
Synthesis and Evaluation of Novel 2-Pyrrolidone-Fused (2-Oxoindolin-3-ylidene)methylpyrrole Derivatives as Potential Multi-Target Tyrosine Kinase Receptor Inhibitors

Abstract
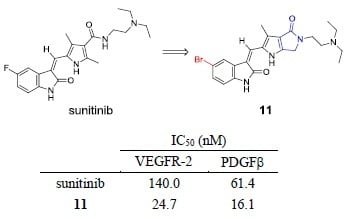
:
1. Introduction
2. Results and Discussion
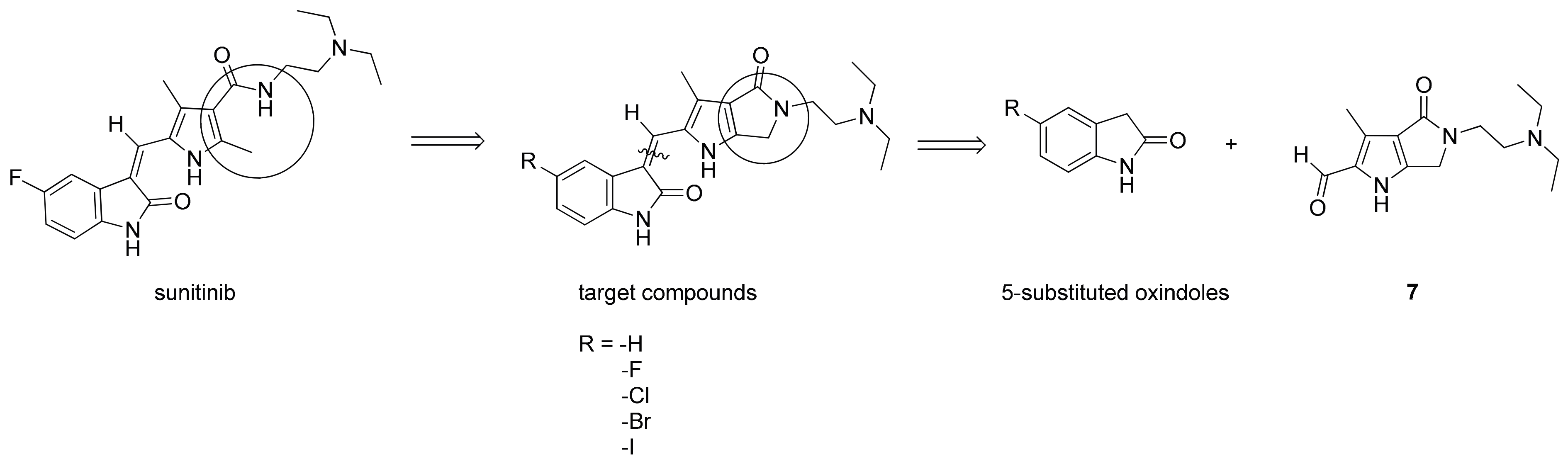
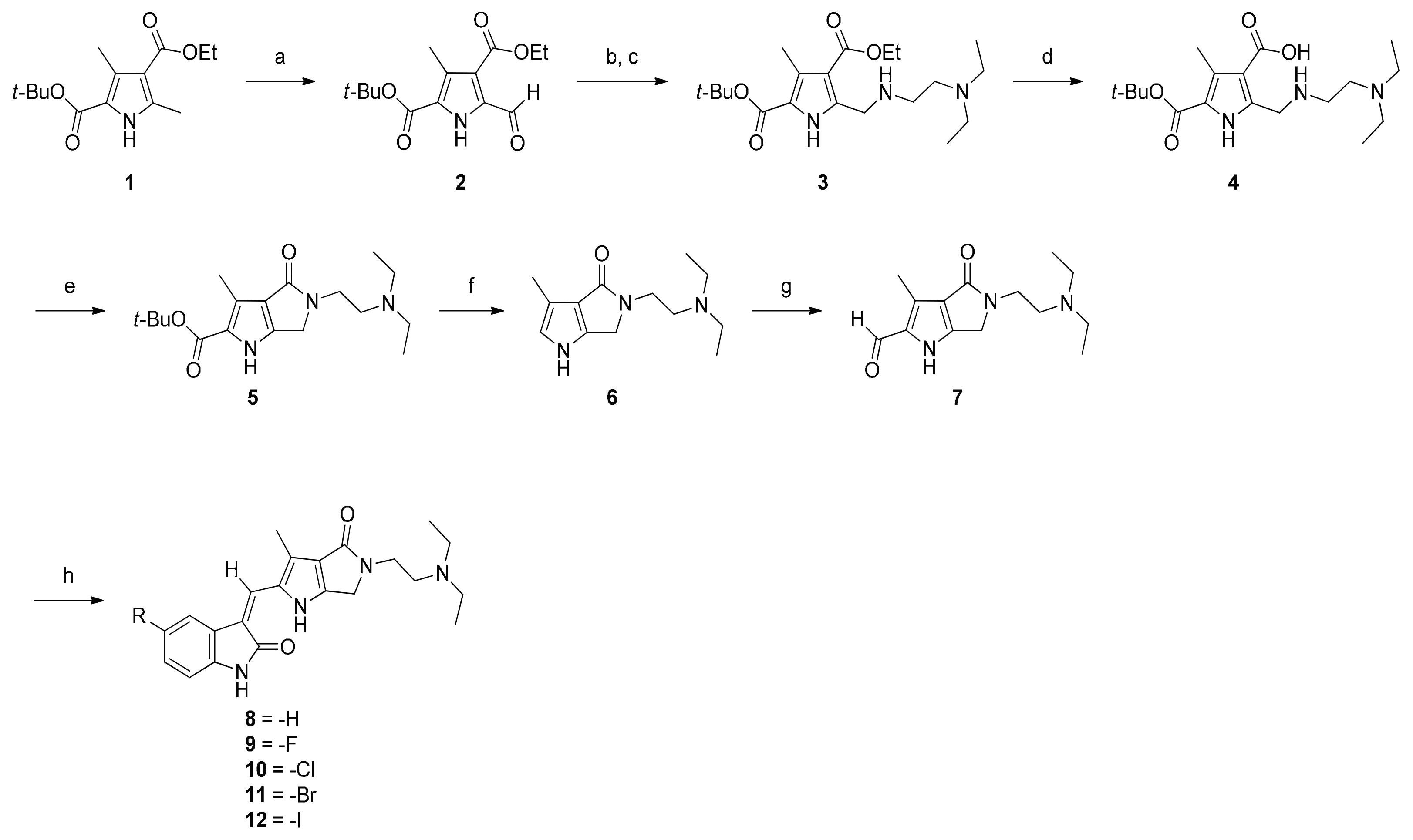
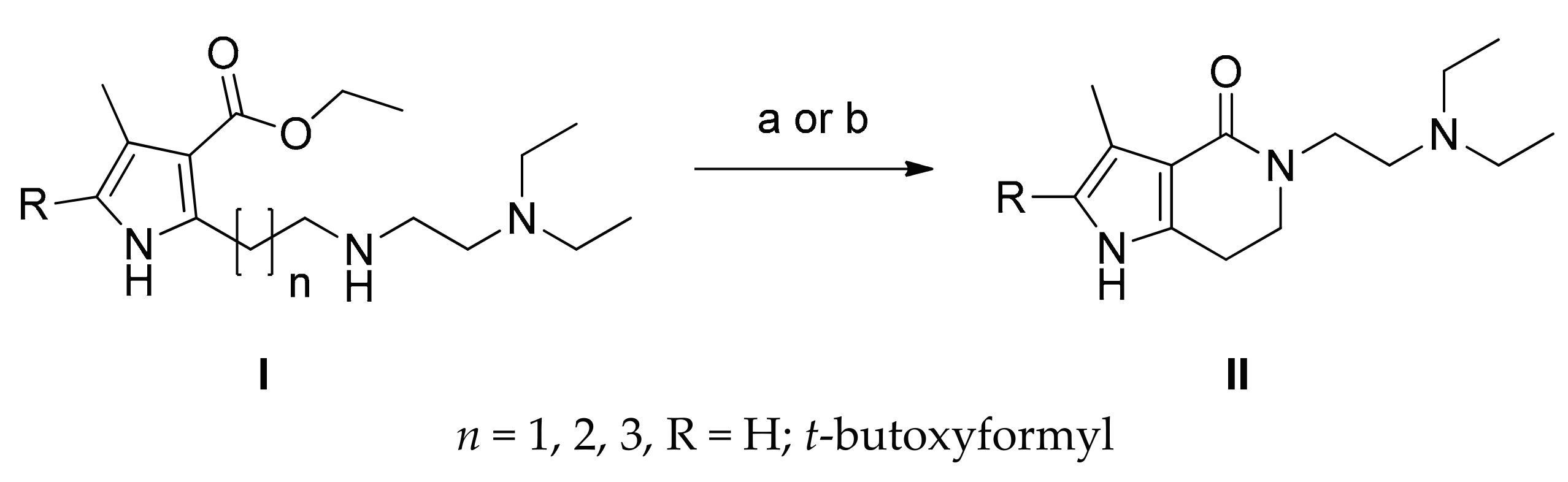
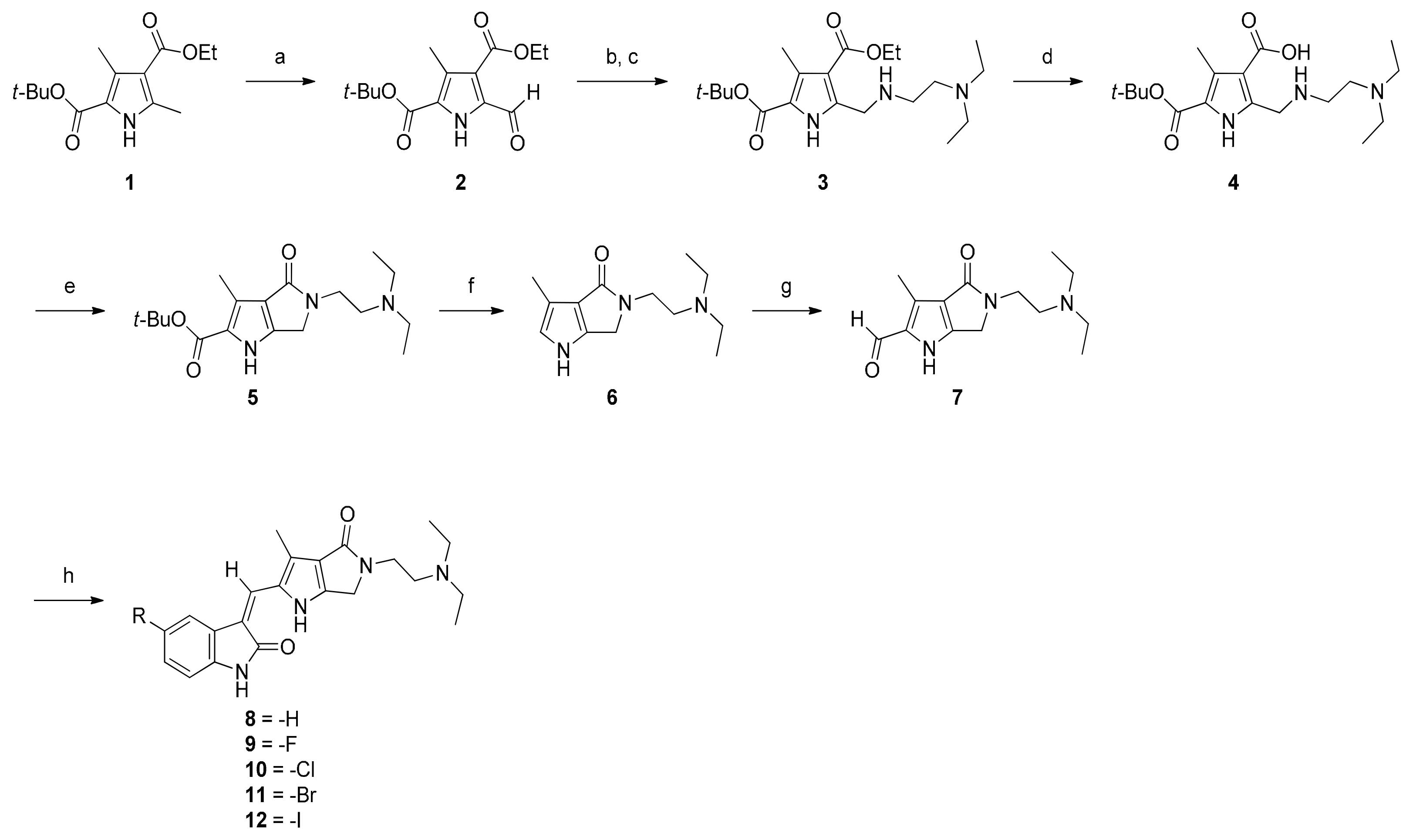
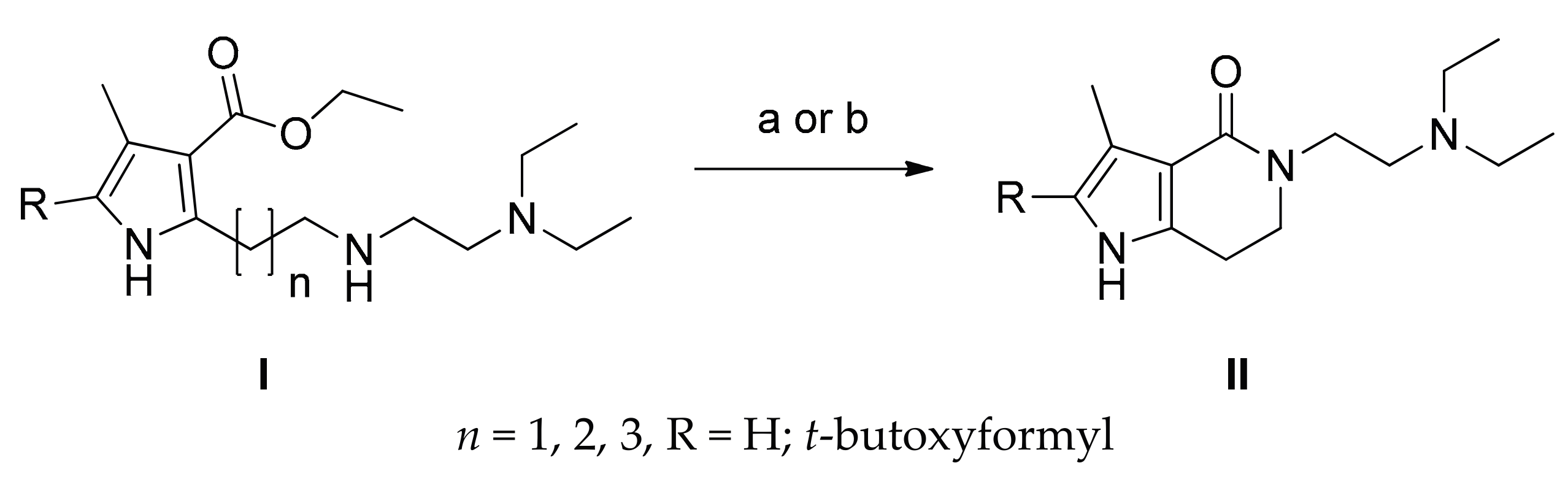
2.1. Chemistry
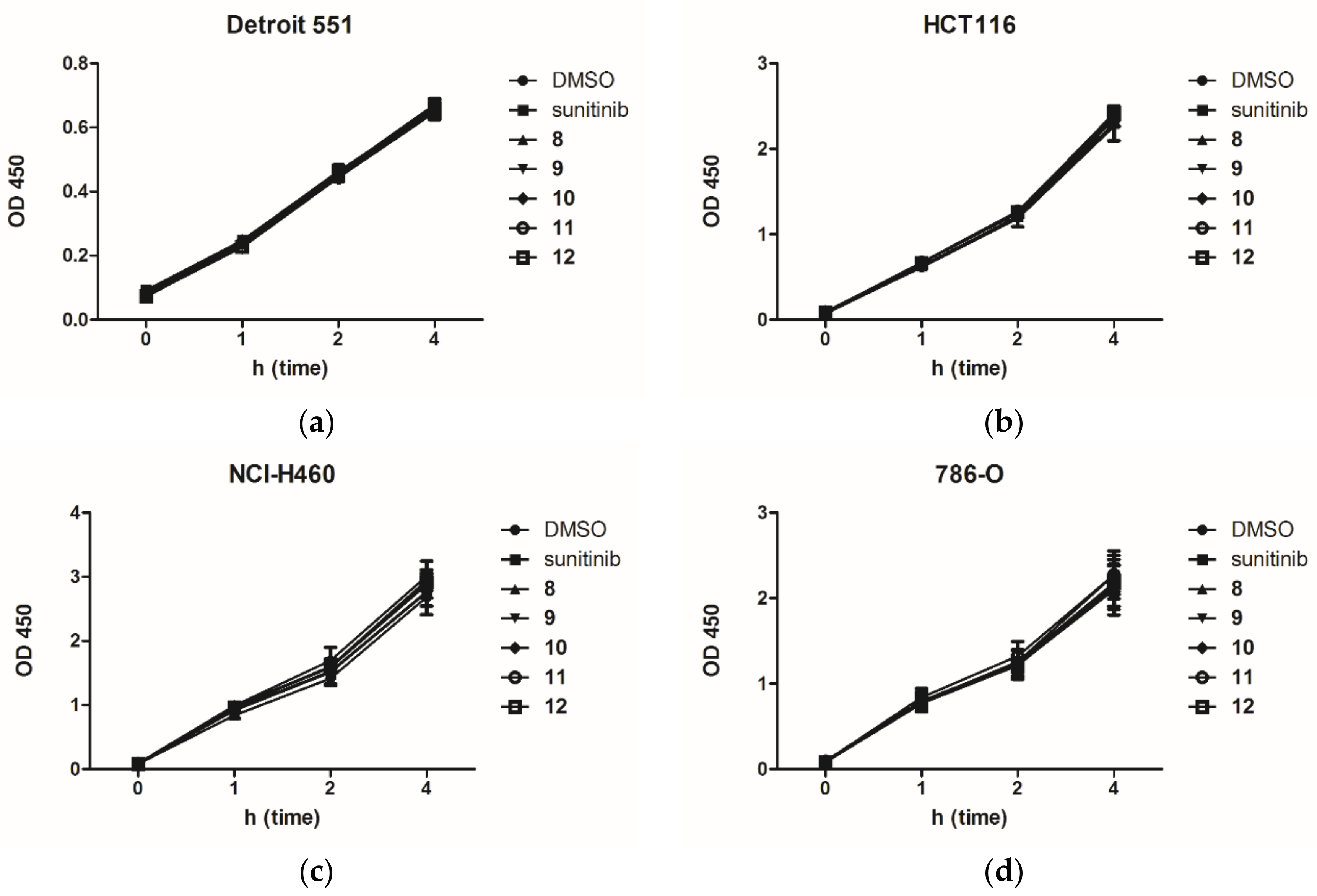
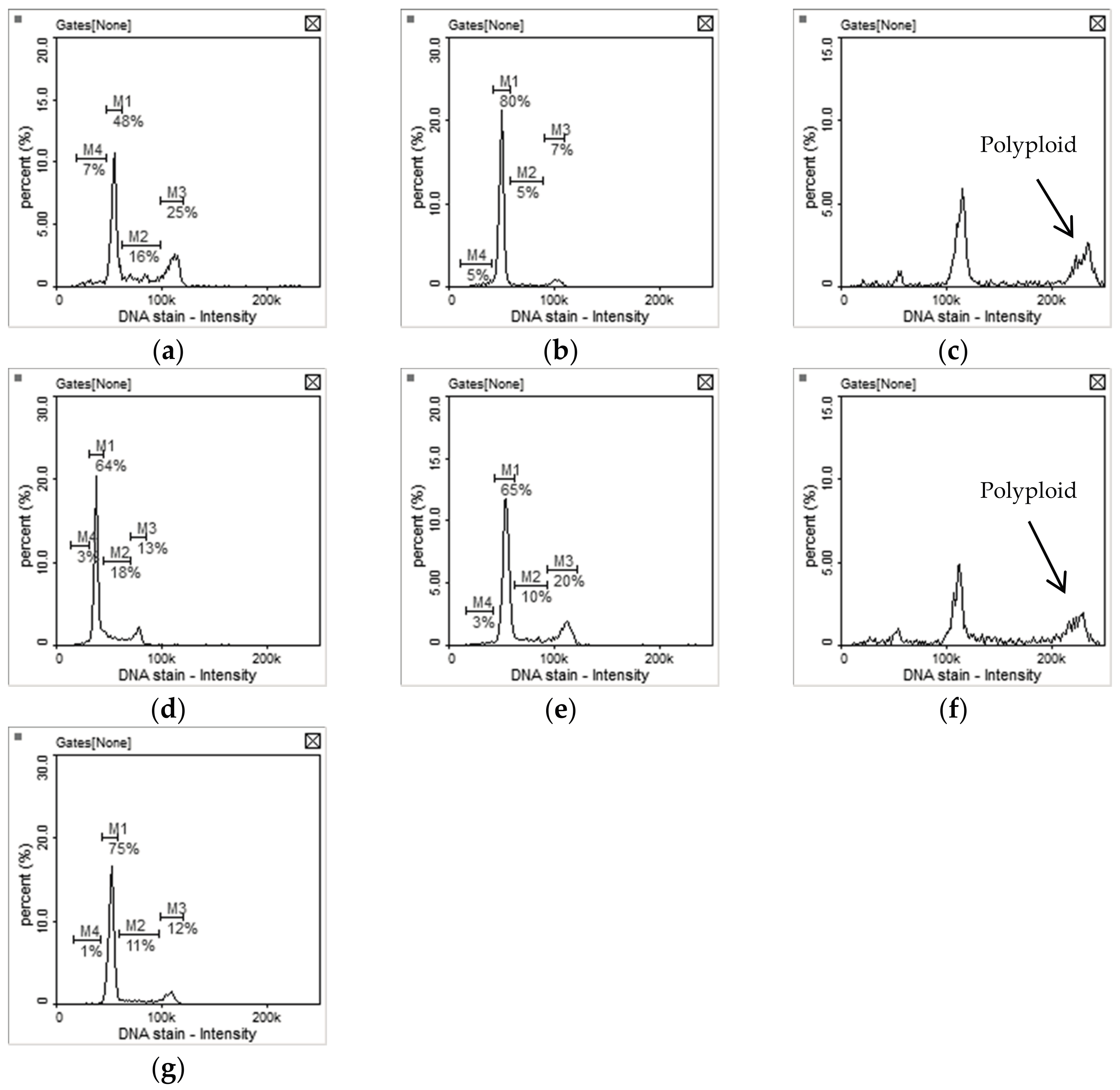
2.2. Anti-Proliferation Activity and Acute Cytotoxicity
2.3. Kinase Inhibitory Assays
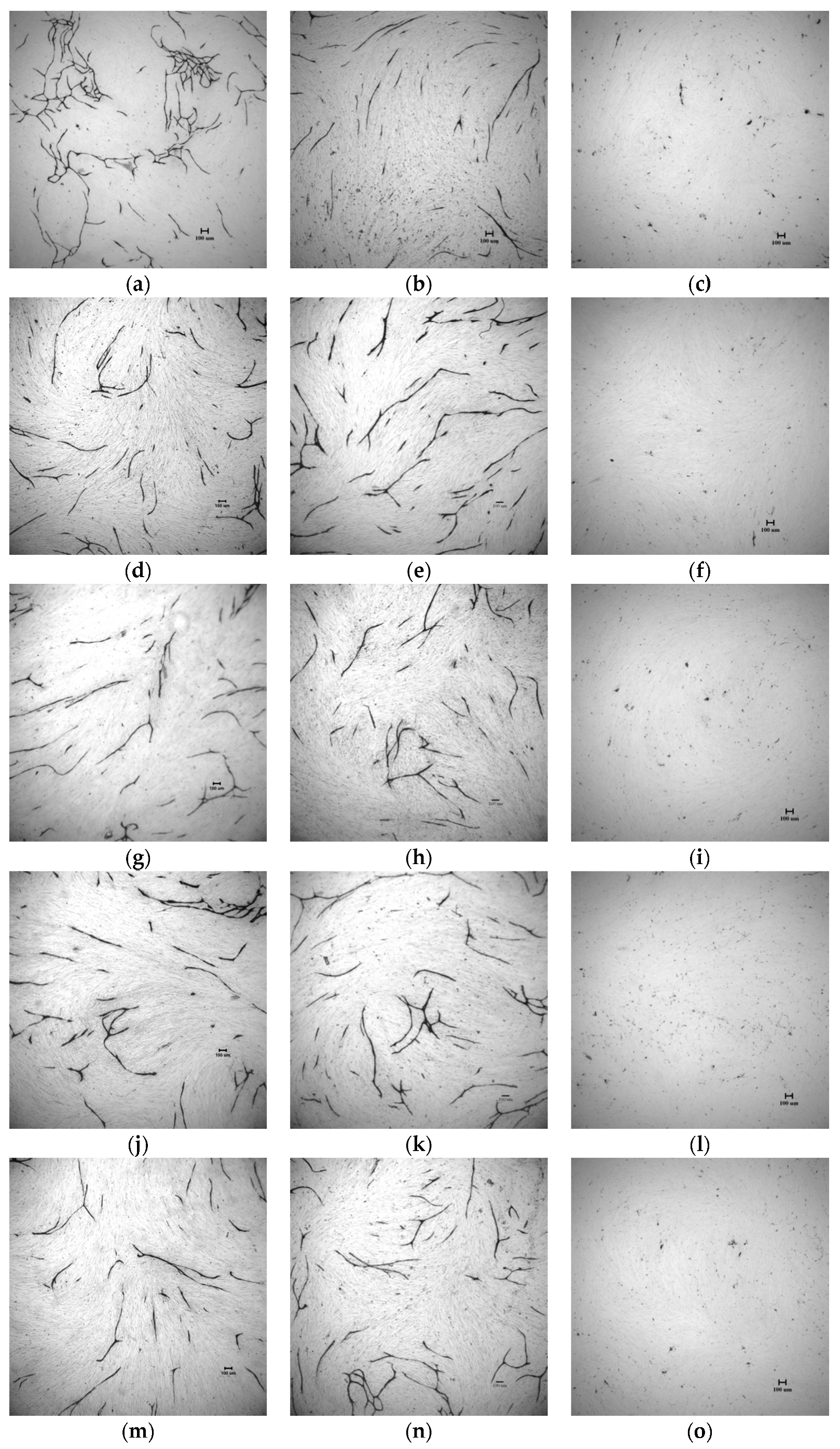
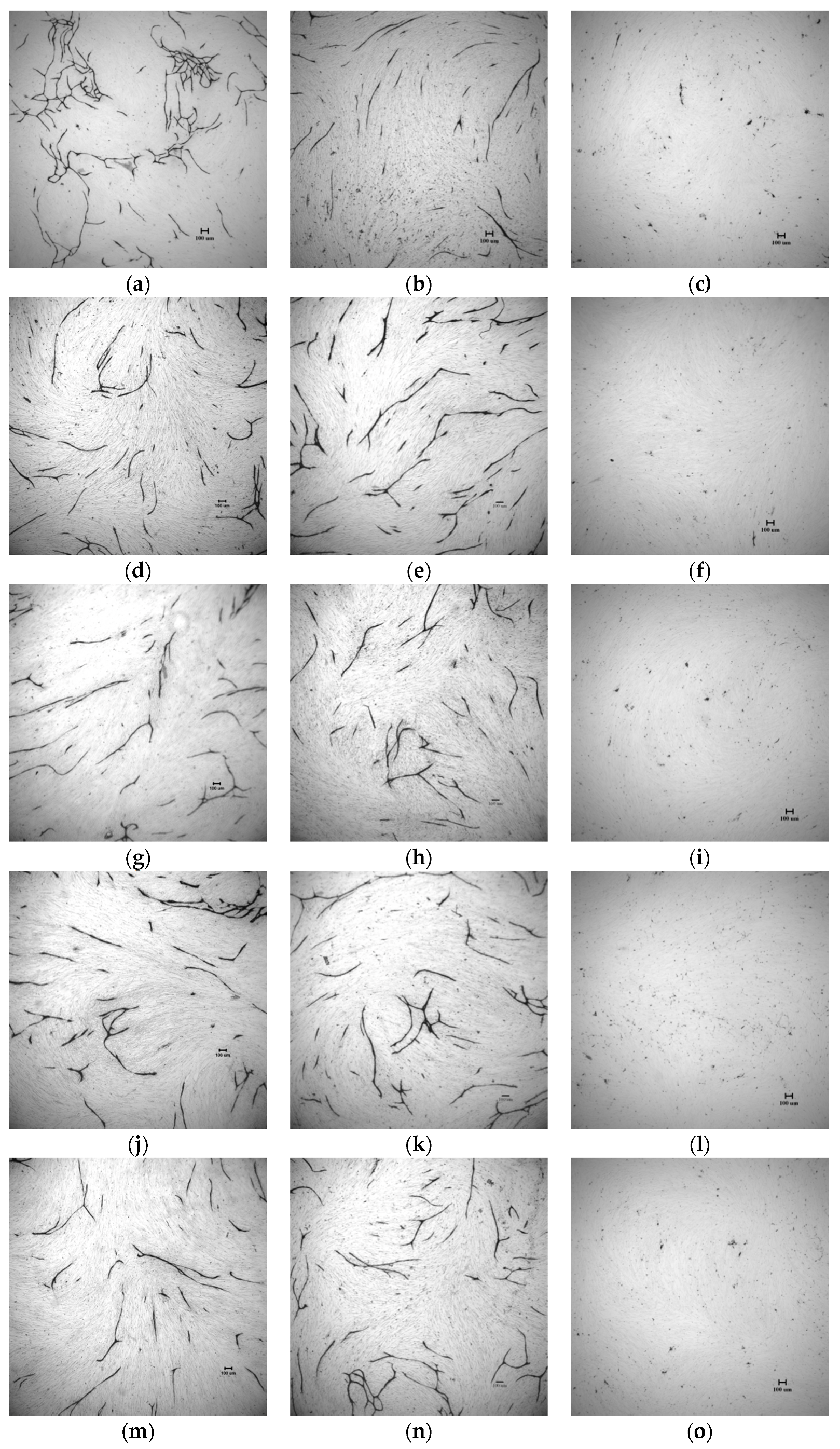
2.4. In Vitro Tube Formation Assay
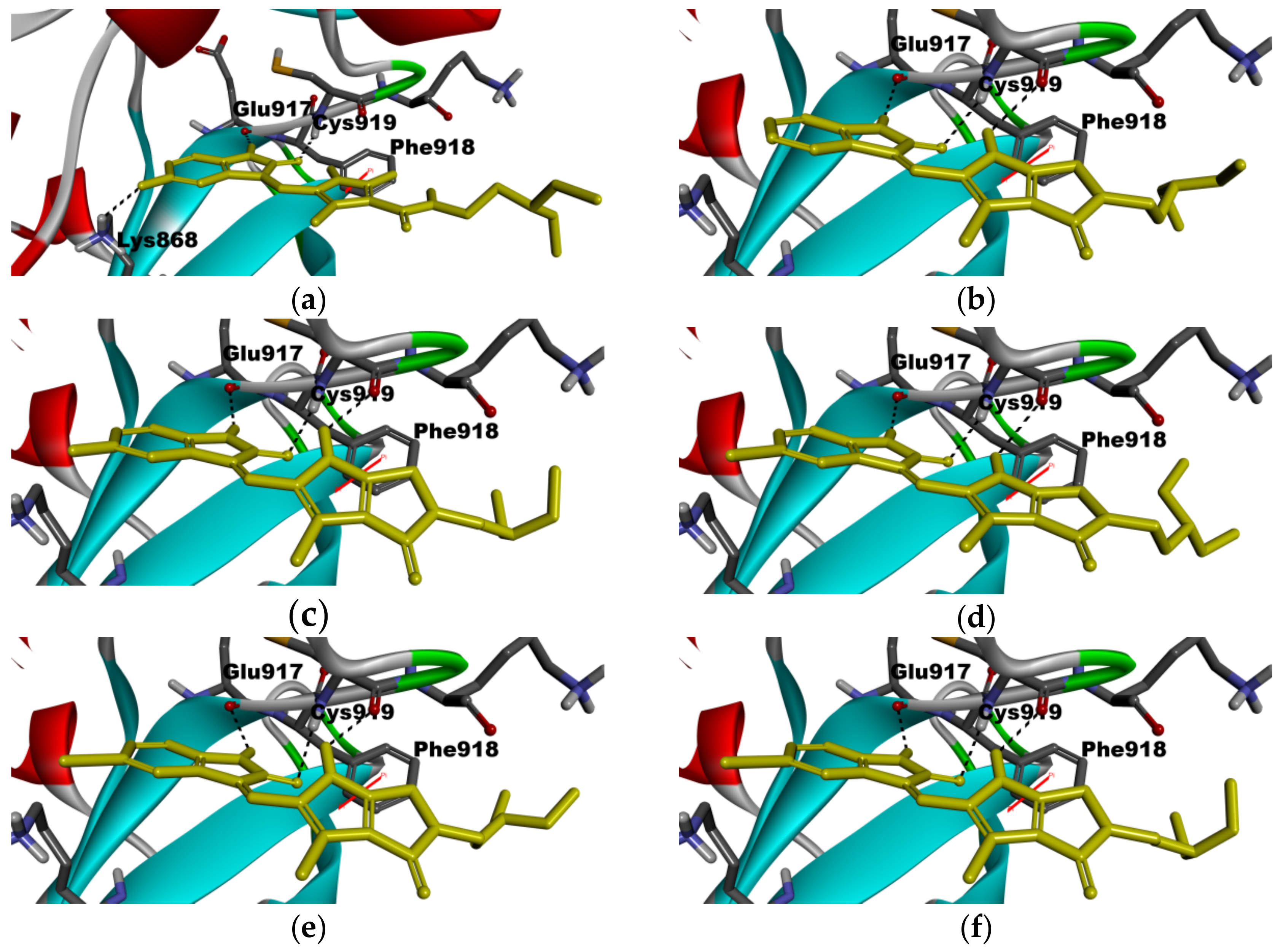
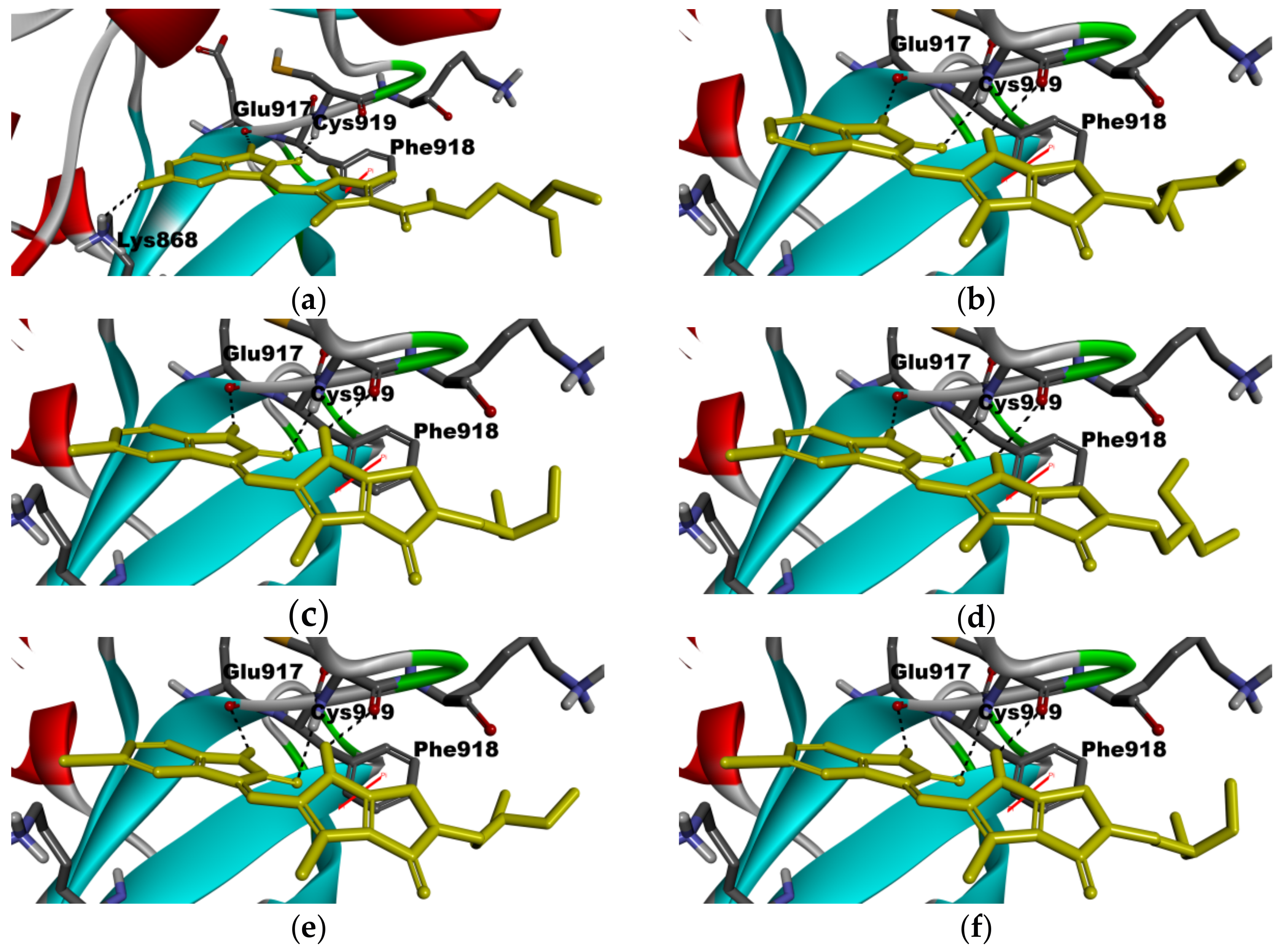
2.5. Molecular Modeling
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of 2–7
5-Formyl-3-methyl-1H-pyrrole-2,4-dicarboxylic acid 2-tert-butyl ester 4-ethyl ester (2)
2-tert-Butyl-4-ethyl 5-(((2-(diethylamino)ethyl)amino)methyl)-3-methyl-1H-pyrrole-2,4-dicarboxylate (3)
2-tert-Butyl 5-(((2-(diethylamino)ethyl)amino)methyl)-3-methyl-1H-pyrrole-2,4-dicarboxylic acid (4)
5-(2-(Diethylamino)ethyl)-3-methyl-4-oxo-1,4,5,6-tetrahydropyrrolo[3,4-b]pyrrole-2-carboxylic acid tert-butyl ester (5)
5-(2-(Diethylamino)ethyl)-3-methyl-4-oxo-1,4,5,6-tetrahydropyrrolo[3,4-b]pyrrole-2-carbaldehyde (7)
3.1.2. Synthesis of 8–12
3.2. Biology
3.2.1. Cell Culture
3.2.2. Cell Proliferation Assay
3.2.3. Acute Cytotoxicity
3.2.4. Image Cytometry
3.2.5. In Vitro Tube Formation Assay
3.2.6. In Vitro Kinase Assay
3.3. Molecular Modeling
