Cell-Free Production of Pentacyclic Triterpenoid Compound Betulinic Acid from Betulin by the Engineered Saccharomyces cerevisiae



Abstract
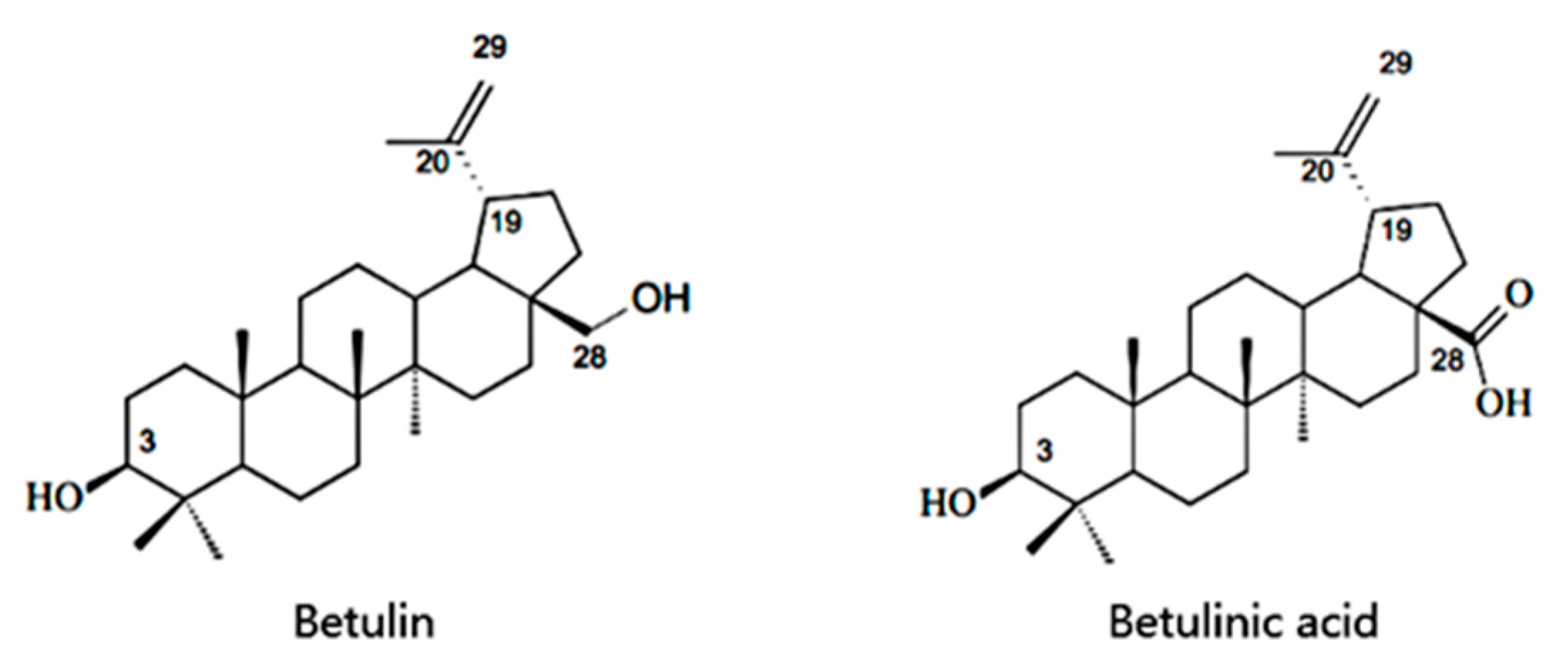
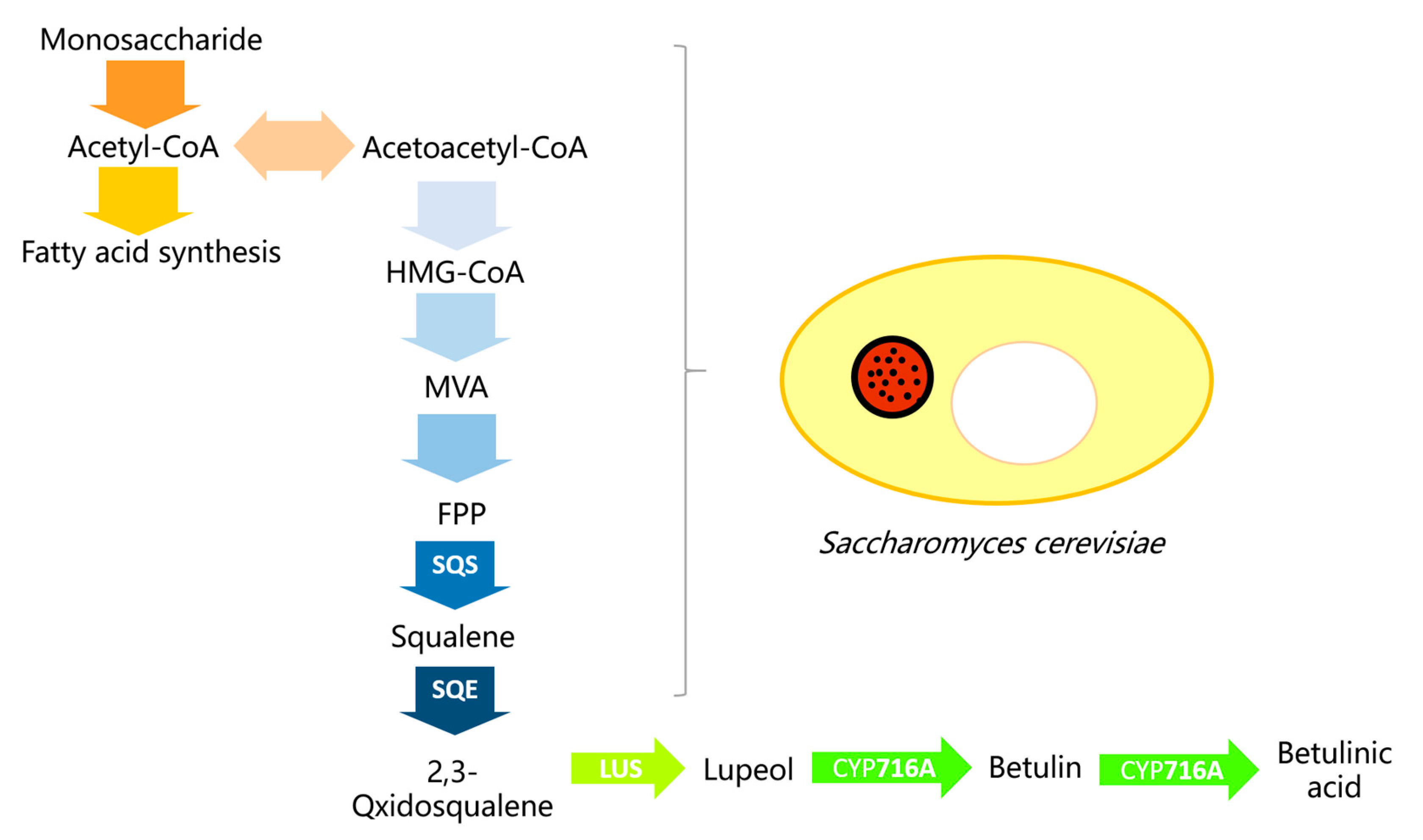
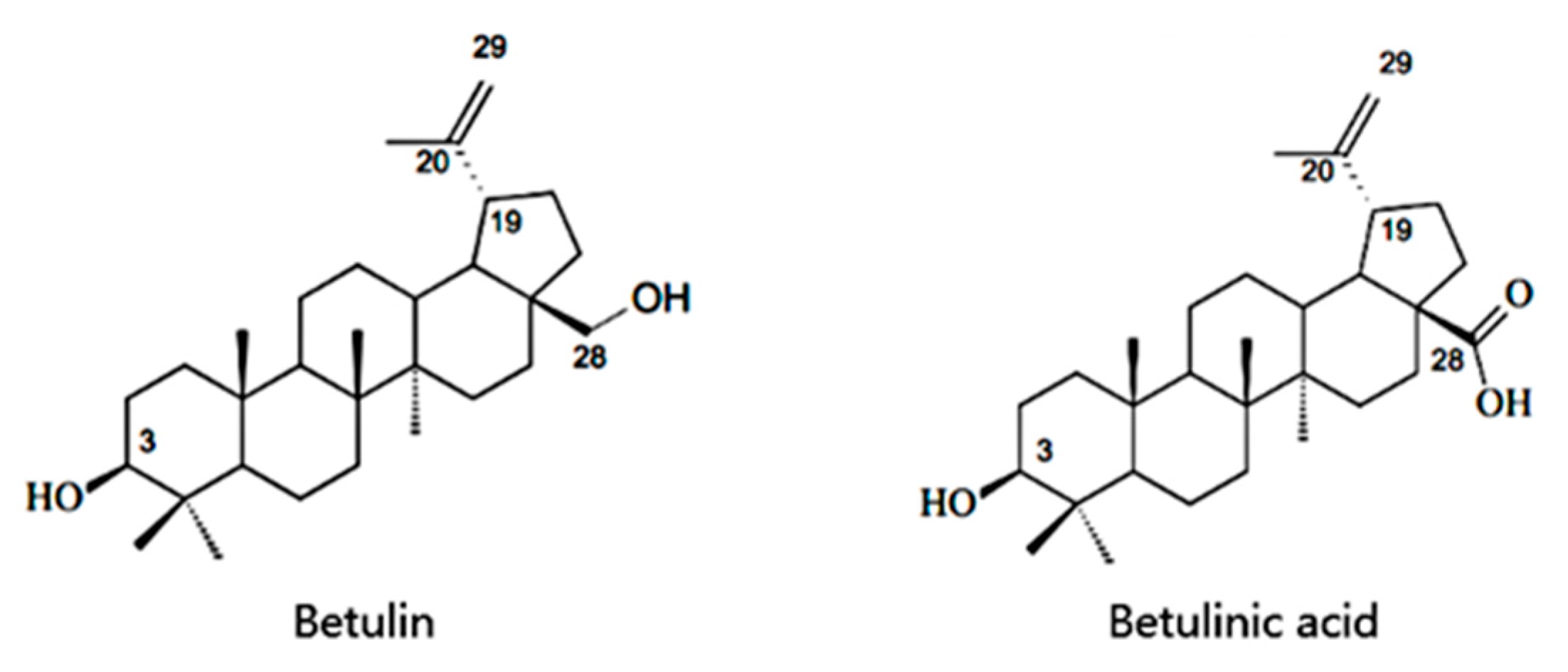
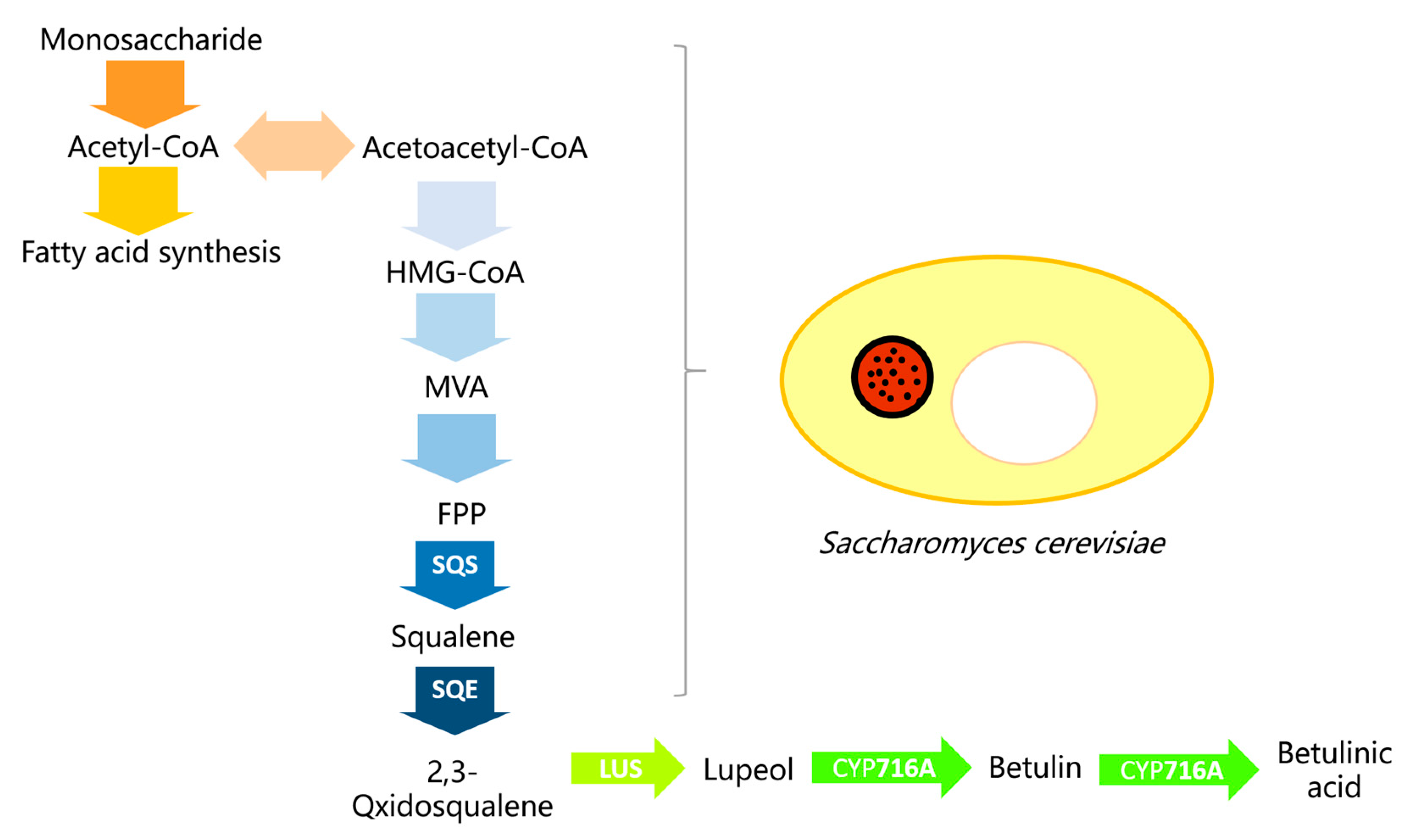
:1. Introduction
2. Results
2.1. Target Genes Cloning and the Construction of a Co-Expression System
2.2. Betulin Transformation and Optimization
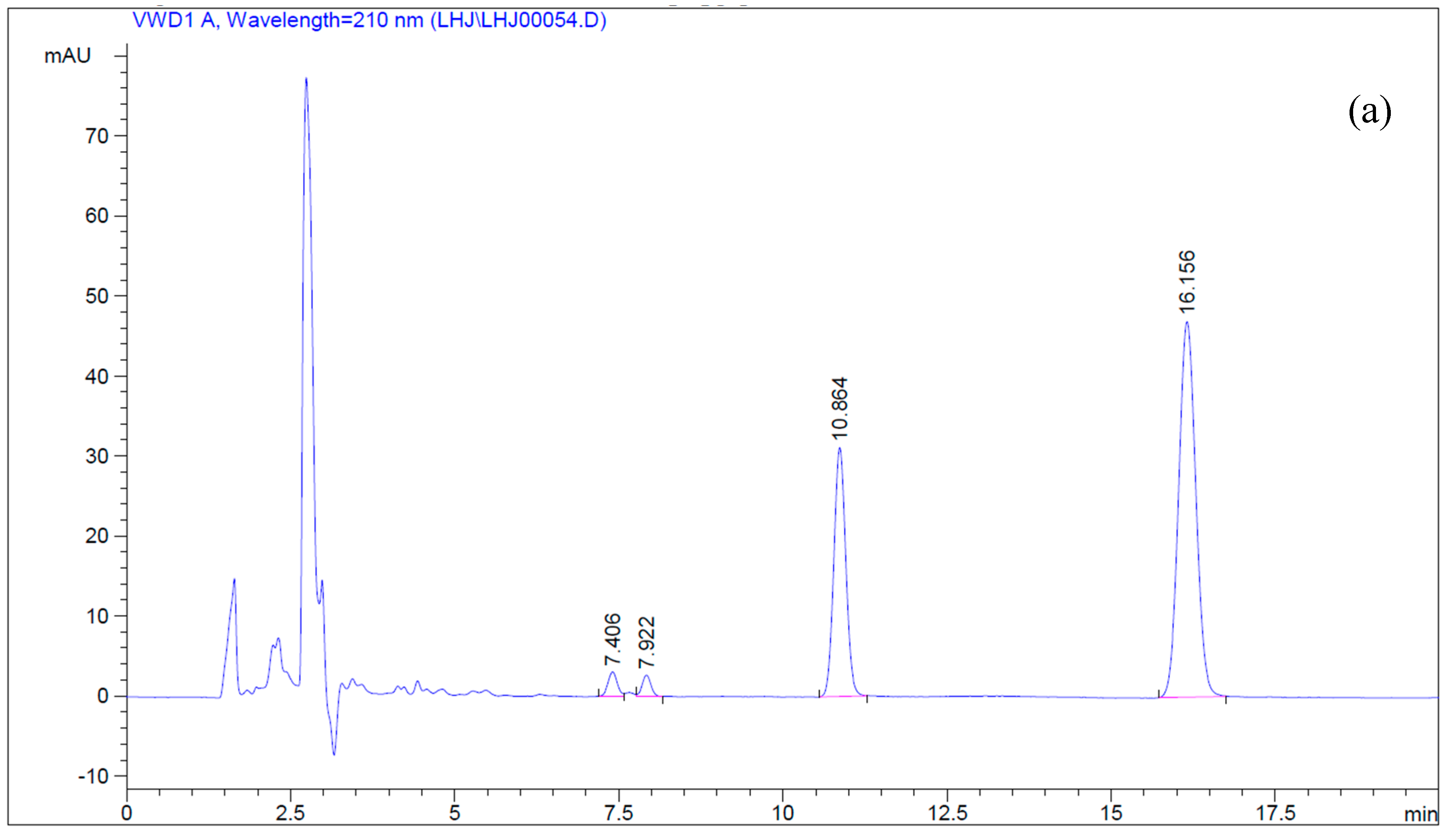
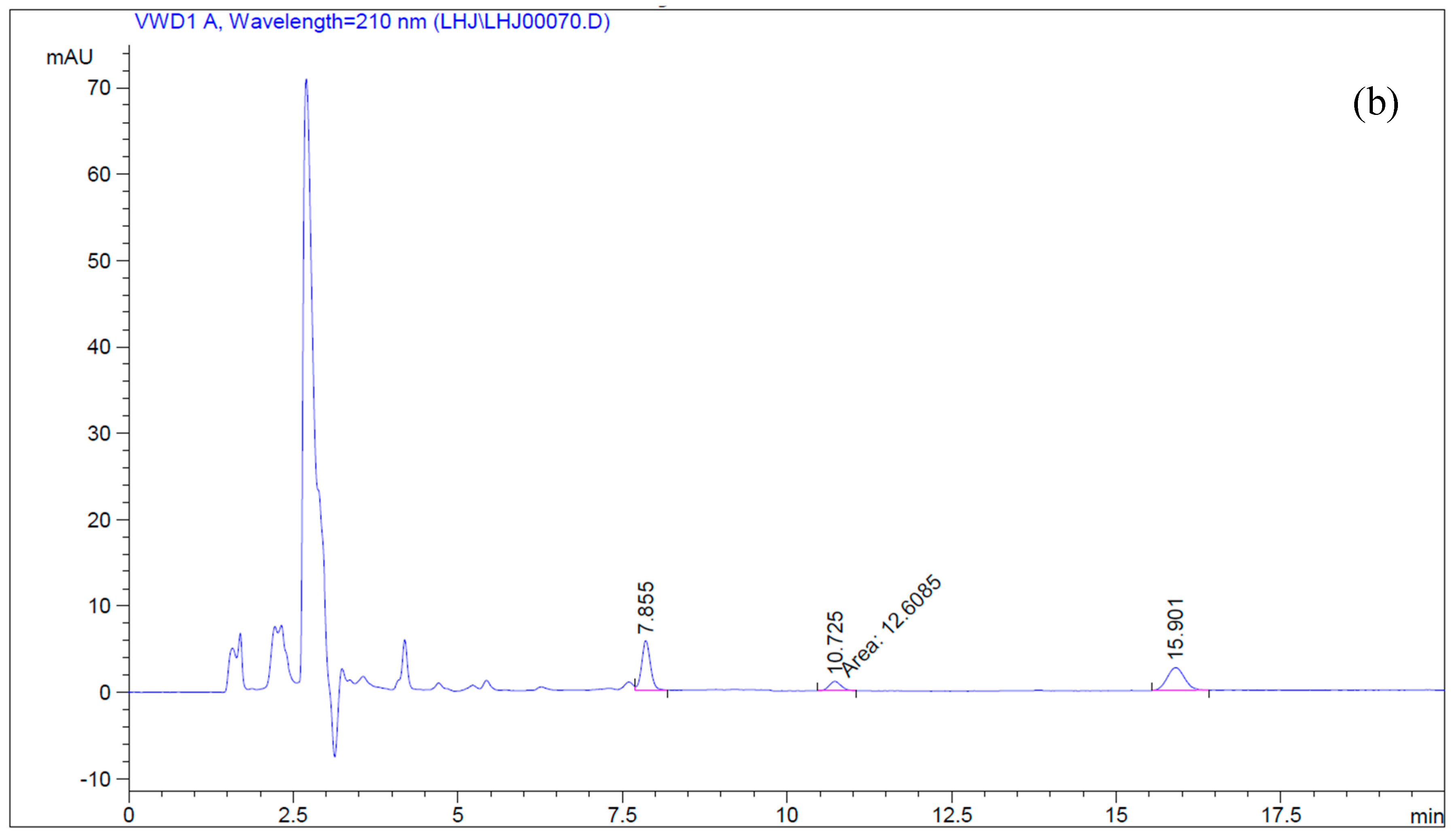
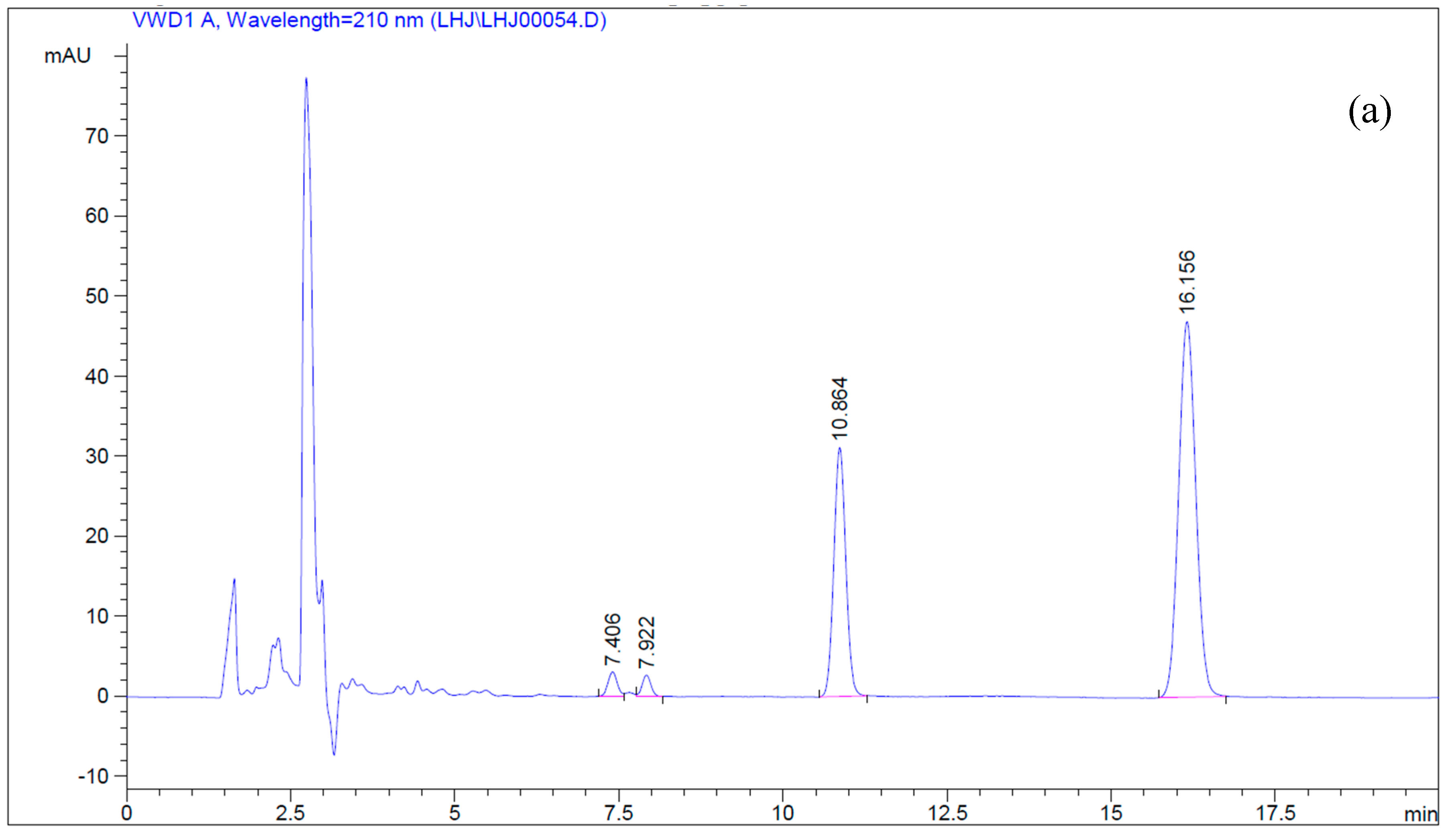
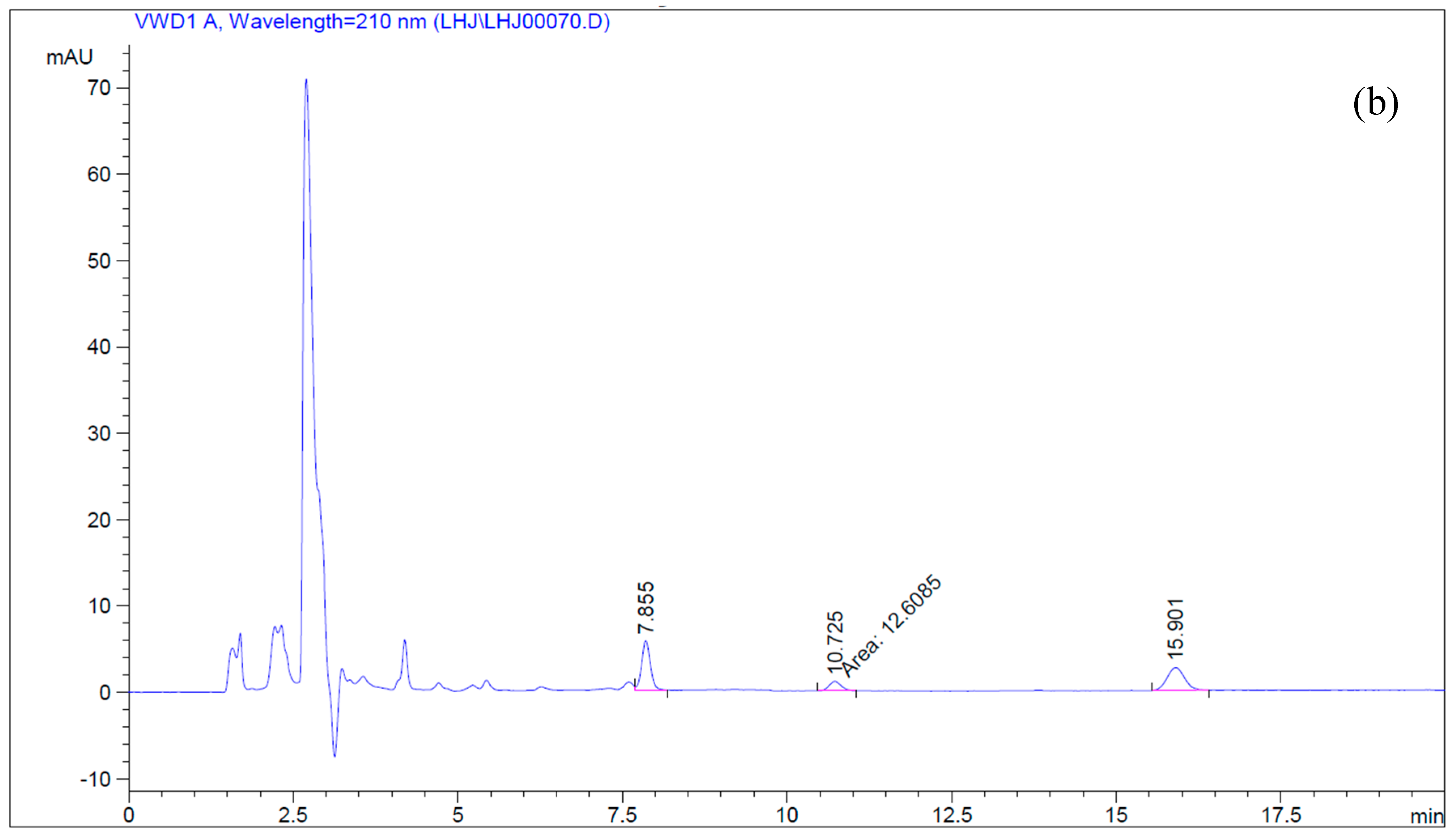
2.2.1. Microsome Preparation and Betulin Transformation
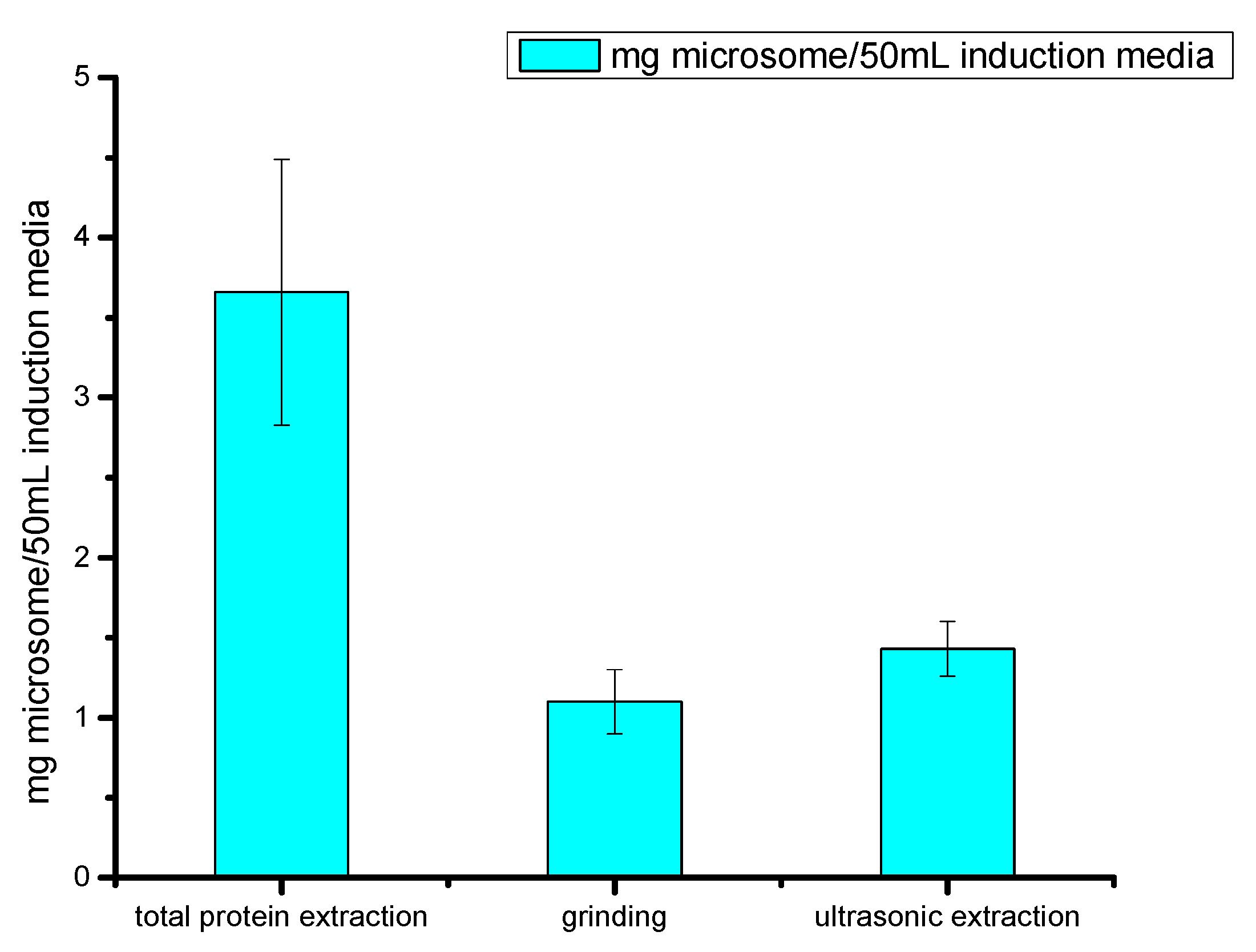
2.2.2. Optimization of Microsome Protein Preparation Methods
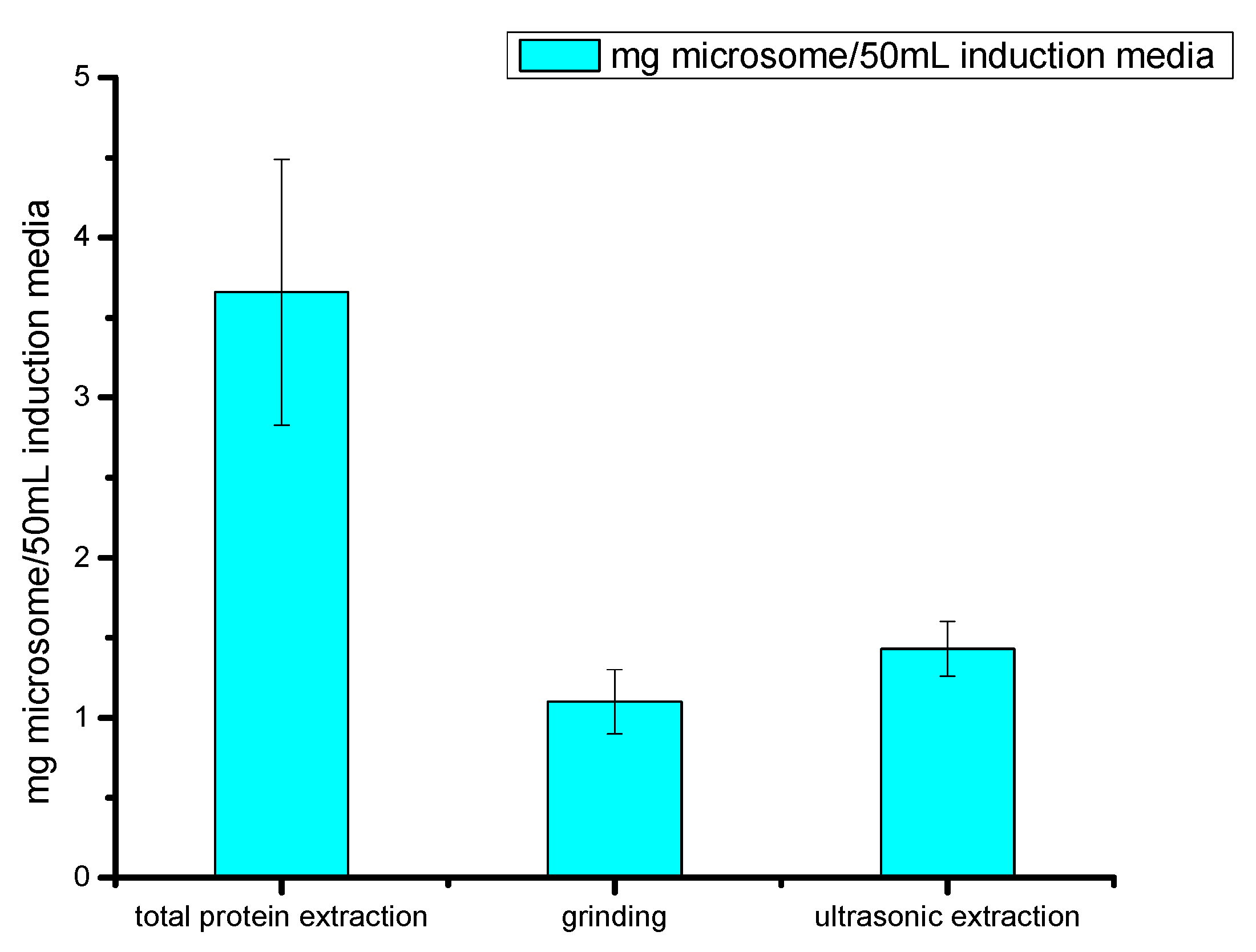
2.2.3. Comparison of Cell Fragmentation Methods
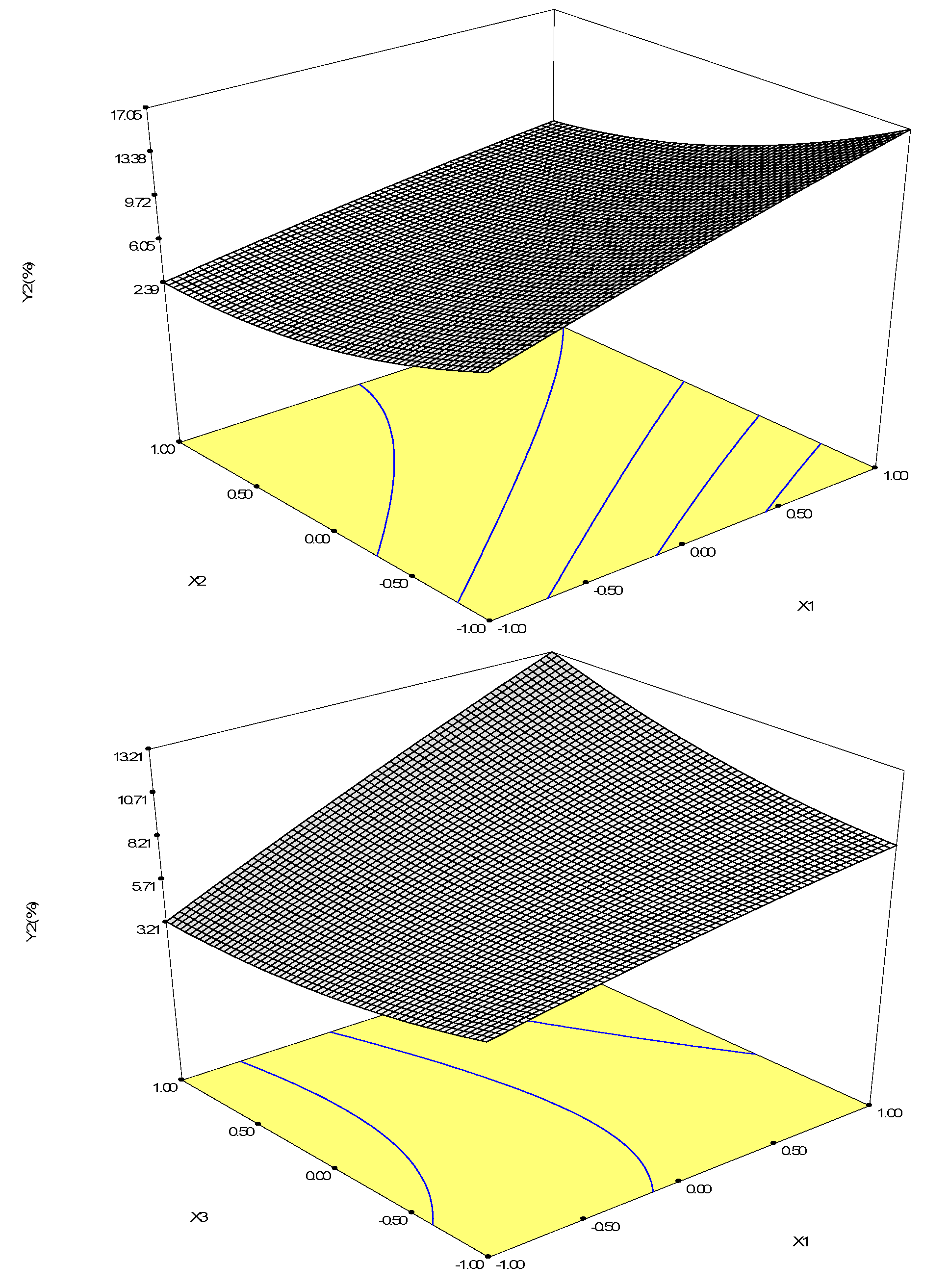
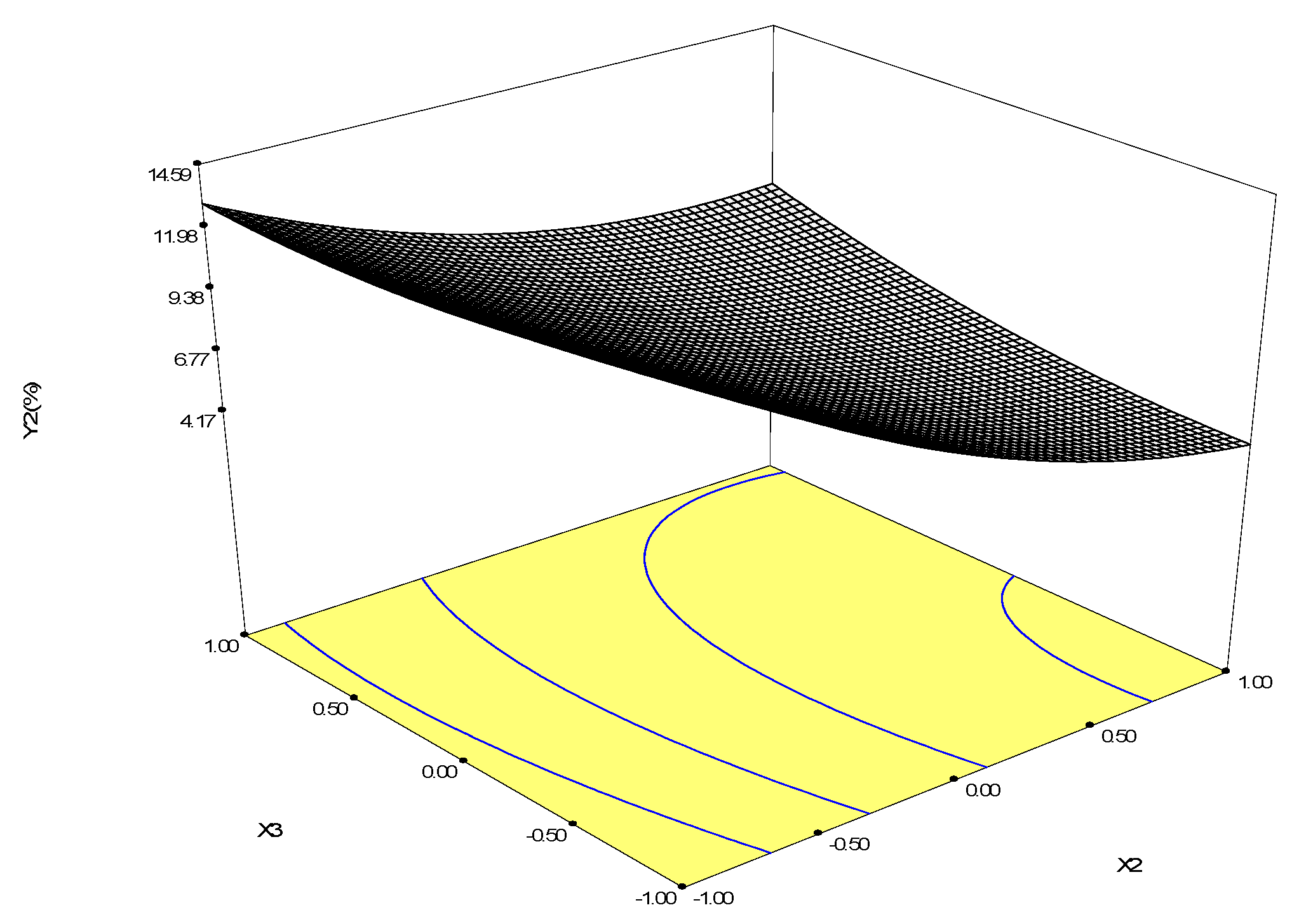
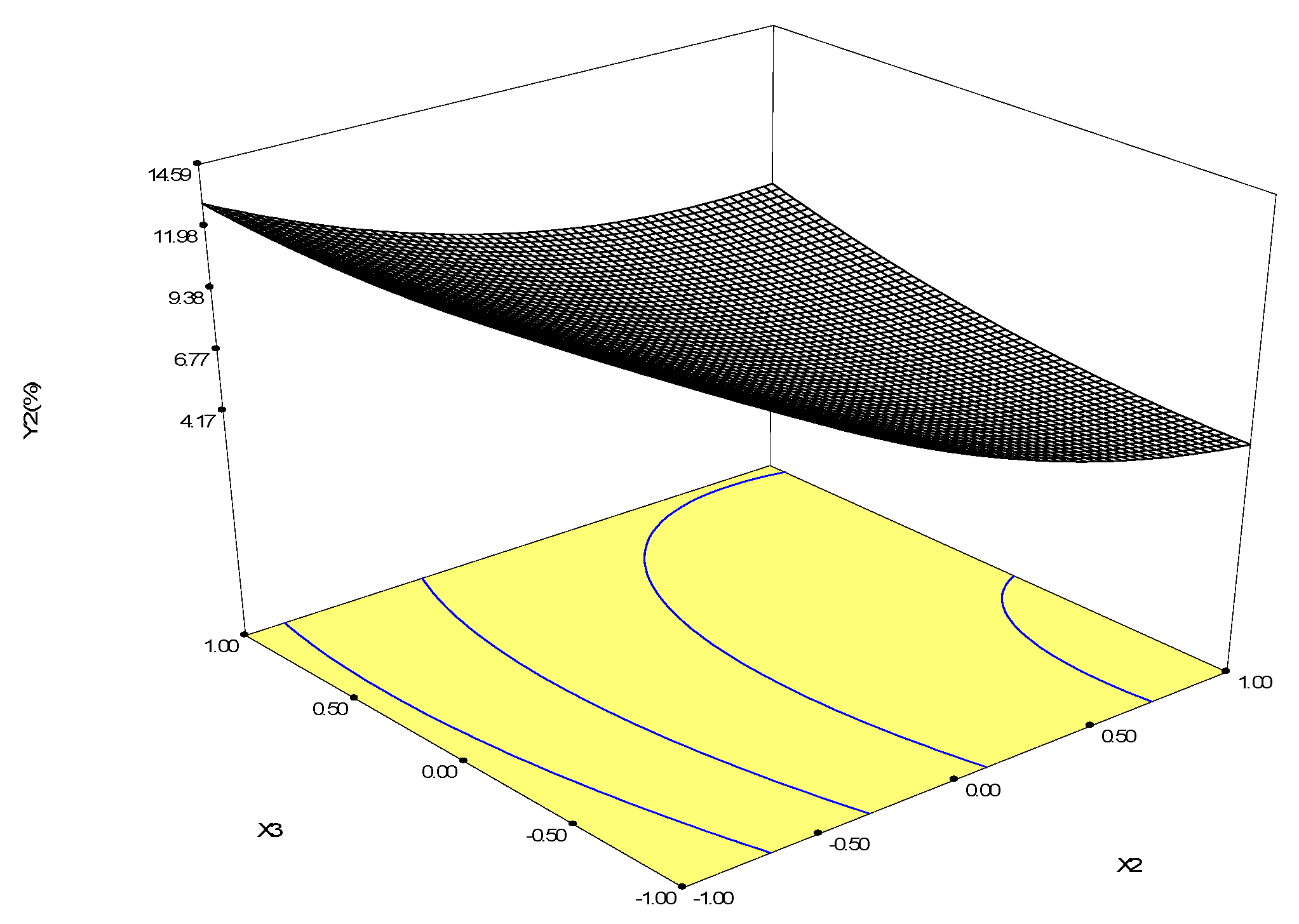
2.2.4. Optimization of Biotransformation Conditions by S. cerevisiae ZJUQH311 Microsome Protein In Vitro
3. Discussion
4. Materials and Methods
4.1. Full-Length cDNA Amplification
4.2. Experimental Strains and Plasmids
4.3. Yeast Expression and Microsome Preparation
4.4. Transformation Procedure and the Isolation of the Main Metabolites
4.5. Simultaneous Determination of Betulin and Betulinic Acid by Reverse-Phase HPLC
4.6. Comparison of Different Cell Disruption Methods
4.7. Response Surface Methodology Design
