Abstract
The involvement of protein kinase CK1δ in the pathogenesis of severe disorders such as Alzheimer’s disease, amyotrophic lateral sclerosis, familial advanced sleep phase syndrome, and cancer has dramatically increased interest in the development of effective small molecule inhibitors for both therapeutic application and basic research. Unfortunately, the design of CK1 isoform-specific compounds has proved to be highly complicated due to the existence of six evolutionarily conserved human CK1 members that possess similar, different, or even opposite physiological and pathophysiological implications. Consequently, only few potent and selective CK1δ inhibitors have been reported so far and structurally divergent approaches are urgently needed in order to establish SAR that might enable complete discrimination of CK1 isoforms and related p38α MAPK. In this study we report on design and characterization of optimized 4,5-diarylimidazoles as highly effective ATP-competitive inhibitors of CK1δ with compounds 11b (IC50 CK1δ = 4 nM, IC50 CK1ε = 25 nM), 12a (IC50 CK1δ = 19 nM, IC50 CK1ε = 227 nM), and 16b (IC50 CK1δ = 8 nM, IC50 CK1ε = 81 nM) being among the most potent CK1δ-targeting agents published to date. Inhibitor compound 11b, displaying potential as a pharmacological tool, has further been profiled over a panel of 321 protein kinases exhibiting high selectivity. Cellular efficacy has been evaluated in human pancreatic cancer cell lines Colo357 (EC50 = 3.5 µM) and Panc89 (EC50 = 1.5 µM). SAR is substantiated by X-ray crystallographic analysis of 16b in CK1δ and 11b in p38α.
1. Introduction
Protein kinase CK1δ is a member of the ubiquitously expressed and constitutively active Ser/Thr-specific CK1 (formerly known as casein kinase 1) family which comprises the six human isoforms α, γ1, γ2, γ3, δ, and ε, together with their closest relatives the tau tubulin kinases 1 and 2 (TTBK1/2) and vaccinia-related kinases 1-3 (VRK1–3) [1,2,3]. Despite CK1 being evolutionarily highly conserved within their catalytic domains, all isoforms differ significantly in the length and the primary structure of their regulatory N- and C-terminal regions. Among them, isoforms δ and ε display the highest consensus, with a 98% sequence identity within their catalytic domain and at least 40% homology within their autoregulatory C-terminal domains [2,3,4,5]. Pathophysiologically, identification of mutations within the coding region of CK1δ as well as deregulation of CK1δ expression and/or activity levels as important determinants in development and progression of severe human disorders such as Alzheimer’s disease (AD) [2,6,7,8], amyotrophic lateral sclerosis (ALS) [9], familial advanced sleep phase syndrome (FASPS) [10], and cancer [2,5,11,12,13,14,15,16,17,18,19] has dramatically increased interest in the development of potent and selective small molecule kinase inhibitors for both therapeutic approaches and basic research. However, the existence of paralogous CK1 isoforms that possess similar, different, or even opposite physiological and pathophysiological implications render the design of suitable candidate molecules that target CK1δ in an ideally isoform-dependent manner enormously difficult. The most extensively used and characterized CK1 inhibitor to date, IC261, moderately inhibits CK1 isoforms δ and ε (IC50 CK1δ/ε = 1 µM, IC50 CK1α = 10 µM) and proved valuable in diverse pharmacological studies [16,20,21,22]. Furthermore, IC261 even revealed therapeutic potential for the treatment of pancreatic cancer in a subcutaneous mouse xeno-transplantation model, despite the fact that the inhibitor is active against several other targets, including tubulin polymerization and ion channels [16,22,23,24]. In addition, few further compounds have been reported as CK1 inhibitors, mainly those which are commonly referred to as “linear” [25,26] and “tear-drop”-like binders [27] with respect to their three-dimensional structure. Among the latter, a promising 4,5-diarylimidazole-based inhibitor 1, originally designed as an inhibitor of p38α MAPK, was revealed in 2009 by Peifer et al. to inhibit CK1δ/ε with IC50 values in the low nanomolar range (1 IC50 CK1δ = 5 nM, 1 IC50 CK1ε = 73 nM) [28]. Interestingly, sulfoxidation of thioether 1 leading to sulfoxide 2 significantly enhanced discrimination of highly related isoforms δ and ε to at least 40-fold while preserving good potency for CK1δ (2 IC50 CK1δ = 11 nM, 2 IC50 CK1ε = 447 nM, Figure 1) [28]. Unfortunately, the reduced chemical stability of 1 and 2 in solution due to E/Z-isomerization and the presence of a Michael acceptor moiety in the cinnamic acid side chain are responsible for their limited usability in vitro and in vivo.
Figure 1.
ATP-competitive dual specific inhibitors 1 and 2 of CK1δ/ε and p38α MAPK.
The present follow-up [28] study reports on the optimization of lead structures 1 and 2, respectively, leading to stable novel inhibitors of CK1δ/ε with IC50 values in the low nanomolar range. The optimization strategy followed a well-established procedure in medicinal chemistry including in silico design, hit synthesis, and in vitro biological evaluation [29,30].
2. Results and Discussion
2.1. Molecular Modeling
The binding modes of ATP-competitive type-I inhibitors 1 and 2 in CK1δ and p38α have been postulated based on structure-based molecular modeling (Figure 2) [28]. Comparable to poses of similar “tear-drop”-like binders (e.g., pdb 3UZP [31]) two hydrogen bonds are formed between the 2-amino-pyridine moiety and CK1δ hinge residue Leu85. The positive mesomeric electron donating effect of the amino group in ortho-position of the pyridine nitrogen positively influences electron density and thus the H-bond acceptor strength at the pyridine-N [32].
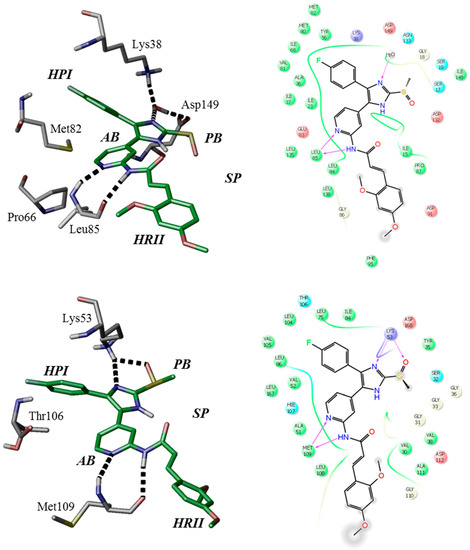
Figure 2.
Modeled binding modes of 2 in CK1δ (top, pdb 3UZP [31]) and p38α (bottom, pdb 1BMK [33]) ATP-binding pockets. Key amino acid residues and ligand-active site interactions are shown. Left: in accordance to Traxler et al. [34], the ATP-binding pocket of protein kinases ought to be subdivided into hydrophobic pocket I (HPI), hydrophobic region II (HRII), adenine-binding region (AB), phosphate-binding region (PB), and sugar pocket (SP). Right: 2D ligand interaction diagrams.
