Assessment of Enzyme Inhibition: A Review with Examples from the Development of Monoamine Oxidase and Cholinesterase Inhibitory Drugs


Abstract
1. Introduction
2. Inhibition of Neurotransmitter Breakdown in Neurodegenerative Disease
2.1. Monoamine Oxidases (MAO A and MAO B) as Drug Targets
2.2. Inhibitors of Monoamine Oxidases (MAO A and MAO B)
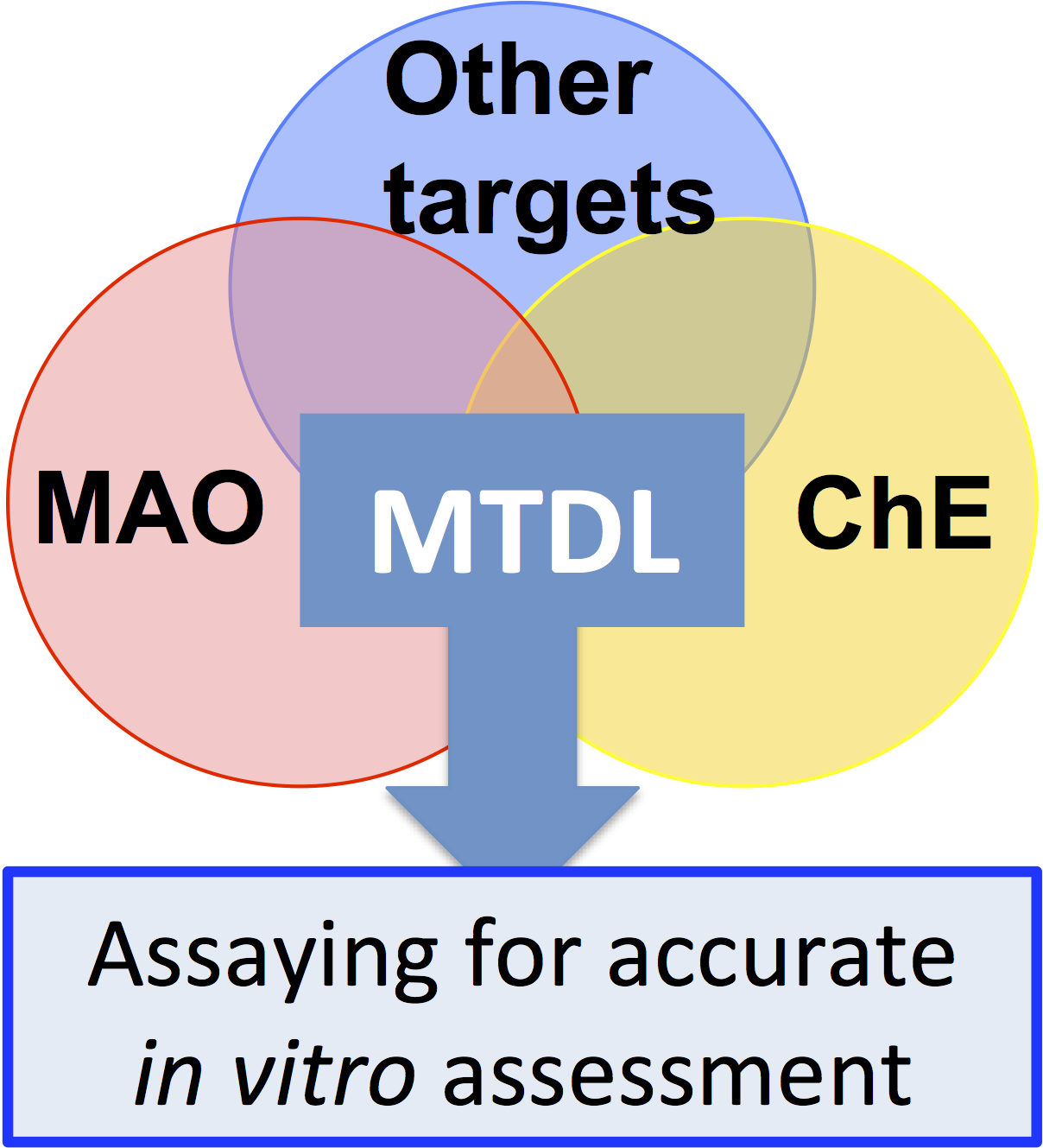
2.2.1. Irreversible Inhibitors
Hydrazines
Cyclopropylamines
Propargylamines
2.2.2. Reversible Inhibitors
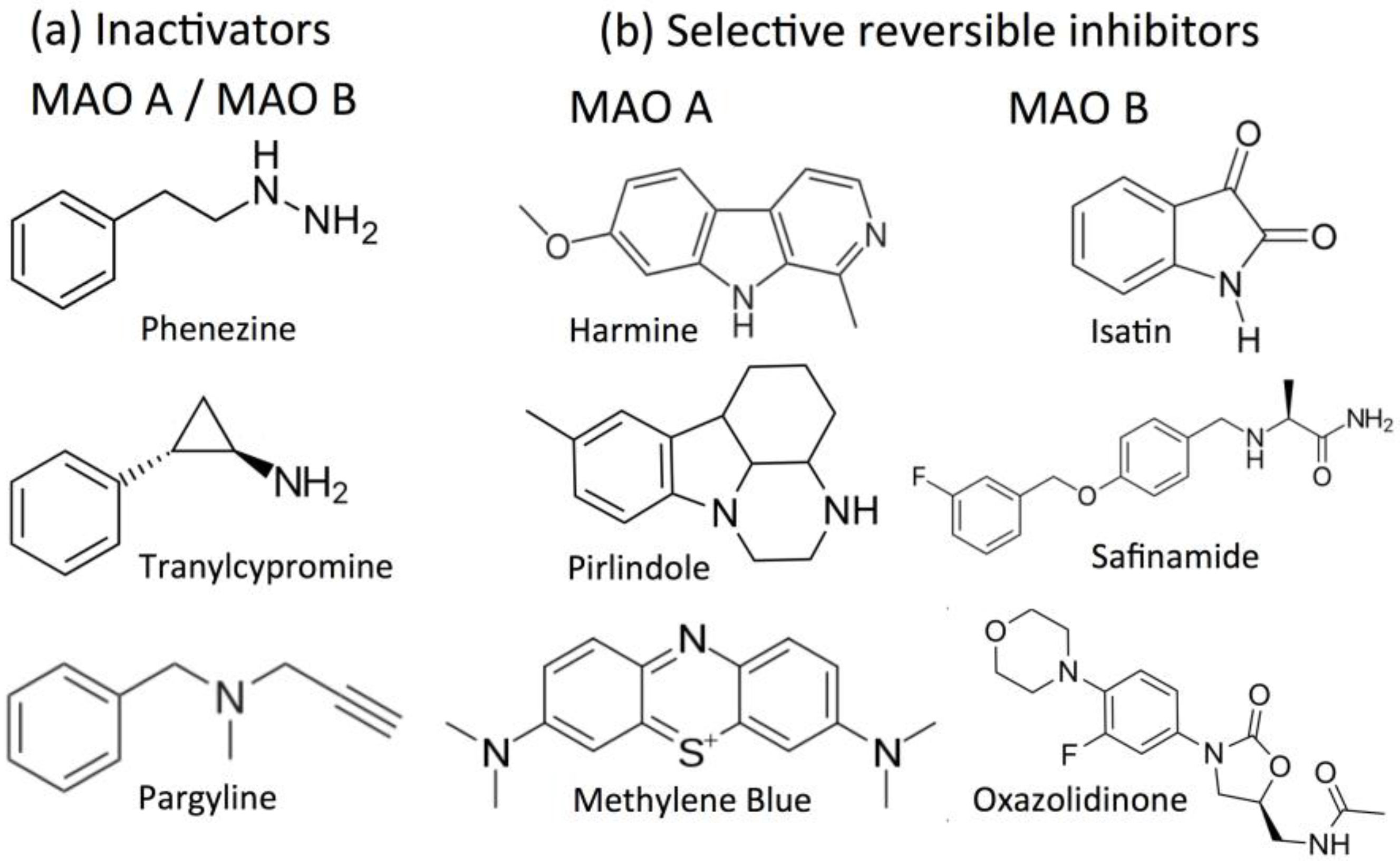
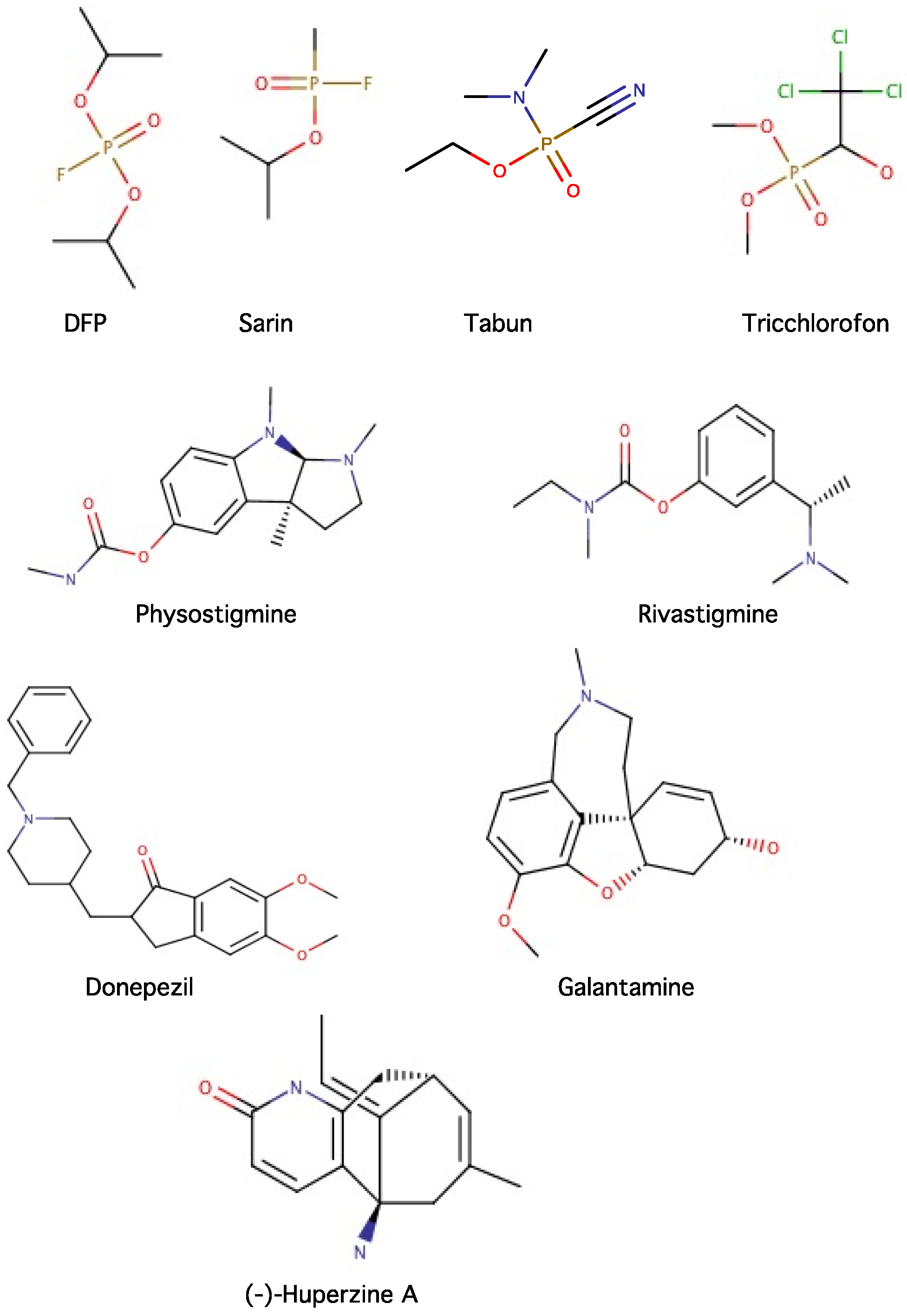
2.3. Acetylcholinesterase (AChE) and Butyrylcholinesterase (BChE) as Drug Targets
2.4. Inhibitors of Acetylcholinesterase (AChE) and Butyrylcholinesterase (BChE)
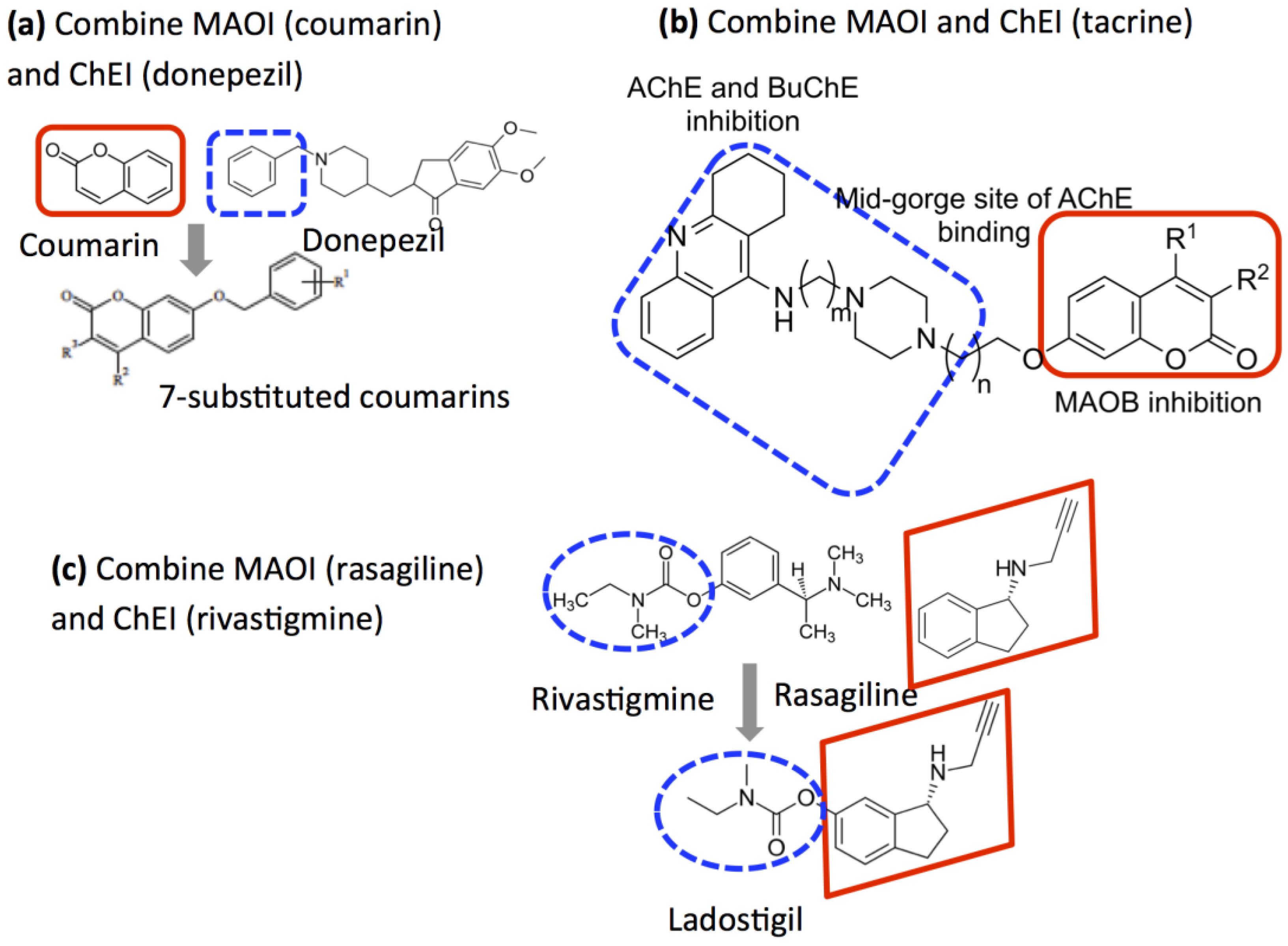
2.5. Design of Compounds that Inhibit Both MAO and ChE
3. Measurement of Enzyme Activity
3.1. Assay Procedures
- Continuous assays that monitor changes in reactant concentrations in real time.
