Chalcone Derivatives: Promising Starting Points for Drug Design

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Synthesis of Chalcone Scaffolds
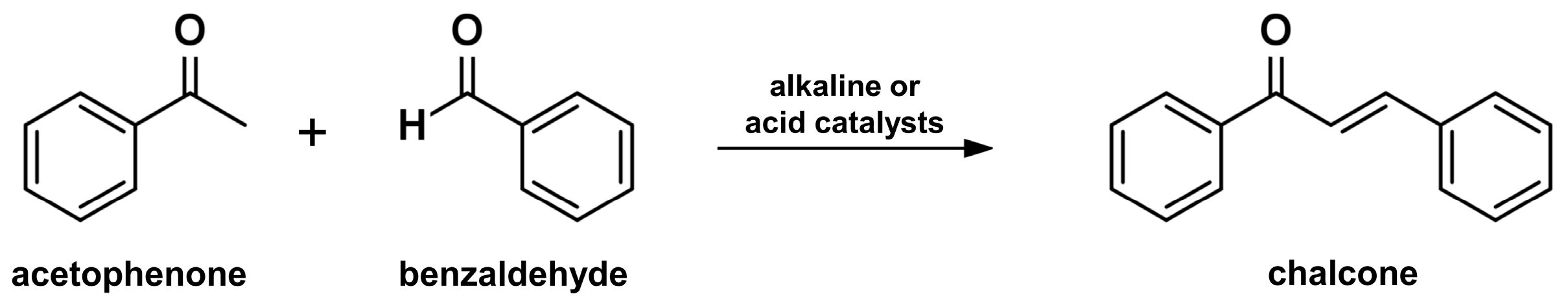
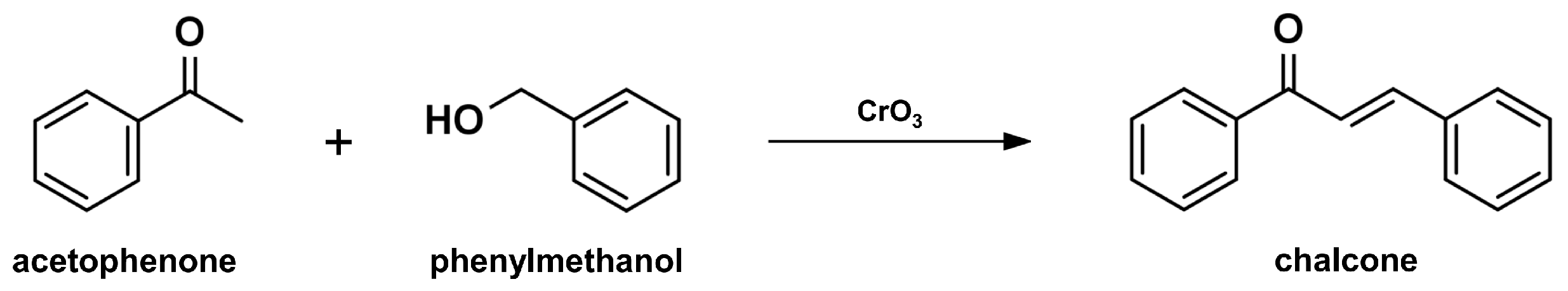
2.1. Claisen-Schmidt Condensation
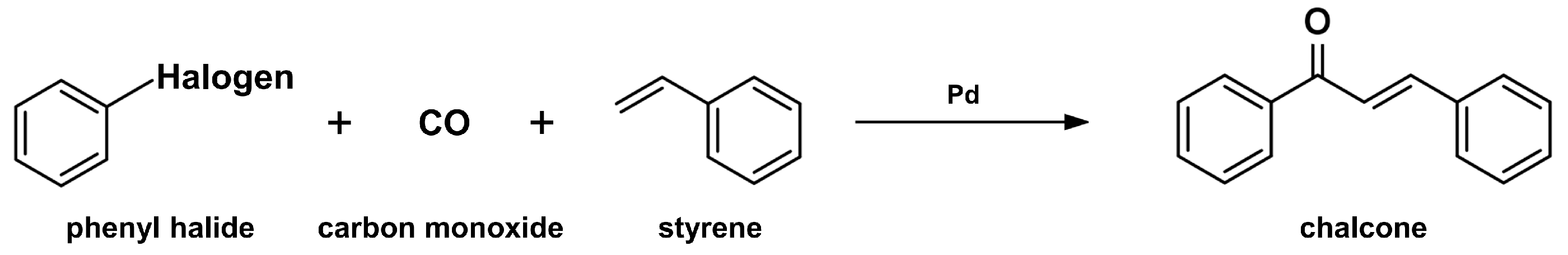
2.2. Carbonylative Heck Coupling Reaction
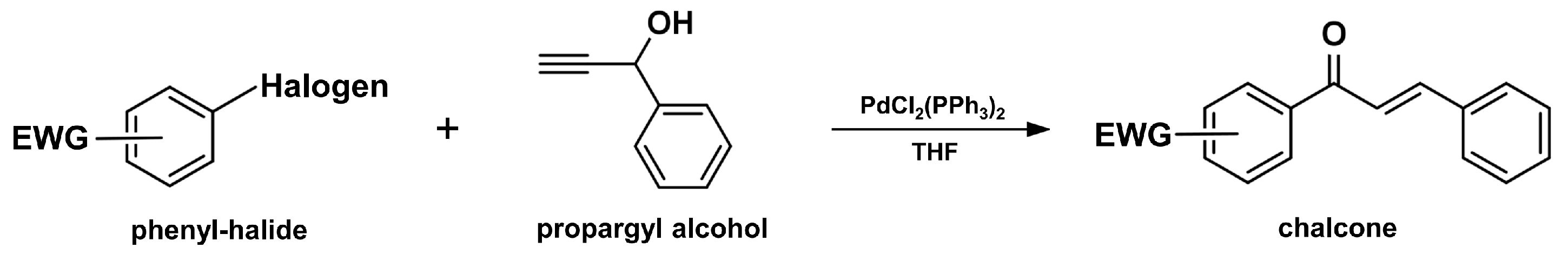
2.3. Coupling Reaction
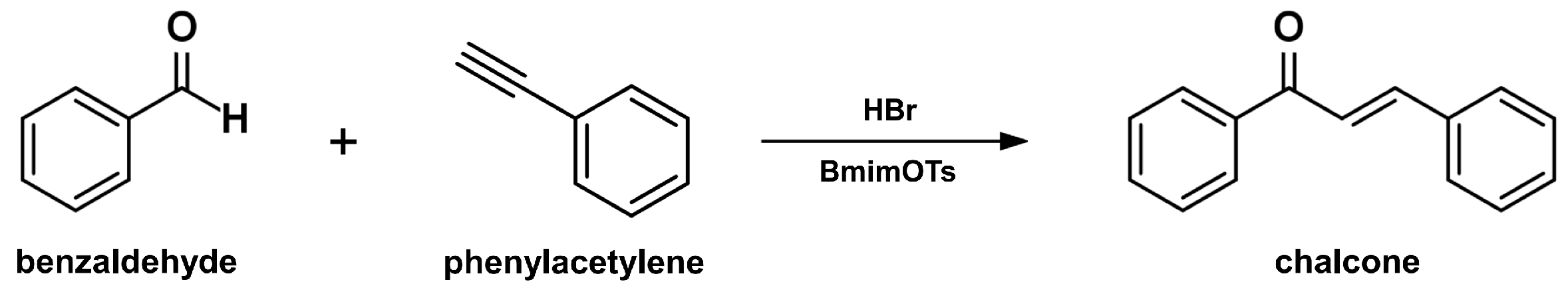
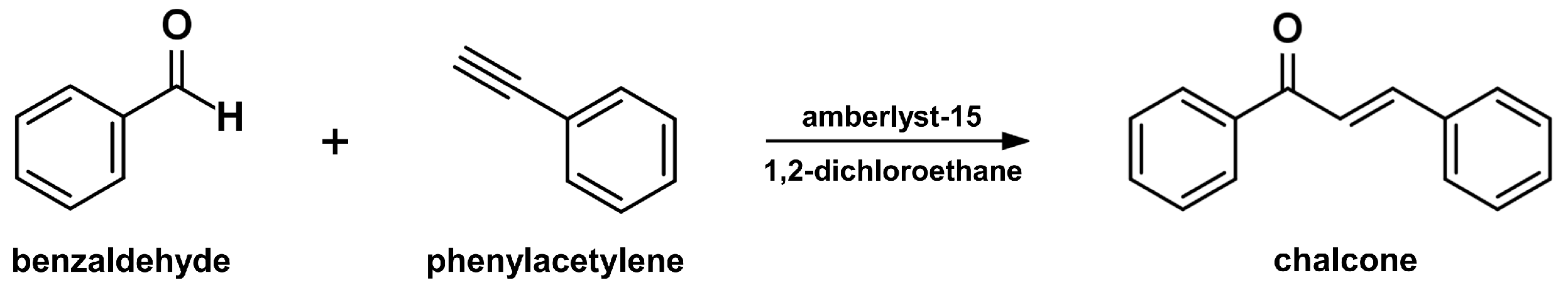
2.4. Sonogashira Isomerization Coupling
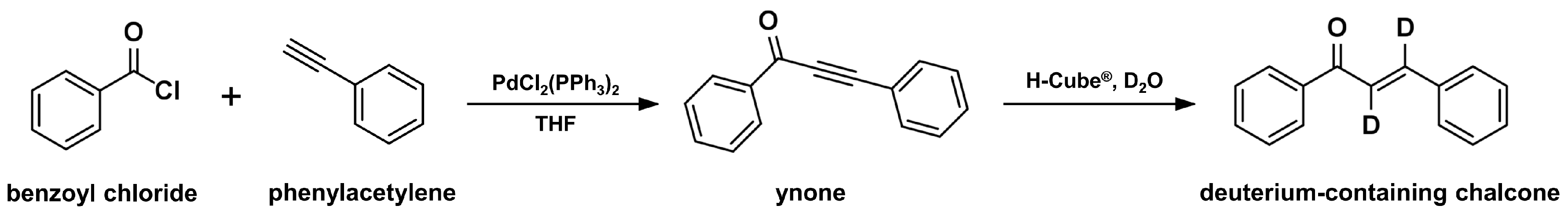
2.5. Continuous-Flow Deuteraction Reaction
2.6. Suzuki–Miyaura Coupling Reaction
2.7. One-Pot Synthesis
2.8. Solid Acid Catalyst Mediated Reaction
3. Design of New Chalcone Derivatives
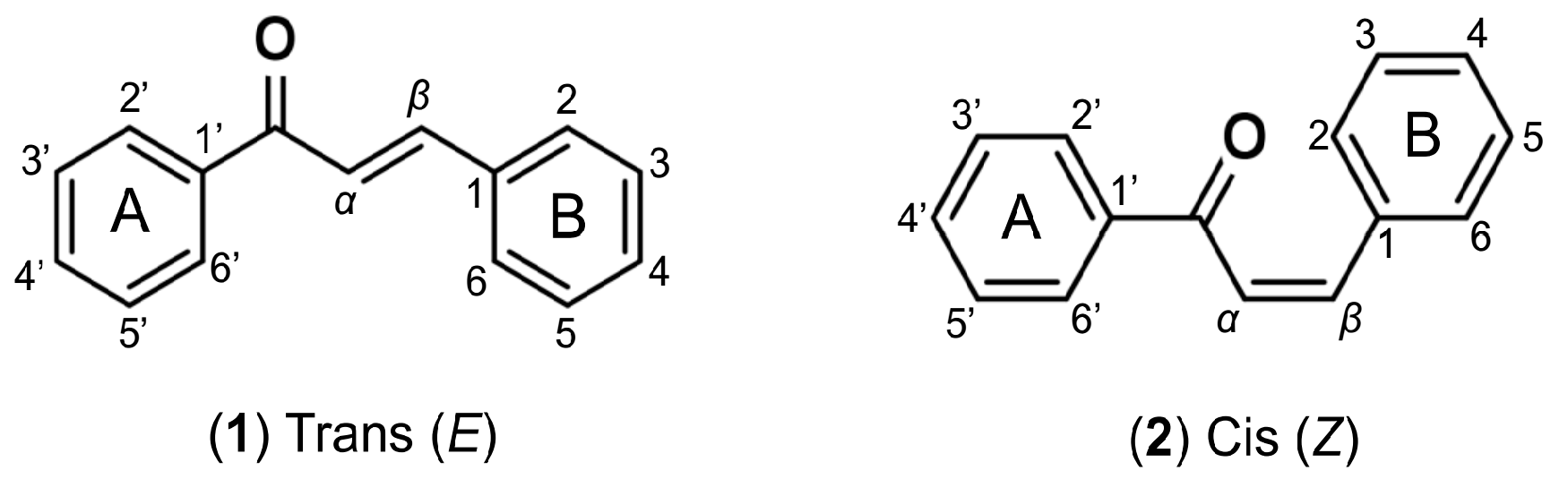
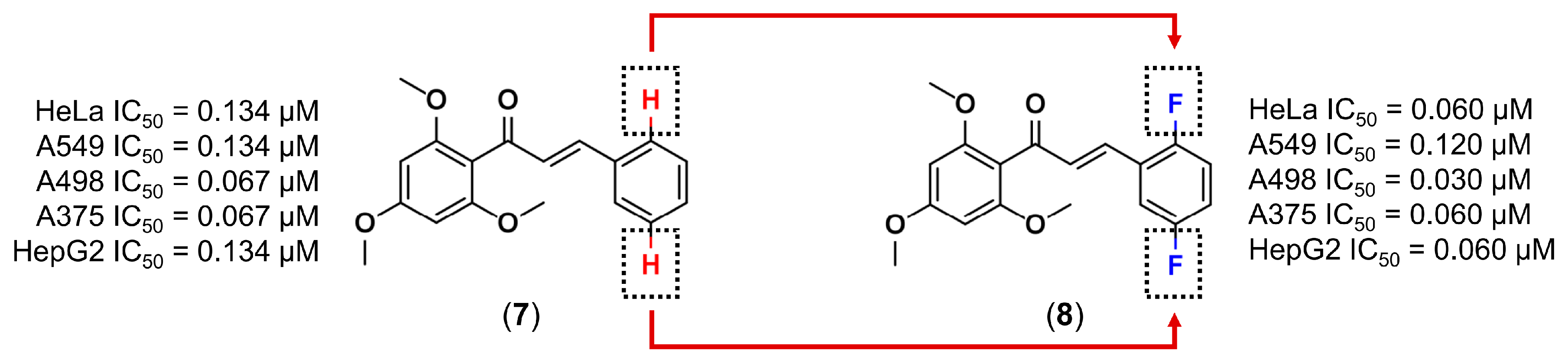
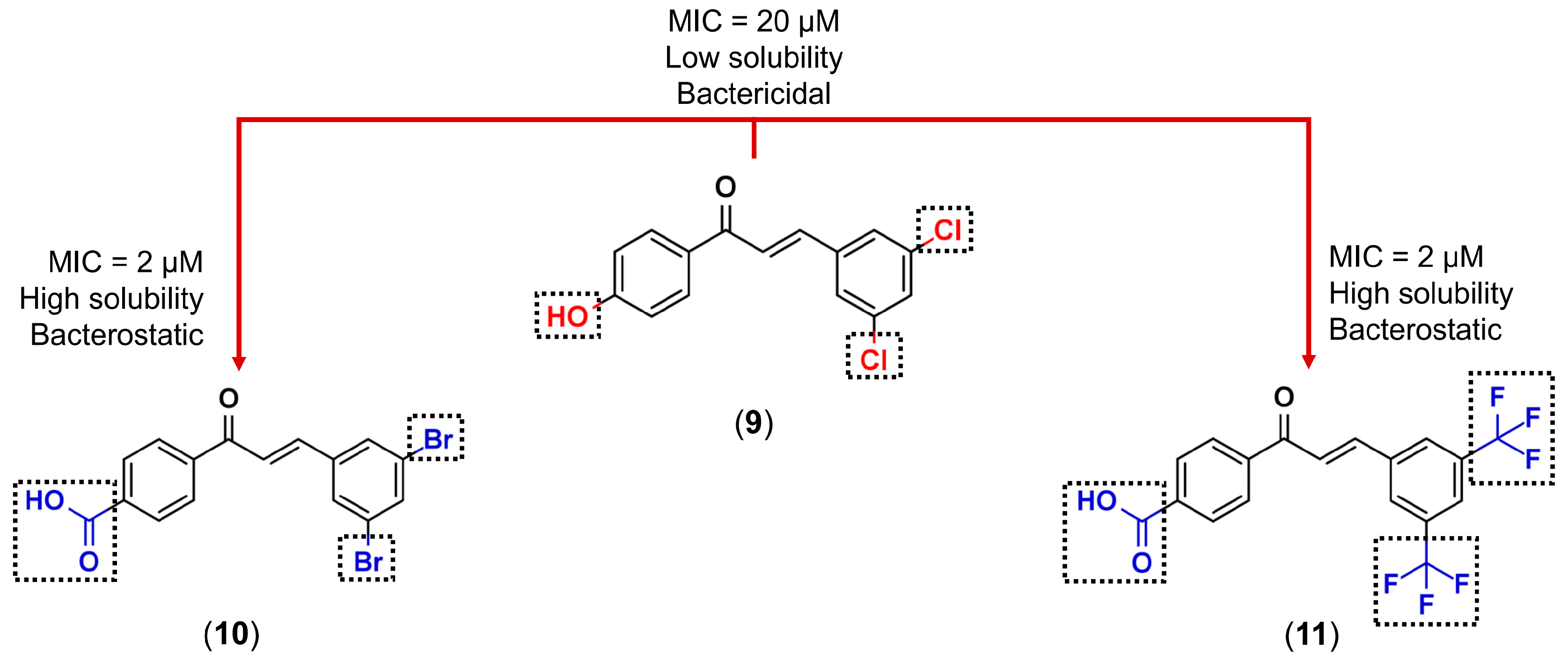
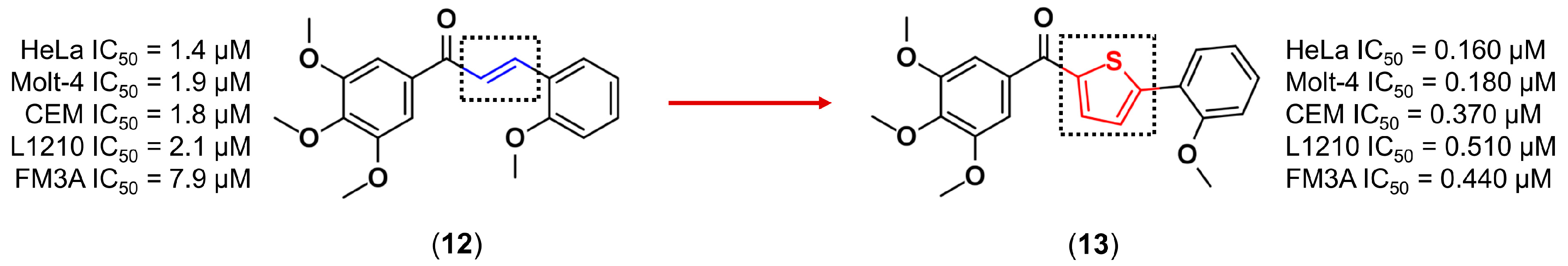
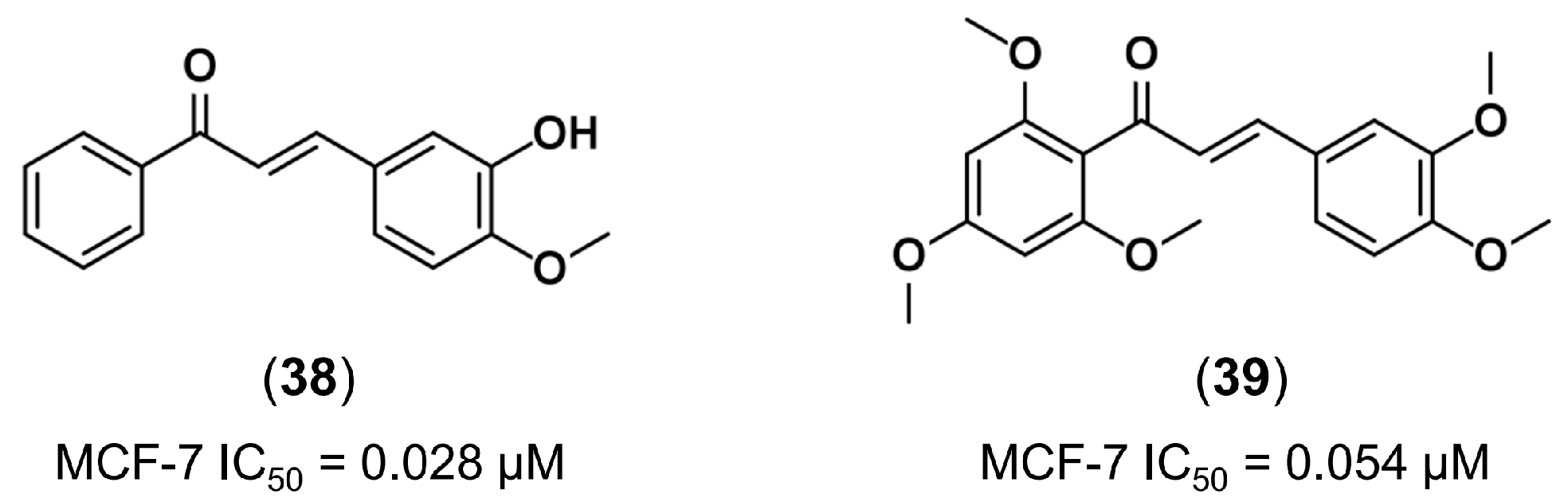
3.1. Bioisosterism
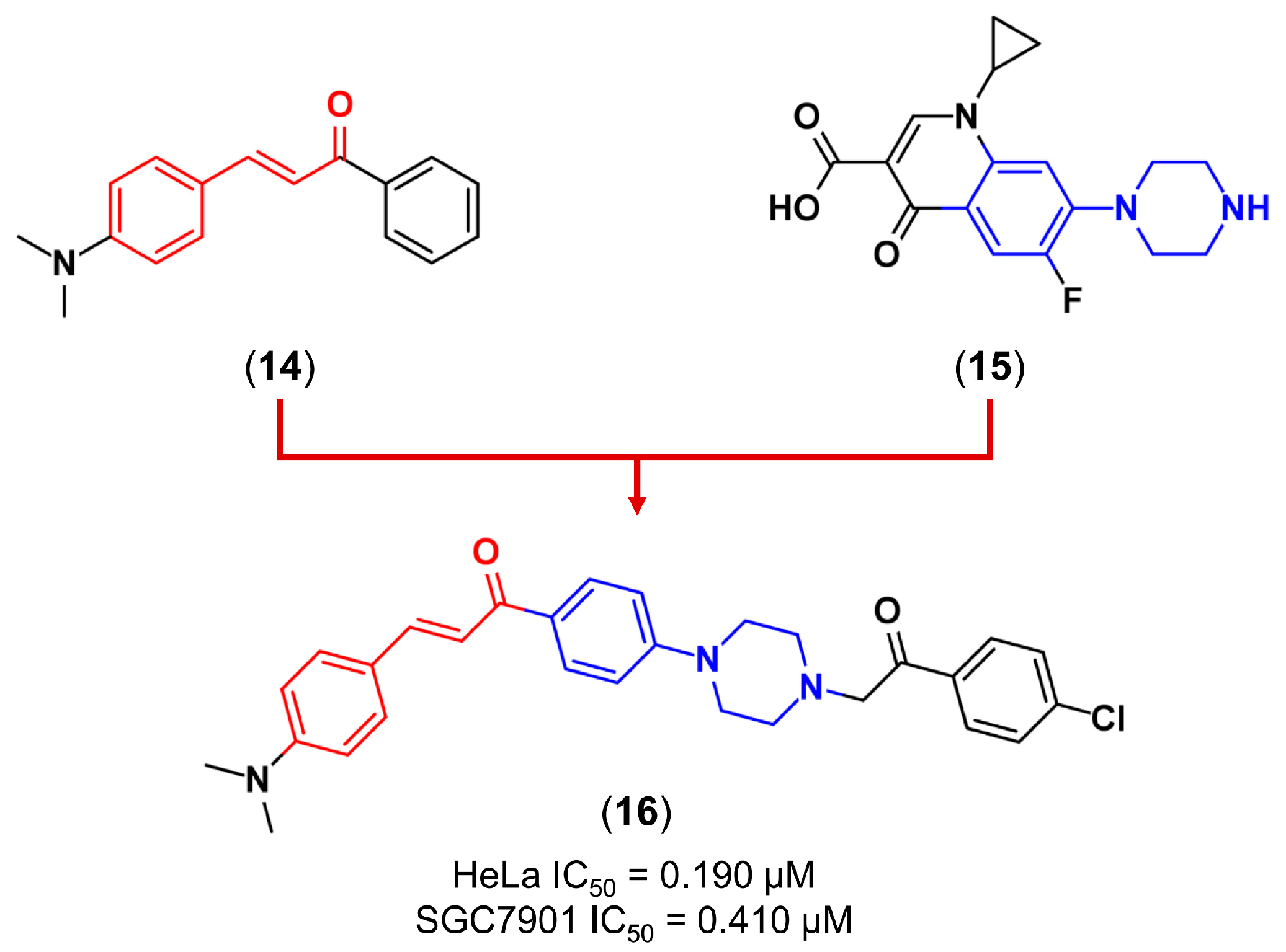
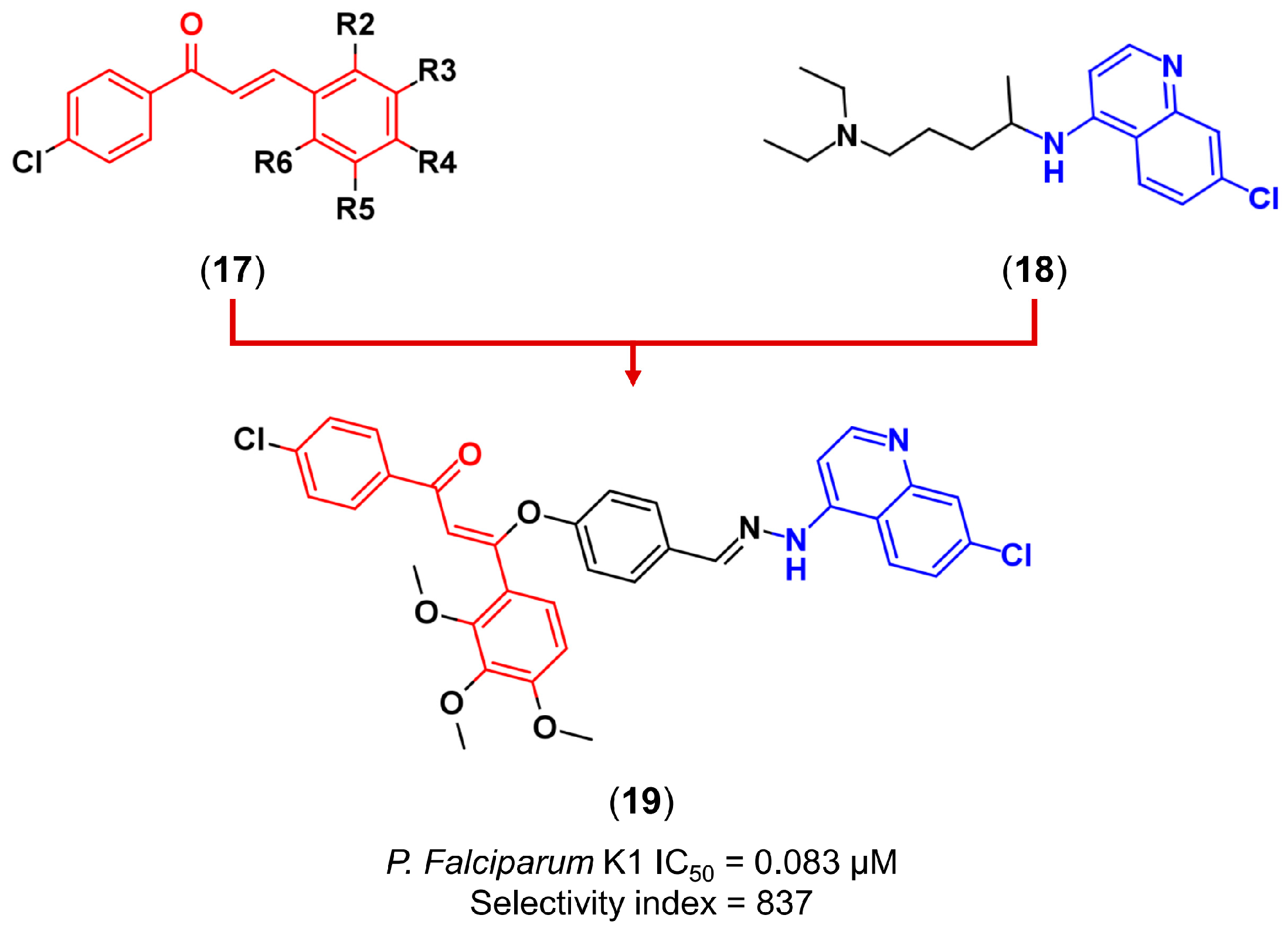
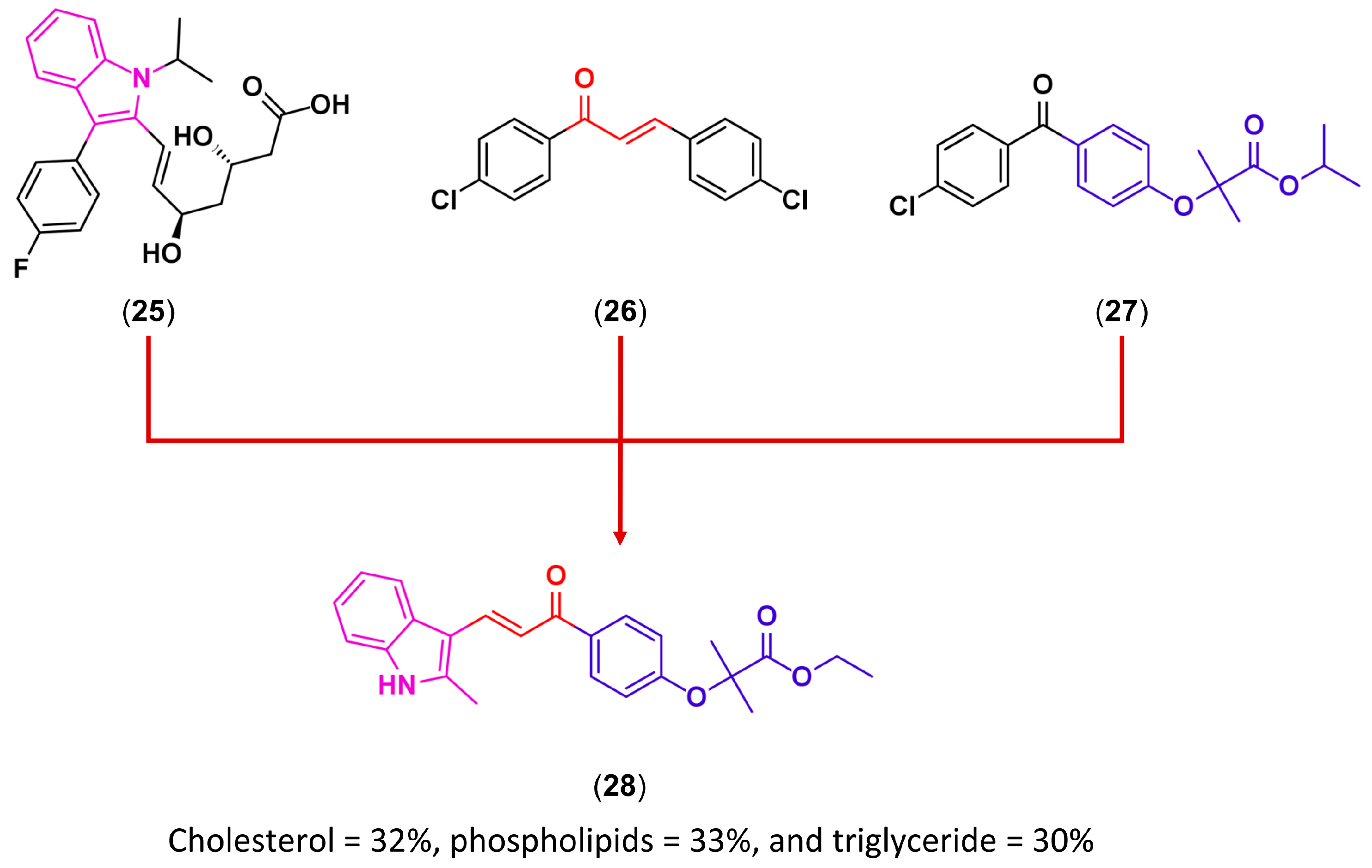
3.2. Molecular Hybridization
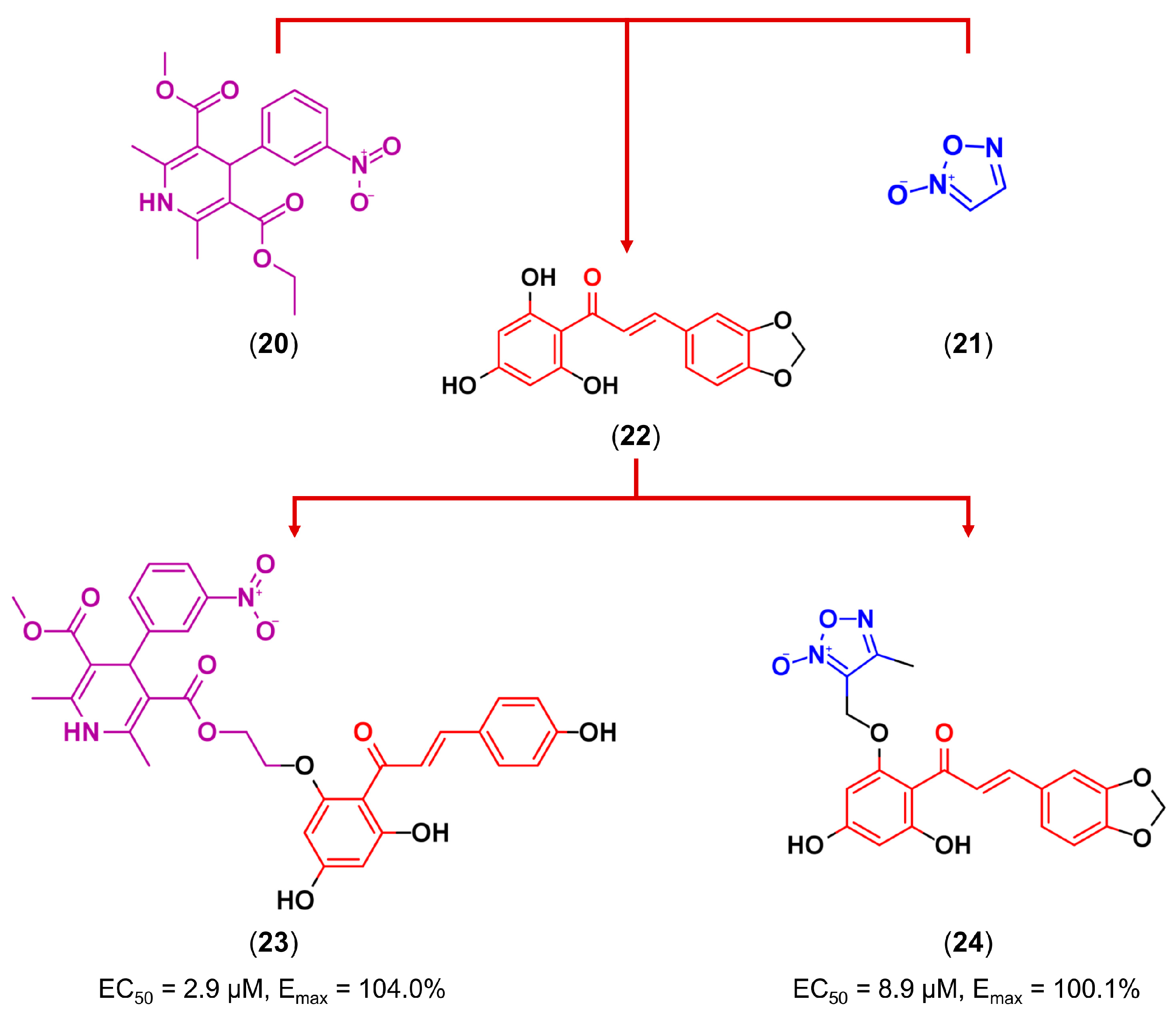
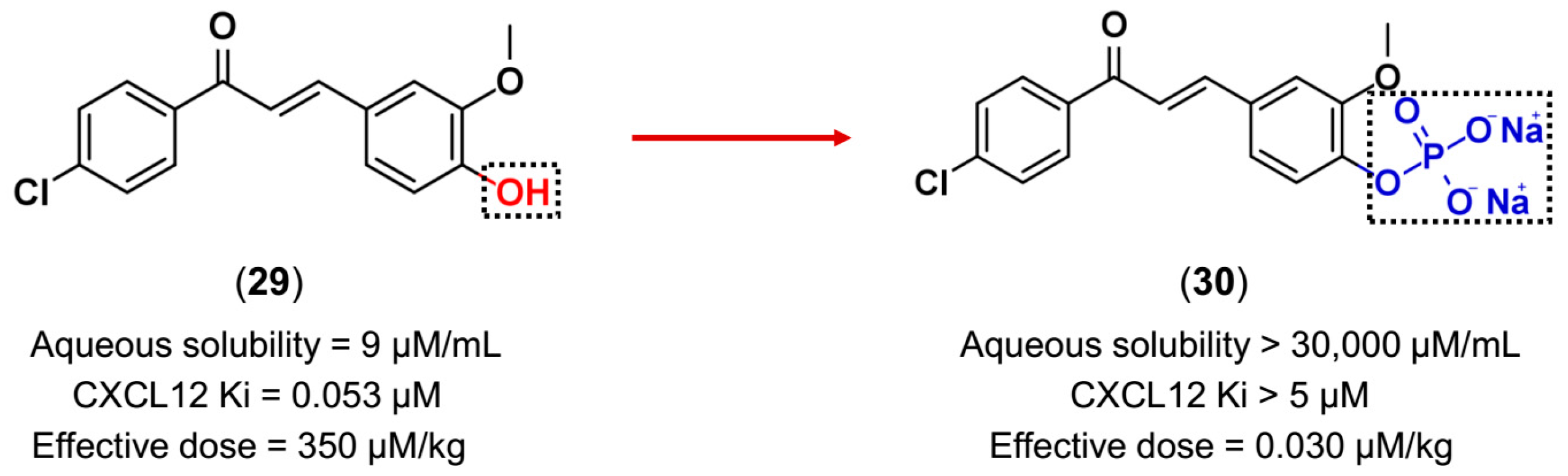
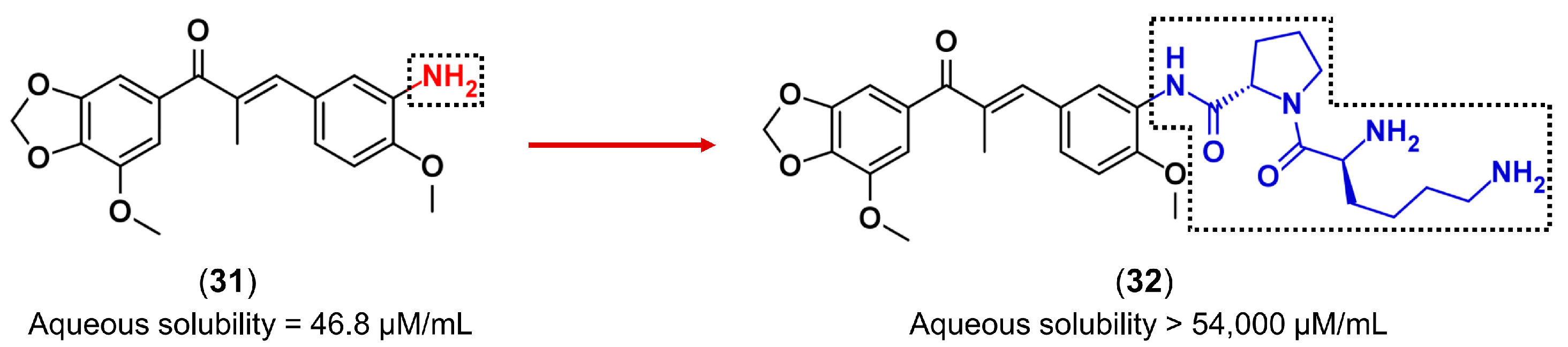
3.3. Drug Latentiation
4. Computer-Assisted Drug Design (CADD)
4.1. Structure-Based Drug Design (SBDD)
4.1.1. Protein-Ligand Docking
4.1.2. Molecular Dynamics (MD) Simulations
4.1.3. Structure-Based Pharmacophores (SBPs)
4.2. Ligand-Based Drug Design (LBDD)
4.2.1. Similarity Search
4.2.2. Ligand-Based Pharmacophores (LBPs)
4.2.3. QSAR
4.2.4. Quantum Mechanical (QM) Methods
4.3. Practical Applications of CADD in Chalcone Field
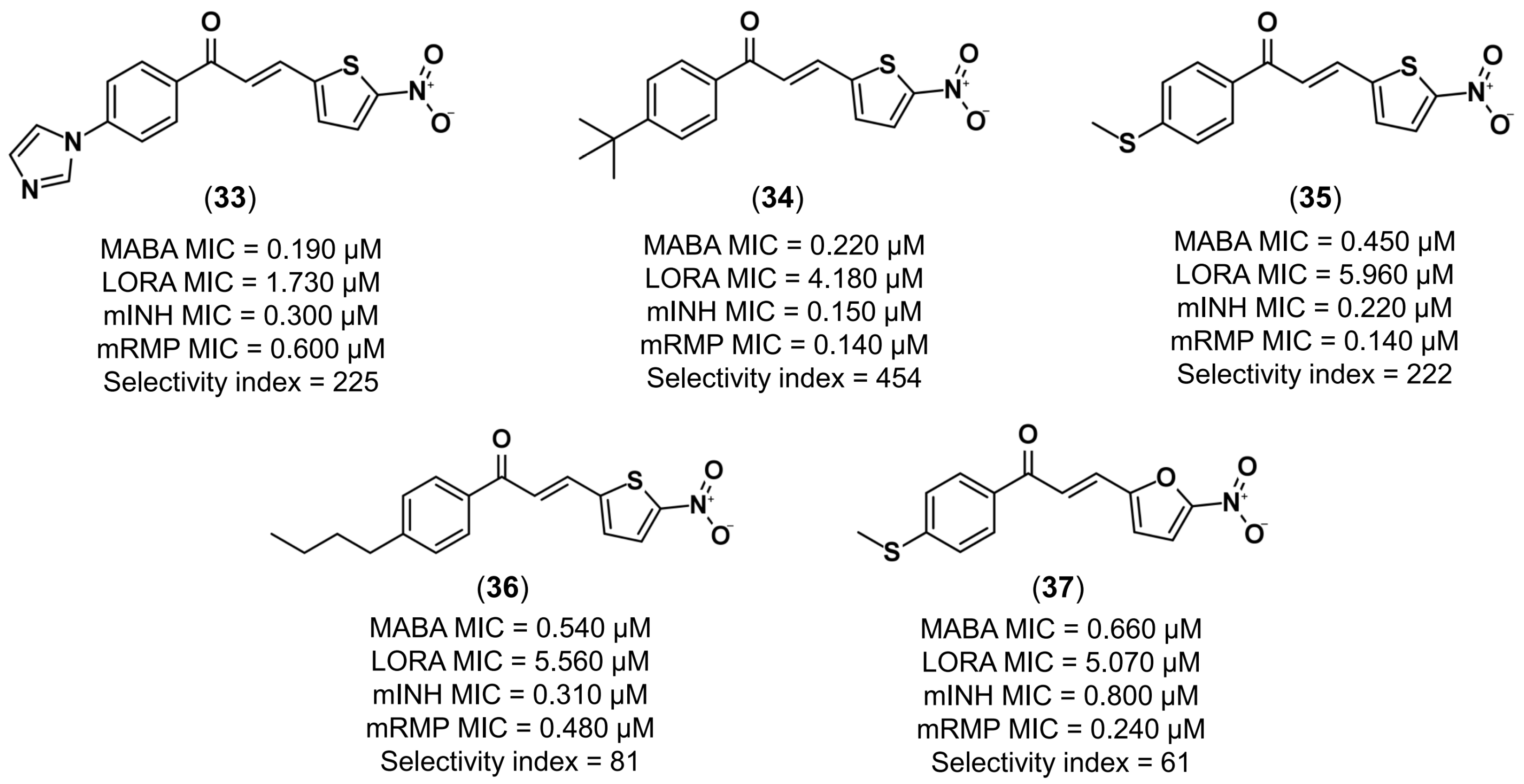
4.3.1. Design of Anti-Tuberculosis Agents
4.3.2. Discovery of New Tubulin Inhibitors
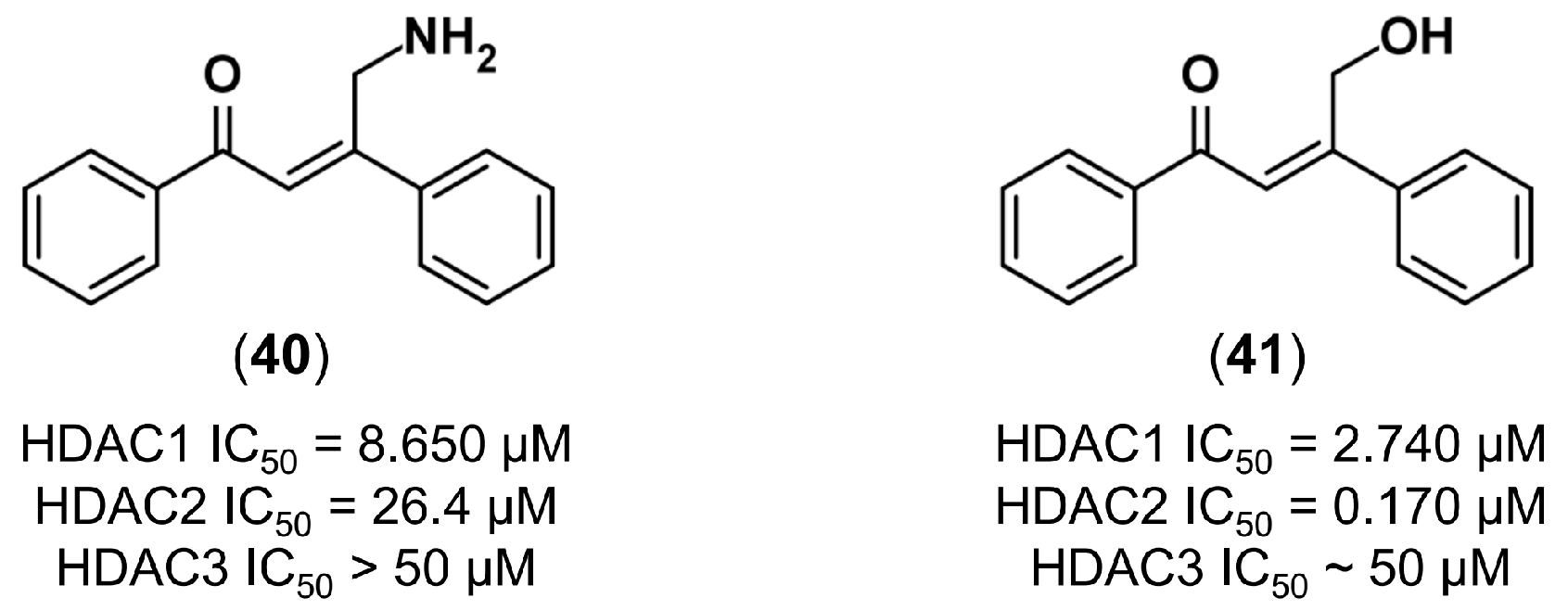
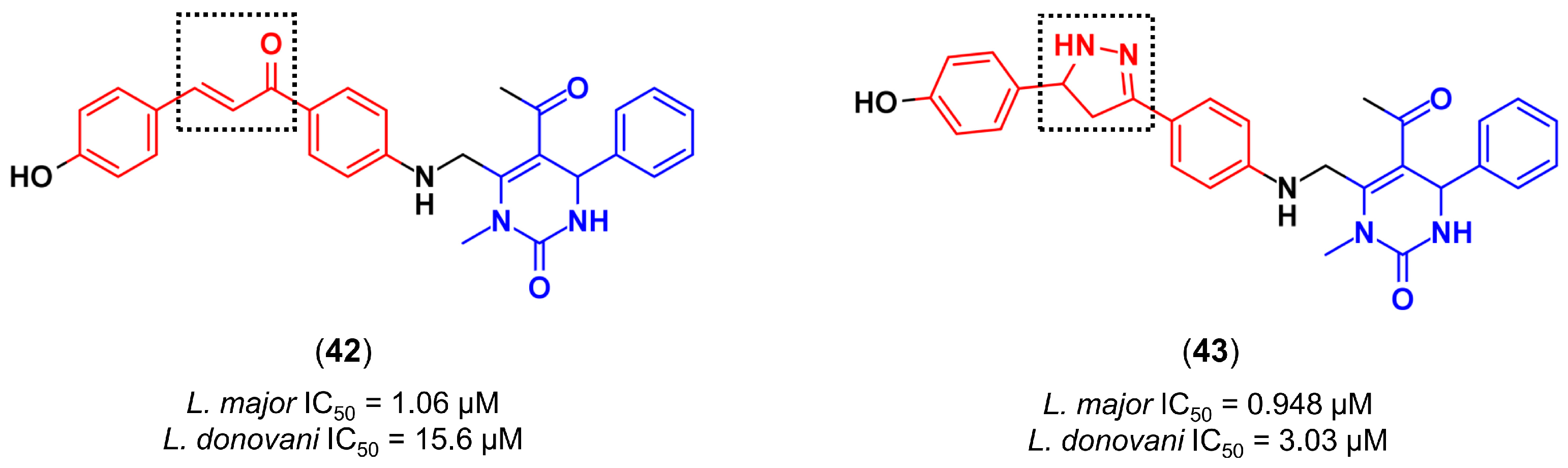
4.3.3. De Novo Design of Histone Deacetylase 2 Inhibitors
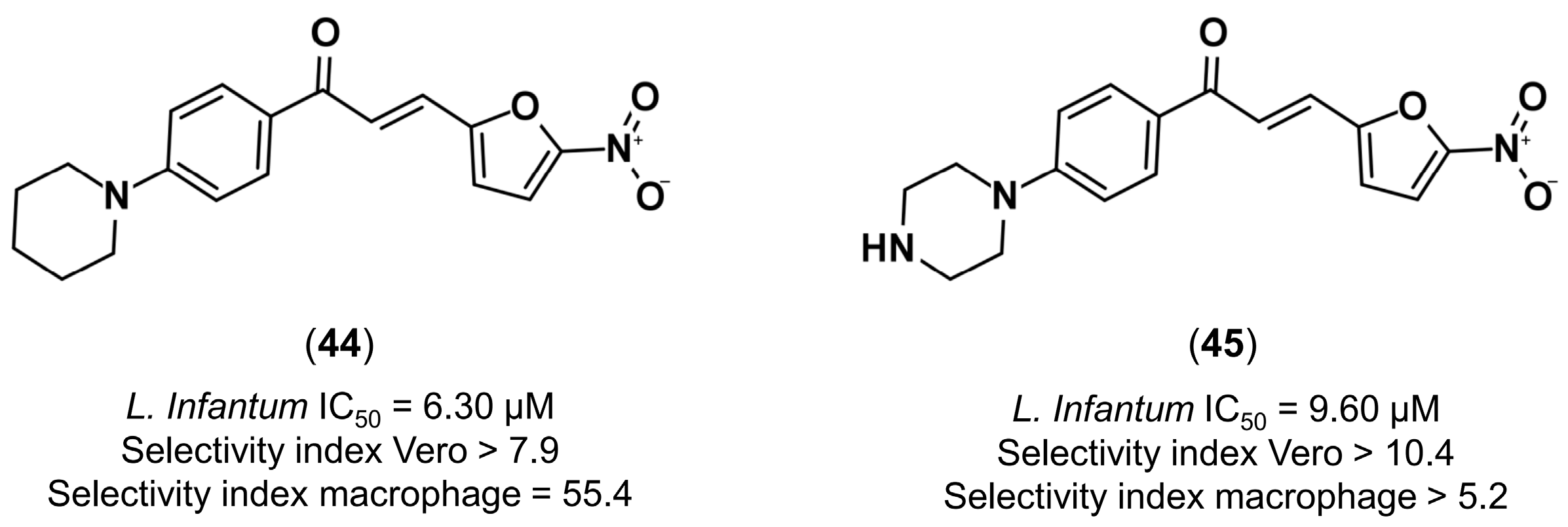
4.3.4. Design of Anti-Leishmanial Agents
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ni, L.; Meng, C.Q.; Sikorski, J.A. Recent advances in therapeutic chalcones. Expert Opin. Ther. Pat. 2004, 14, 1669–1691. [Google Scholar] [CrossRef]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, J.; Kohli, D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Wong, E. The role of chalcones and flavanones in flavonoid biosynthesis. Phytochemistry 1968, 7, 1751–1758. [Google Scholar] [CrossRef]
- Evranos Aksöz, B.; Ertan, R. Chemical and structural properties of chalcones I. FABAD J. Pharm. Sci. 2011, 36, 223–242. [Google Scholar]
- Israf, D.; Khaizurin, T.; Syahida, A.; Lajis, N.; Khozirah, S. Cardamonin inhibits COX and iNOS expression via inhibition of p65NF-κB nuclear translocation and Iκ-B phosphorylation in RAW 264.7 macrophage cells. Mol. Immunol. 2007, 44, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Curtis-Long, M.J.; Yuk, H.J.; Wang, Y.; Song, Y.H.; Jeong, S.H.; Park, K.H. Quantitative analysis of phenolic metabolites from different parts of Angelica keiskei by HPLC–ESI MS/MS and their xanthine oxidase inhibition. Food Chem. 2014, 153, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yoshimura, M.; Yamaguchi, F.; Kouchi, T.; Tsuji, R.; Saito, M.; Obata, A.; Kikuchi, M. Anti-allergic activity of naringenin chalcone from a tomato skin extract. Biosci. Biotechnol. Biochem. 2004, 68, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Muko, M.; Ohta, E.; Ohta, S. C-geranylated chalcones from the stems of Angelica keiskei with superoxide-scavenging activity. J. Nat. Prod. 2008, 71, 1308–1310. [Google Scholar] [CrossRef] [PubMed]