Furthermore, core catalytic residues Lys38 and Asp149 [13,31] coordinate a structural water within the catalytic cleft which donates another hydrogen bond towards an imidazole nitrogen of the respective inhibitor. Gatekeeper residue Met82 is rotated by 180° towards Pro66 [13,31], thereby permitting access to the hydrophobic pocket I (selectivity pocket, HPI) [2,34] which is ideally occupied by the 4-fluorophenyl moiety [31,35]. Molecular modeling further suggests five-membered heterocycles to dictate an ideal angle for positioning of the vicinal aryl moieties within the ATP-binding pocket of CK1δ [36]. The (E)-configured cinnamic acid side chain of 1 and 2 extends into the spacious solvent-exposed hydrophobic region II (affinity pocket, HRII) [2,34]. Modeling calculations further considered different di- or trimethoxyphenyl substitution pattern optimal within this region as they enable flexible occupation of hydrophobic surfaces and shielding deeper cavities of the ATP-binding pocket from surrounding water, thus entailing increased enthalpic contribution of buried hydrogen bonds. Analogous binding poses have been achieved regarding p38α with the bidentate hinge-binding moiety addressing Met109 and the imidazole-N accepting a hydrogen bond from Lys53. Rotation of the smaller gatekeeper residue Thr106, however, does not seem necessary in order to occupy HPI.
The binding modes described above are based on (E)-configured cinnamic moieties of 1 and 2, respectively. In contrast, molecular modeling of the respective (Z)-configurations did not result in plausible binding modes (not shown). Accordingly, computational analysis assume these (Z)-stereoisomers to be less bioactive. Furthermore, the acrylamide Michael acceptor moiety of the cinnamic acid side chain was considered responsible for the observed chemical instability of 1 and 2 in solution. In line with this notion, within a short period of time after preparing a solution of 1 and 2 in DMSO a HPLC analysis showed an increasing number of not identifiable degradation products. Consequently, our primary goal towards an optimized inhibitor was to gain chemical stability. Thus, having identified the cinnamic side chain to be responsible for the chemical instability issue we aimed towards stable side chains attached at the validated 2-aminopyridine core moiety. By these modifications we set out to explore the respective hydrophobic region II formerly occupied by the cinnamic acid moiety. At the same time, both potency and selectivity for CK1δ were taken into account. Therefore, in our systematic approach four structurally divergent series of inhibitors with variable side chains (Scheme 1) have been designed based on the following considerations.
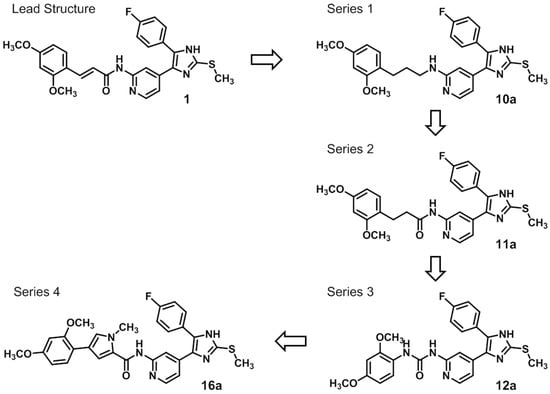
Scheme 1.
Structural considerations based on molecular modeling leading to lead structure 1 derivatives 10a (series 1), 11a (series 2), 12a (series 3), and 16a (series 4).
First, removal of both the planar (sp2) π-bond and carbonyl group in 1 and 2 led to respective sp3 hybridized 3-(2,4-dimethoxyphenyl)propanamine 10a and derivatives (series 1). However, at this position of the ligand, additional degrees of freedom and enhanced conformational flexibility are typically accompanied by losses of both potency and selectivity; Second, maintaining the amide function but formally reducing the π-bond resulted in presumably stable and potent 3-(2,4-dimethoxyphenyl)propionic amide derivatives (e.g., 11a, series 2). Third, a carbamide moiety in 12a and derivatives (series 3) might enable an additional hydrogen bond towards hinge Leu85 and therefore could account for enthalpic binding energy gains. The additional fixation was further suggested to exploit different folding of related CK1δ, CK1ε, and p38α within range of the hinge region and thus to be a key parameter for triggering inhibitor selectivity. And fourth, fixing the (E)-configuration of cinnamic amides 1 and 2 within five-membered heterocycles led to 4-(2,4-dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylic amide (16a, series 4). Taken together, by our compound design concept in the side chain Michael acceptor characteristics and inactive (Z)-isomers were eliminated while potentially conserving beneficial impacts regarding potency and selectivity.
Based on the inhibitor categories described above we generated a virtual set of compounds being subsequently processed in a LigPrep/Glide docking campaign using CK1δ (pdb 3UZP [31]) and p38α (pdb 1BMK [33]) protein structures. Thereby we focused on variation of side chains addressing the HRII while maintaining the fixed 4,5-diaryl-imidazole pharmacophore. This included different sets of substituted lipophilic and mainly sterically demanding moieties in order to exploit this region. As methoxy-substituents were assumed most favorable in this context, efforts have been devoted to methoxy-screenings investigating different substitution patterns.
2.2. Synthesis
In order to effectively synthesize the designed and top ranked hits from docking, a straightforward five-step procedure towards the building blocks 2-fluoro-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridine (6) and 4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridine-2-amine (7) was established in accordance to literature protocols [37,38]. In the final step, by substitution of key compounds 6 and 7, variations of side chains were introduced and thus four compound series were prepared (Scheme 2).
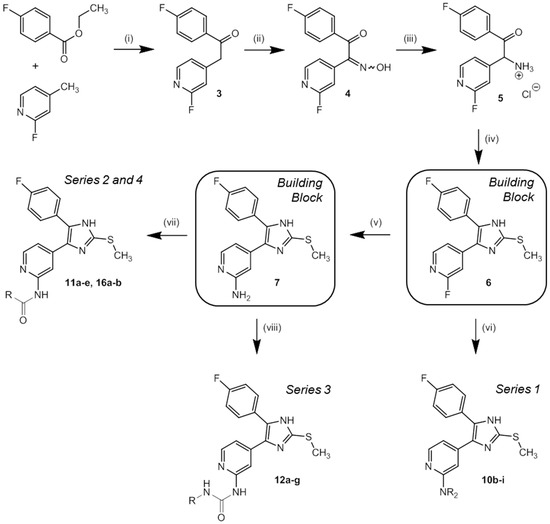
Scheme 2.
Synthesis of key building blocks 6 and 7 as well as inhibitors 10b–i (series 1), 11a–e (series 2), 12a–g (series 3), and 16a–b (series 4). Reagents and Conditions: (i) NaHMDS, THF, 2 h 0 °C, 1 h r.t.; (ii) CH3CHOOH, NaNO2, 1 h 0 °C, 3.5 h r.t.; (iii) H2, Pd/C, HCl-saturated 2-propanol, 12 h r.t.; (iv) methyl thiocyanate, DMF, 45 min reflux, 45 min r.t.; (v) NH3, 20–30 bar, 18 h 180 °C; (vi) HNR2, 12 h 160 °C; (vii) carboxylic acid, CDI or PyBOP/DIPEA, DMF, 12 h 110 °C; (viii) isocyanate, DIPEA, DMF, 12 h r.t.