3.4. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Felmeden, D.C.; Blann, A.D.; Lip, G.Y. Angiogenesis: Basic pathophysiology and implications for disease. Eur. Heart J. 2003, 24, 586–603. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Roskoski, R., Jr. Sunitinib: A vegf and pdgf receptor protein kinase and angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 2007, 356, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Charnsangavej, C.; Hortobagyi, G.N. Angiogenesis modulation in cancer research: Novel clinical approaches. Nat. Rev. Drug Discov. 2002, 1, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Chouhan, J.D.; Zamarripa, D.E.; Lai, P.H.; Oramasionwu, C.U.; Grabinski, J.L. Sunitinib (sutent): A novel agent for the treatment of metastatic renal cell carcinoma. J. Oncol. Pharm. Pract. 2007, 13, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, W.; Jia, Q.; Chen, J.; Zhang, S.; Yao, W.; Wei, F.; Zhang, Y.; Yang, F.; Huang, W.; et al. Efficient extravasation of tumor-repopulating cells depends on cell deformability. Sci. Rep. 2016, 6, 19304. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.H.; Furukawa, T.; Claron, M.; Boturyn, D.; Coll, J.L.; Fukumura, T.; Fujibayashi, Y.; Dumy, P.; Saga, T. Positron emission tomography imaging of tumor angiogenesis and monitoring of antiangiogenic efficacy using the novel tetrameric peptide probe 64Cu-cyclam-RAFT-c(-RGDfK-)4. Angiogenesis 2012, 15, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Fong, T.A.; Shawver, L.K.; Sun, L.; Tang, C.; App, H.; Powell, T.J.; Kim, Y.H.; Schreck, R.; Wang, X.; Risau, W.; et al. Su5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 1999, 59, 99–106. [Google Scholar] [PubMed]