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yogeeswari, P.; Sriram, D. Betulinic acid and its derivatives: A review on their biological properties. Curr. Med. Chem. 2005, 12, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef]
- Jager, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants—Rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.L.; Chen, Q.H. Biotransformation optimization of betulin into betulinic acid production catalysed by cultured Armillaria luteo-virens Sacc ZJUQH100–6 cells. J. Appl. Microbiol. 2011, 110, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Ro, D.K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent Antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Ajikumar, P.K.; Xiao, W.-H.; Tyo, K.E.J.; Wang, Y.; Simeon, F.; Leonard, E.; Mucha, O.; Phon, T.H.; Pfeifer, B.; Stephanopoulos, G. Isoprenoid Pathway Optimization for Taxol Precursor Overproduction in Escherichia coli. Science 2010, 330, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, E.O.; Seki, H.; Ohyama, K.; Ono, E.; Umemoto, N.; Mizutani, M.; Saito, K.; Muranaka, T. CYP716A subfamily members are multifunctional oxidases in triterpenoid biosynthesis. Plant Cell Physiol. 2011, 52, 2050–2061. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Kim, H.J.; Kwon, Y.S.; Choi, Y.E. The Cyt P450 enzyme CYP716A47 catalyzes the formation of protopanaxadiol from dammarenediol-II during ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol. 2011, 52, 2062–2073. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, J.; Ye, H.; Li, C.; Wang, H.; Liu, B.; Zhang, Y. Molecular characterization of the pentacyclic triterpenoid biosynthetic pathway in Catharanthus roseus. Planta 2012, 236, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, J.; Li, C.; Zhang, Y. Improvement of betulinic acid biosynthesis in yeast employing multiple strategies. BMC Biotechnol. 2016, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.-W.; Zhang, J.; Liu, J.-H.; Yu, B.-Y. Direct microbial-catalyzed asymmetric α-hydroxylation of betulonic acid by Nocardia sp. NRRL 5646. Tetrahedron Lett. 2009, 50, 2193–2195. [Google Scholar] [CrossRef]
- Fu, M.-L.; Liu, J.; Dong, Y.-C.; Feng, Y.; Fang, R.-S.; Chen, Q.-H.; Liu, X.-J. Effect of ionic liquid-containing system on betulinic acid production from betulin biotransformation by cultured Armillaria luteo-virens Sacc cells. Eur. Food Res. Technol. 2011, 233, 507–515. [Google Scholar]
- Tarasova, E.V.; Grishko, V.V.; Ivshina, I.B. Cell adaptations of Rhodococcus rhodochrous IEGM 66 to betulin biotransformation. Process Biochem. 2017, 52, 1–9. [Google Scholar] [CrossRef]
- Bezerra, M.A.; Santelli, R.E.; Oliveira, E.P.; Villar, L.S.; Escaleira, L.A. Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta 2008, 76, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Galgon, T.; Hoke, D.; Drager, B. Identification and quantification of betulinic acid. Phytochem. Anal. 1999, 10, 187–190. [Google Scholar] [CrossRef]