Slightly deviating from the procedure depicted in Scheme 2, pyridine-2-amine 10a has been synthesized by a SN2 reaction of single Boc-protected 2-amino-4-methylpyridine and 1-(3-bromopropyl)-2,4-dimethoxybenzene (9) followed by subsequent formation of the 4,5-diaryl-imidazole scaffold using the procedure developed for synthesis of 6. Alkyl halide 9 was synthesized from 3-(2,4-dimethoxyphenyl)propionic acid by reduction [39] followed by an Appel reaction using triphenylphosphine and N-bromosuccinimide (Scheme 3) [40]. Detailed information about the synthesis of 10a is presented in the Supporting Information.
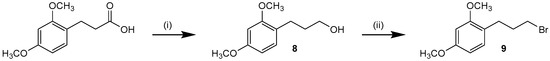
Scheme 3.
Synthesis of 1-(3-bromopropyl)-2,4-dimethoxybenzene (9). Reagents and Conditions: (i) LiAlH4, THF, 1 h r.t.; (ii) NBS, PPh3, DCM, 2 h r.t.
Series 1 pyridine-2-amines 10b–f and piperazines 10g–i were prepared by a Tschitschibabin-like nucleophilic substitution [32,38]. Therefore, 2-fluoropyridine 6 was dissolved or suspended in an excess of the appropriate amine or piperazine and heated to 160 °C (Scheme 2). In general, in this reaction primary amines accounted for better yields.
Syntheses of series 2 included the reaction of building block 7 with CDI-activated differently di- or trimethoxy-substituted 3-phenylpropionic acids to afford amides 11a–e. The poor nucleophilic character of 7, however, required heating to 110 °C in order to achieve suitable reactivity (Scheme 2) [28].
In contrast, highly reactive isocyanates readily acylated 7 at room temperature in terms of a Wöhler-like synthesis leading to series 3 carbamide derivatives 12a–g (Scheme 2) [41]. Noteworthy, addition of Hünig’s base led to increased yields and reactions had to be performed under protective gas atmosphere in order to prevent formation of carbamide dimers.
Series 4 precursor 4-(2,4-dimethoxy-phenyl)pyrrole-2-carboxylic acid was prepared in a three step synthesis starting from ethyl 4-bromo-1H-pyrrole-2-caboxylate [42]. Methylation of the pyrrole nitrogen using methyl iodide was followed by Suzuki cross-coupling with (2,4-dimethoxyphenyl)boronic acid and subsequent simple ester hydrolysis in diluted alkali. The 2,5-dimethoxyphenyl side chain derivative 15b has been synthesized analogously. Finally, 16a–b were obtained by PyBOP-supported amide coupling with building block 7 under elevated temperature (Scheme 4).
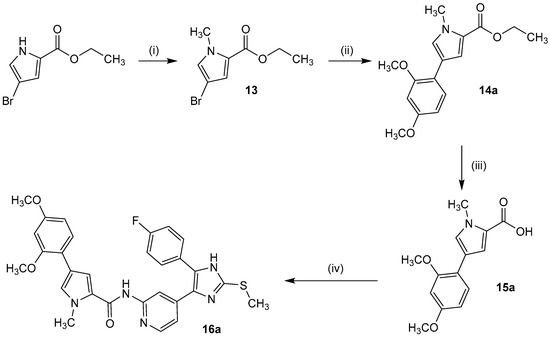
Scheme 4.
Synthesis of 4-(2,4-dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylic acid (15a). Reagents and Conditions: (i) NaH, DMF, 20 min 0 °C, CH3I, 15 min 0 °C, 2.5 h r.t.; (ii) 2,4-dimethoxyphenyl boronic acid, Pd(PPh3)4, aq. NaHCO3, DMF, 4 h reflux, 12 h r.t.; (iii) aq. NaOH, THF/methanol, 5 h 50 °C, 12 h r.t., (iv) DIPEA, DMF, 30 min r.t., 7, 12 h 110 °C.
As mentioned above, the respective sulfoxides might possess interesting potential regarding selectivity. Therefore, oxidation of selected compounds was performed in accordance to literature using Oxone® at 0 °C or at ambient temperature (Scheme 5). During thioether oxidation it was essential to strictly monitor reaction progress for quenching at the sulfoxide level [28]. Accordingly, sulfoxidation was performed for compounds 10c–d, 11a–b, 12a, 12c–g, and 16a leading to 10j–k (series 1), 11f–g (series 2), 12h–m (series 3), and 16c (series 4). It has to be noted that sulfoxides are chiral compounds and we are always referring to the racemate. However, docking analysis did not reveal differences between the enantiomers. In the literature the racemate is reported for this inhibitor class with only one exception, where the R-configuration was observed to possess increased affinity for p38α MAPK [43].
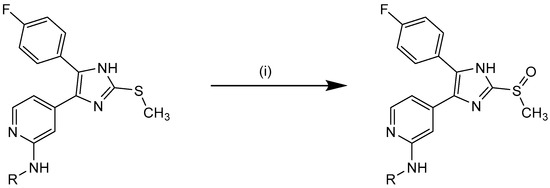
Scheme 5.
Synthesis of sulfoxides. Reagents and Conditions: (i) Oxone® (potassium peroxomonosulfate), THF/H2O, 30 min–2 h 0 °C.
All synthesized target compounds 10a–k, 11a–g, 12a–m, 16a–c are listed in Table 1. They were stable in DMSO solution at room temperature without detectable degradation over a period of 72 h by HPLC analysis.

Table 1.
Synthesized test compounds of series 1, 2, 3, and 4. R refers to the side chains indicated in Scheme 5. Only a limited selection of thioether compounds has been oxidized leading to sulfoxide compounds (12h–12m, 16c). Abbreviation: # compound number.
2.3. Kinase Assays and IC50 Determination
Compounds 10a–k (series 1), 11a–g (series 2), 12a–m (series 3), and 16a–c (series 4) have initially been screened for their ability to inhibit the activity of CK1δ and CK1ε in an in vitro kinase assay at a concentration of 10 µM. Inhibitors exhibiting promising effects were further subjected to IC50 value determination (Table 2). In addition, IC50 values regarding inhibition of CK1δ and p38α MAPK have commercially been obtained from ProQinase GmbH (Freiburg, Germany) for 11b, 12a, and 16b as these were the most potent representatives of their respective series (Table 3). In comparison to our data, IC50 values measured by ProQinase appear slightly lower due to the different assay setup such as the ATP concentration used (compare Experimental Section/Supporting Information and www.proqinase.com).
Table 2.
IC50 values of promising inhibitors for CK1δ and CK1ε. All compounds have initially been screened in an in vitro assay against CK1δ and CK1ε at a concentration of 10 µM. The most promising agents were subjected to IC50 value determination. Results are presented as mean ± SD from experiments performed in triplicate (n = 3). Abbreviation: # compound number.
Table 3.
IC50 values of key inhibitors for CK1δ and p38α MAPK. IC50 values of the most promising agents of series 2 (11b), series 3 (12a), and series 4 (16b) have commercially been obtained from ProQinase GmbH (Freiburg, Germany). Single experiments have been performed (n = 1). Abbreviation: # compound number.