- (a)
- Direct, in which the decrease in substrate or increase in product is measured, e.g., the spectrophotometric determination of benzaldehyde production from benzylamine, or the use of the oxygen electrode in MAO assays.
- (b)
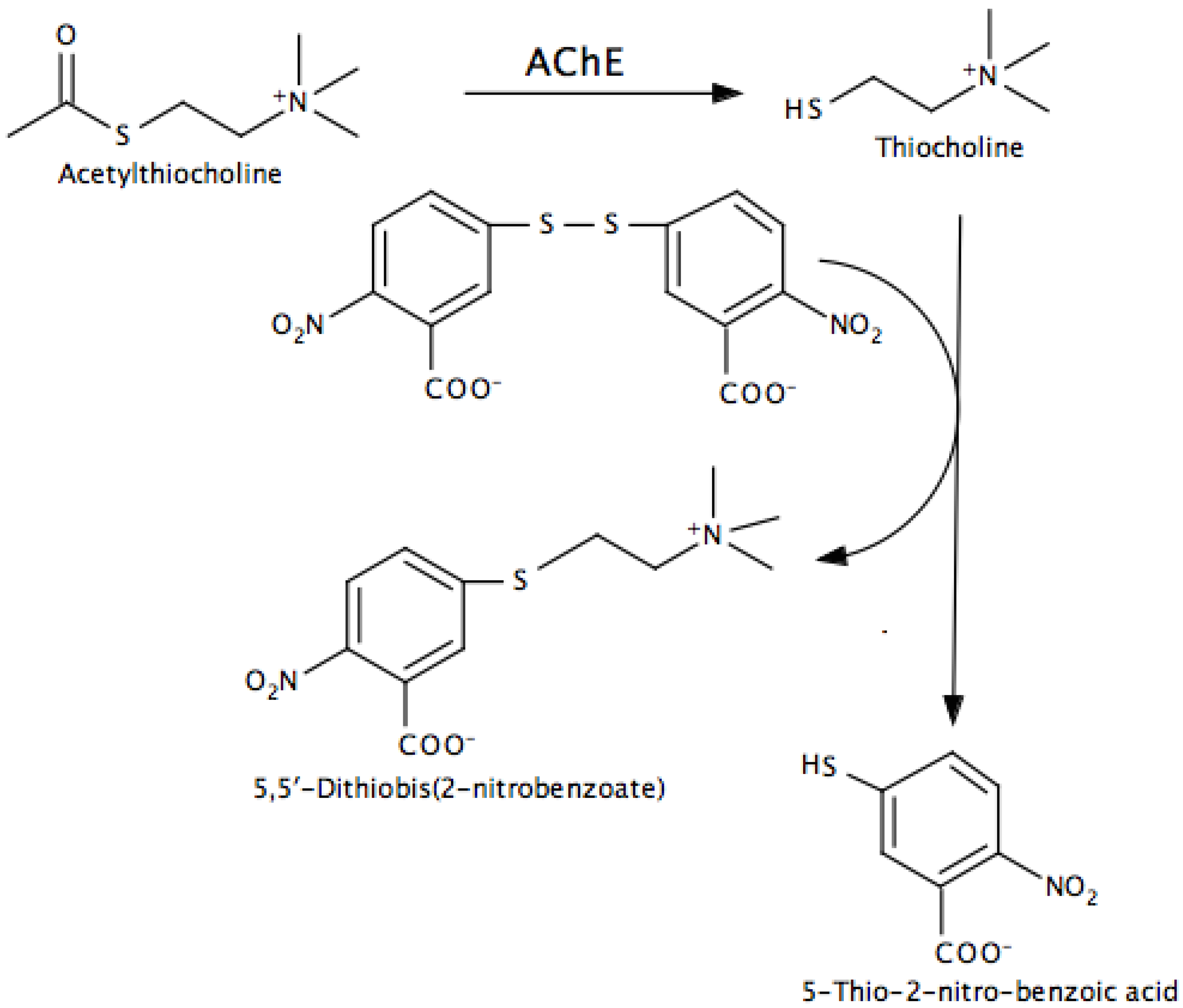
- Indirect, in which additional reactions are used to convert a product into something that can be easily monitored. The Ellman assay for AChE and peroxidase-coupled assays for MAO and AChE are examples.
- Discontinuous (sampling) assays. These involve stopping the reaction after fixed time(s) before separating the product for quantification. Radiochemical assays and those based on HPLC for AChE and MAO fall into this class.
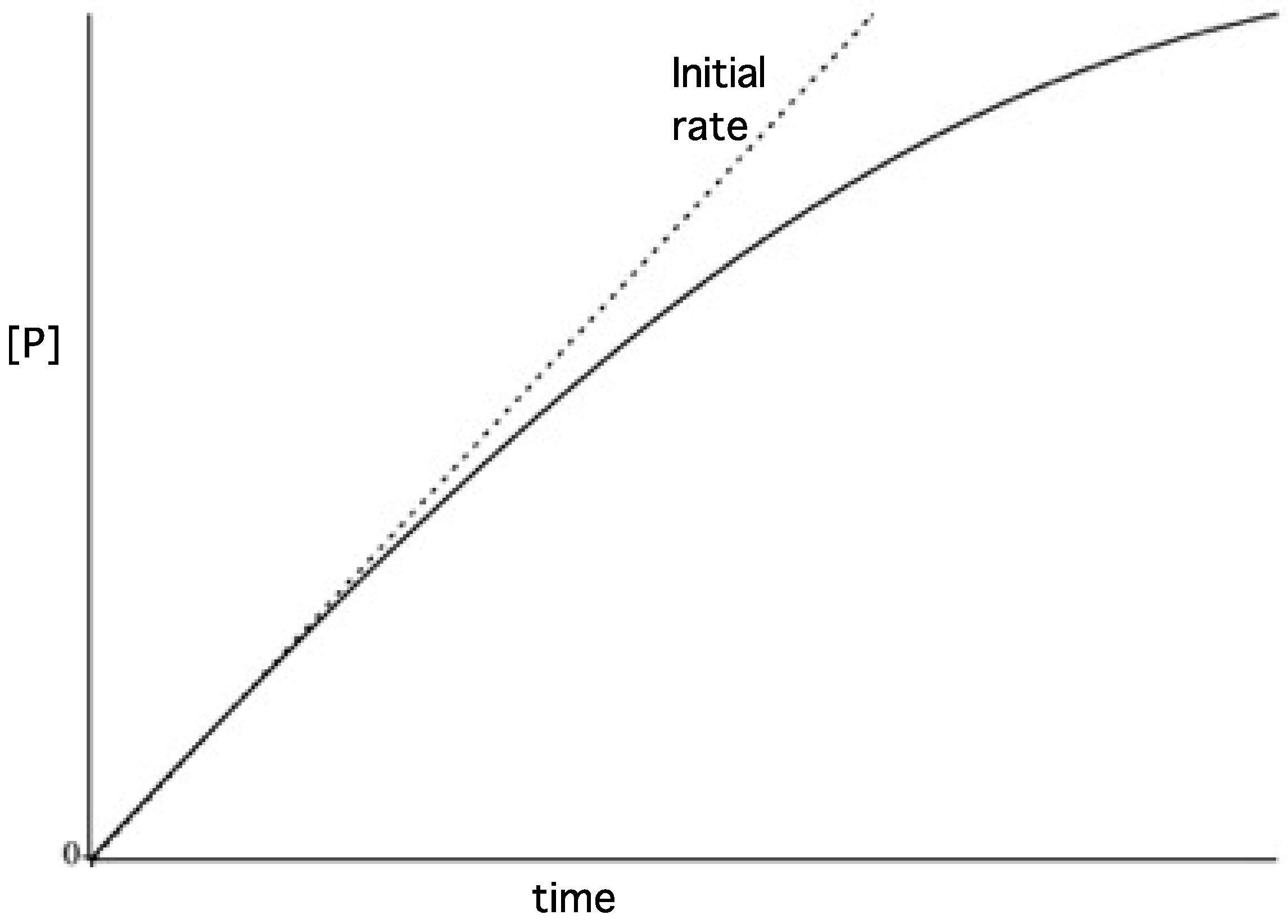
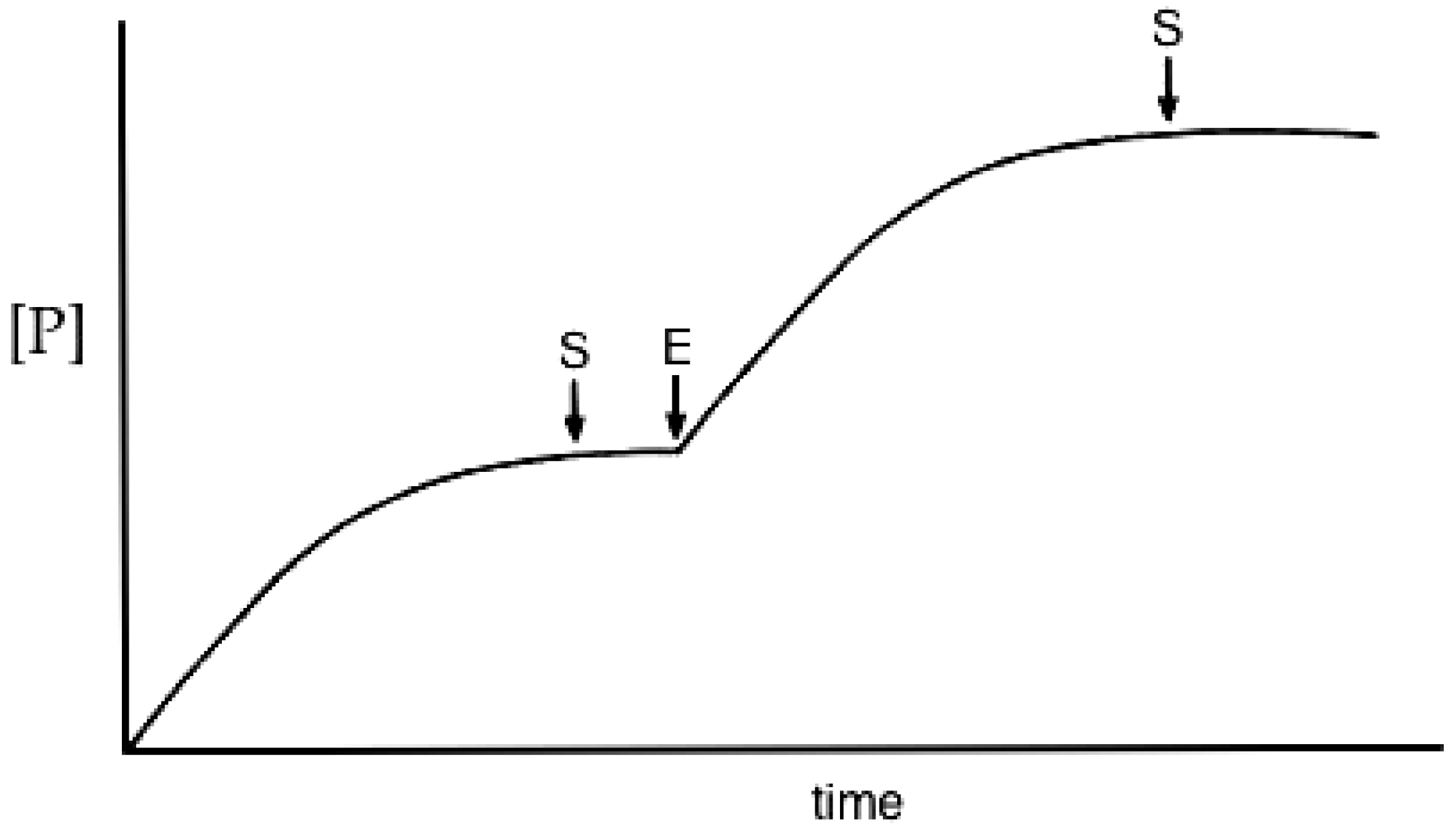
3.2. Reaction Progress Curves
- Substrate depletion: The reaction may be slowing down because the substrate is being used up. As the substrate concentration falls the enzyme will become less and less saturated and the velocity will fall, tending to zero when all the substrate is used.
- Approaching equilibrium: A reversible reaction may be slowing down because it is approaching equilibrium, where the rate of the backward reaction (converting product to substrate) will increase until, at equilibrium, it is equal to the rate of the forward (substrate to product) reaction and no net rate will be observed.
- Product inhibition: Products of enzyme-catalysed reactions are frequently reversible inhibitors and their accumulation can result in a decreasing reaction rate.
- Instability: One of the components of the assay system may be unstable, losing activity or breaking down during the assay. This may be the enzyme itself or one of the substrates.
- Time-dependent inhibition: Some enzyme substrates are also time-dependent irreversible inhibitors, sometimes referred to as ‘suicide-substrates’ (see Section 3.8 for discussion).
- Assay method artifacts: If the specific detection procedure used ceases to respond linearly to increasing product concentrations, this can lead to a decline in the measured rate of the reaction with time (see [132]).
- Change in assay conditions: If the assay conditions are not constant the rate of product formation might be expected to change. If, for example, the reaction involves the formation or consumption of hydrogen ions, the pH of the reaction mixture may change during the reaction, unless it is adequately buffered. If this resulted in a change of pH away from the optimum pH of the reaction this could lead to a decrease in the rate of the reaction.