- Scagliotti, G.; Govindan, R. Targeting angiogenesis with multitargeted tyrosine kinase inhibitors in the treatment of non-small cell lung cancer. Oncologist 2010, 15, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Godl, K.; Gruss, O.J.; Eickhoff, J.; Wissing, J.; Blencke, S.; Weber, M.; Degen, H.; Brehmer, D.; Orfi, L.; Horvath, Z.; et al. Proteomic characterization of the angiogenesis inhibitor SU6668 reveals multiple impacts on cellular kinase signaling. Cancer Res. 2005, 65, 6919–6926. [Google Scholar] [CrossRef] [PubMed]
- Kogan, M.; Fischer-Smith, T.; Kaminsky, R.; Lehmicke, G.; Rappaport, J. CSF-1R up-regulation is associated with response to pharmacotherapy targeting tyrosine kinase activity in amL cell lines. Anticancer Res. 2012, 32, 893–899. [Google Scholar] [PubMed]
- Jeong, W.J.; Mo, J.H.; Park, M.W.; Choi, I.J.; An, S.Y.; Jeon, E.H.; Ahn, S.H. Sunitinib inhibits papillary thyroid carcinoma with RET/PTC rearrangement but not braf mutation. Cancer Biol. Ther. 2011, 12, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Rock, E.P.; Goodman, V.; Jiang, J.X.; Mahjoob, K.; Verbois, S.L.; Morse, D.; Dagher, R.; Justice, R.; Pazdur, R. Food and drug administration drug approval summary: Sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma. Oncologist 2007, 12, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Tran, N.; Liang, C.; Hubbard, S.; Tang, F.; Lipson, K.; Schreck, R.; Zhou, Y.; McMahon, G.; Tang, C. Identification of substituted 3-[(4,5,6,7-tetrahydro-1H-indol-2-yl)methylene]-1,3-dihydroindol-2-ones as growth factor receptor inhibitors for VEGF-R2 (Flk-1/KDR), FGF-R1, and PDGF-Rβ tyrosine kinases. J. Med. Chem. 2000, 43, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liang, C.; Shirazian, S.; Zhou, Y.; Miller, T.; Cui, J.; Fukuda, J.Y.; Chu, J.Y.; Nematalla, A.; Wang, X.; et al. Discovery of 5-[5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J. Med. Chem. 2003, 46, 1116–1119. [Google Scholar] [PubMed]