Fortunately, potent CK1δ inhibition and appropriate SAR correlated with the calculated binding modes for the majority of compounds. Especially 11b, 16a, and 16b exhibited excellent efficacy with IC50 values in the single-digit nanomolar range in standardized kinase assays and are therefore among the most potent CK1δ inhibitors reported to date. In general, loss of potency for series 1 inhibitors when compared to lead structure 1 has been observed in vitro: while 1 has an IC50 for CK1δ of 5 nM [28] a side chain without both carbonyl group and π-bond resulted in considerably decreased inhibition (e.g., series 1, 10a IC50 CK1δ = 386 nM). In contrast, compounds possessing the carbonyl group but without π-bond afforded chemically stable series 2 (e.g., 11a IC50 CK1δ = 20 nM, IC50 CK1ε = 129 nM) and thereby restored potency almost to the level reported for lead 1 (IC50 CK1δ = 5 nM, IC50 CK1ε = 73 nM). Isoform selectivity regarding CK1ε, however, seemed to be slightly decreased. Consequently, the π-bond of lead 1 can be identified to be responsible for compound instability, while the carbonyl depicts an important determinant concerning potency. In addition, series 3 and 4 both provided potent inhibitors of CK1δ with IC50 values in the low nanomolar range. However, the aimed impact on CK1δ/ε isoform selectivity by rigidification of the double bond in the (E)-configuration has not been fully achieved. Unfortunately, tridentate hinge binding carbamide derivatives 12a–m (series 3) were actually limited by their poor solubility in vitro.
Oxidation of the exocyclic sulfur at the imidazole-2-position of thioether 1 to afford sulfoxide 2 reflects in vivo metabolization by phase I enzymes [44]. Interestingly, this sulfoxidation increased CK1δ/ε isoform selectivity in one reported previously study [28]. In our hands, however, this effect could not be confirmed for the newly designed and synthesized set of inhibitors as oxidation only slightly altered potency, though without significantly affecting isoform selectivity. Nevertheless, sulfoxidized compounds remained potent inhibitors of CK1δ with IC50 values in the low nanomolar range. This indicates that these agents will retain activity despite metabolization.
2.4. X-ray Analysis of Binding Modes in CK1δ and p38α
In order to verify modeled binding modes and to obtain further insights regarding ligand-protein interactions determining selectivity we set out to co-crystallize potent inhibitors of series 2, 3, and 4 (11b, 12a, and 16b) in CK1δ. In line with this notion, a ligand-protein structure of 16b could be co-crystallized with a C-terminally truncated CK1δ construct and the complex structure was determined at 2.0 Å resolution. Unfortunately, co-crystallization of 11b and 12a failed experimentally.
Additionally, overall most active agent 11b has been co-crystallized with p38α MAPK at 1.9 Å resolution. In fact, crystallographic data largely confirmed the calculated binding pose of 16b within the active site of CK1δ (Figure 3). As determined by X-ray analysis the 4-fluorophenyl moiety deeply penetrates into HPI, enabled by rotation of gatekeeper Met82 towards Pro66. The bidentate hinge-binding 2-aminopyridine motif forms two hydrogen bonds towards Leu85. Furthermore, Lys38 and Asp149 coordinate a water molecule which donates another hydrogen bond accepted by an imidazole nitrogen of 16b. HRII, however, is addressed by bulky 4-(2,5-dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxamide, probably displacing energetically unfavorable water molecules and shielding deeper hydrophobic cavities of the binding pocket. Although the 2,5-dimethoxyphenyl moiety herein mainly interacts with Pro87 by π-aliphatic-stacking, electron density has also been observed within range of Phe95 which can be interpreted by different conformers of 16b, thus indicating additional interaction. The complex of 11b in p38α MAPK predominantly confirmed the expected 4,5-diarylimidazole core fitting, similar to CK1δ, with gatekeeper Thr106-lined HPI occupied by the 4-fluorophenyl moiety and the 2-aminopyridine acting as bidentate hinge binder addressing Met109. The ATP-binding pocket in this structure does not contain structural water molecules and Lys53 directly interacts with 11b imidazole nitrogen by formation of a hydrogen bond. Interestingly, DFG-motif Phe169 is coordinated between 11b imidazole-N and Lys53 by π–π- and, most likely, π-cation-stacking, respectively, thereby interrupting the hydrophobic spine consisting of Leu75, Leu86, Phe169, and His148 [45,46,47]. This pose is stabilizing the kinase in an intermediate conformation between DFG-in and DFG-out (DFG-in-between) [48], thereby showing several characteristics similar to type-I½-like inhibition of p38α MAPK [49,50], although molecular alignMent studies may suggest the DFG-in state most likely to be inhibited by 11b as well. However, the DFG-out conformation can be assumed to be sterically prohibited (Figure 4, left). Interestingly, the 3-(2,5-dimethoxyphenyl)propionic amide side chain of 11b addressing HRII has not been definitely resolvable by X-ray analysis in terms of electron density (Figure 4, right). In fact, this is in agreement with our hypothesis postulating high flexibility of this ligand part within HRII. In line with this notion, we originally designed bulky inhibitor side chains to suboptimal fit into less extensive hydrophobic region II of p38α MAPK, in turn allowing higher affinity for CK1δ. Thus, in that way paying enthalpic and entropic penalty should result in decreased affinity for p38α MAPK when compared to the situation in CK1δ [51]. However, in contrast to our hypothesis inhibitors actually showing the bulky side chain, e.g., 11b, 12a, and 16b, were determined to be nanomolar inhibitors of p38α MAPK as well as of CK1δ. This finding suggests that—despite bulky side chains—the core 4,5-diaryl-imidazole scaffold addressing both ATP sites of CK1δ and p38α is able to determine high affinity for these kinases.
Figure 3.
Similar binding modes of 16b in CK1δ have been gained by co-crystallization (top, pdb 5MQV) and molecular modeling (bottom, pdb 3UZP [31]). The poses are presented by key residues and hydrogen bond interactions (left) and Connolly molecular surface (right).
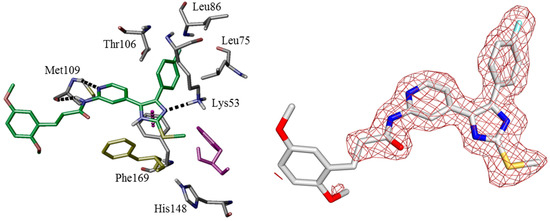
Figure 4.
Co-crystallization of 11b in p38α MAPK (pdb 5ML5). The binding mode is presented by key residues and hydrogen bond interactions as well as π-π-stacking (left). DFG Phe169 is stabilized in an intermediate state between active DFG-in (purple, pdb 1BMK [33]) and inactive DFG-out (yellow, pdb 1WBT [52]) conformation. DFG-in/out states were represented by structure alignment using Schrödinger software. The inhibitor side chain addressing HRII has not been definitely resolvable in terms of electron density (right, generated with PyMOL).
2.5. Selectivity Profiling of 11b
In order to further characterize potent inhibitor 11b selectivity profiling has been performed at a concentration of 100 nM in a panel of 321 protein kinases (ProQinase GmbH). These data revealed high selectivity of 11b for CK1δ hitting only six additional kinases with residual activities less than 50% apart from CK1δ (residual activity = 3%) and CK1ε (residual activity = 7%, CK1 isoforms γ1–3 remained unaffected). Among these, CK1α (residual activity = 26%) and p38α MAPK (residual activity = 22%) were found as prominent targets. Also the highly related kinases JNK2 (residual activity = 21%), JNK3 (residual activity = 37%), and RIPK2 (residual activity = 45%) were identified as additional hits. Furthermore, Tyr-specific kinase LCK (residual activity = 26%) was inhibited (Figure 5 and Supporting Information). Consequently, 11b represents an agent highly selective for CK1δ within the range of its IC50 value of 4 nM with an overall selectivity score S(50) of 0.027. The S-Score(50) has been calculated in accordance to Karaman et al. [53] describing the portion of kinases with a residual activity >50% in relation to all tested kinases included in this project.