- Birari, R.B.; Gupta, S.; Mohan, C.G.; Bhutani, K.K. Antiobesity and lipid lowering effects of Glycyrrhiza chalcones: Experimental and computational studies. Phytomedicine 2011, 18, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Christensen, S.B.; Blom, J.; Lemmich, E.; Nadelmann, L.; Fich, K.; Theander, T.G.; Kharazmi, A. Licochalcone A, a novel antiparasitic agent with potent activity against human pathogenic protozoan species of Leishmania. Antimicrob. Agents Chemother. 1993, 37, 2550–2556. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Kim, S.; Jin, Z.; Yang, H.; Han, D.; Baek, N.I.; Jo, J.; Cho, C.W.; Park, J.H.; Shimizu, M.; et al. Isoliquiritigenin, a chalcone compound, is a positive allosteric modulator of GABA A receptors and shows hypnotic effects. Biochem. Biophys. Res. Commun. 2011, 413, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; He, J.-X.; Nagai, H.; Tani, T.; Akao, T. Isoliquiritigenin, one of the antispasmodic principles of Glycyrrhiza ularensis roots, acts in the lower part of intestine. Biol. Pharm. Bull. 2007, 30, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, K.; Watanabe, T.; Tanigawa, T.; Tominaga, K.; Fujiwara, Y.; Arakawa, T. Sofalcone, a gastroprotective drug, promotes gastric ulcer healing following eradication therapy for Helicobacter pylori: A randomized controlled comparative trial with cimetidine, an H2-receptor antagonist. J. Gastroenterol. Hepatol. 2010, 25, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Beltramino, R.; Penenory, A.; Buceta, A.M. An open-label, randomized multicenter study comparing the efficacy and safety of Cyclo 3 Fort® versus hydroxyethyl rutoside in chronic venous lymphatic insufficiency. Angiology 2000, 51, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Beltramino, R.; Penenory, A.; Buceta, A.M. An open-label, randomised multicentre study comparing the efficacy and safety of CYCLO 3 FORT versus hydroxyethyl rutoside in chronic venous lymphatic insufficiency. Int. Angiol. A J. Int. Union Angiol. 1999, 18, 337–342. [Google Scholar]
- Weindorf, N.; Schultz-Ehrenburg, U. Controlled study of increasing venous tone in primary varicose veins by oral administration of Ruscus aculeatus and trimethylhespiridinchalcone. Z. Hautkrankh. 1987, 62, 28–38. [Google Scholar]
- Zhou, B.; Xing, C. Diverse molecular targets for chalcones with varied bioactivities. Med. Chem. 2015, 5, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Schwöbel, J.A.H.; Wondrousch, D.; Koleva, Y.K.; Madden, J.C.; Cronin, M.T.D.; Schüürmann, G. Prediction of michael-type acceptor reactivity toward glutathione. Chem. Res. Toxicol. 2010, 23, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Amslinger, S.; Al-Rifai, N.; Winter, K.; Wörmann, K.; Scholz, R.; Baumeister, P.; Wild, M. Reactivity assessment of chalcones by a kinetic thiol assay. Org. Biomol. Chem. 2013, 11, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Comprehensive Organic Name Reactions and Reagents, 3nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Bukhari, S.N.A.; Jasamai, M.; Jantan, I. Synthesis and biological evaluation of chalcone derivatives (mini review). Mini Rev. Med. Chem. 2012, 12, 1394–1403. [Google Scholar] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Chalcone scaffolds as anti-infective agents: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 101, 496–524. [Google Scholar] [CrossRef] [PubMed]
- Fringuelli, F.; Pizzo, F.; Vittoriani, C.; Vaccaro, L. Polystyryl-supported TBD as an efficient and reusable catalyst under solvent-free conditions. Chem. Commun. 2004, 2756–2757. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, Y.K. Ecofriendly microwave assisted synthesis of some chalcones. Rasayan J. Chem. 2008, 1, 884–886. [Google Scholar]