- Kammasud, N.; Boonyarat, C.; Sanphanya, K.; Utsintong, M.; Tsunoda, S.; Sakurai, H.; Saiki, I.; Andre, I.; Grierson, D.S.; Vajragupta, O. 5-Substituted pyrido[2,3-d]pyrimidine, an inhibitor against three receptor tyrosine kinases. Bioorg. Med. Chem. Lett. 2009, 19, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.C.; Lin, Y.H.; Lin, S.F.; Lai, C.L.; Liu, C.; Wei, W.Y.; Yang, S.C.; Wang, R.W.; Teng, L.W.; Chuang, S.H.; et al. Discovery of pyrrole-indoline-2-ones as aurora kinase inhibitors with a different inhibition profile. J. Med. Chem. 2010, 53, 5929–5941. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.P.; Dong, S.Y.; Jun, F.; Hong, F.J.; Liang, Y.J.; Lu, X.; Hua, P.J.; Li, L.Y.; Lei, Z.; Bing, H.; et al. Novel potent orally active multitargeted receptor tyrosine kinase inhibitors: Synthesis, structure-activity relationships, and antitumor activities of 2-indolinone derivatives. J. Med. Chem. 2010, 53, 8140–8149. [Google Scholar] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Fda-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Gajiwala, K.S.; Wu, J.C.; Christensen, J.; Deshmukh, G.D.; Diehl, W.; DiNitto, J.P.; English, J.M.; Greig, M.J.; He, Y.A.; Jacques, S.L.; et al. Kit kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. USA 2009, 106, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Ha, B.H.; Lou, H.J.; Morse, E.M.; Zhang, R.; Calderwood, D.A.; Turk, B.E.; Boggon, T.J. Substrate and inhibitor specificity of the type II p21-activated kinase, PAK6. PLoS ONE 2013, 8, e77818. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.P.; Kormos, C.M.; Burdette, S.C. Ferribright: A rationally designed fluorescent probe for redox active metals. J. Am. Chem. Soc. 2009, 131, 8578–8586. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.T.; Kang, S.K. Direct and indirect reductive amination of aldehydes and ketones with solid acid-activated sodium borohydride under solvent-free conditions. Tetrahedron 2005, 61, 5725–5734. [Google Scholar] [CrossRef]
- Li, J.; Subramaniam, K.; Smith, D.; Qiao, J.X.; Li, J.J.; Qian-Cutrone, J.; Kadow, J.F.; Vite, G.D.; Chen, B.-C. AlMe3-promoted formation of amides from acids and amines. Org. Lett. 2012, 14, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Humphreys, L.D.; Walker, M.D.; Woodward, S. Amide bond formation using an air-stable source of AlMe3. Tetrahedron Lett. 2006, 47, 5767–5769. [Google Scholar] [CrossRef]
- Basha, A.; Lipton, M.; Weinreb, S.M. A mild, general method for conversion of esters to amides. Tetrahedron Lett. 1977, 18, 4171–4172. [Google Scholar] [CrossRef]
- Dubois, N.; Glynn, D.; McInally, T.; Rhodes, B.; Woodward, S.; Irvine, D.J.; Dodds, C. On DABAL-Me3 promoted formation of amides. Tetrahedron 2013, 69, 9890–9897. [Google Scholar] [CrossRef]
- Kuchar, M.; Oliveira, M.C.; Gano, L.; Santos, I.; Kniess, T. Radioiodinated sunitinib as a potential radiotracer for imaging angiogenesis—radiosynthesis and first radiopharmacological evaluation of 5-[125i]iodo-sunitinib. Bioorg. Med. Chem. Lett. 2012, 22, 2850–2855. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.Z.; Fu, D.X.; Ma, N.; Li, Z.C.; Liu, Q.H.; Xiao, L.; Zhang, R.H. Synthesis and biological evaluation of 3-substituted-indolin-2-one derivatives containing chloropyrrole moieties. Molecules 2011, 16, 9368–9385. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wei, L.; Yu, J.; Zhang, L. Regorafenib inhibits colorectal tumor growth through PUMA-mediated apoptosis. Clin. Cancer Res. 2014, 20, 3472–3484. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; He, K.; Zhang, L.; Yu, J. Crizotinib induces puma-dependent apoptosis in colon cancer cells. Mol. Cancer Ther. 2013, 12, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, C.; Peng, R.; Wang, P.; Sebastiani, A.; Yu, J.; Zhang, L. Inhibiting oncogenic signaling by sorafenib activates PUMA via GSK3β and NF-κB to suppress tumor cell growth. Oncogene 2012, 31, 4848–4858. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Sun, Q.; Brown, M.F.; Dudgeon, C.; Chandler, J.; Xu, X.; Shu, Y.; Zhang, L.; Yu, J. The multi-targeted kinase inhibitor sunitinib induces apoptosis in colon cancer cells via PUMA. PLoS ONE 2012, 7, e43158. [Google Scholar]
- Sun, J.; Knickelbein, K.; He, K.; Chen, D.; Dudgeon, C.; Shu, Y.; Yu, J.; Zhang, L. Aurora kinase inhibition induces puma via NF-κB to kill colon cancer cells. Mol. Cancer Ther. 2014, 13, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Fancelli, D.; Berta, D.; Bindi, S.; Cameron, A.; Cappella, P.; Carpinelli, P.; Catana, C.; Forte, B.; Giordano, P.; Giorgini, M.L.; et al. Potent and selective aurora inhibitors identified by the expansion of a novel scaffold for protein kinase inhibition. J. Med. Chem. 2005, 48, 3080–3084. [Google Scholar] [CrossRef] [PubMed]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.; Jha, M. Concise syntheses of the cruciferous phytoalexins brassilexin, sinalexin, wasalexins, and analogues: Expanding the scope of the vilsmeier formylation. J. Org. Chem. 2005, 70, 1828–1834. [Google Scholar] [CrossRef] [PubMed]