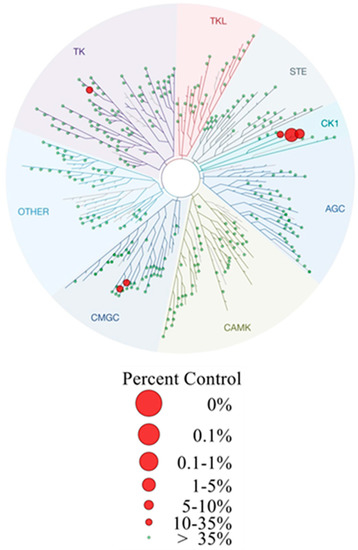
Figure 5.
Dendrogram representation of selectivity profiling of 11b screened over 321 protein kinases at a concentration of 100 nM (ProQinase GmbH, Freiburg, Germany). Residual activity was determined compared to DMSO control. The S-Score(50) of 11b is 0.027. The dendrogram was generated utilizing TREEspot Software Tool, DISCOVERX CORPORATION 2010. Corresponding raw data are given in Supplementary Table S5.
2.6. Cellular Assays and EC50 Determination
Synthesized compounds have further been evaluated regarding their efficacy in cellular systems in MTT viability assays using colorectal HT-29 or pancreatic Colo357, Panc89, Panc-1, and MiaPaCa-2 carcinoma cell lines. The cell lines were chosen as they are reportedly overexpressing CK1δ and CK1ε and exhibit resistance against a variety of chemotherapeutic agents [12,15,16]. In general, amines 10a–k (series 1) were rather ineffective, even at an elevated concentration of 20 µM with 10e being most active exhibiting an EC50 value of 9.3 µM in HT-29 cells (data can be found in Supporting Information). For the subsequently designed compound series EC50 data were only obtained for compounds which showed the most promising inhibition of CK1δ in vitro. Inhibitors from series 2 (11a–e) showed significant increases of potency, presumably referable to the amide carbonyl group which has already been reported to beneficially affect metabolic stability [44]. As expected, 16a–b (series 4) were only slightly less active. In contrast, carbamides 12a–k (series 3) were only moderately active, presumably suffering from poor solubility. Among all series, 11b again proved the most effective agent with EC50 values of 3.5 µM and 1.5 µM in Colo357 and Panc89 cells, respectively (determined EC50 values can be found in Table 4).
Table 4.
EC50 values of selected inhibitor compounds as determined for Colo357, Panc89, Panc-1, and MiaPaCa-2 cell lines. Results are presented as mean ± SD from experiments performed in triplicate (n = 3). Abbreviations: # compound number; n.d., not determined.
If the EC50 values determined in cell-based assays are compared with in vitro determined IC50 data, massive differences can be observed and compounds which appeared to be extremely potent are apparently less efficient in the treatment of living cells. This difference is compound and cell line dependent, can be due to different reasons, and has already been documented in previous reports [26,54]. Firstly, limited cell permeability of small molecule inhibitors may limit their cellular uptake resulting in lower potency; Secondly, once compounds successfully crossed the cell membrane, ATP-competitive inhibitors must compete with high intracellular ATP levels leading to a discrepancy between IC50 values determined by enzymatic versus cellular assays. Additionally, time of binding in the ATP pocket and drug export systems can reduce the inhibitory effect. Therefore, cell-based assays are essential in order to validate the inhibitory effects of newlx identified small molecule inhibitors.
3. Materials and Methods
3.1. Molecular Modeling
Molecular modeling was performed on a DELL Precision T5500 eight core workstation. For visualization Maestro, version 10.6, 2016 (Schrödinger LLC, New York, NY, USA) was used. Protein crystal structures were prepared prior to docking by the Protein Preparation Wizard [55] synchronizing the following modules: Epik, version 3.6, 2016 [56]; Impact, version 7.1, 2016; Prime, version 4.4, 2016 [57]. In order to achieve high Enrichment-factors, the common refinement protocol by Sastry et al. [55] has been adjusted: the process involved assignment of bond orders, addition of hydrogen atoms, identification of disulfide bonds, and the conversion of artifical selenomethionines to methionines (default settings). Missing side chains were filled in using Prime. Missing loops have not been detected. Water molecules beyond 5 Å from hetero atoms have been deleted automatically. H-bond optimization was performed in a standard sampling, the Root-mean-square deviation for atomic positions cutoff for heavy atoms in subsequent protein minimization was set to 0.3 Å
Ligands were prepared to generate energetically minimized three-dimensional coordinates with an extended cutoff by MacroModel, version 11.2, 2016 (Schrödinger LLC). Ionization and tautomeric states were estimated at pH 7 ± 2 by LigPrep, version 3.8, 2016 (Schrödinger LLC) [55], utilizing Hammet and Taft methodology-based Epik [56]. Additionally, Epik state penalties (kcal∙mol−1) were calculated for each ligand to quantify the energetic cost for state transition in solution [55]. In order to indicate ligand flexibility, up to 50 bioactive conformers per ligand were identified and prioritized utilizing the conformational search module in the fast mode (ConfGen, version 3.6, 2016, Schrödinger LLC) [58]. Receptor grid generation was generated with Glide, version 7.1, 2016 (Schrödinger LLC) [59]. For ligand docking and screening the Glide XP workflow was used [59]. Energetically minimized ligand conformations were docked into the active site of the protein; possible binding poses were determined and subsequently ranked based on their calculated binding affinities.