- Kakati, D.; Sarma, J.C. Microwave assisted solvent free synthesis of 1,3-diphenylpropenones. Chem. Cent. J. 2011, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-F.; Neumann, H.; Spannenberg, A.; Schulz, T.; Jiao, H.; Beller, M. Development of a general palladium-catalyzed carbonylative heck reaction of aryl halides. J. Am. Chem. Soc. 2010, 132, 14596–14602. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-W.; Li, L.; Xia, C.-G.; Zhao, P.-Q. Efficient coupling reactions of arylalkynes and aldehydes leading to the synthesis of enones. Helv. Chim. Acta 2004, 87, 3080–3084. [Google Scholar] [CrossRef]
- Takahashi, S.; Kuroyama, Y.; Sonogashira, K.; Hagihara, N. A convenient synthesis of ethynylarenes and diethynylarenes. Synthesis 1980, 1980, 627–630. [Google Scholar] [CrossRef]
- Braun, R.U.; Ansorge, M.; Muller, T.J.J. Coupling-isomerization synthesis of chalcones. Chem. A Eur. J. 2006, 12, 9081–9094. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-T.; Ötvös, S.B.; Wu, Y.-C.; Mándity, I.M.; Chang, F.-R.; Fülöp, F. Highly selective continuous-flow synthesis of potentially bioactive deuterated chalcone derivatives. Chempluschem 2015, 80, 859–864. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Hsieh, C.T.; Wu, Y.C.; Li, J.H.; Chang, F.R.; Fülöp, F.; Gilmore, K. Continuous-flow synthesis of deuterium-labeled antidiabetic chalcones: Studies towards the selective deuteration of the alkynone core. Molecules 2016, 21, 318. [Google Scholar] [CrossRef] [PubMed]
- Selepe, M.A.; Van Heerden, F.R. Application of the Suzuki-Miyaura reaction in the synthesis of flavonoids. Molecules 2013, 18, 4739–4765. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Bootwicha, T.; Baars, H.; Sugiono, E. Continuous-flow hydration–condensation reaction: Synthesis of α,β-unsaturated ketones from alkynes and aldehydes by using a heterogeneous solid acid catalyst. Beilstein J. Org. Chem. 2011, 7, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Senger, M.R.; Fraga, C.A. M.; Dantas, R.F.; Silva-Júnior, F.P. Filtering promiscuous compounds in early drug discovery: Is it a good idea? Drug Discov. Today 2016, 21, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the utility of alerts for pan—Assay interference compounds. J. Chem. Inf. Model. 2017, 57, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Oliveira, N.; Miteva, M.A.; Villoutreix, B.O. Pan-assay interference compounds (PAINS) that may not be too painful for chemical biology projects. Drug Discov. Today 2017, 6446. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Patani, G.A.; LaVoie, E.J. Bioisosterism: A rational approach in drug design. Chem. Rev. 1996, 96, 3147–3176. [Google Scholar] [CrossRef] [PubMed]
- Burger, A. Isosterism and bioisosterism in drug design. Prog. Drug Res. 1991, 37, 287–371. [Google Scholar] [PubMed]
- Meanwell, N.A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.R.; Ertl, P.; Brown, N. Bioisosteric replacement and scaffold hopping in lead generation and optimization. Mol. Inform. 2010, 29, 366–385. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Burmaoglu, S.; Algul, O.; Aktas, D.; Gobek, A.; Gulbol, G. Bioorganic & medicinal chemistry letters synthesis and anti-proliferative activity of fluoro-substituted chalcones. Bioorg. Med. Chem. Lett. 2016, 26, 3172–3176. [Google Scholar] [PubMed]
- Nielsen, S.F.; Boesen, T.; Larsen, M.; Schønning, K.; Kromann, H. Antibacterial chalcones—Bioisosteric replacement of the 4′-hydroxy group. Bioorg. Med. Chem. 2004, 12, 3047–3054. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Cruz-Lopez, O.; Preti, D.; Tolomeo, M.; Grimaudo, S.; Cristina, A.D.; Zonta, N.; et al. Design, synthesis, and biological evaluation of thiophene analogues of chalcones. Bioorg. Med. Chem. 2008, 16, 5367–5376. [Google Scholar] [CrossRef] [PubMed]
- Fraga, C.A.M. Drug hybridization strategies: Before or after lead identification? Expert Opin. Drug Discov. 2009, 4, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreiro, E.J.; Fraga, C.A.M. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Zheng, X.; Qi, Y.; Zhang, M.; Huang, Y.; Wan, C.; Rao, G. Synthesis and biological evaluation of novel hybrid compounds between chalcone and piperazine as potential antitumor agents. RSC Adv. 2016, 6, 7723–7727. [Google Scholar] [CrossRef]