- Tezuka, Y.; Stampoulis, P.; Banskota, A.H.; Awale, S.; Tran, K.Q.; Saiki, I.; Kadota, S. Constituents of the Vietnamese medicinal plant Orthosiphon stamineus. Chem. Pharm. Bull. 2000, 48, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Machado, D.G.; Cunha, M.P.; Neis, V.B.; Balen, G.O.; Colla, A.; Bettio, L.E.; Oliveira, A.; Pazini, F.L.; Dalmarco, J.B.; Simionatto, E.L.; et al. Antidepressant-like effects of fractions, essential oil, carnosol and betulinic acid isolated from Rosmarinus officinalis L. Food Chem. 2013, 136, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Kroemer, G. Targeting mitochondrial apoptosis by betulinic acid in human cancers. Drug Dis. Today 2009, 14, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, T.; Kashiwada, Y.; Kilkuskie, R.E.; Cosentino, L.M.; Ballas, L.M.; Jiang, J.B.; Janzen, W.P.; Chen, I.S.; Lee, K.H. Anti-aids agents, 11. betulinic acid and platanic acid as anti-hiv principles from syzigium-claviflorum, and the anti-HIV activity of structurally related triterpenoids. J. Nat. Prod. 1994, 57, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Kashiwada, Y.; Hashimoto, F.; Cosentino, L.M.; Chen, C.H.; Garrett, P.E.; Lee, K.H. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J. Med. Chem. 1996, 39, 1016–1017. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, V.; Kovacs, J.; Koczka, I. Occurrence of betulinic acid in the bark of the plane tree. J. Chem. Soc. 1948, 1, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R. Plant cytochrome P450s from moss to poplar. Phytochem. Rev. 2006, 5, 193–204. [Google Scholar] [CrossRef]