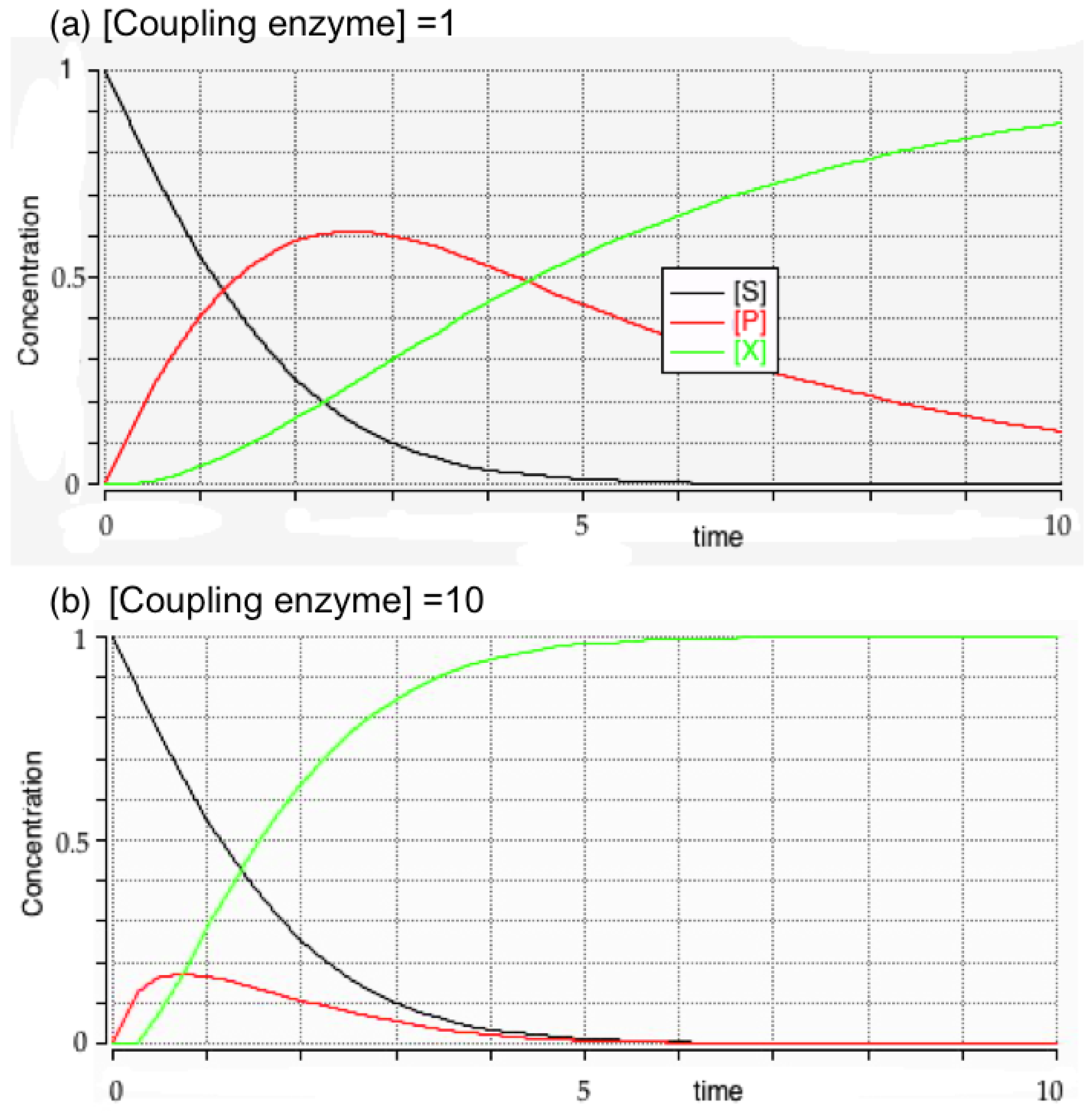
3.3. Initial Rates and Coupled Assays
3.4. Expression of Enzyme Activity
3.5. Inhibition
3.5.1. Competitive Inhibitors
3.5.2. Uncompetitive Inhibitors
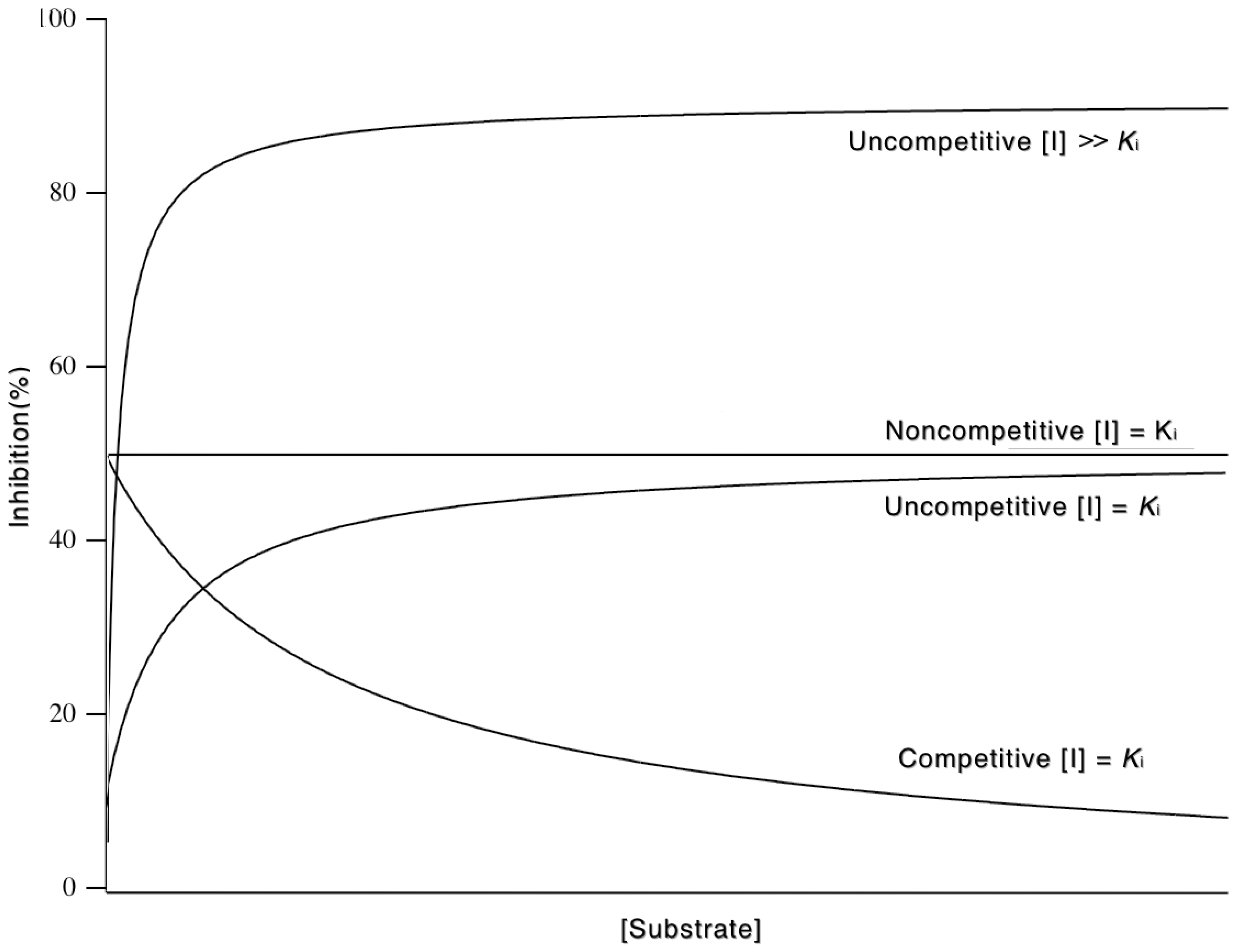
3.5.3. Mixed and Noncompetitive Inhibition
3.6. More Complex Reversible Inhibitor Behavior
3.6.1. Partial Inhibition
3.6.2. Tight-Binding Inhibitors
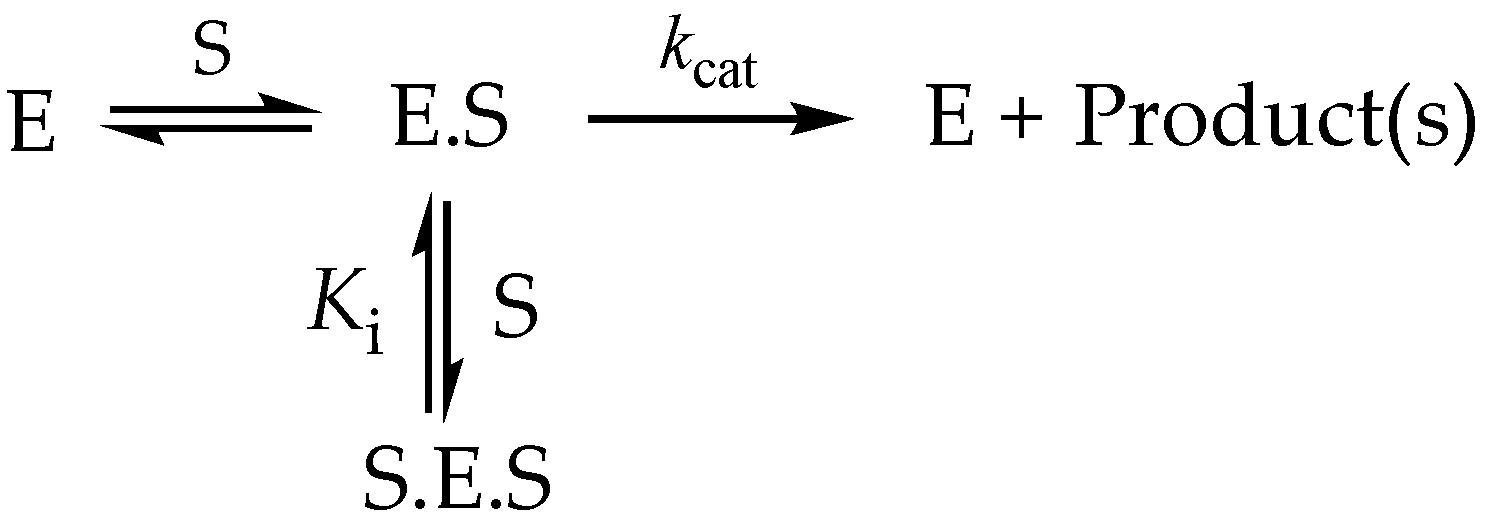
3.6.3. High-Substrate Inhibition
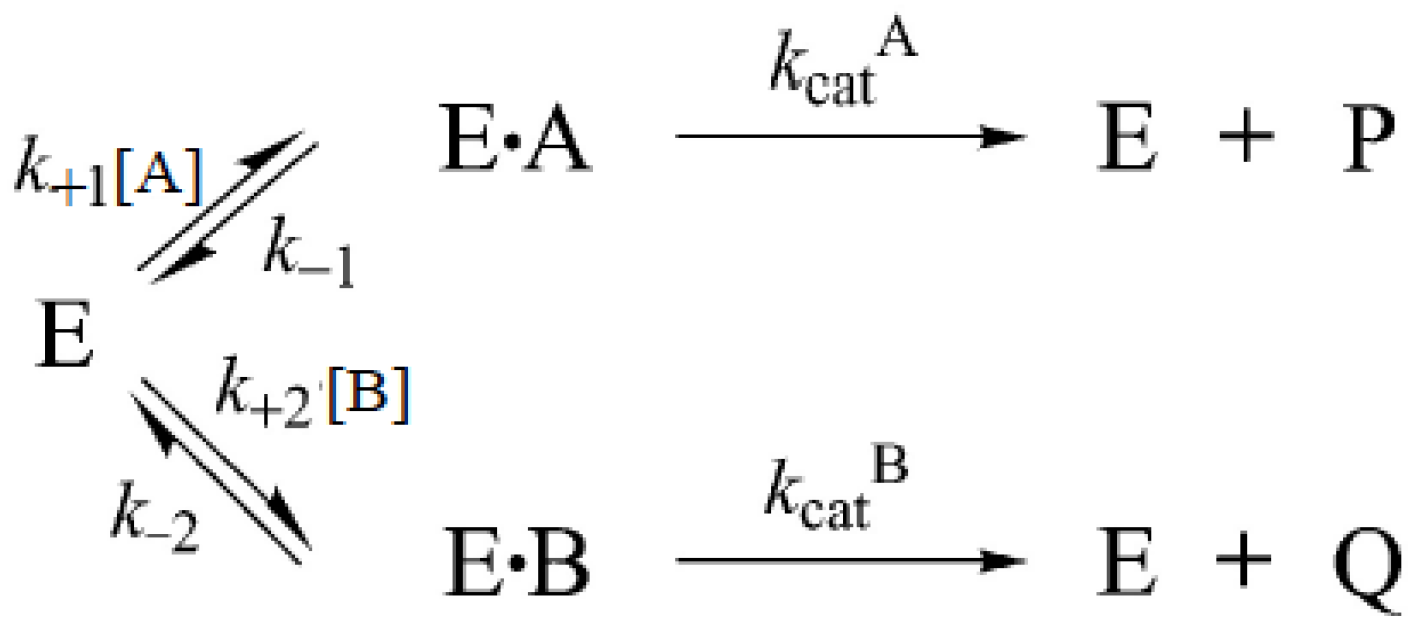
3.6.4. Reactions Involving More than One Substrate
3.7. Competition Between Substrates
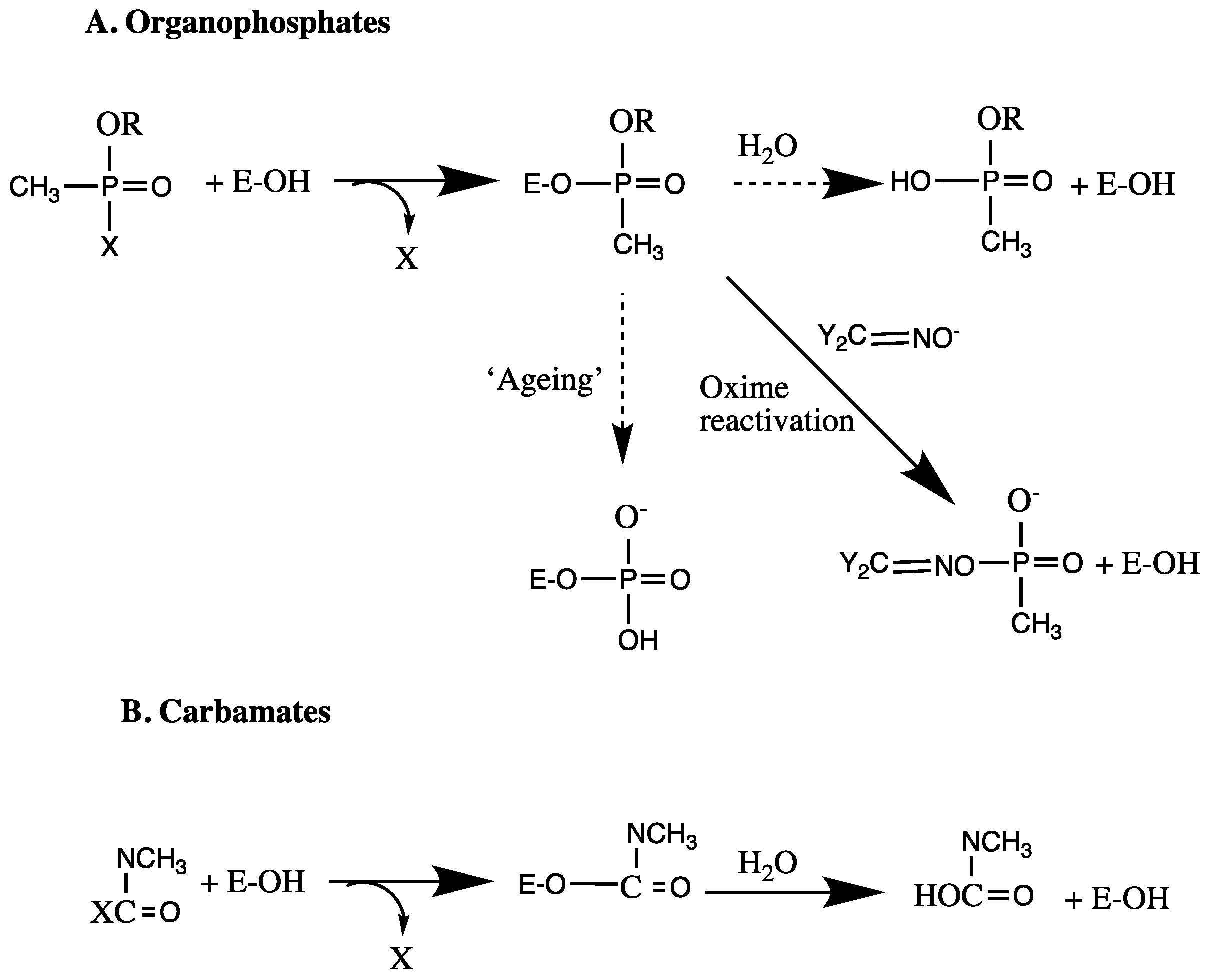
3.8. Irreversible Inhibition
3.9. Irreversible Inhibitors as Substrates
3.10. IC50 Values
4. MAO Assays
4.1. Direct Assays for MAO Activity
4.2. Coupled Assays for MAO Activity
4.3. Controls for Coupled Fluorescence Assays of MAO Activity
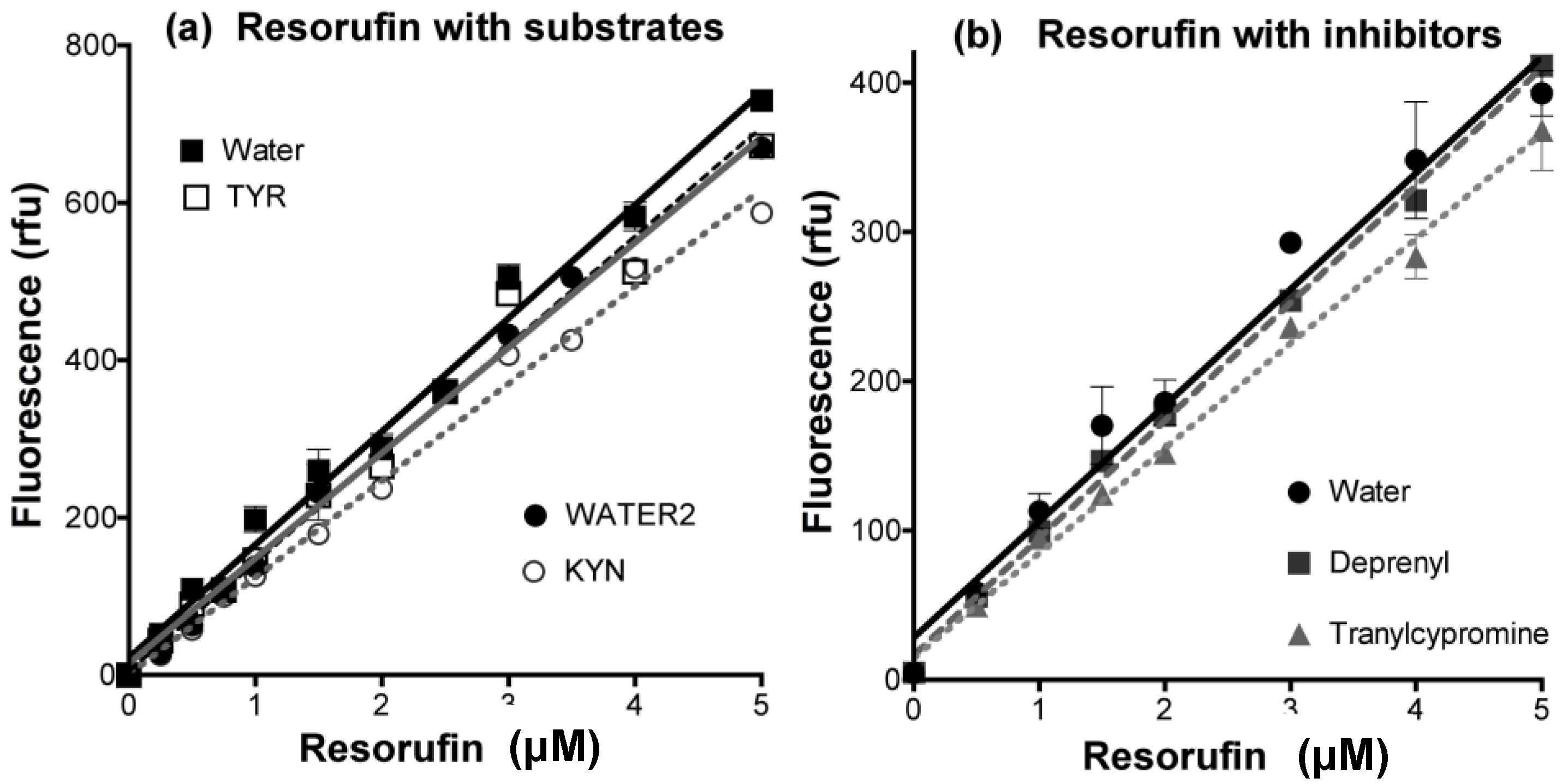
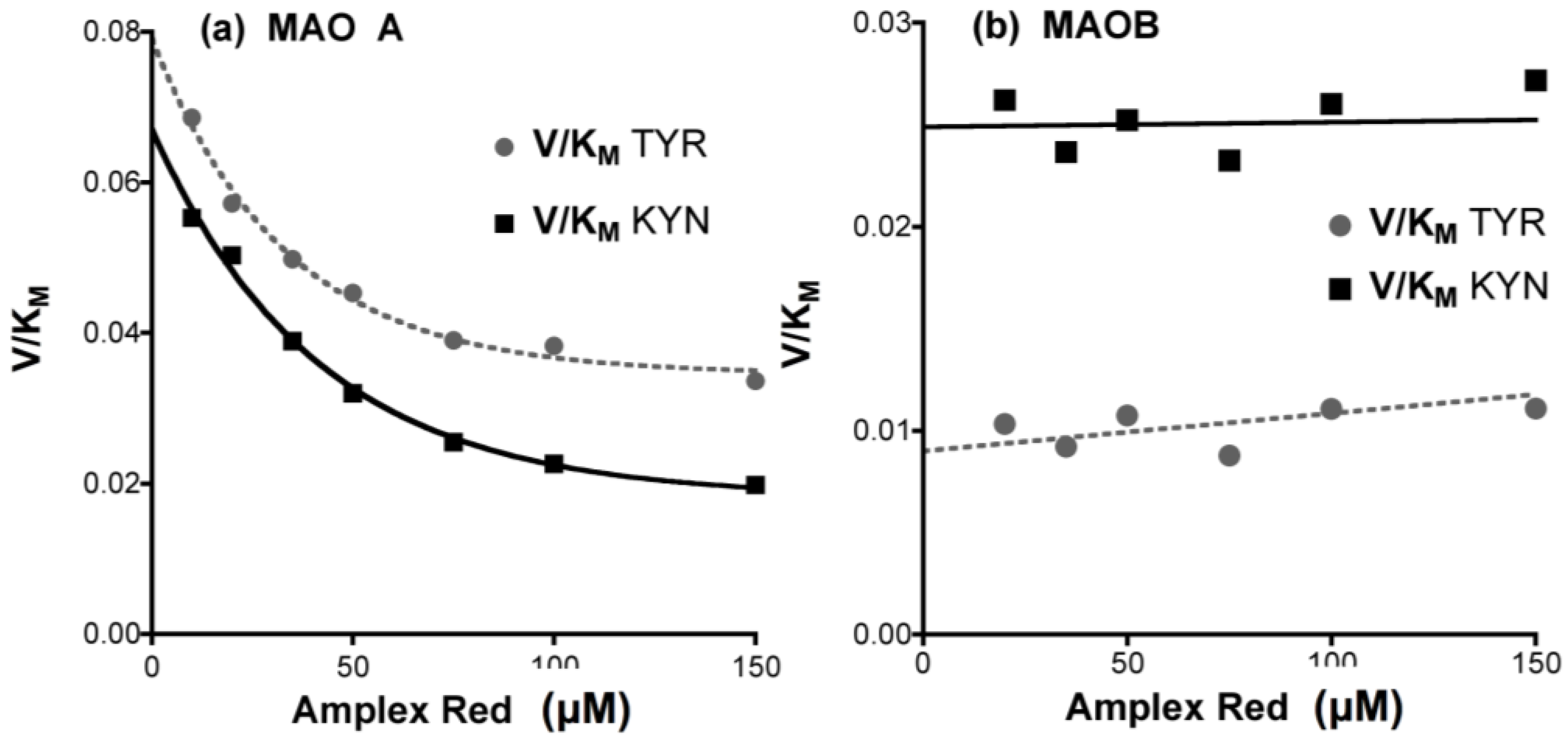
- Determine any effect of the new compounds on resorufin fluorescence. Measure the fluorescence of fresh solutions of resorufin at 0–5 μM (excitation at 535 nm, emission at 595 nm) in the absence (solvent alone) or presence of any chemical compound at the highest concentration to be tested. Quenching of fluorescence by amines and inhibitors of MAO is frequently observed, even (slightly) for routine substrates or inhibitors (Figure 12).
- Check that the new compounds do not inhibit the coupling enzyme, horseradish peroxidase (HRP). The assay mixture in a final volume of 200 μL 50 mM potassium phosphate buffer (pH 7.4) should contain: H2O2 (50 μM), Amplex Red (200 μM), HRP (0.02 U/mL) and a single compound of interest or its solvent as the control. The HRP activity is determined by the fluorescence of the resorufin formed with time at 30 °C [176].
- Check the linearity of product generation with enzyme and time, and verify that less than 10% of substrate is consumed.
- When comparing two enzymes, determine the Km for substrate to ensure that the substrate concentrations used in the inhibitor screen gives similar enzyme saturation for each enzyme.
- If the inhibition increases with time, check whether it is reversible (and, for a full investigation, determine the mechanism).
- If the inhibition is irreversible, the IC50 value will depend on time. This should be checked by preincubating the enzyme and inhibitor for various times before the addition of substrate to measure the activity remaining.
- Some irreversible inhibitors of MAO are substrates with high partition ratios, so can generate H2O2 in the absence of the normal substrate (for example, phenelzine [177]). This further detracts from the meaning of IC50 results.
4.4. Optimized Conditions for MAO Inhibition Screening Using the Amplex Red Coupled Assay
4.5. Species Differences in Inhibition of MAO
5. Determination of Acetylcholinesterase Activity
5.1. Radiochemical Assays
5.2. Hydrogen Ion Liberation
5.3. Assays Based on Artificial Substrates
5.4. Enzyme Coupled Assays
6. Virtual Screening
7. Inhibition for Effective Drugs
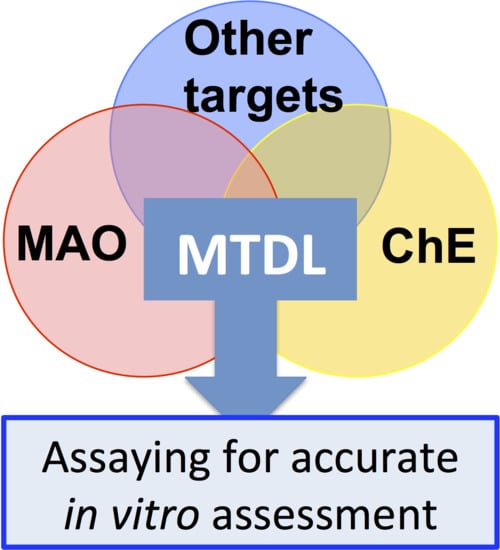
7.1. Inhibiting Multiple Enzyme Targets
7.2. Adding Value to MTDL
8. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ACh | acetylcholine |
| AChE | acetylcholinesterase |
| ASDIN | active-site-directed inhibitor |
| BChE | butyrylcholinesterase |
| AChEIs | acetylcholinesterase inhibitor |
| AD | Alzheimer’s disease |
| ChE | cholinesterase |
| COMT | catechol-O-methyltransferase |
| DA | dopamine |
| DFP | diisopropylfluoro phosphate (diisopropylphosphofluoridate) |
| L-DOPA | L-3,4-dihydroxyphenylalanine (dopamine precursor) |
| DTNB | 5,5’-dithio-bis-[2-nitrobenzoic acid] |
| FAD | flavin adenine dinucleotide |
| 5-HT | 5-hydroxytryptamine (serotonin) |
| MAO | monoamine oxidase |
| MAOI | monoamine oxidase inhibitor |
| MTDL | multitarget-directed ligand |
| NTE | neuropathy-target esterase |
| OP | organophosphate |
| PAS | peripheral anionic site |
| PD | Parkinson’s disease |
| PS | presenilin |
| QSAR | quantitative structure-activity relationship |
| TNB | 2-nitro-5-thiobenzoate |
References
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Biava, M.; Poce, G. COX inhibitors: A patent review (2011–2014). Expert Opin. Ther. Pat. 2015, 25, 1357–1371. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.L.; Chen, S.Y.; Chew, E.H.; Chui, W.K. Applying the designed multiple ligands approach to inhibit dihydrofolate reductase and thioredoxin reductase for antiproliferative activity. Eur. J. Med. Chem. 2016, 115, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, K.; Mavridis, L.; Djikic, T.; Vucicevic, J.; Agbaba, D.; Yelekci, K.; Mitchell, J.B.O. Drug design for CNS diseases: Polypharmacological profiling of compounds using cheminformatic, 3D-QSAR and virtual screening methodologies. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.E.; Nikolic, K.; Ramsay, R.R. One for all? hitting multiple Alzheimer’s disease targets with one drug. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, K.; Mavridis, L.; Bautista-Aguilera, O.M.; Marco-Contelles, J.; Stark, H.; Carreiras, M.D.; Rossi, I.; Massarelli, P.; Agbaba, D.; Ramsay, R.R.; et al. Predicting targets of compounds against neurological diseases using cheminformatic methodology. J. Comput.-Aided Mol. Des. 2015, 29, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Ramos, K.; Moreno-Castilla, P.; Castro-Cruz, M.; McGaugh, J.L.; Martinez-Coria, H.; LaFerla, F.M.; Bermudez-Rattoni, F. Restoration of dopamine release deficits during object recognition memory acquisition attenuates cognitive impairment in a triple transgenic mice model of Alzheimer’s disease. Learn. Mem. 2012, 19, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Yogev-Falach, M.; Youdim, M.B.H. The neuroprotective mechanism of action of the multimodal drug ladostigil. Front. Biosci.-Landmark 2008, 13, 5131–5137. [Google Scholar] [CrossRef][Green Version]
- Bar-Am, O.; Weinreb, O.; Amit, T.; Youdim, M.B.H. The novel cholinesterase-monoamine oxidase inhibitor and antioxidant, ladostigil, confers neuroprotection in neuroblastoma cells and aged rats. J. Mol. Neurosci. 2009, 37, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Bolea, I.; Juarez-Jimenez, J.; de los Rios, C.; Chioua, M.; Pouplana, R.; Javier Luque, F.; Unzeta, M.; Marco-Contelles, J.; Samadi, A. Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2011, 54, 8251–8270. [Google Scholar] [PubMed]
- Marco-Contelles, J.; Unzeta, M.; Bolea, I.; Esteban, G.; Ramsay, R.R.; Romero, A.; Martnez-Murillo, R.; Carreiras, M.C.; Ismaili, L. ASS234, As a new multi-target directed propargylamine for Alzheimer’s disease therapy. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.-S.; Wang, X.; Jiang, N.; Yu, W.; Wang, K.D.G.; Lan, J.-S.; Li, Z.-R.; Kong, L.-Y. Multi-target tacrine-coumarin hybrids: Cholinesterase and monoamine oxidase B inhibition properties against Alzheimer’s disease. Eur. J. Med. Chem. 2015, 95, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Farina, R.; Pisani, L.; Catto, M.; Nicolotti, O.; Gadaleta, D.; Denora, N.; Soto-Otero, R.; Mendez-Alvarez, E.; Passos, C.S.; Muncipinto, G.; et al. Structure-based design and optimization of multitarget-directed 2h-chromen-2-one derivatives as potent inhibitors of monoamine oxidase b and cholinesterases. J. Med. Chem. 2015, 58, 5561–5578. [Google Scholar] [CrossRef] [PubMed]
- Joubert, J.; Foka, G.B.; Repsold, B.P.; Oliver, D.W.; Kapp, E.; Malan, S.F. Synthesis and evaluation of 7-substituted coumarin derivatives as multimodal monoamine oxidase-B and cholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 125, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Westlund, K.N.; Denney, R.M.; Kochersperger, L.M.; Rose, R.M.; Abell, C.W. Distinct monoamine oxidase-A and oxidase-B populations in primate brain. Science 1985, 230, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Saura, J.; Kettler, R.; Daprada, M.; Richards, J.G. Quantitative enzyme autoradiography with H-3 Ro 41–1049 and H-3 Ro 19–6327 invitro—Localization and abundance of MAO-A and MAO-B in Rat CNS, peripheral organs, and human brain. J. Neurosci. 1992, 12, 1977–1999. [Google Scholar] [PubMed]
- Saura, J.; Bleuel, Z.; Ulrich, J.; Mendelowitsch, A.; Chen, K.; Shih, J.C.; Malherbe, P.; DaPrada, M.; Richards, J.G. Molecular neuroanatomy of human monoamine oxidases A and B revealed by quantitative enzyme radioautography and in situ hybridization histochemistry. Neuroscience 1996, 70, 755–774. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Binda, C.; Mattevi, A. Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Arch. Biochem. Biophys. 2007, 464, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.K.; Ramsay, R.R. Substrate-specific enhancement of the oxidative half-reaction of Monoamine-Oxidase. Biochemistry 1993, 32, 2137–2143. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Newton-Vinson, P.; Hubalek, F.; Edmondson, D.E.; Mattevi, A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat. Struct. Biol. 2002, 9, 22–26. [Google Scholar] [CrossRef] [PubMed]
- De Colibus, L.; Binda, C.; Edmondson, D.E.; Mattevi, A. 2BXR: Human monoamine oxidase A in complex with clorgyline, crystal form A. Protein Data Bank 2005. [Google Scholar] [CrossRef]
- Ma, J.C.; Yoshimura, M.; Yamashita, E.; Nakagawa, A.; Ito, A.; Tsukihara, T. Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. J. Mol. Biol. 2004, 338, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.H.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Tan, A.K.; Weyler, W. Kinetic-properties of cloned human liver monoamine-oxidase-A. J. Neural Transm.-Suppl. 1994, 41, 17–26. [Google Scholar] [PubMed]