- Koziel, R.; Szczepanowska, J.; Magalska, A.; Piwocka, K.; Duszynski, J.; Zablocki, K. Ciprofloxacin inhibits proliferation and promotes generation of aneuploidy in Jurkat cells. J. Physiol. Pharmacol. 2010, 61, 233–239. [Google Scholar] [PubMed]
- Aranha, O.; Grignon, R.; Fernandes, N.; McDonnell, T.J.; Wood, D.P.; Sarkar, F.H. Suppression of human prostate cancer cell growth by ciprofloxacin is associated with cell cycle arrest and apoptosis. Int. J. Oncol. 2003, 22, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Miclau, T.; Edin, M.L.; Lester, G.E.; Lindsey, R.W.; Dahners, L.E. Effect of ciprofloxacin on the proliferation of osteoblast-like MG-63 human osteosarcoma cells in vitro. J. Orthop. Res. 1998, 16, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Somekh, E.; Douer, D.; Shaked, N.; Rubinstein, E. In vitro effects of ciprofloxacin and pefloxacin on growth of normal human hematopoietic progenitor cells and on leukemic cell lines. J. Pharmacol. Exp. Ther. 1989, 248, 415–418. [Google Scholar] [PubMed]
- Lawrence, N.J.; McGown, A.T.; Ducki, S.; Hadfield, J.A. The interaction of chalcones with tubulin. Anticancer Drug Des. 2000, 15, 135–141. [Google Scholar] [PubMed]
- Ducki, S. Antimitotic chalcones and related compounds as inhibitors of tubulin assembly. Anticancer Agents Med. Chem. 2009, 9, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Avula, S.R.; Palnati, G.R.; Singh, S.V.; Srivastava, K.; Puri, S.K.; Saxena, J.K. Synthesis and in vitro evaluation of new chloroquine-chalcone hybrids against chloroquine-resistant strain of Plasmodium falciparum. Bioorg. Med. Chem. Lett. 2012, 22, 5455–5459. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.V.; Bisht, H.; Babbarwal, V.K.; Srivastava, J.; Pandey, K.C.; Chauhan, V.S. Mechanism of malarial haem detoxification inhibition by chloroquine. Biochem. J. 2001, 355, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Coronado, L.M.; Nadovich, C.T.; Spadafora, C. Malaria hemozoin: From target to toll. Biochim. Biophys. Acta 2014, 1840, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, X.; Gong, B.; Selzer, P.M.; Li, Z.; Davidson, E.; Kurzban, G.; Miller, R.E.; Nuzum, E.O.; McKerrow, J.H.; et al. Structure-based design of parasitic protease inhibitors. Bioorg. Med. Chem. 1996, 4, 1421–1427. [Google Scholar] [CrossRef]
- Dong, X.; Du, L.; Pan, Z.; Liu, T.; Yang, B.; Hu, Y. Synthesis and biological evaluation of novel hybrid chalcone derivatives as vasorelaxant agents. Eur. J. Med. Chem. 2010, 45, 3986–3992. [Google Scholar] [CrossRef] [PubMed]
- Bellemann, P.; Ferry, D.; Lübbecke, F.; Glossman, H. [3H]-Nitrendipine, a potent calcium antagonist, binds with high affinity to cardiac membranes. Arzneimittelforschung 1981, 31, 2064–2067. [Google Scholar] [PubMed]
- Stoepel, K.; Heise, A.; Kazda, S. Pharmacological studies of the antihypertensive effect of nitrendipine. Arzneimittelforschung 1981, 31, 2056–2061. [Google Scholar] [PubMed]
- Bohn, H.; Brendel, J.; Martorana, P.A.; Schönafinger, K. Cardiovascular actions of the furoxan CAS 1609, a novel nitric oxide donor. Br. J. Pharmacol. 1995, 114, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Ferioli, R.; Folco, G.C.; Ferretti, C.; Gasco, A.M.; Medana, C.; Fruttero, R.; Civelli, M.; Gasco, A. A new class of furoxan derivatives as NO donors: Mechanism of action and biological activity. Br. J. Pharmacol. 1995, 114, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Chen, J.; Jiang, C.; Liu, T.; Hu, Y. Design, synthesis, and biological evaluation of prenylated chalcones as vasorelaxant agents. Arch. Pharm. 