- Schuler, M.A. Plant cytochrome P450 monooxygenases. Crit. Rev. Plant Sci. 1996, 15, 235–284. [Google Scholar] [CrossRef]
- Seki, H.; Ohyama, K.; Sawai, S.; Mizutani, M.; Ohnishi, T.; Sudo, H.; Akashi, T.; Aoki, T.; Saito, K.; Muranaka, T. Licorice beta-amyrin 11-oxidase, a cytochrome P450 with a key role in the biosynthesis of the triterpene sweetener glycyrrhizin. Proc. Natl. Acad. Sci. USA 2008, 105, 14204–14209. [Google Scholar] [CrossRef] [PubMed]
- Pompon, D.; Louerat, B.; Bronine, A.; Urban, P. Yeast expression of animal and plant P450s in optimized redox environments. Methods Enzymol. 1996, 272, 51–64. [Google Scholar] [PubMed]
- Urban, P.; Mignotte, C.; Kazmaier, M.; Delorme, F.; Pompon, D. Cloning, yeast expression, and characterization of the coupling of two distantly related Arabidopsis thaliana NADPH-Cytochrome P450 reductases with P450 CYP73A5. J. Biol. Chem. 1997, 272, 19176–19186. [Google Scholar] [CrossRef] [PubMed]
- Kizer, L.; Pitera, D.J.; Pfleger, B.F.; Keasling, J.D. Application of functional genomics to pathway optimization for increased isoprenoid production. Appl. Environ. Microbiol. 2008, 74, 3229–3241. [Google Scholar] [CrossRef] [PubMed]
- Ehsani, M.; Fernandez, M.R.; Biosca, J.A.; Dequin, S. Reversal of coenzyme specificity of 2,3-butanediol dehydrogenase from Saccharomyces cerevisae and in vivo functional analysis. Biotechnol. Bioeng. 