3.2. Chemistry
Infrared spectra were recorded on an IRAffinity-1S FTIR-spectrometer (Shimadzu Europa GmbH, Hannover, Germany). NMR spectra were recorded on an Avance III 300 spectrometer, tempered at 298 K: 1H (300 MHz), 13C-NMR (75 MHz) (Bruker Daltonik GmbH, Bremen, Germany). The data is reported as follows: chemical shift in ppm from tetramethylsilane (TMS) as external standard, multiplicity and coupling constant J (Hz). Spectra were either referenced to TMS or internal DMSO-d6 (1H-NMR δ 2.50) and internal DMSO-d6 (13C-NMR δ 39.52) or internal CHCl3 (1H-NMR δ 7.26) and internal CDCl3 (13C-NMR δ 77.00). The following NMR abbreviations have been used: b (broad), s (singlet), d (doublet), t (triplet), m (unresolved multiplet). Several target compounds show spectra of at least two isomers in DMSO-d6 with a maximal ratio of 1:3. These are due to atropisomers and tautomers as already reported for similar 4,5-diaryl-imidazoles [32]. Although such effects have not been observed whenever CDCl3 was used, DMSO-d6 was the most frequented solvent with respect to its favorable solubility-mediating properties. For reasons of clarity, signals are only given for the main isomer. The labelling scheme of structures to correlate NMR signals can be found in Supporting Information. LC-MS was performed with an 1100 HPLC system (Agilent Technologies, Santa Clara, CA, USA) over an Agilent Eclipse XDB-C8 column (150 × 4.6 mm, 5 µm) using a 0.1% acetic acid/acetonitrile gradient for mobile phase (flow rate = 1 mL∙min−1). Mass spectra with nominal solution were recorded on a Bruker Esquire ~LC ion trap mass spectrometer with electron spray ionization (ESI) operating in the positive ion mode, with the following parameters: drying gas nitrogen 8 L∙min−1, nebulizer 35 psi, drying temperature 350 °C). HRMS spectra were recorded on an AccuTOF™ GCv 4G electron ionization (EI)/field desorption (FD) mass spectrometer (JEOL Germany, Freising, Germany). For clarity, only the highest measured peak is given for mass spectra. Melting points/decomposition temperatures were determined on a SMP3 Melting Point Apparatus (Stuart Scientific, Keison Products, Chelmsford, Essex, UK) and are uncorrected. Column chromatography was performed using a LaFlash system (VWR International GmbH, Darmstadt, Germany). The crude product was loaded on silica gel 60 (63–200 µm) (Macherey-Nagel, Düren, Germany) or PuriFlash IR-50 C18 modified silica gel (50 µm) (Interchim Deutschland GmbH, Mannheim, Germany) and packed in Interchim PuriFlash-DLE/12G dry-load precolumns. Pre-packed Interchim PuriFlash-30SIHP silica gel columns (30 µm, 40 g) and Interchim PuriFlash-15C18HP modified silica gel columns (15 µm, 55 g) were used for separation with flow rates usually adjusted to 30 mL∙min−1 or 20.5 mL∙min−1. Progress of reactions was monitored by thin-layer chromatography (TLC) performed with Macherey-Nagel 0.2 mm Polygram® SIL G/UV254 pre-coated silica gel polyester sheets and Silicagel 60 RP-18 F254 modified silica gel aluminum plates (Merck Millipore, Darmstadt, Germany). Where necessary, reactions were carried out in a nitrogen atmosphere using 4 Å molecular sieves. All reagents and solvents were obtained from commercial sources (abcr GmbH, Karlsruhe, Germany; Sigma-Aldrich Chemie GmbH, Munich, Germany; Merck Group, Munich, Germany; Merck Millipore; Acros Organics Thermo Fisher Scientific, Geel, Belgium; VWR International GmbH, Hannover, Germany and used as received: THF was used after distillation over Na/benzophenone. HPLC analysis was performed on a Hewlett Packard HP 1050 Series using either a ZORBAX® Eclipse XDB-C8 (150 mm × 4.6 mm, 5 µm) or a Kinetex® C8 (150 × 4.6 mm, 5 µm) column (mobile phase flow 1.5 mL·min−1, gradient KH2PO4 buffer 10 mM, pH 2.3/methanol, UV-detection 254 nm). All key compounds submitted to biological assays were proven by this method to show ≥98% purity. Syntheses under elevated pressure were performed in Berghof highpreactor™ BR-25 with corresponding heating block on a MR Hei-Standard laboratory heating plate (Heidolph, Schwabach, Germany). Microwave syntheses were performed in a CEM Discover Microwave Synthesizer (CEM GmbH, Kamp-Lintfort, Germany) under air cooling and high stirring with a maximal power of 100 W.
3.2.1. Syntheses of Key Building Blocks 3–7
1-(4-Fluorophenyl)-2-(2-fluoropyridin-4-yl)-ethan-1-one (3). NaHMDS (66.7 mL 2 M solution in THF, 133 mmol) was slowly added to a stirred solution of 2-fluoro-4-methylpyridine (10.6 mL, 103 mmol) and ethyl 4-fluorobenzoate (18.1 mL, 123 mmol) in 40 mL anhyd. THF at 0 °C under a nitrogen atmosphere. After stirring at 0 °C for 2 h the reaction was allowed to reach rt and stirring continued for 1 h. The mixture was diluted with ethyl acetate and washed twice with 10% aq. HCl. The organic layer was dried over anhyd. Na2SO4 and solvent was removed under reduced pressure. Recrystallization from ethyl acetate afforded 3 as colorless solid. Yield 23.9 g (quant.); C13H9F2NO (Mr 233.22); m.p. 102 °C; 1H-NMR (DMSO-d6): δ = 4.59 (s, 2H, CH2), 7.10 (s, 1H, C3H, Pyr), 7.25–7.27 (m, 1H, C5H, Pyr), 7.37–7.43 (m, 2H, C3/5H, F-Phe), 8.11–8.19 (m, 3H, C2/6H, F-Phe, C6H, Pyr) ppm; 13C-NMR (DMSO-d6): δ = 43.6 (d, 4JCF = 2.8 Hz, CH2), 110.8 (d, 2JCF = 37.6 Hz, C3H, Pyr), 115.8 (d, 2JCF = 22.0 Hz, C3/5H, F-Phe), 123.8 (d, 4JCF = 3.8 Hz, C5H, Pyr), 131.3 (d, 3JCF = 9.6 Hz, C2/6H, F-Phe), 132.9 (d, 4JCF = 2.7 Hz, C1, F-Phe), 147.0 (d, 5JCF = 15.5 Hz, C6H, Pyr), 150.7 (d, 3JCF = 8.5 Hz, C1, Pyr), 163.1 (d, 1JCF = 234.3 Hz, C2F, Pyr), 165.2 (d, 1JCF = 252.0 Hz, C4F, F-Phe), 194.6 (CO) ppm; MS (ESI, 70 eV) m/z 234 [MH]+.
1-(4-Fluorophenyl)-2-(2-fluoropyridin-4-yl)-2-(hydroximino)ethan-1-one (4). NaNO2 (2.06 g, 29.8 mmol) in 12 mL H2O was slowly added to a stirred solution of 3 (2.30 g, 9.86 mmol) in 17 mL glacial acetic acid at 10 °C. After stirring at r.t. for 1 h, 30 mL H2O were added and stirring continued for 3.5 h. The suspension was cooled to 8 °C, filtered, and the residue was washed with H2O and dried under reduced pressure to afford 4 as colorless solid. Yield 2.46 g (95%); C13H8F2N2O2 (Mr 262.22); m.p. 185 °C; 1H-NMR (DMSO-d6): δ = 7.19 (bs, 1H, C3H, Pyr), 7.37–7.46 (m, 3H, C5H, Pyr and C3/5H, F-Phe), 7.92–7.98 (m, 2H, C2/6H, F-Phe), 8.30 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 12.67 (s, 1H, OH) ppm; 13C-NMR (DMSO-d6): δ = 105.4 (d, 2JCF = 38.9 Hz, C3H, Pyr), 116.7 (d, 2JCF = 22.5 Hz, C3/5H, F-Phe), 118.2 (d, 4JCF = 4.2 Hz, C5H, Pyr), 130.8 (d, 4JCF = 2.7 Hz, C1, F-Phe), 132.2 (d, 3JCF = 10.1 Hz, C2/6H, F-Phe), 144.3 (d, 3JCF = 8.4 Hz, C4, Pyr), 148.8 (d, 5JCF = 15.6 Hz, C6H, Pyr), 151.9 (d, 4JCF = 4.0 Hz, CNOH), 163.5 (d, 1JCF = 235.6 Hz, C2F, Pyr), 166.0 (d, 1JCF = 254.8 Hz, C4F, F-Phe), 192.04 (CO) ppm; MS (ESI, 70 eV) m/z = 263 [MH]+.