- Boiadjiev, S.E.; Lightner, D.A. Readily synthesized novel fluorescent dipyrrinones. J. Org. Chem. 2005, 70, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yu, D.; Wu, H.; Liu, H.; Zhou, H.; Gu, R.; Zhang, R.; Zhang, S.; Wu, G. Anticancer activity of SAHA, a potent histone deacetylase inhibitor, in NCI-H460 human large-cell lung carcinoma cells in vitro and in vivo. Int. J. Oncol. 2014, 44, 451–458. [Google Scholar] [PubMed]
- Tomita, S.; Ishibashi, K.; Hashimoto, K.; Sugino, T.; Yanagida, T.; Kushida, N.; Shishido, K.; Aikawa, K.; Sato, Y.; Suzutani, T.; et al. Suppression of SOCS3 increases susceptibility of renal cell carcinoma to interferon-α. Cancer Sci. 2011, 102, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Chiang, S.H.; Wang, H.Y.; Wu, P.S.; Lin, C.C. Curcumin enhances the production of major structural components of elastic fibers, elastin, and fibrillin-1, in normal human fibroblast cells. Biosci. Biotechnol. Biochem. 2015, 79, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
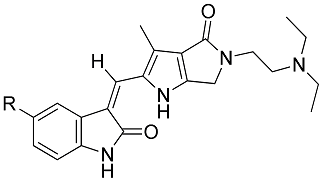
| Compd. | R | IC50 (μM)/Selective Index (SI) | |||
|---|---|---|---|---|---|
| HCT116 | NCI-H460 | 786-O | Detroit 551 | ||
| Sunitinib | - | 3.42 ± 0.57 | 6.23 ± 0.57 | 6.27 ± 0.67 | 9.48 ± 0.18 |
| 2.77 | 1.52 | 1.51 | |||
| 8 | -H | 4.25 ± 1.88 | >10 | >10 | >10 |
| >2.35 | nd | nd | |||
| 9 | -F | 2.94 ± 0.66 | >10 | >10 | >10 |
| >3.4 | nd | nd | |||
| 10 | -Cl | 3.09 ± 0.70 | >10 | 7.30 ± 1.20 | >10 |
| >3.23 | nd | >1.37 | |||
| 11 | -Br | 1.05 ± 0.18 | 6.57 ± 1.64 | 7.06 ± 0.30 | >10 |
| >9.52 | >1.52 | >1.42 | |||
| 12 | -I | 0.42 ± 0.16 | 2.95 ± 0.83 | 7.76 ± 0.29 | 9.21 ± 1.61 |
| 21.93 | 3.12 | 1.19 | |||
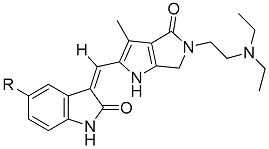
| Compd. | R | % Inhibition at 50 nM | IC50 (nM) | % Inhibition of Aurora A at 1.0 μM | LibDock Score of VEGFR-2 Binding | ||
|---|---|---|---|---|---|---|---|
| VEGFR-2 | PDGFRβ | VEGFR-2 | PDGFRβ | ||||