2009, 104, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Y. Modulating betulinic acid production in Saccharomyces cerevisiae by managing the intracellular supplies of the co-factor NADPH and oxygen. J. Biosci. Bioeng. 2015, 119, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Celton, M.; Goelzer, A.; Camarasa, C.; Fromion, V.; Dequin, S. A constraint-based model analysis of the metabolic consequences of increased NADPH oxidation in Saccharomyces cerevisiae. Met. Eng. 2012, 14, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Bhat, P.J.; Oh, D.; Hopper, J.E. Analysis of the GAL3 signal transduction pathway activating GAL4 protein-dependent transcription in Saccharomyces-cerevisiae. Genetics 1990, 125, 281–291. [Google Scholar] [PubMed]
- Olsen, K.M.; Hehn, A.; Jugde, H.; Slimestad, R.; Larbat, R.; Bourgaud, F.; Lillo, C. Identification and characterisation of CYP75A31, a new flavonoid 3′5′-hydroxylase, isolated from Solanum lycopersicum. BMC Plant Biol. 2010, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Ohta, D. Two isoforms of NADPH: Cytochrome P450 reductase in Arabidopsis thaliana—Gene structure, heterologous expression in insect cells, and differential regulation. Plant Physiol. 1998, 116, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.L.C.; Bruns, R.E.; Ferreira, H.S.; Matos, G.D.; David, J.M.; Brandao, G.C.; da Silva, E.G.P.; Portugal, L.A.; dos Reis, P.S.; Souza, A.S.; et al. Box-Behnken design: An alternative for the optimization of analytical methods. Anal. Chim. Acta 2007, 597, 179–186. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Runs | X1 | X2 | X3 | Y1 (Betulin Conversion Rate, %) | Y2 (Betulinic Acid Yield, %) |