2009, 342, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Dodda, R.P.; Sonkar, R.; Reddy Palnati, G.; Bhatia, G. Design and synthesis of novel indole-chalcone fibrates as lipid lowering agents. Eur. J. Med. Chem. 2014, 81, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Kumar, A.; Kumar, M.; Srivastava, A.; Puri, A. Synthesis and antihyperlipidemic activity of novel coumarin bisindole derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 6504–6507. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.J.; Jardine, A.G. The use of fluvastatin in cardiovascular risk management. Expert Opin. Pharmacother. 2008, 9, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Deslypere, J.P. Clinical implications of the biopharmaceutical properties of fluvastatin. Am. J. Cardiol. 1994, 73, 12D–17D. [Google Scholar] [CrossRef]
- Santos, L.; Curi Pedrosa, R.; Correa, R.; Cechinel Filho, V.; Nunes, R.J.; Yunes, R.A. Biological evaluation of chalcones and analogues as hypolipidemic agents. Arch. Pharm. 2006, 339, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Katsiki, N.; Nikolic, D.; Montalto, G.; Banach, M.; Mikhailidis, D.P.; Rizzo, M. The role of fibrate treatment in dyslipidemia: An overview. Curr. Pharm. Des. 2013, 19, 3124–3131. [Google Scholar] [CrossRef] [PubMed]
- Jornada, D.; dos Santos Fernandes, G.; Chiba, D.; de Melo, T.; dos Santos, J.; Chung, M. The Prodrug approach: A successful tool for improving drug solubility. Molecules 2016, 21, 42. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Hachet-Haas, M.; Balabanian, K.; Rohmer, F.; Pons, F.; Franchet, C.; Lecat, S.; Chow, K.Y.C.; Dagher, R.; Gizzi, P.; Didier, B.; et al. Small neutralizing molecules to inhibit actions of the chemokine CXCL12. J. Biol. Chem. 2008, 283, 23189–23199. [Google Scholar] [CrossRef] [PubMed]
- Gasparik, V.; Daubeuf, F.; Hachet-Haas, M.; Rohmer, F.; Gizzi, P.; Haiech, J.; Galzi, J.-L.; Hibert, M.; Bonnet, D.; Frossard, N. Prodrugs of a CXC chemokine-12 (CXCL12) neutraligand prevent inflammatory reactions in an asthma model in vivo. ACS Med. Chem. Lett. 2012, 3, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Canela, M.-D.; Noppen, S.; Bueno, O.; Prota, A.E.; Bargsten, K.; Sáez-Calvo, G.; Jimeno, M.-L.; Benkheil, M.; Ribatti, D.; Velázquez, S.; et al. Antivascular and antitumor properties of the tubulin-binding chalcone TUB091. Oncotarget 2017, 8, 14325–14342. [Google Scholar] [CrossRef] [PubMed]
- Joy Macalino, S.Y.; Vijayakumar Gosu, B.; Sunhye Hong, B.; Sun Choi, B. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Roy, K. How far can virtual screening take us in drug discovery? Expert Opin. Drug Discov. 2013, 8, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.H. Impact of computational structure-based methods on drug discovery. Curr. Pharm. Des. 2014, 20, 3380–3386. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.D.; Salum, L.L.B.; Abraham, D.J. Structure-based drug design strategies in medicinal chemistry. Curr. Top. Med. Chem. 2009, 9, 771–790. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Westbrook, J.D.; Woo, J.; et al. The RCSB Protein Data Bank: Views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015, 43, D345–D356. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.-P.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Petukh, M.; Stefl, S.; Alexov, E. The role of protonation states in ligand-receptor recognition and binding. Curr. Pharm. Des. 2013, 19, 4182–4190. [Google Scholar] [CrossRef] [PubMed]