| Sunitinib | - | 29 | 44 | 140.0 | 61.4 | 50.7 | 110.303 |
| 8 | -H | 56 | 64 | 30.2 | 22.4 | 95.9 | 134.754 |
| 9 | -F | 35 | 36 | 110.7 | 97.5 | 93.1 | 137.901 |
| 10 | -Cl | 58 | 64 | 35.0 | 24.9 | 93.7 | 130.921 |
| 11 | -Br | 64 | 68 | 24.7 | 16.1 | 93.1 | 129.433 |
| 12 | -I | 58 | 62 | 35.1 | 29.3 | 92.9 | 125.031 |
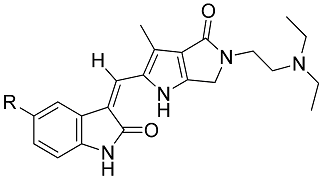
| Compd. | R | IC50 of Tube Area (nM) |
|---|---|---|
| Sunitinib | - | 387 ± 16 |
| 8 | -H | 310 ± 15 |
| 9 | -F | 367 ± 16 |
| 10 | -Cl | 299 ± 70 |
| 11 | -Br | 179 ± 29 |
| 12 | -I | 313 ± 37 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, T.-H.; Lee, C.-I.; Huang, W.-H.; Lee, A.-R. Synthesis and Evaluation of Novel 2-Pyrrolidone-Fused (2-Oxoindolin-3-ylidene)methylpyrrole Derivatives as Potential Multi-Target Tyrosine Kinase Receptor Inhibitors. Molecules 2017, 22, 913. https://doi.org/10.3390/molecules22060913
Yang T-H, Lee C-I, Huang W-H, Lee A-R. Synthesis and Evaluation of Novel 2-Pyrrolidone-Fused (2-Oxoindolin-3-ylidene)methylpyrrole Derivatives as Potential Multi-Target Tyrosine Kinase Receptor Inhibitors. Molecules. 2017; 22(6):913. https://doi.org/10.3390/molecules22060913
Chicago/Turabian StyleYang, Ting-Hsuan, Chun-I Lee, Wen-Hsin Huang, and An-Rong Lee. 2017. "Synthesis and Evaluation of Novel 2-Pyrrolidone-Fused (2-Oxoindolin-3-ylidene)methylpyrrole Derivatives as Potential Multi-Target Tyrosine Kinase Receptor Inhibitors" Molecules 22, no. 6: 913. https://doi.org/10.3390/molecules22060913
APA StyleYang, T.-H., Lee, C.-I., Huang, W.-H., & Lee, A.-R. (2017). Synthesis and Evaluation of Novel 2-Pyrrolidone-Fused (2-Oxoindolin-3-ylidene)methylpyrrole Derivatives as Potential Multi-Target Tyrosine Kinase Receptor Inhibitors. Molecules, 22(6), 913. https://doi.org/10.3390/molecules22060913