|---|---|---|---|---|---|
| 1 | 0 | 1 | −1 | 21.58 | 3.33 |
| 2 | −1 | 0 | −1 | 61.76 | 4.14 |
| 3 | 0 | −1 | −1 | 20.06 | 16.93 |
| 4 | 0 | 0 | 0 | 30.28 | 6.33 |
| 5 | 1 | 0 | −1 | 35.87 | 9.10 |
| 6 | −1 | 1 | 0 | 57.02 | 4.81 |
| 7 | −1 | −1 | 0 | 58.74 | 7.63 |
| 8 | 0 | 1 | 1 | 33.48 | 5.16 |
| 9 | 1 | −1 | 0 | 45.52 | 14.67 |
| 10 | −1 | 0 | 1 | 57.25 | 3.23 |
| 11 | 0 | −1 | 1 | 38.83 | 13.83 |
| 12 | 1 | 1 | 0 | 41.66 | 7.96 |
| 13 | 0 | 0 | 0 | 37.10 | 7.87 |
| 14 | 0 | 0 | 0 | 32.05 | 7.56 |
| 15 | 0 | 0 | 0 | 36.89 | 7.06 |
| 16 | 0 | 0 | 0 | 35.12 | 7.32 |
| 17 | 1 | 0 | 1 | 39.87 | 14.75 |
| Source | Sum of Squares | DF | Mean Square | F Value | Prob > F |
|---|---|---|---|---|---|
| Model | 254.21 | 9 | 28.25 | 6.36 | 0.0117 |
| X1 | 88.97 | 1 | 88.97 | 20.05 | 0.0029 |
| X2 | 126.38 | 1 | 126.38 | 28.48 | 0.0011 |
| X3 | 1.50 | 1 | 1.50 | 0.34 | 0.5793 |
| X12 | 0.23 | 1 | 0.23 | 0.05 | 0.8247 |
| X22 | 13.25 | 1 | 13.25 | 2.99 | 0.1276 |
| X32 | 2.76 | 1 | 2.76 | 0.62 | 0.4564 |
| X1X2 | 3.80 | 1 | 3.80 | 0.86 | 0.3856 |
| X1X3 | 10.76 | 1 | 10.76 | 2.42 | 0.1634 |
| X2X3 | 6.07 | 1 | 6.07 | 1.37 | 0.2805 |
| Residual | 31.07 | 7 | 4.44 | ||
| Lack of Fit | 29.71 | 3 | 9.90 | 29.18 | 0.0035 |
| Pure error | 1.36 | 4 | 0.34 | ||
| Cor Total | 285.27 | 16 |
| Name | Sequence (5′-3′) | Restriction Enzyme |