- Fowler, C.J.; Oreland, L. The nature of the substrate-selective interaction between rat-liver mitochondrial monoamine-oxidase and oxygen. Biochem. Pharmacol. 1980, 29, 2225–2233. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Olivieri, A.; Holt, A. An improved approach to steady-state analysis of monoamine oxidases. J. Neural Transm. 2011, 118, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, F.; Maccioni, E.; Secci, D.; Bolasco, A.; Chimenti, P.; Granese, A.; Carradori, S.; Alcaro, S.; Ortuso, F.; Yanez, M.; et al. Synthesis, stereochemical identification, and selective inhibitory activity against human monoamine oxidase-B of 2-methylcyclohexylidene-(4-arylthiazol-2-yl)hydrazones. J. Med. Chem. 2008, 51, 4874–4880. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.S.; Volkow, N.D.; Logan, J.; Wang, G.J.; Macgregor, R.R.; Schlyer, D.; Wolf, A.P.; Pappas, N.; Alexoff, D.; Shea, C.; et al. Slow recovery of human brain MAO-B after L-Deprenyl (selegeline) withdrawal. Synapse 1994, 18, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.S.; Logan, J.; Volkow, N.D.; Shumay, E.; McCall-Perez, F.; Jayne, M.; Wang, G.-J.; Alexoff, D.L.; Apelskog-Torees, K.; Hubbard, B.; et al. Evidence that formulations of the selective MAO-B inhibitor, selegiline, which bypass irst-pass metabolism, also inhibit MAO-A in the human brain. Neuropsychopharmacology 2015, 40, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, S.; Rascol, O. The safety and efficacy of safinamide mesylate for the treatment of Parkinson’s disease. Expert Rev. Neurother. 2016, 16, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Rojas, R.J.; Edmondson, D.E.; Almos, T.; Scott, R.; Massari, M.E. Reversible and irreversible small molecule inhibitors of monoamine oxidase B (MAO-B) investigated by biophysical techniques. Bioorg. Med. Chem. 2015, 23, 770–778. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.R.; Olivieri, A.; Ramsay, R.R.; Holt, A. On the formation and nature of the imidazoline I (2) binding site on human monoamine oxidase-B. Pharmacol. Res. 2010, 62, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanni, G.; Svob Strac, D.; Sole, M.; Unzeta, M.; Tipton, K.F.; Mück-Šeler, D.; Bolea, I.; Della Corte, L.; Nikolac Perkovic, M.; Pivac, N.; et al. Monoaminergic and histaminergic strategies and treatments in brain diseases. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, M.; Shih, J.C. Behavioral outcomes of monoamine oxidase deficiency: Preclinical and clinical evidence. Int. Rev. Neurobiol. 2011, 100, 13–42. [Google Scholar] [PubMed]
- Meyer, J.H.; Ginovart, N.; Boovariwala, A.; Sagrati, S.; Hussey, D.; Garcia, A.; Young, T.; Praschak-Rieder, N.; Wilson, A.A.; Houle, S. Elevated monoamine oxidase A levels in the brain—An explanation for the monoamine imbalance of major depression. Arch. Gen. Psychiatry 2006, 63, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.S.; Logan, J.; Shumay, E.; Alia-Klein, N.; Wang, G.-J.; Volkow, N.D. Monoamine oxidase: Radiotracer chemistry and human studies. J. Label. Compd. Radiopharm. 2015, 58, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Seidler, F.J.; Ritchie, J.C. Effects of aging and glucocorticoid treatment on monoamine oxidase subtypes in rat cerebral cortex: Therapeutic implications. Brain Res. Bull. 1998, 47, 345–348. [Google Scholar] [CrossRef]
- Finberg, J.P.M. Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: Focus on modulation of CNS monoamine neurotransmitter release. Pharmacol. Ther. 2014, 143, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Scott, A.L.; Ladenheim, B.; Chen, K.; Ouyang, X.; Lathia, J.D.; Mughal, M.; Cadet, J.L.; Mattson, M.P.; Shih, J.C. Monoamine oxidases regulate telencephalic neural progenitors in late embryonic and early postnatal development. J. Neurosci. 2010, 30, 10752–10762. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Borchert, A.; Ugun-Klusek, A.; Tang, L.Y.; Lui, W.T.; Chu, C.Y.; Billett, E.; Kuhn, H.; Ufer, C. Monoamine oxidase a expression is vital for embryonic brain development by modulating developmental apoptosis. J. Biol. Chem. 2011, 286, 28322–28330. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.C.; Ufer, C.; Billett, E.E. A link between monoamine oxidase-A and apoptosis in serum deprived human SH-SY5Y neuroblastoma cells. J. Neural Transm. 2007, 114, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.-M.; Chen, K.; Shih, J.C. Monoarnine oxidase A and repressor R1 are involved in apoptotic signaling pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 10923–10928. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Akao, Y.; Maruyama, W.; Chen, K.; Shih, J.; Naoi, M. Type A monoamine oxidase is the target of an endogenous dopaminergic neurotoxin, N-methyl(R)salsolinol, leading to apoptosis in SH-SY5Y cells. J. Neurochem. 2006, 96, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Pennington, P.R.; Wei, Z.; Rui, L.; Doig, J.A.; Graham, B.; Kuski, K.; Gabriel, G.G.; Mousseau, D.D. Alzheimer disease-related presenilin-1 variants exert distinct effects on monoamine oxidase—A activity in vitro. J. Neural Transm. 2011, 118, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Gabriel, G.G.; Rui, L.; Cao, X.; Pennington, P.R.; Chlan-Fourney, J.; Nazarali, A.J.; Baker, G.B.; Mousseau, D.D. Monoamine oxidase—A physically interacts with presenilin-1(m146v) in the mouse cortex. J. Alzheimers Dis. 2012, 28, 403–422. [Google Scholar] [PubMed]
- Simic, G.; Babic Leko, M.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buee, L.; de Silva, R.; Di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017. [Google Scholar] [CrossRef]
- De Deurwaerdere, P.; Di Giovanni, G. Serotonergic modulation of the activity of mesencephalic dopaminergic systems: Therapeutic implications. Prog. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.M.; Brambilla, R.; et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B.H. Ladostigil: A novel multimodal neuroprotective drug with cholinesterase and brain-selective Monoamine Oxidase inhibitory activities for Alzheimer’s Disease treatment. Curr. Drug Targets 2012, 13, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Wang, J.; Li, M.; Hubalek, F.; Mattevi, A.; Edmondson, D.E. Structural and mechanistic studies of arylalkylhydrazine inhibition of human monoamine oxidases A and B. Biochemistry 2008, 47, 5616–5625. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.H.; Tipton, K.F. Deuterium-isotope effect of phenelzine on the inhibition of rat-liver mitochondrial monoamine-oxidase Activity. Biochem. Pharmacol. 1989, 38, 4245–4251. [Google Scholar] [CrossRef]
- Finberg, J.P.M.; Gillman, K. Selective inhibitors of monoamine oxidase type B and the “cheese effect”. Int. Rev. Neurobiol. 2011, 100, 169–190. [Google Scholar] [PubMed]
- Silverman, R.B. Radical ideas about monoamine-oxidase. Acc. Chem. Res. 1995, 28, 335–342. [Google Scholar] [CrossRef]
- Vintem, A.P.B.; Price, N.T.; Silverman, R.B.; Ramsay, R.R. Mutation of surface cysteine 374 to alanine in monoamine oxidase A alters substrate turnover and inactivation by cyclopropylamines. Bioorg. Med. Chem. 2005, 13, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Malcomson, T.; Yelekci, K.; Borrello, M.T.; Ganesan, A.; Semina, E.; De Kimpe, N.; Mangelinckx, S.; Ramsay, R.R. Cis-cyclopropylamines as mechanism-based inhibitors of monoamine oxidases. FEBS J. 2015, 282, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Valente, S.; Romanenghi, M.; Pilotto, S.; Cirilli, R.; Karytinos, A.; Ciossani, G.; Botrugno, O.A.; Forneris, F.; Tardugno, M.; et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 2010, 132, 6827–6833. [Google Scholar] [CrossRef] [PubMed]
- Shepard, E.M.; Heggem, H.; Juda, G.A.; Dooley, D.M. Inhibition of six copper-containing amine oxidases by the antidepressant drug tranylcypromine. BBA-Proteins Proteom. 2003, 1647, 252–259. [Google Scholar] [CrossRef]
- Polasek, T.M.; Elliot, D.J.; Somogyi, A.A.; Gillam, E.M.J.; Lewis, B.C.; Miners, J.O. An evaluation of potential mechanism-based inactivation of human drug metabolizing cytochromes P450 by monoamine oxidase inhibitors, including isoniazid. Br. J. Clin. Pharmacol. 2006, 61, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Salsali, M.; Holt, A.; Baker, G.B. Inhibitory effects of the monoamine oxidase inhibitor tranylcypromine on the cytochrome P450 enzymes CYP2C19, CYP2C9, and CYP2D6. Cell. Mol. Neurobiol. 2004, 24, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Frieling, H.; Bleich, S. Tranylcypromine—New perspectives on an “old’’ drug. Eur. Arch. Psychiatry Clin Neurosci 2006, 256, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Borstnar, R.; Repic, M.; Krzan, M.; Mavri, J.; Vianello, R. Irreversible inhibition of monoamine oxidase B by the antiparkinsonian medicines rasagiline and selegiline: A computational study. Eur. J. Org. Chem. 2011, 6419–6433. [Google Scholar] [CrossRef]
- Youdim, M.B.H. The path from anti Parkinson drug selegiline and rasagiline to multi-functional neuroprotective anti Alzheimer drugs ladostigil and M30. Curr. Alzheimer Res. 2006, 3, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.; Clow, A.; Glover, V.; Halket, J.; Przyborowska, A.; Sandler, M. Isatin, regional distribution in rat-brain and tissues. Neurochem. Int. 1990, 17, 321–323. [Google Scholar] [CrossRef]
- Bonnet, U. Moclobemide: Therapeutic use and clinical studies. CNS Drug Rev. 2003, 9, 97–140. [Google Scholar] [CrossRef] [PubMed]
- Reck, F.; Zhou, F.; Girardot, M.; Kern, G.; Eyermann, C.J.; Hales, N.J.; Ramsay, R.R.; Gravestock, M.B. Identification of 4-substituted 1,2,3-triazoles as novel oxazolidinone antibacterial agents with reduced activity against monoamine oxidase A. J. Med. Chem. 2005, 48, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Mazouz, F.; Lebreton, L.; Milcent, R.; Burstein, C. 5-Aryl-1,3,4-oxadiazol-2(3H)-one derivatives and sulfur analogs as new selective and competitive monoamine-oxidase type-B inhibitors. Eur. J. Med. Chem. 1990, 25, 659–671. [Google Scholar] [CrossRef]
