.; Lozano, G. p53 mutation heterogeneity in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.M.; Siu, W.Y.; Lau, A.; Poon, R.Y.C. How many mutant p53 molecules are needed to inactivate a tetramer? Mol. Cell. Biol. 2004, 24, 3536–3551. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.; Fersht, A. Structure–function–rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Friedler, A.; DeDecker, B.S.; Freund, S.M.V.; Blair, C.; Rüdiger, S.; Fersht, A.R. Structural Distortion of p53 by the Mutation R249S and its Rescue by a Designed Peptide: Implications for “mutant Conformation”. J. Mol. Biol. 2004, 336, 187–196. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Lezon, T.R.; Yang, L.W.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Lukman, S.; Lane, D.P.; Verma, C.S. Mapping the structural and dynamical features of multiple p53 DNA binding domains: Insights into loop 1 intrinsic dynamics. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, D.; Ano Bom, A.P.D.; Lima, L.M.T.R.; Quesado, P.A.; Oyama, M.F.C.; De Moura Gallo, C.V.; Cordeiro, Y.; Silva, J.L. Cognate DNA stabilizes the tumor suppressor p53 and prevents misfolding and aggregation. Biochemistry 2009, 48, 6126–6135. [Google Scholar] [CrossRef] [PubMed]
- Lambrughi, M.; De Gioia, L.; Gervasio, F.L.; Lindorff-Larsen, K.; Nussinov, R.; Urani, C.; Bruschi, M.; Papaleo, E. DNA-binding protects p53 from interactions with cofactors involved in transcription-independent functions. Nucleic Acids Res. 2016, 44, 9096–9109. [Google Scholar] [CrossRef] [PubMed]
- Selivanova, G.; Wiman, K.G. Reactivation of mutant p53: Molecular mechanisms and therapeutic potential. Oncogene 2007, 26, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Chène, P.; Che, P. The role of tetramerization in p53 function. Oncogene 2001, 393, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Kamada, R.; Nomura, T.; Anderson, C.W.; Sakaguchi, K. Cancer-associated p53 tetramerization domain mutants: Quantitative analysis reveals a low threshold for tumor suppressor inactivation. J. Biol. Chem. 2011, 286, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.C.; Fitzgerald, M.X.; Marmorstein, R. Structure of the p53 core domain dimer bound to DNA. J. Biol. Chem. 2006, 281, 20494–20502. [Google Scholar] [CrossRef] [PubMed]
- Paciello, G.; Acquaviva, A.; Ficarra, E.; Deriu, M.A.; Macii, E.A. molecular dynamics study of a miRNA:mRNA interaction. J. Mol. Model. 2011, 17, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Tuszynski, J.A.; Morbiducci, U.; Licandro, G.; Danani, A.; Deriu, M.A. Thermodynamic and kinetic stability of the Josephin Domain closed arrangement: Evidences from replica exchange molecular dynamics. Biol. Direct 2017, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Deriu, M.A.; Grasso, G.; Tuszynski, J.A.; Gallo, D.; Morbiducci, U.; Danani, A. Josephin Domain Structural Conformations Explored by Metadynamics in Essential Coordinates. PLoS Comput. Biol. 2016, 12, e1004699. [Google Scholar] [CrossRef] [PubMed]
- Deriu, M.A.; Grasso, G.; Tuszynski, J.A.; Massai, D.; Gallo, D.; Morbiducci, U.; Danani, A. Characterization of the AXH domain of Ataxin-1 using enhanced sampling and functional mode analysis. Proteins Struct. Funct. Bioinform. 2016, 84, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Deriu, M.A.; Tuszynski, J.A.; Gallo, D.; Morbiducci, U.; Danani, A. Conformational fluctuations of the AXH monomer of Ataxin-1. Proteins Struct. Funct. Bioinform. 2016, 84, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Grasso, G.; Deriu, M.A.; Prat, M.; Rimondini, L.; Vernè, E.; Follenzi, A.; Danani, A. Cell Penetrating Peptide Adsorption on Magnetite and Silica Surfaces: A Computational Investigation. J. Phys. Chem. B 2015, 119, 8239–8246. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 1.8; Schrödinger, LLC: San Carlos, CA, USA, 2017.
- Chemical Computing Group Inc. Molecular Operating Environment (MOE). Sci. Comput. Instrum. 2004, 22, 32. [Google Scholar] [CrossRef]
- Labute, P. The generalized born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber14 Reference Manual; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Omar, S.I.; Tuszynski, J. Ranking the Binding Energies of p53 Mutant Activators and Their ADMET Properties. Chem. Biol. Drug Des. 2015, 86, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; De Groot, B.L. Detection of functional modes in protein dynamics. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.L.; Bouldin, D.W. A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell. 1979, 1, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Caliński, T.; Harabasz, J. A dendrite method for cluster analysis. Commun. Stat. Methods 2007, 3, 1–27. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born model suitable for macromolecules. J. Phys. Chem. B 2000, 104, 3712–3720. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BE of the p53 Dimer to DNA | BE of p53 Monomer A to B | ||||
| BE (kcal/mol) | SD (kcal/mol) | BE (kcal/mol) | SD (kcal/mol) | ||
| wt-p53 | −100 | 17 | wt-p53 | −4 | 7 |
| G245S-mp53 | −129 | 22 | G245S-mp53 | −2 | 7 |
| BE of p53 Monomer A to DNA | BE of p53 Monomer B to DNA | ||||
| BE (kcal/mol) | SD (kcal/mol) | BE (kcal/mol) | SD (kcal/mol) | ||
| wt-p53 | −60 | 15 | wt-p53 | −33 | 12 |
| G245S-mp53 | −55 | 13 | G245S-mp53 | −70 | 20 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lepre, M.G.; Omar, S.I.; Grasso, G.; Morbiducci, U.; Deriu, M.A.; Tuszynski, J.A. Insights into the Effect of the G245S Single Point Mutation on the Structure of p53 and the Binding of the Protein to DNA. Molecules 2017, 22, 1358. https://doi.org/10.3390/molecules22081358
Lepre MG, Omar SI, Grasso G, Morbiducci U, Deriu MA, Tuszynski JA. Insights into the Effect of the G245S Single Point Mutation on the Structure of p53 and the Binding of the Protein to DNA. Molecules. 2017; 22(8):1358. https://doi.org/10.3390/molecules22081358
Chicago/Turabian StyleLepre, Marco Gaetano, Sara Ibrahim Omar, Gianvito Grasso, Umberto Morbiducci, Marco Agostino Deriu, and Jack A. Tuszynski. 2017. "Insights into the Effect of the G245S Single Point Mutation on the Structure of p53 and the Binding of the Protein to DNA" Molecules 22, no. 8: 1358. https://doi.org/10.3390/molecules22081358
APA StyleLepre, M. G., Omar, S. I., Grasso, G., Morbiducci, U., Deriu, M. A., & Tuszynski, J. A. (2017). Insights into the Effect of the G245S Single Point Mutation on the Structure of p53 and the Binding of the Protein to DNA. Molecules, 22(8), 1358. https://doi.org/10.3390/molecules22081358