|---|---|---|
| C-F | AGGAGAAAAAACCCCGGATCCATGGAGCCTAATTTCTATCTCTCCCT | BamHI |
| C-R | TTAGAGCGGATCTTAGCTAGCTTAAGCTTTGTGTGGATAAAGGCGA | NheI |
| A-F | AACCCTCACTAAAGGGCGGCCGCATGACTTCTGCTTTGTATGCTTCC | NotI |
| A-R | GTTAATTAAGAGCTCAGATCTTCACCAGACATCTCTGAGGTATC | BglII |
| GAL1-F | GGTAATTAATCAGCGAAGCGATG | |
| GAL1-R | CGAGTCAGTGAGCGAGGAA | |
| GAL10-F | GGTGGTAATGCCATGTAATATG | |
| GAL1-R | GGCAAGGTAGACAAGCCGACAAC |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Niu, Y.; Bakur, A.; Li, H.; Chen, Q. Cell-Free Production of Pentacyclic Triterpenoid Compound Betulinic Acid from Betulin by the Engineered Saccharomyces cerevisiae. Molecules 2017, 22, 1075. https://doi.org/10.3390/molecules22071075
Wu J, Niu Y, Bakur A, Li H, Chen Q. Cell-Free Production of Pentacyclic Triterpenoid Compound Betulinic Acid from Betulin by the Engineered Saccharomyces cerevisiae. Molecules. 2017; 22(7):1075. https://doi.org/10.3390/molecules22071075
Chicago/Turabian StyleWu, Jianan, Yongwu Niu, Abdelmoneim Bakur, Hao Li, and Qihe Chen. 2017. "Cell-Free Production of Pentacyclic Triterpenoid Compound Betulinic Acid from Betulin by the Engineered Saccharomyces cerevisiae" Molecules 22, no. 7: 1075. https://doi.org/10.3390/molecules22071075
APA StyleWu, J., Niu, Y., Bakur, A., Li, H., & Chen, Q. (2017). Cell-Free Production of Pentacyclic Triterpenoid Compound Betulinic Acid from Betulin by the Engineered Saccharomyces cerevisiae. Molecules, 22(7), 1075. https://doi.org/10.3390/molecules22071075