2-(4-Fluorophenyl)-1-(2-fluoropyridin-4-yl)-2-oxoethan-1-aminium chloride (5). Pd/C 10% (279 mg) was added in one portion under a nitrogen atmosphere to an intensely stirred solution of 4 (1.60 g, 6.10 mmol) in 15 mL 2-propanol and 20 mL HCl sat. 2-propanol and stirring continued for 12 h at r.t. The crude product was obtained by filtration, the residue was resuspended in methanol, filtered again, and the solvent was removed under reduced pressure. Recrystallization from methanol/diethyl ether afforded 5 as beige solid. Yield 1.43 g (82%); C13H11ClF2N2O (Mr 284.69); m.p. 216 °C; 1H-NMR (DMSO-d6): δ = 6.56 (s, 1H, CH), 7.35–7.41 (m, 2H, C3/5H, F-Phe), 7.52–7.54 (m, 2H, C3/5H, Pyr), 8.21 (dd, 3J = 8.4 Hz, 4J = 5.6 Hz, 2H, C2/6H, F-Phe), 8.31 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 9.36 (bs, 3H, NH3+) ppm; 13C-NMR (DMSO-d6): δ = 56.8 (CNH3+), 110.5 (d, 2JCF = 39.3 Hz, C3H, Pyr), 116.9 (d, 2JCF = 22.1 Hz, C3/5H, F-Phe), 122.3 (d, 4JCF = 4.2 Hz, C5H, Pyr), 130.0 (d, 4JCF = 2.7 Hz, C1, F-Phe), 132.9 (d, 3JCF = 9.8 Hz, C2/6H, F-Phe), 147.4 (d, 3JCF = 8.0 Hz, C4, Pyr), 149.4 (d, 5JCF = 15.0 Hz, C6H, Pyr), 163.5 (d, 1JCF = 236.7 Hz, C2F, Pyr), 166.3 (d, 1JCF = 255.1 Hz, C4F, F-Phe), 191.4 (CO) ppm; MS (ESI, 70 eV) m/z 249 [MCl]+.
2-Fluoro-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridine (6). A mixture of 5 (4.41 g, 15.5 mmol) and methyl thiocyanate (3.18 mL, 46.5 mmol) was refluxed under a nitrogen atmosphere for 45 min in 140 mL anhyd. DMF and stirred 45 min at r.t. before ice-cold H2O (400 mL) was added. The suspension was cooled to 8 °C, filtered, the residue was washed with H2O, and dried under reduced pressure to afford 6 as fine yellow solid. Yield 3.59 g (76%); C15H11F2N3S (Mr 303.33); m.p. 205 °C; 1H-NMR (DMSO-d6): δ = 2.63 (s, 3H, SCH3), 7.09 (bs, 1H, C3H, Pyr), 7.26–7.34 (m, 3H, C5H, Pyr and C3/5H, F-Phe), 7.49–7.54 (m, 2H, C2/6H, F-Phe), 8.09 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 12.84 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 14.9 (SCH3), 105.0 (d, 2JCF = 39.5 Hz, C3H, Pyr), 115.9 (d, 2JCF = 21.6 Hz, C3/5H, F-Phe), 118.5 (d, 4JCF = 2.5 Hz, C5H, Pyr), 126.5 (C1, F-Phe), 130.9 (C2/6H, F Phe), 132.9 (d, 3JCF = 8.6 Hz, C4, Pyr), 143.0 (C2, Imdz), 147.6 (d, 3JCF = 16.1 Hz, C6H, Pyr), 162.1 (d, 1JCF = 246.4 Hz, CF, F-Phe), 163.7 (d, 1JCF = 233.3 Hz, CF, Pyr) ppm; MS (ESI, 70 eV) m/z 304 [MH]+.
4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-amine (7). A solution of 6 (1.00 g, 3.30 mmol) in 15 mL 32% aq. ammonia solution was heated in a high pressure reactor at 180 °C for 18 h. The reactor was allowed to reach r.t. and H2O was added. The crude product was obtained by filtration, washed with H2O and diisopropyl ether and purified by flash chromatography (SiO2, 50%–100% ethyl acetate/petrol ether) to afford 7 as beige solid. Yield 852 mg (86%); C15H13FN4S (Mr 300.36); 1H-NMR (DMSO-d6): δ = 2.60 (s, 1H, SCH3), 5.79–5.95 (m, 2H, NH2), 6.42–6.67 (m, 2H, C3/5H, Pyr), 7.17–7.27 (m, 2H, C3/5H, F-Phe), 7.48 (bs, 2H, C2/6H, F-Phe), 7.74–7.86 (m, 1H, C6H, Pyr), 12.58 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 105.1 (C5H, Pyr), 110.0 (C3H, Pyr), 115.7 (d, 3JCF = 21.1 Hz, C3/5H, F-Phe), 127.0 (s, C4/C5, Imdz), 129.2 (s, C1, F-Phe), 130.5 (d, 4JCF = 6.6 Hz, C2/6H, F-Phe), 147.5 (C6H, Pyr), 160.1 (CF) ppm; MS (ESI, 70 eV) m/z 301 [MH]+.
3.2.2. Synthesis of 4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-amines and -piperazines 8, 9, 10a–i
3-(2,4-Dimethoxyphenyl)propan-1-ol (8). Under a nitrogen atmosphere a solution of LiAlH4 (120 mg, 3.16 mmol) in 3 mL anhyd. THF was slowly added to 3-(2,4-dimethoxyphenyl)propionic acid (508 mg, 2.42 mmol) in 6 mL anhyd. THF at r.t. with intense stirring that continued for 1 h. After completion, the reaction was cooled down in an ice-bath and quenched by successive addition of H2O (120 µL), 15% aq. NaOH solution (120 µL), and H2O (360 µL). The precipitate was filtered off, washed with THF, and the filtrate was concentrated under reduced pressure. The crude product was purified by flash chromatography (SiO2, 20%–30% ethyl acetate/petrol ether) to afford 8 as colorless oil. Yield 457 mg (96%); C11H16O3 (Mr 196.25); 1H-NMR (CDCl3): δ = 1.78–1.83 (m, 2H, CH2CH2CH2OH), 2.65 (t, 3J = 7.3 Hz, 2H, CH2CH2CH2OH), 3.59 (t, 3J = 6.3 Hz, 1H, CH2CH2CH2OH), 3.78 (s, 1H, OH), 3.79 (s, 3H, C4OCH3), 3.81 (s, 3H, C2OCH3), 6.42–6.46 (m, 2H, C3/5H, (OCH3)2-Phe), 7.03 (dd, 3J = 7.7 Hz, 5J = 0.6 Hz, 1H, C6H, (OCH3)2-Phe) ppm; 13C-NMR (CDCl3): δ = 25.4 (CH2CH2CH2OH), 33.2 (CH2CH2CH2OH), 55.5 (C2/4OCH3), 62.1 (CH2CH2CH2OH), 98.6 (C3H, (OCH3)2-Phe), 104.3 (C5H, (OCH3)2-Phe), 122.4 (C1, (OCH3)2-Phe), 130.4 (C6H, (OCH3)2-Phe), 158.3 (C2OCH3), 159.3 (C4OCH3) ppm.