- Silverman, R.B. Oxazolidinones, dihydrofuranones, and pyrrolidinones as inactivators and substrates of monoamine oxidase B: Approaches to the design of antiparkinsonian agents. Farmaco 1997, 52, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Curet, O.; Damoiseau, G.; Aubin, N.; Sontag, N.; Rovei, V.; Jarreau, F.X. Befloxatone, a new reversible and selective monoamine oxidase-A inhibitor. 1. Biochemical profile. J. Pharmacol. Exp. Ther. 1996, 277, 253–264. [Google Scholar] [PubMed]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Carradori, S.; Petzer, J.P. Novel monoamine oxidase inhibitors: A patent review (2012–2014). Expert Opin. Ther. Pat. 2015, 25, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Farina, R.; Catto, M.; Iacobazzi, R.M.; Nicolotti, O.; Cellamare, S.; Mangiatordi, G.F.; Denora, N.; Soto-Otero, R.; Siragusa, L.; et al. Exploring basic tail modifications of coumarin-based dual acetylcholinesterase-monoamine oxidase B inhibitors: Identification of water-soluble, brain-permeant neuroprotective multitarget agents. J. Med. Chem. 2016, 59, 6791–6806. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, G.; Soreq, H. Termination and beyond: Acetylcholinesterase as a modulator of synaptic transmission. Cell Tissue Res. 2006, 326, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Massoulie, J. The origin of the molecular diversity and functional anchoring of cholinesterases. Neurosignals 2002, 11, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Perrier, A.L.; Massoulie, J.; Krejci, E. PRiMA: The membrane anchor of acetylcholinesterase in the brain. Neuron 2002, 33, 275–285. [Google Scholar] [CrossRef]
- Haas, R.; Jackson, B.C.; Reinhold, B.; Foster, J.D.; Rosenberry, T.L. Glycoinositol phospholipid anchor and protein C-terminus of bovine erythrocyte acetylcholinesterase: Analysis by mass spectrometry and by protein and DNA sequencing. Biochem. J. 1996, 314, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic-structure of acetylcholinesterase from torpedo-californica—A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Chatonnet, A.; Lockridge, O. Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem. J. 1989, 260, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- George, S.T.; Balasubramanian, A.S. The aryl acylamidases and their relationship to cholinesterases in human-serum, erythrocyte and liver. Eur. J. Biochem. 1981, 121, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Grantham, D.L.; Hopkins, D.A. Distribution of butyrylcholinesterase in the human amygdala and hippocampal formation. J. Comp. Neurol. 1998, 393, 374–390. [Google Scholar] [CrossRef]
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.; Whittaker, M.; Lehmann, H.; Silk, E. The pseudocholinesterase variants. Esterase levels and dibucaine numbers in families selected through suxamethonium sensitive individuals. Acta Genet. Stat. Med. 1960, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shafferman, A.; Velan, B.; Ordentlich, A.; Kronman, C.; Grosfeld, H.; Leitner, M.; Flashner, Y.; Cohen, S.; Barak, D.; Ariel, N. Substrate-inhibition of acetylcholinesterase - residues affecting signal transduction from the surface to the catalytic center. EMBO J. 1992, 11, 3561–3568. [Google Scholar] [PubMed]
- Arendt, T.; Bruckner, M.K.; Lange, M.; Bigl, V. Changes in acetylcholinesterase and butyrylcholinesterase in Alzheimers-disease resemble embryonic-development—A study of molecular-forms. Neurochem. Int. 1992, 21, 381–396. [Google Scholar] [CrossRef]
- Ballard, C.; Greig, N.; Guillozet-Bongaarts, A.; Enz, A.; Darvesh, S. Cholinesterases: Roles in the brain during health and disease. Curr. Alzheimer Res. 2005, 2, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int. J. Neuropsychopharmacol. 2006, 9, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Inhibitors of acetylcholinesterase and butyrylcholinesterase meet immunity. Int. J. Mol. Sci. 2014, 15, 9809–9825. [Google Scholar] [CrossRef] [PubMed]
- Grisaru, D.; Pick, M.; Perry, C.; Sklan, E.H.; Almog, R.; Goldberg, I.; Naparstek, E.; Lessing, J.B.; Soreq, H.; Deutsch, V. Hydrolytic and nonenzymatic functions of acetylcholinesterase comodulate hemopoietic stress responses. J. Immunol. 2006, 176, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Turner, A.J. AChE and the amyloid precursor protein (APP)—Cross-talk in Alzheimer’s disease. Chem.-Biol. Interact. 2016, 259, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Branduardi, D.; Gervasio, F.L.; Cavalli, A.; Recanatini, M.; Parrinello, M. The role of the peripheral anionic site and cation-pi interactions in the ligand penetration of the human AChE gorge. J. Am. Chem. Soc. 2005, 127, 9147–9155. [Google Scholar] [CrossRef] [PubMed]
- Silman, I.; Sussman, J.L. Acetylcholinesterase: How is structure related to function? Chem.-Biol. Interact. 2008, 175, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions—Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.; Moore, S.W. The peripheral anionic site of acetylcholinesterase: Structure, functions and potential role in rational drug design. Curr. Pharm. Des. 2006, 12, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Soreq, H. It all starts at the ends: multifaceted involvement of C- and N-terminally modified cholinesterases in Alzheimer’s disease. Rambam Maimonides Med. J. 2010, 1, e0014. [Google Scholar] [CrossRef] [PubMed]
- Podoly, E.; Shalev, D.E.; Shenhar-Tsarfaty, S.; Bennett, E.R.; Ben Assayag, E.; Wilgus, H.; Livnah, O.; Soreq, H. The butyrylcholinesterase K variant confers structurally derived risks for Alzheimer pathology. J. Biol. Chem. 2009, 284, 17170–17179. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.C.; Jiang, Y.R.; Wang, X.; Du, Y.S.; Xiao, D.D.; Deng, Y.C.; Wang, J.L. Butyrylcholinesterase K variant and Alzheimer’s disease risk: A meta-analysis. Med. Sci. Monit. 2015, 21, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Soreq, H.; Seidman, S. Acetylcholinesterase—New roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Shaw, F.H.; Bentley, G. Some aspects of the pharmacology of morphine with special reference to its antagonism by 5-amino-acridine and other chemically related compounds. Med. J. Aust. 1949, 2, 868–874. [Google Scholar] [PubMed]
- Heilbronn, E. Inhibition of cholinesterases by tetrahydroaminacrin. Acta Chem. Scand. 1961, 15, 1386–1390. [Google Scholar] [CrossRef]
- Berman, H.A.; Leonard, K. Interaction of tetrahydroaminoacridine with acetylcholinesterase and butyrylcholinesterase. Mol. Pharmacol. 1992, 41, 412–418. [Google Scholar] [PubMed]
- TR, F. Mechanism of action of organophosphorus and carbamate insecticides. Environ. Health Perspect. 1990, 87, 245–254. [Google Scholar]
- Aldridge, W.N. Some properties of specific cholinesterase with particular reference to the mechanism of inhibition by diethyl para-nitrophenyl thiophosphate (E 605) and analogues. Biochem. J. 1950, 46, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.K. A phosphorylation site in brain and delayed neurotoxic effect of some organophosphorus compounds. Biochem. J. 1969, 111, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Mangas, I.; Vilanova, E.; Estévez, J. Phenyl valerate esterase activity of human butyrylcholinesterase. Arch. Toxicol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.J.; Hein, N.D.; Wijeyesakere, S.J.; Fink, J.K.; Makhaeva, G.F. Neuropathy target esterase (NTE): Overview and future. Chem.-Biol. Interact. 2013, 203, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, P.; Sun, Y.-J.; Xu, M.-Y.; Wu, Y.-J. Disturbed phospholipid homeostasis in endoplasmic reticulum initiates tri-o-cresyl phosphate-induced delayed neurotoxicity. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Quilliam, J.P. DI-ISO Propyleluorophosphonate (DFP): Its pharmacology and its therapeutic uses in glaucoma and myasthenia gravis. Postgrad. Med. J. 1947, 23, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Arrieta, J.M.; Schneider, L. Metrifonate for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Jewsbury, J.M. Metrifonate in schistosomiasis—Therapy and prophylaxis. Acta Pharmacol. Toxicol. 1981, 49, 123–130. [Google Scholar] [CrossRef]
- Darvesh, S.; Darvesh, K.V.; McDonald, R.S.; Mataija, D.; Walsh, R.; Mothana, S.; Lockridge, O.; Martin, E. Carbamates with differential mechanism of inhibition toward acetylcholinesterase and butyrylcholinesterase. J. Med. Chem. 2008, 51, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Gunderson, C.H.; Lehmann, C.R.; Sidell, F.R.; Jabbari, B. Nerve agents—A review. Neurology 1992, 42, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A. The early toxicology of physostigmine. Toxicol. Rev. 2006, 25, 99–138. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Adem, A.; Sabbagh, M. New acetylcholinesterase inhibitors for alzheimer’s disease. Int. J. Alzheimers Dis. 2012, 2012, 728983. [Google Scholar] [CrossRef] [PubMed]
- Braida, D.; Sala, M. Eptastigmine: Ten years of pharmacology, toxicology, pharmacokinetic, and clinical studies. CNS Drug Rev. 2001, 7, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Emre, M.; Cummings, J.L.; Lane, R.M. Rivastigmine in dementia associated with Parkinson’s disease and Alzheimer’s disease: Similarities and differences. J. Alzheimers Dis. 2007, 11, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Ogura, H.; Arai, Y.; Iimura, Y.; Yamanishi, Y. Research and development of donepezil hydrochloride, a new type of acetylcholinesterase inhibitor. Jpn. J. Pharmacol. 2002, 89, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.A.; Qosa, H.; Kaddoumi, A. Age-related decline in brain and hepatic clearance of amyloid-beta is rectified by the cholinesterase inhibitors donepezil and rivastigmine in rats. ACS Chem. Neurosci. 2015, 6, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Bartolucci, C.; Perola, E.; Pilger, C.; Fels, G.; Lamba, D. Three-dimensional structure of a complex of galanthamine (Nivalin (R)) with acetylcholinesterase from Torpedo californica: Implications for the design of new anti-Alzheimer drugs. Proteins 2001, 42, 182–191. [Google Scholar] [CrossRef]