- Ten Brink, T.; Exner, T.E. pK(a) based protonation states and microspecies for protein-ligand docking. J. Comput. Aided Mol. Des. 2010, 24, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.C.; Mancera, R.L. Ligand-protein docking with water molecules. J. Chem. Inf. Model. 2008, 48, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.J.; Tidor, B. Systematic placement of structural water molecules for improved scoring of protein-ligand interactions. Protein Eng. Des. Sel. 2011, 24, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; BidonChanal, A.; Barril, X.; Luque, F.J. Protein flexibility and ligand recognition: Challenges for molecular modeling. Curr. Top. Med. Chem. 2011, 11, 192–210. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-H. Accommodating protein flexibility for structure-based drug design. Curr. Top. Med. Chem. 2011, 11, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.F.S.A.; Gesto, D.; Oliveira, E.F.; Santos-Martins, D.; Brás, N.F.; Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Receptor-based virtual screening protocol for drug discovery. Arch. Biochem. Biophys. 2015, 582, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.; dos Santos, R.; Oliva, G.; Andricopulo, A. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lill, M.A. Induced fit docking, and the use of QM/MM methods in docking. Drug Discov. Today Technol. 2013, 10, e411–e418. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual Screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Li, K.; Yu, X.; Sun, J.; Fang, H. Protein flexibility in docking-based virtual screening: Discovery of Novel lymphoid-specific tyrosine phosphatase inhibitors using multiple crystal structures. J. Chem. Inf. Model. 2015, 55, 1973–1983. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Sitzmann, M.; Pugliese, A.; Nicklaus, M.C. Software and resources for computational medicinal chemistry. Future Med. Chem. 2011, 3, 1057–1085. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhang, Y.; Xiu, Z. Rescoring ligand docking poses. Curr. Opin. Drug Discov. Dev. 2010, 13, 326–334. [Google Scholar]
- Brown, S.P.; Muchmore, S.W. High-throughput calculation of protein-ligand binding affinities: Modification and adaptation of the MM-PBSA protocol to enterprise grid computing. J. Chem. Inf. Model. 2006, 46, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, C.R.W.; Cardozo, M. MM-GB/SA rescoring of docking poses in structure-based lead optimization. J. Chem. Inf. Model. 2008, 48, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 2017, 22, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.-P.P. Modelling and enhanced molecular dynamics to steer structure-based drug discovery. Prog. Biophys. Mol. Biol. 2014, 114, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Mark, A.E.; Van Gunsteren, W.F. Parametrization of aliphatic CHn united atoms of GROMOS96 force field. J. Comput. Chem. 1998, 19, 535–547. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry. Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Pirhadi, S.; Shiri, F.; Ghasemi, J.B. Methods and applications of structure based pharmacophores in drug discovery. Curr. Top. Med. Chem. 2013, 13, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Löwer, M.; Proschak, E. Structure-based pharmacophores for virtual screening. Mol. Inform. 2011, 30, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Caporuscio, F.; Tafi, A. Pharmacophore modelling: A forty year old approach and its modern synergies. Curr. Med. Chem. 2011, 18, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Glaab, E. Building a virtual ligand screening pipeline using free software: A survey. Brief. Bioinform. 2016, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Mukherjee, P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin. Drug Discov. 2016, 11, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V.; Xiang, H.; Holliday, J.; Buscema, M.; Willett, P. Similarity coefficients for binary chemoinformatics data: Overview and extended comparison using simulated and real data sets. J. Chem. Inf. Model. 2012, 52, 2884–2901. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Schuster, D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.C.; Andrade, C.H. Assessing the performance of 3D pharmacophore models in virtual screening: How good are they? Curr. Top. Med. Chem. 2013, 13, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.J.; Dantas, R.F.; Senger, M.R.; Melo-Filho, C.C.; Valente, W.C.G.; Almeida, A.C.M.; Rezende-Neto, J.M.; Lima, E.F.C.; Paveley, R.; Furnham, N.; et al. Discovery of new anti-schistosomal hits by integration of QSAR-based virtual screening and high content screening. J. Med. Chem. 2016, 59, 7075–7088. [Google Scholar] [CrossRef] [PubMed]