- Wessler, I.; Kirkpatrick, C.J. Acetylcholine beyond neurons: The non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008, 154, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Xu, Y.; Chen, J.; Liu, Q.; Gu, J.; Wang, X.; Ma, J.; Li, H.; Onuchic, J.N.; Jiang, H. Free energy landscape for the binding process of Huperzine A to acetylcholinesterase. Proc. Natl. Acad. Sci. USA 2013, 110, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.-X.; Huang, X.-T.; Chen, Y.-T.; Tang, X.-C.; Zhang, H.-Y. Acetylcholinesterase-independent protective effects of huperzine A against iron overload-induced oxidative damage and aberrant iron metabolism signaling in rat cortical neurons. Acta Pharmacol. Sin. 2016, 37, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Walsh, S.; Little, J.T.; Behan, K.; Reynolds, B.; Ward, C.; Jin, S.; Thomas, R.; Aisen, P.S.; Alzheimer’s Dis Cooperative, S. A phase II trial of huperzine A in mild to moderate Alzheimer disease. Neurology 2011, 76, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wang, Y.; Tian, J.; Liu, J.-P. Huperzine A for Alzheimer’s disease: A systematic review and meta-analysis of randomized clinical trials. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Hiremathad, A.; Chand, K.; Esteves, A.R.; Cardoso, S.M.; Ramsay, R.R.; Chaves, S.; Keri, R.S.; Santos, M.A. Tacrine-allyl/propargylcysteine-benzothiazole trihybrids as potential anti-Alzheimer’s drug candidates. RSC Adv. 2016, 6, 53519–53532. [Google Scholar] [CrossRef]
- Weinstock, M.; Goren, T.; Youdim, M.B.H. Development of a novel neuroprotective drug (TV3326) for the treatment of Alzheimer’s disease, with cholinesterase and monoamine oxidase inhibitory activities. Drug Dev. Res. 2000, 50, 216–222. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons, Inc.: Chichester, UK, 2005; Volume 46. [Google Scholar]
- Tipton, K. Enzyme Assay and Kinetic Studies; Elsevier/North-Holland: Shannon, Ireland, 1985; p. 56. ISBN 9780471227618. [Google Scholar] [CrossRef]
- Tipton, K.F. Principles of enzyme assay and kinetic studies. In Enzyme Assays: A Practical Approach; Eisenthal, R., Danson, M.J., Eds.; Oxford University Press: London, UK, 2002; Volume 257, pp. 1–47. [Google Scholar]
- McDonald, A.; Tipton, K. Kinetics of catalyzed reactions—Biological. In Encyclopedia of Catalysis; Horváth, I.T., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2002. [Google Scholar]
- Tipton, K.F.; Davey, G.; Motherway, M. Monoamine oxidase assays. Curr. Protoc. Toxicol. 2006, 30, 1–42. [Google Scholar] [CrossRef]
- Miao, Y.Q.; He, N.Y.; Zhu, J.J. History and new developments of assays for cholinesterase activity and inhibition. Chem. Rev. 2010, 110, 5216–5234. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, F.B.; Baugher, B.W.; Beissner, R.S. Techniques in coupled enzyme assays. Methods Enzymol. 1979, 63, 22–42. [Google Scholar] [PubMed]
- Komersová, A.; Komers, K.; Cegan, A. New findings about Ellman’s method to determine cholinesterase activity. Naturforsch. C 2007, 62, 150–154. [Google Scholar] [CrossRef]
- Tipton, K.; Armstrong, R.N.; Bakker, B.M.; Bairoch, A.; Cornish-Bowden, A.; Halling, P.J.; Hofmeyr, J.-H.; Leyh, T.; Kettner, C.; Raushel, F.M.; et al. Standards for reporting enzyme data: The STRENDA Consortium: What it aims to do and why it should be helpful. Perspect. Sci. 2014, 1, 131–137. [Google Scholar] [CrossRef]
- Dowd, J.; Riggs, D. A comparison of estimates of Michaelis—Menten kinetic constants from various linear transformations. J. Biol. Chem. 1965, 240, 863–869. [Google Scholar] [PubMed]
- Mooser, G.; Sigman, D. Ligand binding properties of acetylcholinesterase determined with fluorescent probes. Biochemistry 1974, 13, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Taylor, P.; Radić, Z.; Marchot, P. Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. EMBO J. 2003, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tipton, K. Patterns of enzyme inhibition. In Enzymology LabFax; Engel, P.C., Ed.; Bios Scientific Publishers: Oxford, UK, 1996; pp. 115–174. [Google Scholar]
- Tipton, K.F.; Davey, G.P.; McDonald, A.G. Kinetic behavior and reversible inhibition of monoamine oxidases-enzymes that many want dead. Int. Rev. Neurobiol. 2011, 100, 43–64. [Google Scholar] [PubMed]
- Dixon, M. The graphical determination of Km and Ki. Biochem. J. 1972, 129, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Henderson, P.J. Steady-state enzyme-kinetics with high-affinity substrates or inhibitors—Statistical treatment of dose-response curves. Biochem. J. 1973, 135, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.F. Kinetics of reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 1968, 185, 269–286. [Google Scholar] [CrossRef]
- Fowler, C.J.; Benedetti, M.S. Cimoxatone is a reversible tight-binding inhibitor of the a form of rat-brain monoamine-oxidase. J. Neurochem. 1983, 40, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Herbet, A.; Pétillot, Y.; Pichat, L.; Glowinski, J.; Hamon, M. [3H] Harmaline as a specific ligand of MAO A–I. Properties of the active site of MAO A from rat and bovine brains. J. Neurochem. 1979, 32, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Dunford, C.; Gillman, P.K. Methylene blue and serotonin toxicity: Inhibition of monoamine oxidase A (MAO A) confirms a theoretical prediction. Br. J. Pharmacol. 2007, 152, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Kinemuchi, H.; Arai, Y.; Oreland, L.; Tipton, K.F.; Fowler, C.J. Time-dependent inhibition of monoamine-oxidase by beta—Phenethylamine. Biochem. Pharmacol. 1982, 31, 959–964. [Google Scholar] [PubMed]
- Houslay, M.D.; Garrett, N.J.; Tipton, K.F. Mixed Substrate experiments with human brain monoamine-oxidase. Biochem. Pharmacol. 1974, 23, 1937–1944. [Google Scholar] [CrossRef]
- Kitz, R.; Wilson, I.B. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 1962, 237, 3245–3249. [Google Scholar] [PubMed]
- Fowler, C.J.; Mantle, T.J.; Tipton, K.F. The nature of the inhibition of rat-liver monoamine-oxidase type-A and type-B by the acetylenic inhibitors clorgyline, l-deprenyl and pargyline. Biochem. Pharmacol. 1982, 31, 3555–3561. [Google Scholar] [CrossRef]
- Forsberg, A.; Puu, G. Kinetics for the inhibition of acetylcholinesterase from the electric eel by some organophosphates and carbamates. Eur. J. Biochem. 1984, 140, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, P.; Millard, C.B.; Harel, M.; Dvir, H.; Enz, A.; Sussman, J.L.; Silman, I. Kinetic and structural studies on the interaction of cholinesterases with the anti-Alzheimer drug rivastigmine. Biochemistry 2002, 41, 3555–3564. [Google Scholar] [CrossRef] [PubMed]
- Tipton, K.F.; McCrodden, J.M.; Youdim, M.B.H. Oxidation and enzyme-activated irreversible inhibition of rat-liver monoamine oxidase-b by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Biochem. J. 1986, 240, 379–383. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.M.; Tipton, K.F.; McCrodden, J.M.; Youdim, M.B.H. The interactions of milacemide with monoamine-oxidase. Biochem. Pharmacol. 1994, 47, 617–623. [Google Scholar] [CrossRef]
- Tipton, K.F.; Fowler, C.J.; McCrodden, J.M.; Benedetti, M.S. The enzyme-activated irreversible inhibition of type-B monoamine-oxidase by 3-(4-(3-chlorophenyl)methoxy phenyl)-5-(methylamino)methyl -2-oxazolidinone methanesulfonate (compound MD 780236) and the enzyme-catalyzed oxidation of this compound as competing reactions. Biochem. J. 1983, 209, 235–242. [Google Scholar] [PubMed]
- Tatsunami, S.; Yago, N.; Hosoe, M. Kinetics of suicide substrates—Steady-state treatments and computer-aided exact-solutions. Biochim. Biophys. Acta 1981, 662, 226–235. [Google Scholar] [CrossRef]
- Waley, S.G. Kinetics of suicide substrates—Practical procedures for determining parameters. Biochem. J. 1985, 227, 843–849. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.M.; Dostert, P.; Tipton, K.F. Species-differences in the interactions of the anticonvulsant milacemide and some analogs with monoamine oxidase-B. Biochem. Pharmacol. 1995, 50, 317–324. [Google Scholar] [CrossRef]
- Inoue, H.; Castagnoli, K.; Van der Schyf, C.; Mabic, S.; Igarashi, K.; Castagnoli, N. Species-dependent differences in monoamine oxidase A and B-catalyzed oxidation of various C4 substituted 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridinyl derivatives. J. Pharmacol. Exp. Ther. 1999, 291, 856–864. [Google Scholar] [PubMed]
- Weissbach, H.; Smith, T.E.; Daly, J.W.; Witkop, B.; Udenfriend, S. A rapid spectrophotometric assay of Monoamine Oxidase based on the rate of disappearance of kynuramine. J. Biol. Chem. 1960, 235, 1160–1163. [Google Scholar] [PubMed]
- Meyerson, L.R.; McMurtrey, K.D.; Davis, V.E. A rapid and sensitive potentiometric assay for monoamine oxidase using an ammonia-selective electrode. Anal. Biochem. 1978, 86, 287–297. [Google Scholar] [CrossRef]
- Krueger, M.J.; Singer, T.P. An Examination of the reliability of the radiochemical assay for monoamine oxidase-A and oxidase-B. Anal. Biochem. 1993, 214, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.J.; Zhong, B.; Silverman, R. Direct continuous fluorometric assay for monoamine oxidase B. Anal. Biochem. 1996, 234, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.Y.; Caldwell, G.W.; Zhao, B.Y.; Reitz, A.B. A high-throughput monoamine oxidase inhibition assay using liquid chromatography with tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Holt, A.; Palcic, M.M. A peroxidase-coupled continuous absorbance plate-reader assay for flavin monoamine oxidases, copper-containing amine oxidases and related enzymes. Nat. Protoc. 2006, 1, 2498–2505. [Google Scholar] [CrossRef] [PubMed]
- Valley, M.P.; Zhou, W.; Hawkins, E.M.; Shultz, J.; Cali, J.J.; Worzella, T.; Bernad, L.; Good, T.; Good, D.; Riss, T.L.; et al. A bioluminescent assay for monoamine oxidase activity. Anal. Biochem. 2006, 359, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.H.; Valley, M.P.; Shultz, J.; Hawkins, E.M.; Bernad, L.; Good, T.; Good, D.; Riss, T.L.; Klaubert, D.H.; Wood, K.V. New bioluminogenic substrates for monoamine oxidase assays. J. Am. Chem. Soc. 2006, 128, 3122–3123. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Zhang, G.; Zhang, D.; Wang, Y.; Zhu, D. A direct continuous fluorometric turn-on assay for monoamine oxidase B and its inhibitor-screening based on the abnormal fluorescent behavior of silole. Analyst 2010, 135, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Y.; Wang, Y.G.; Dai, B.; Dai, Y.Q.; Wang, Z.; Fu, Z.W.; Zhu, Q. A novel fluorogenic probe for monoamine oxidase assays. Chin. Chem. Lett. 2008, 19, 947–950. [Google Scholar] [CrossRef]
- Huang, G.L.; Zhu, F.; Chen, Y.H.; Chen, S.Q.; Liu, Z.H.; Li, X.; Gan, L.L.; Zhang, L.; Yu, Y. A spectrophotometric assay for monoamine oxidase activity with 2,4-dinitrophenylhydrazine as a derivatized reagent. Anal. Biochem. 2016, 512, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Li, L.H.; Shi, W.; Gong, Q.Y.; Li, X.H.; Ma, H.M. Sensitive and selective ratiometric fluorescence probes for detection of intracellular endogenous monoamine oxidase A. Anal. Chem. 2016, 88, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Hroch, L.; Guest, P.; Benek, O.; Soukup, O.; Janockova, J.; Dolezal, R.; Kuca, K.; Aitken, L.; Smith, T.K.; Gunn-Moore, F.; et al. Synthesis and evaluation of frentizole-based indolyl thiourea analogues as MAO/ABAD inhibitors for Alzheimer’s disease treatment. Bioorg. Med. Chem. 2016, 25, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Tipton, K.F.; Spires, I.P.C. Kinetics of phenethylhydrazine oxidation by monoamine oxidase. Biochem. J. 1971, 125, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Bach, A.W.J.; Lan, N.C.; Johnson, D.L.; Abell, C.W.; Bembenek, M.E.; Kwan, S.W.; Seeburg, P.H.; Shih, J.C. cDNA cloning of human-liver monoamine oxidase-A and oxidase-B—Molecular basis of differences in enzymatic properties. Proc. Natl. Acad. Sci. USA 1988, 85, 4934–4938. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.K.; Wang, J.; Edmondson, D.E. Comparison of the structural properties of the active site cavities of human and rat monoamine oxidase A and B in their soluble and membrane-bound forms. Biochemistry 2008, 47, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Aldeco, M.; Arslan, B.K.; Edmondson, D.E. Catalytic and inhibitor binding properties of zebrafish monoamine oxidase (zMAO): Comparisons with human MAO A and MAO B. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2011, 159, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Krueger, M.J.; Mazouz, F.; Ramsay, R.R.; Milcent, R.; Singer, T.P. Dramatic species differences in the susceptibility of Monoamine-Oxidase-B to a group of powerful inhibitors. Biochem. Biophys. Res. Commun. 1995, 206, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Fierro, A.; Osorio-Olivares, M.; Cassels, B.K.; Edmondson, D.E.; Sepulveda-Boza, S.; Reyes-Parada, M. Human and rat monoamine oxidase-A are differentially inhibited by (S)-4-alkylthioamphetamine derivatives: Insights from molecular modeling studies. Bioorg. Med. Chem. 2007, 15, 5198–5206. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bautista-Aguilera, O.M.; Esteban, G.; Bolea, I.; Nikolic, K.; Agbaba, D.; Moraleda, I.; Iriepa, I.; Samadi, A.; Soriano, E.; Unzeta, M.; et al. Design, synthesis, pharmacological evaluation, QSAR analysis, molecular modeling and ADMET of novel donepezil-indolyl hybrids as multipotent cholinesterase/monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 75, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Esteban, G.; Allan, J.; Samadi, A.; Mattevi, A.; Unzeta, M.; Marco-Contelles, J.; Binda, C.; Ramsay, R.R. Kinetic and structural analysis of the irreversible inhibition of human monoamine oxidases by ASS234, a multi-target compound designed for use in Alzheimer’s disease. BBA-Proteins Proteom. 2014, 1844, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Silk, E.; King, J.; Whittaker, M. Assay of cholinesterase in clinical chemistry. Ann. Clin. Biochem. 1979, 16, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Holas, O.; Musilek, K.; Pohanka, M.; Kuca, K. The progress in the cholinesterase quantification methods. Expert Opin. Drug Discov. 2012, 7, 1207–1223. [Google Scholar] [CrossRef] [PubMed]
- Fonnum, F. Radiochemical micro assays for determination of choline acetyltransferase and acetylcholinesterase activities. Biochem. J. 1969, 115, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Michel, H.O. An electrometric method for the determination of red blood cell and plasma cholinesterase activity. J. Lab. Clin. Med. 1949, 34, 1564–1568. [Google Scholar]
- Timur, S.; Telefoncu, A. Acetylcholinesterase (AChE) electrodes based on gelatin and chitosan matrices for the pesticide detection. Artif. Cells Blood Substit. Biotechnol. 2004, 32, 427–442. [Google Scholar] [CrossRef]
- Zdrazilova, P.; Stepankova, S.; Vranova, M.; Komers, K.; Komersova, A.; Cegan, A. Kinetics of total enzymatic hydrolysis of acetylcholine and acetylthiocholine. Z. Naturforsch. C 2006, 61, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Moris-Varas, F.; Shah, A.; Aikens, J.; Nadkarni, N.P.; Rozzell, J.D.; Demirjian, D.C. Visualization of enzyme-catalyzed reactions using pH indicators: Rapid screening of hydrolase libraries and estimation of the enantioselectivity. Bioorg. Med. Chem. 1999, 7, 2183–2188. [Google Scholar] [CrossRef]
- Pohanka, M.; Karasova, J.Z.; Kuca, K.; Pikula, J.; Holas, O.; Korabecny, J.; Cabal, J. Colorimetric dipstick for assay of organophosphate pesticides and nerve agents represented by paraoxon, sarin and VX. Talanta 2010, 81, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Eyer, P.; Worek, F.; Kiderlen, D.; Sinko, G.; Stuglin, A.; Simeon-Rudolf, V.; Reiner, E. Molar absorption coefficients for the reduced Ellman reagent: Reassessment. Anal. Biochem. 2003, 312, 224–227. [Google Scholar] [CrossRef]
- Benabent, M.; Vilanova, E.; Sogorb, M.; Estévez, J. Cholinesterase assay by an efficient fixed time endpoint method. MethodsX 2014, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Wille, T.; Thiermann, H.; Worek, F. Development of a high-throughput screening for nerve agent detoxifying materials using a fully-automated robot-assisted biological assay. Toxicol. Vitro 2010, 24, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Worek, F.; Mast, U.; Kiderlen, D.; Diepold, C.; Eyer, P. Improved determination of acetylcholinesterase activity in human whole blood. Clin. Chim. Acta 1999, 288, 73–90. [Google Scholar] [CrossRef]
- Sinko, G.; Calic, M.; Bosak, A.; Kovarik, Z. Limitation of the Ellman method: Cholinesterase activity measurement in the presence of oximes. Anal. Biochem. 2007, 370, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Worek, F.; Eyer, P.; Thiermann, H. Determination of acetylcholinesterase activity by the Ellman assay: A versatile tool for in vitro research on medical countermeasures against organophosphate poisoning. Drug Test. Anal. 2012, 4, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Liddell, J. Value of butyrylthiocholine assay for identification of cholinesterase variants. J. Med. Genet. 1970, 7, 351–355. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Main, A.R.; Miles, K.E.; Braid, P.E. Determination of human-serum-cholinesterase activity with o-nitrophenyl butyrate. Biochem. J. 1961, 78, 769. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M.; Hrabinova, M.; Kuca, K.; Simonato, J.P. Assessment of acetylcholinesterase activity using indoxylacetate and comparison with the standard Ellman’s method. Int. J. Mol. Sci. 2011, 12, 2631–2640. [Google Scholar] [CrossRef] [PubMed]
- Gehauf, B.; Goldenson, J. Detection and estimation of nerve gases by fluorescence reaction. Anal. Chem. 1957, 29, 276–278. [Google Scholar] [CrossRef]
- Guilbault, G.G.; Kramer, D.N. Resorufin butyrate + indoxyl acetate as fluorogenic substrates for cholinesterase. Anal. Chem. 1965, 37, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Villatte, F.; Bachman, T.T.; Hussein, A.S.; Schmid, R.D. Acetylcholinesterase assay for rapid expression screening in liquid and solid media. Biotechniques 2001, 30, 81–84. [Google Scholar] [PubMed]
- Pohanka, M.; Drtinova, L. Spectrophotometric methods based on 2,6-dichloroindophenol acetate and indoxylacetate for butyrylcholinesterase activity assay in plasma. Talanta 2013, 106, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Okabe, H.; Sagesaka, K.; Nakajima, N.; Noma, A. New enzymatic assay of cholinesterase activity. Clin. Chim. Acta 1977, 80, 87–94. [Google Scholar] [PubMed]
- Birman, S. Determination of acetylcholinesterase activity by a new chemi-luminescence assay with the natural substrate. Biochem. J. 1985, 225, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Santillo, M.F.; Liu, Y.T. A fluorescence assay for measuring acetylcholinesterase activity in rat blood and a human neuroblastoma cell line (SH-SY5Y). J. Pharmacol. Toxicol. Methods 2015, 76, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, D.; Caballero, J. Is it reliable to use common molecular docking methods for comparing the binding affinities of enantiomer pairs for their protein target? Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chadha, N.; Tiwari, A.K.; Sehgal, N.; Mishra, A.K. Prospective atom-based 3D-QSAR model prediction, pharmacophore generation, and molecular docking study of carbamate derivatives as dual inhibitors of AChE and MAO-B for Alzheimer’s disease. Med. Chem. Res. 2014, 23, 1114–1122. [Google Scholar] [CrossRef]