- Melo-Filho, C.C.; Dantas, R.F.; Braga, R.C.; Neves, B.J.; Senger, M.R.; Valente, W.C.G.; Rezende-Neto, J.M.; Chaves, W.T.; Muratov, E.N.; Paveley, R.A.; et al. QSAR-driven discovery of novel chemical scaffolds active against schistosoma mansoni. J. Chem. Inf. Model. 2016, 56, 1357–1372. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.O. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Vapnik, V.N. The Nature of Statistical Learning Theory; Springer: New York, NY, USA, 2000. [Google Scholar]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Muratov, E.N.; Artemenko, A.G.; Varlamova, E.V.; Polischuk, P.G.; Lozitsky, V.P.; Fedchuk, A.S.; Lozitska, R.L.; Gridina, T.L.; Koroleva, L.S.; Sil’nikov, V.N.; et al. Per aspera ad astra: Application of simplex QSAR approach in antiviral research. Future Med. Chem. 2010, 2, 1205–1226. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Curation of chemogenomics data. Nat. Chem. Biol. 2015, 11, 535. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.N.; Tropsha, A. Trust, but verify II: A practical guide to chemogenomics data curation. J. Chem. Inf. Model. 2016, 56, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.J.; Muratov, E.; Machado, R.B.; Andrade, C.H.; Cravo, P.V.L. Modern approaches to accelerate discovery of new antischistosomal drugs. Expert Opin. Drug Discov. 2016, 11, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Huang, D.; Caflisch, A. Quantum mechanical methods for drug design. Curr. Top. Med. Chem. 2010, 10, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Fang, D.; Ito, S.; Okamoto, Y.; Ovchinnikov, V.; Cui, Q. QM/MM free energy simulations: Recent progress and challenges. Mol. Simul. 2016, 42, 1056–1078. [Google Scholar] [CrossRef] [PubMed]
- Kamerlin, S.C.L.; Haranczyk, M.; Warshel, A. Progress in ab initio QM/MM free-energy simulations of electrostatic energies in proteins: Accelerated QM/MM studies of pKa, redox reactions and solvation free energies. J. Phys. Chem. B 2009, 113, 1253–1272. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.C.; Andrade, C.H. QSAR and QM/MM approaches applied to drug metabolism prediction. Mini Rev. Med. Chem. 2012, 12, 573–582. [Google Scholar] [CrossRef] [PubMed]
- AlMatar, M.; AlMandeal, H.; Var, I.; Kayar, B.; Köksal, F. New drugs for the treatment of Mycobacterium tuberculosis infection. Biomed. Pharmacother. 2017, 91, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.N.; Braga, R.C.; Grzelak, E.M.; Neves, B.J.; Muratov, E.N.; Ma, R.; Klein, L.K.; Cho, S.; Oliveira, G.R.; Franzblau, S.G.; et al. QSAR-driven design, synthesis and discovery of potent and selective chalcone derivatives with antitubercular activity. Eur. J. Med. Chem. 2017, 137, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C. Mechanisms of action and resistance to tubulin-binding agents. Expert Opin. Investig. Drugs 2000, 9, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Qin, J.; Tian, C.; Yan, X.; Dong, F.; Cheng, Z.; Fida, G.; Yang, M.; Chen, H.; Gu, Y. Tubulin inhibitors: Pharmacophore modeling, virtual screening and molecular docking. Acta Pharmacol. Sin. 2014, 35, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; Davakis, S.; et al. Histone deacetylase inhibitors: An attractive therapeutic strategy against breast cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, A.J.; Horwitz, S.M. Targeting histone deacetylases in T-cell lymphoma. Leuk. Lymphoma 2017, 58, 1306–1319. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting class I histone deacetylases in a “Complex” environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, S.; Zhou, N.; Cao, Z.; Zhang, Y. A proton-shuttle reaction mechanism for histone deacetylase 8 and the catalytic role of metal ions. J. Am. Chem. Soc. 2010, 132, 9471–9479. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, M.; Chen, N.; Wang, S.; Luo, H.-B.; Zhang, Y.; Wu, R. Computational design of a time-dependent histone deacetylase 2 selective inhibitor. ACS Chem. Biol. 2015, 10, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.W.; Berman, J.D.; Davies, C.R.; Saravia, N.G. Advances in leishmaniasis. Lancet 2005, 366, 1561–1577. [Google Scholar] [CrossRef]
- Tiuman, T.S.; Santos, A.O.; Ueda-Nakamura, T.; Filho, B.P.D.; Nakamura, C.V. Recent advances in leishmaniasis treatment. Int. J. Infect. Dis. 2011, 15, e525–e532. [Google Scholar] [CrossRef] [PubMed]
- Yasinzai, M.; Khan, M.; Nadhman, A.; Shahnaz, G. Drug resistance in leishmaniasis: Current drug-delivery systems and future perspectives. Future Med. Chem. 2013, 5, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Rashid, U.; Sultana, R.; Shaheen, N.; Hassan, S.F.; Yaqoob, F.; Ahmad, M.J.; Iftikhar, F.; Sultana, N.; Asghar, S.; Yasinzai, M.; et al. Structure based medicinal chemistry-driven strategy to design substituted dihydropyrimidines as potential antileishmanial agents. Eur. J. Med. Chem. 2016, 115, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kumar, A.; Verma, S.S.; Dwivedi, N.; Singh, N.; Siddiqi, M.I.; Tripathi, R.P.; Dube, A.; Singh, N. Leishmania donovani pteridine reductase 1: Biochemical properties and structure-modeling studies. Exp. Parasitol. 2008, 120, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Sundar, S.; Singh, N. Molecular docking, structure–activity relationship and biological evaluation of the anticancer drug monastrol as a pteridine reductase inhibitor in a clinical isolate of Leishmania donovani. J. Antimicrob. Chemother. 2010, 65, 1742–1748. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.N.; Alcântara, L.M.; Neves, B.J.; Melo-Filho, C.C.; Freitas-Junior, L.H.; Moraes, C.B.; Ma, R.; Franzblau, S.G.; Muratov, E.; Andrade, C.H. Computer-aided discovery of two novel chalcone-like compounds active and selective against Leishmania infantum. Bioorg. Med. Chem. Lett. 2017, 27, 2459–2464. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Rognan, D. Chemogenomic approaches to rational drug design. Br. J. Pharmacol. 2007, 152, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Klabunde, T. Chemogenomic approaches to drug discovery: Similar receptors bind similar ligands. Br. J. Pharmacol. 2007, 152, 5–7. [Google Scholar] [CrossRef] [PubMed]
























© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, M.N.; Muratov, E.N.; Pereira, M.; Peixoto, J.C.; Rosseto, L.P.; Cravo, P.V.L.; Andrade, C.H.; Neves, B.J. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. https://doi.org/10.3390/molecules22081210
Gomes MN, Muratov EN, Pereira M, Peixoto JC, Rosseto LP, Cravo PVL, Andrade CH, Neves BJ. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules. 2017; 22(8):1210. https://doi.org/10.3390/molecules22081210
Chicago/Turabian StyleGomes, Marcelo N., Eugene N. Muratov, Maristela Pereira, Josana C. Peixoto, Lucimar P. Rosseto, Pedro V. L. Cravo, Carolina H. Andrade, and Bruno J. Neves. 2017. "Chalcone Derivatives: Promising Starting Points for Drug Design" Molecules 22, no. 8: 1210. https://doi.org/10.3390/molecules22081210
APA StyleGomes, M. N., Muratov, E. N., Pereira, M., Peixoto, J. C., Rosseto, L. P., Cravo, P. V. L., Andrade, C. H., & Neves, B. J. (2017). Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules, 22(8), 1210. https://doi.org/10.3390/molecules22081210