- Page, M.I.; Jencks, W.P. Entropic contributions to rate accelerations in enzymic and intramolecular reactions and chelate effect. Proc. Natl. Acad. Sci. USA 1971, 68, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Koca, M.; Yerdelen, K.O.; Anil, B.; Kasap, Z.; Sevindik, H.; Ozyurek, I.; Gunesacar, G.; Turkaydin, K. Design, synthesis and biological activity of 1H-indene-2-carboxamides as multi-targeted anti-Alzheimer agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 13–23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pisani, L.; Farina, R.; Soto-Otero, R.; Denora, N.; Mangiatordi, G.F.; Nicolotti, O.; Mendez-Alvarez, E.; Altomare, C.D.; Catto, M.; Carotti, A. Searching for multi-targeting neurotherapeutics against Alzheimer’s: Discovery of potent AChE-MAO B inhibitors through the decoration of the 2H-chromen-2-one structural motif. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Unzeta, M.; Esteban, G.; Bolea, I.; Fogel, W.A.; Ramsay, R.R.; Youdim, M.B.H.; Tipton, K.F.; Marco-Contelles, J. Multi-target directed donepezil-like ligands for Alzheimer’s disease. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.S.; Lan, J.S.; Wang, X.B.; Wang, Z.M.; Jiang, N.; Li, F.; Wu, J.J.; Wang, J.; Kong, L.Y. Design, synthesis and biological evaluation of novel donepezil-coumarin hybrids as multi-target agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2016, 24, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Kaasinen, V.; Nagren, K.; Jarvenpaa, T.; Roivainen, A.; Yu, M.X.; Oikonen, V.; Kurki, T.; Rinne, J.O. Regional effects of donepezil and rivastigmine on cortical acetylcholinesterase activity in Alzheimer’s disease. J. Clin. Psychopharmacol. 2002, 22, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Chiuccariello, L.; Cooke, R.G.; Miler, L.; Levitan, R.D.; Baker, G.B.; Kish, S.J.; Kolla, N.J.; Rusjan, P.M.; Houle, S.; Wilson, A.A.; et al. Monoamine oxidase-A occupancy by moclobemide and phenelzine: implications for the development of monoamine oxidase inhibitors. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.C.; Hasan, F.; McCrodden, J.M.; Tipton, K.F. Monoamine oxidase inhibitors and the cheese effect. Neurochem. Res. 1993, 18, 1145–1149. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.D.; Mitchell, J.R.; Timbrell, J.A.; Snodgrass, W.R.; Corcoran, G.B. Isoniazid and iproniazid—Activation of metabolites to toxic intermediates in man and rat. Science 1976, 193, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Schlappi, B. The lack of hepatotoxicity in the rat with the new and reversible MAO-A inhibitor moclobemide in contrast to iproniazid. Arzneim.-Forsch./Drug Res. 1985, 35, 800–803. [Google Scholar]
- Magyar, K.; Szende, B.; Jenei, V.; Tabi, T.; Palfi, M.; Szoko, E. R-deprenyl: Pharmacological spectrum of its activity. Neurochem. Res. 2010, 35, 1922–1932. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.B.; Urichuk, L.J.; McKenna, K.F.; Kennedy, S.H. Metabolism of monoamine oxidase inhibitors. Cell. Mol. Neurobiol. 1999, 19, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.H. Multi target neuroprotective and neurorestorative anti-Parkinson and anti-Alzheimer drugs ladostigil and m30 derived from rasagiline. Exp. Neurobiol. 2013, 22, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Moradov, D.; Finkin-Groner, E.; Bejar, C.; Sunita, P.; Schorer-Apelbaum, D.; Barasch, D.; Nemirovski, A.; Cohen, M.; Weinstock, M. Dose-limiting inhibition of acetylcholinesterase by ladostigil results from the rapid formation and fast hydrolysis of the drug-enzyme complex formed by its major metabolite, R-MCPAI. Biochem. Pharmacol. 2015, 94, 164–172. [Google Scholar] [CrossRef] [PubMed]
- De Deurwaerdère, P.; Binda, C.; Corne, R.; Leone, C.; Valeri, A.; Valoti, M.; Ramsay, R.R.; Fall, Y.; Marco-Contelles, J. Comparative analysis of the neurochemical profile and mao inhibition properties of N-(furan-2-ylmethyl)-n-methylprop-2-yn-1-amine. ACS Chem. Neurosci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kupershmidt, L.; Amit, T.; Bar-Am, O.; Youdim, M.B.H.; Weinreb, O. The novel multi-target iron chelating-radical scavenging compound m30 possesses beneficial effects on major hallmarks of Alzheimer’s disease. Antioxid. Redox Signal. 2012, 17, 860–877. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Li, Y.; Qiang, X.M.; Cao, Z.C.; Xu, R.; Yang, X.; Xiao, G.Y.; Song, Q.; Tan, Z.H.; Deng, Y. Multifunctional thioxanthone derivatives with acetylcholinesterase, monoamine oxidases and beta-amyloid aggregation inhibitory activities as potential agents against Alzheimer’s disease. Bioorg. Med. Chem. 2017, 25, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Rabinovich, A.; Nash, Y.; Frenkel, D.; Wang, Y.Q.; Youdim, M.B.H.; Weinreb, O. Anti-inflammatory and protective effects of MT-031, a novel multitarget MAO-A and AChE/BuChE inhibitor in scopolamine mouse model and inflammatory cells. Neuropharmacology 2017, 113, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.S.; Chen, J.; Li, X.R.; Su, T.; Wang, Y.L.; Wang, Z.R.; Huang, L.; Li, X.S. Synthesis and evaluation of selegiline derivatives as monoamine oxidase inhibitor, antioxidant and metal chelator against Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 3722–3729. [Google Scholar] [CrossRef] [PubMed]
- Ismaili, L.; Refouvelet, B.; Benchekroun, M.; Brogi, S.; Brindisi, M.; Gemma, S.; Campiani, G.; Filipic, S.; Agbaba, D.; Esteban, G.; et al. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 4–34. [Google Scholar] [CrossRef] [PubMed]
- Benek, O.; Aitken, L.; Hroch, L.; Kuca, K.; Gunn-Moore, F.; Musilek, K. A direct interaction between mitochondrial proteins and amyloid-beta peptide and its significance for the progression and treatment of Alzheimer’s disease. Curr. Med. Chem. 2015, 22, 1056–1085. [Google Scholar] [CrossRef]
- Hroudova, J.; Singh, N.; Fisar, Z.; Ghosh, K.K. Progress in drug development for Alzheimer’s disease: An overview in relation to mitochondria energy metabolism. Eur. J. Med. Chem. 2016, 121, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Majekova, M.; Medina, M.; Valoti, M. Key targets for multi-target ligands designed to combat neurodegeneration. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.B.; Mothersill, C.; Mooney, R.; Moriarty, M.; Tipton, K.F. Monoamine oxidase inhibitors l-deprenyl and clorgyline protect nonmalignant human cells from ionising radiation and chemotherapy toxicity. Br. J. Cancer 2003, 89, 1979–1986. [Google Scholar] [CrossRef] [PubMed][Green Version]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
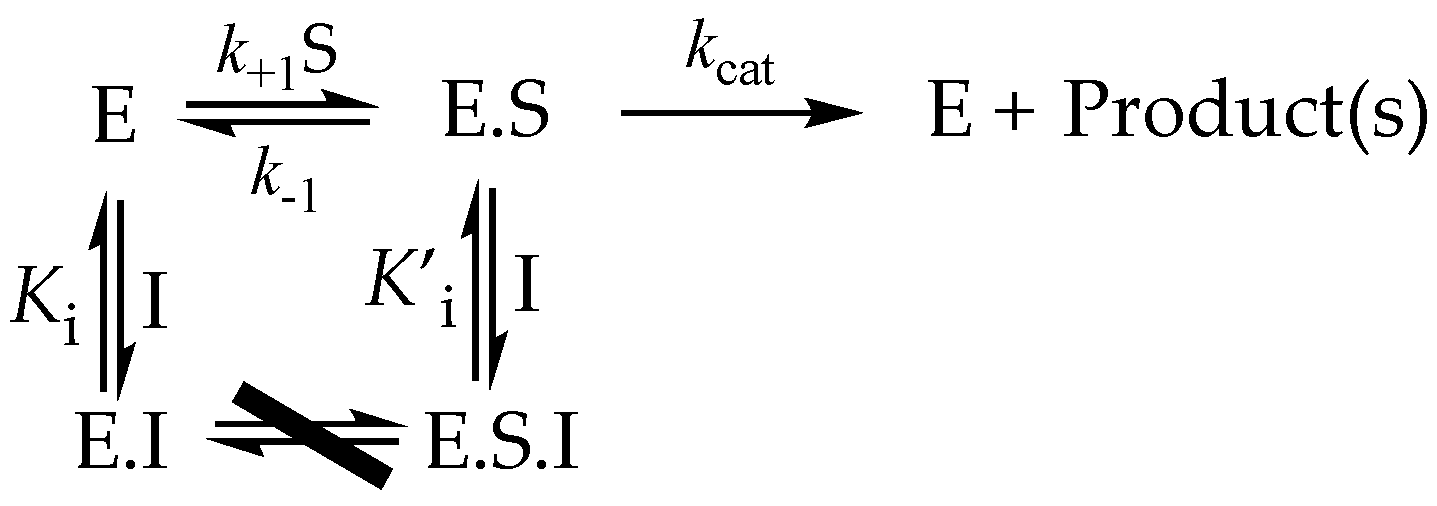{kind=link}
{kind=link}
{kind=link}
{kind=link}
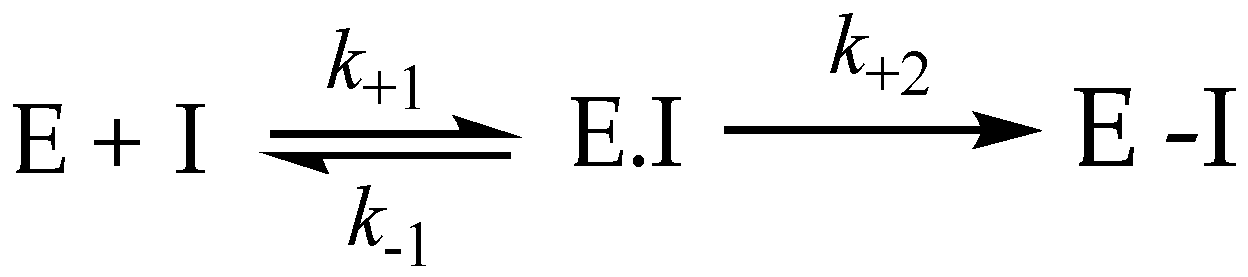{kind=link}
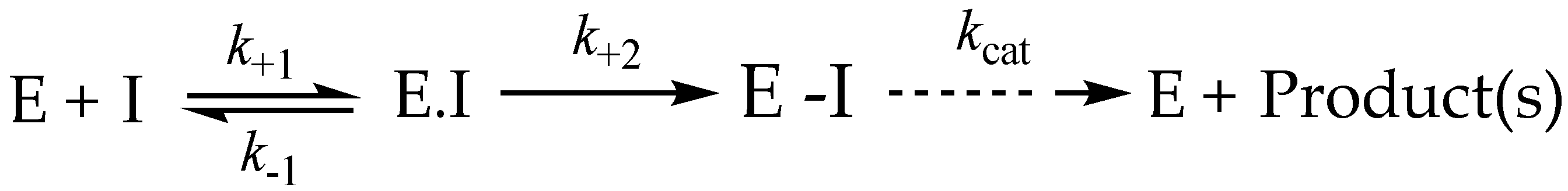{kind=link}
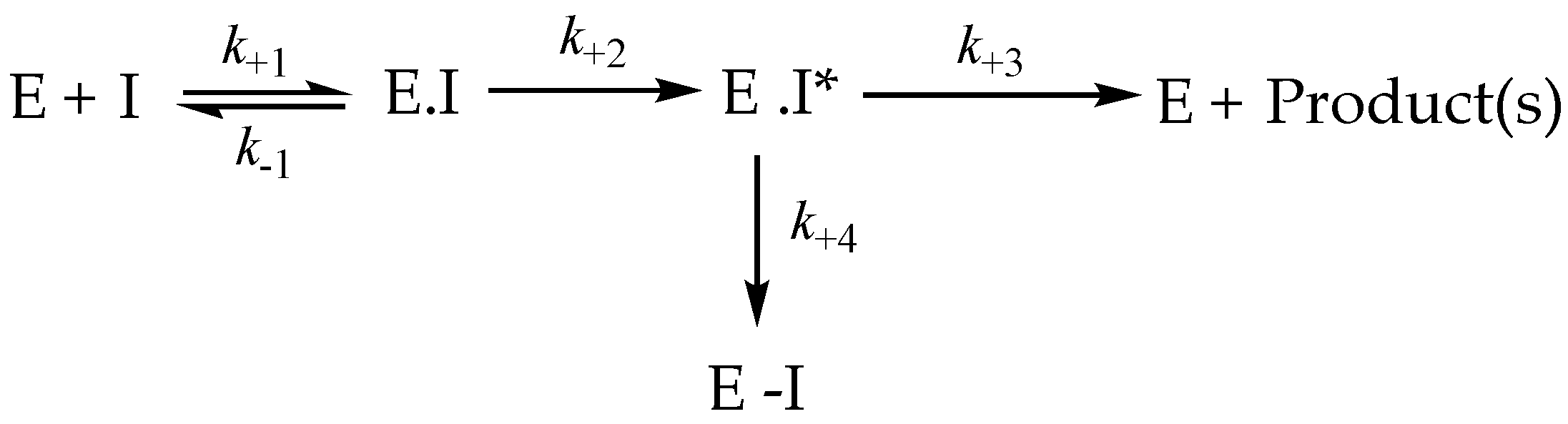{kind=link}
| MAO A | MAO B | |||||
|---|---|---|---|---|---|---|
| Substrate | kcat (s−1) | Km (mM) | kcat/Km | kcat (s−1) | Km (mM) | kcat/Km |
| Benzylamine a | 0.02 | 0.90 | 0.02 | 10 | 0.36 | 27.8 |
| Phenylethylamine a | 0.75 | 0.50 | 1.50 | 3.6 | 0.067 | 53.7 |
| Serotonin (5HT) a | 2.80 | 0.40 | 7.00 | 0.077 | 0.28 | 0.28 |
| Dopamine b | 1.83 | 0.23 | 7.96 | |||
| MPTP a | 0.2 | 0.09 | 2.22 | 0.16 | 0.04 | 4 |
| Oxygen c | 0.06 | 0.28 | ||||
| Tyramine d | 0.45 | 0.22 | ||||
| Substrate | Enzyme | Application * |
|---|---|---|
| Acetylcholine | AChE > BChE | |
| Acetylthiocholine | AChE > BChE | Substrate used in DTNB assay |
| Acetyl-β-methyl-choline | AChE | |
| Acetyl-β-methyl-thiocholine | AChE | Substrate used in DTNB assay |
| Acetyl-β-methyl-choline | AChE | |
| Succinylcholine | BChE | |
| Succinylthiocholine | BChE | Substrate used DTNB assay |
| Adipoylcholine | BChE | |
| Benzoylcholine | BChE | |
| Butyrylcholine | BuChE > AChE | |
| Butyrylthiocholine | BChE > AChE | Substrate used DTNB assay |
| Propionylcholine | AChE & BChE | Substrate used DTNB assay |
| Propionylthiocholine | AChE & BChE | |
| Phenyl valerate | NTE † > BChE | Substrate used in NTE assay |
| Type | Reaction | Kinetic Constant(s) | Rate of Inhibition | Reversibility In Vitro | Reversibility In Vivo |
|---|---|---|---|---|---|
| Reversible | Ki = k−1/k+1 | Rapid | Dialysis, dilution | Elimination of free I | |
| Irreversible | k | Slow | None | Synthesis of new E | |
| Specific irreversible | Ki and k+3 | Slow | None | Synthesis of new E | |
| Poor substrate | Ki and kcat | Slow | Slow | Substrate depletion | |
| Tight-binding | k+1 and k−1 | Slow | Slow | Elimination of free I |
| Type | Effect on Km | Effect on Vmax | Double-Reciprocal Plots at Different [I] |
|---|---|---|---|
| Competitive | Increased to Km (1 + [I]/Ki) | NONE | Intersect on 1/v axis |
| Uncompetitive | Decreased to Km/(1 + [I]/K’i) | Decreased to Km/(1 + [I]/K’i) | Lines parallel |
| Noncompetitive | NONE | Decreased to Km/(1 + [I]/K’i) | Intersect on −1/[S] axis |
| Mixed | Increased to Km (1 + [I]/Ki) | Decreased to Km/(1 + [I]/K’i) | Intersect above 1/v axis if Ki > K’i Intersect below 1/v axis if Ki < K’i |
| Inhibitor Type | IC50 | % Inhibition |
|---|---|---|
| Competitive | ||
| Uncompetitive | ||
| Noncompetitive | Ki | |
| Mixed |
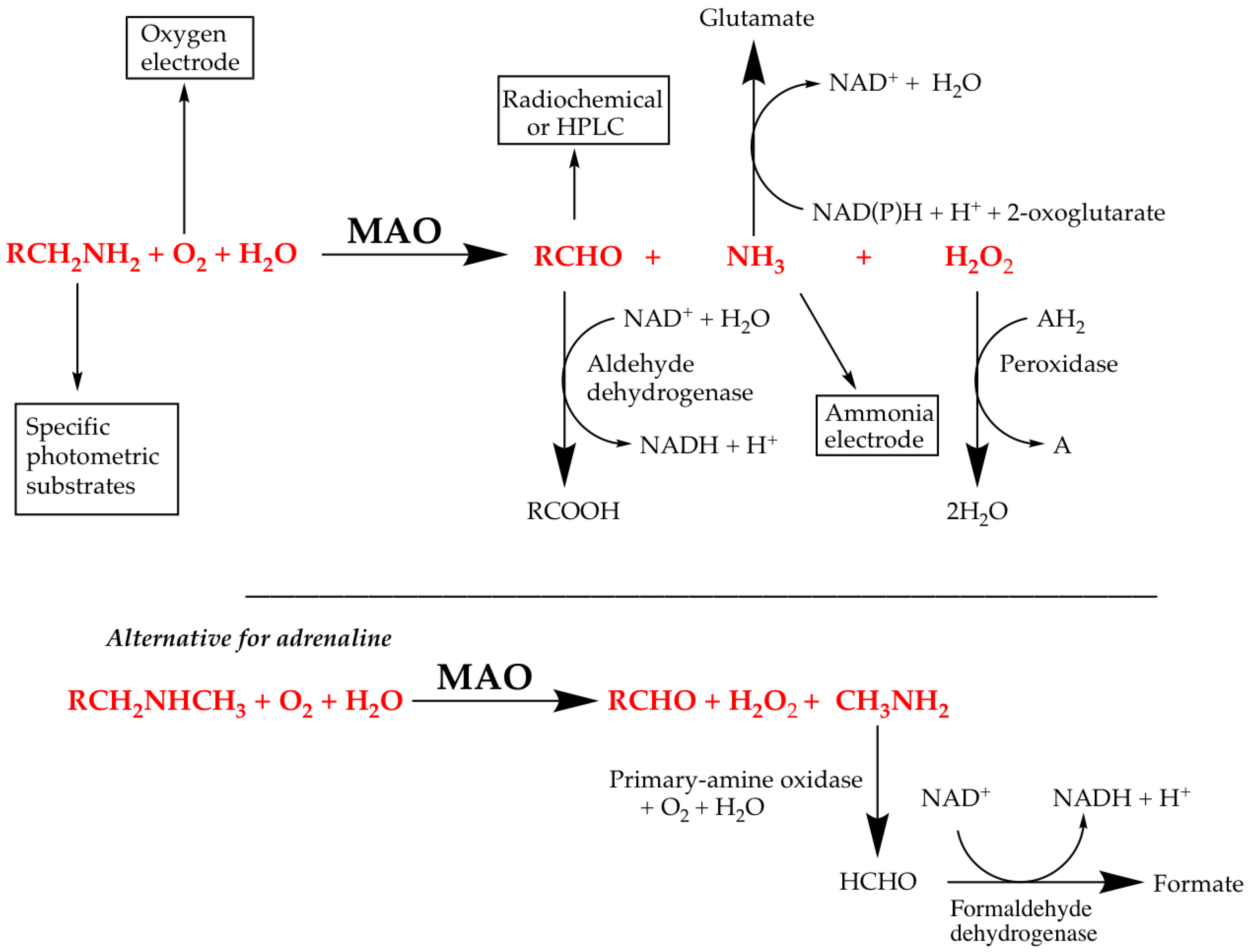
| Assay Type | Substrate | Coupling System | Detected Component | Detection |
|---|---|---|---|---|
| Direct - continuous | Any amine | - | Oxygen | Polarographic (oxygen electrode) |
| Benzylamine | - | Benzaldehyde | Absorbance (250 nm) | |
| Kynuramine | - | 4-Hydroxyquinoline | Absorbance (314 nm) | |
| Stopped | Radiolabelled amine | - | Radiolabelled aldehyde | Scintillation counter— after separation |
| Most Amines | Aldehyde - derivatization | Aldehyde derivative | HPLC | |
| Most Amines | Peroxidase | H2O2 → Dye (e.g., Luminol) | Luminescence | |
| Coupled | Most Amines | Aldehyde dehydrogenase | Aldehyde → NADH2 | Absorbance (340 nm) |
| Most Amines | Peroxidase | H2O2 → Dye (e.g., Amplex Red) | Fluorescence (e.g., resorufin; λex = 535 nm, λem = 595 nm) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramsay, R.R.; Tipton, K.F. Assessment of Enzyme Inhibition: A Review with Examples from the Development of Monoamine Oxidase and Cholinesterase Inhibitory Drugs. Molecules 2017, 22, 1192. https://doi.org/10.3390/molecules22071192
Ramsay RR, Tipton KF. Assessment of Enzyme Inhibition: A Review with Examples from the Development of Monoamine Oxidase and Cholinesterase Inhibitory Drugs. Molecules. 2017; 22(7):1192. https://doi.org/10.3390/molecules22071192
Chicago/Turabian StyleRamsay, Rona R., and Keith F. Tipton. 2017. "Assessment of Enzyme Inhibition: A Review with Examples from the Development of Monoamine Oxidase and Cholinesterase Inhibitory Drugs" Molecules 22, no. 7: 1192. https://doi.org/10.3390/molecules22071192
APA StyleRamsay, R. R., & Tipton, K. F. (2017). Assessment of Enzyme Inhibition: A Review with Examples from the Development of Monoamine Oxidase and Cholinesterase Inhibitory Drugs. Molecules, 22(7), 1192. https://doi.org/10.3390/molecules22071192