1-(3-Bromopropyl)-2,4-dimethoxybenzene (9). To an ice-cold solution of 8 (260 mg, 1.33 mmol) in 4 mL anhyd. DCM, triphenylphosphine (412 mg, 1.57 mmol) and N-bromosuccinimide (263 mg, 1.48 mmol) were added under a nitrogen atmosphere and the reaction was stirred for 2 h. The mixture was washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. Diethyl ether was added, the precipitate filtered off, and the filtrate was concentrated and purified by flash chromatography (SiO2, 2%–10% ethyl acetate/petrol ether) to afford 9 as colorless oil. Yield 205 mg (60%); C11H15BrO (Mr 259.14); 1H-NMR (CDCl3): δ = 2.11 (quint, 3J = 5.6 Hz, 2H, CH2CH2CH2Br), 2.70 (t, 3J = 7.2 Hz, 2H, CH2CH2CH2Br), 3.39 (t, 3J = 6.8 Hz, 2H, CH2CH2CH2Br), 3.80 (s, 6H, C2/4OCH3), 6.42 (dd, 3J = 8.0 Hz, 4J = 2.5 Hz, 1H, C5H, (OCH3)2-Phe), 6.45 (d, 4J = 2.3 Hz, 1H, C3H, (OCH3)2-Phe), 7.05 (dd, 3J = 8.0 Hz, 5J = 0.5 Hz, 1H, C6H, (OCH3)2-Phe) ppm; 13C-NMR (CDCl3): δ = 28.4 (CH2CH2CH2Br), 33.0 (CH2CH2CH2Br), 33.9 (CH2CH2CH2Br), 55.3 (C4OCH3), 55.5 (C2OCH3), 98.7 (C3H, (OCH3)2-Phe), 103.9 (C5H, (OCH3)2-Phe), 121.4 (C1, (OCH3)2-Phe), 130.5 (C6H, (OCH3)2-Phe), 158.5 (C2OCH3), 159.6 (C4OCH3) ppm.
N-(3-(2,4-Dimethoxyphenyl)propyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-amine (10a). The synthetic protocol starting from Boc-protected 2-amino-4-methylpyridine and 9 predominantly matches the procedure described for 7 and can be found in the Supporting Information. 1H-NMR (DMSO-d6): δ = 1.67–1.72 (m, 2H, CH2CH2CH2NH), 2.48–2.52 (m, 2H, CH2CH2CH2NH), 2.60 (s, 3H, SCH3), 3.13–3.17 (m, 2H, CH2CH2CH2NH), 3.72 (s, 3H, C4OCH3), 3.74 (s, 3H, C2OCH3), 6.41–6.50 (m, 5H, C3/5H, Pyr and C3/5H, (OCH3)2-Phe and CH2CH2CH2NH), 7.01 (d, 3J = 8.2 Hz, 1H, C6H, (OCH3)2-Phe), 7.22 (m, 2H, C3/5H, F-Phe), 7.48 (m, 2H, C2/6H, F-Phe), 7.85 (bs, C6H, Pyr), 12.58 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 26.6 (CH2CH2CH2NH), 29.3 (CH2CH2CH2NH), 40.7 (CH2CH2CH2NH), 55.1 (C4OCH3), 55.2 (C2OCH3), 98.3 (C3H, (OCH3)2-Phe), 104.3 (C5H, (OCH3)2-Phe), 104.9 (C3H, Pyr), 109.7 (C5H, Pyr), 115.4 (C3/5H, F-Phe), 121.8 (C1, (OCH3)2-Phe), 126.5 (C4, Imdz), 127.0 (C5, Imdz), 129.7 (C1, F-Phe), 129.7 (C6H, (OCH3)2-Phe), 130.1 (C2/6H, F-Phe), 142.1 (C2, Imdz), 147.5 (C6H, Pyr), 157.9 (C2OCH3), 158.8 (C4OCH3), 159.3 (C2, Pyr) ppm; MS (ESI, 70 eV) m/z 479 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C26H27FN4O2S, 478.1839; found, 478.1839.
General Procedure for the Preparation Compounds 10b–i
Compound 6 (1.0 equiv) was suspended in the appropriate amine/piperazine (4.0 to 6.0 equiv) and the intensely stirred mixture was heated to 160 °C. Progress of the reaction was monitored by HPLC control. After complete conversion the mixture was diluted with ethyl acetate, washed with sat. aq. NaHCO3 solution and H2O, dried over anhyd. Na2SO4, and concentrated under reduced pressure. The crude product was purified by flash chromatography (stationary phase, eluent, and mixing ratio given for each compound, respectively) to afford the particular compound.
N-(2,4-Dimethoxyphenyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-amine (10b). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and 2,4-dimethoxyaniline (808 mg, 5.27 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10b as greyish crystals. Yield 312 mg (54%); C23H21FN4O2S (Mr 436.51); m.p. 206 °C; 1H-NMR (DMSO-d6): δ = 2.60 (s, 3H, SCH3), 3.75 (s, 6H, 2 OCH3), 6.39–6.44 (m, 1H, C5H, (OCH3)2-Phe), 6.56–6.58 (m, 1H, C3H, (OCH3)2-Phe), 6.63 (dd, 3J = 5.3 Hz, 4J = 1.2 Hz, 1H, C5H, Pyr), 6.82 (s, 1H, C3H, Pyr), 7.18–7.28 (m, 2H, C3/5H, F-Phe), 7.41–7.46 (m, 2H, C2/6H, F-Phe), 7.52 (d, 3J = 8.7 Hz, 1H, C6H, (OCH3)2-Phe), 7.71 (s, 1H, NH), 7.95 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 12.60 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 55.2 (OCH3), 55.2 (OCH3), 99.0 (C3H, (OCH3)2-Phe), 104.0 (C5H, (OCH3)2-Phe), 105.4 (C3H, Pyr), 111.5 (C5H, Pyr), 115.4 (dd, 2JCF = 21.8 Hz, C3/5H, F-Phe), 122.5 (C, Imdz), 123.6 (C6H, (OCH3)2-Phe), 126.8 (d, 4JCF = 3.1 Hz, C1, F-Phe), 130.0 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 134.9 (C1, (OCH3)2-Phe), 138.8 (C, Imdz), 141.7 (C2OCH3), 142.7 (C2, Imdz), 147.6 (C6H, Pyr), 152.2 (C4OCH3), 155.9 (C4, Pyr), 157.4 (C2, Pyr), 161.8 (d, 1JCF = 245.0 Hz, CF) ppm; MS (ESI, 70 eV) m/z 437 [MH]+.
N-(2-Ethoxyphenyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-amine (10c). Synthesis was performed according to the general procedure from 6 (1.50 g, 4.95 mmol) and 2-ethoxyaniline (3.88 mL, 29.7 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10c as dark red crystals. Yield 1.10 g (52%); C23H21FN4OS (Mr 420.51); m.p. 179 °C; 1H-NMR (CDCl3): δ = 1.39 (t, 3J = 7.0 Hz, 3H, CH2CH3), 2.63 (s, 3H, SCH3), 4.03 (q, 3J = 7.0 Hz, 2H, CH2CH3), 6.76 (t, 3J = 7.7 Hz, 1H, C5H, EtO-Phe), 6.81–6.84 (m, 2H, C5H, Pyr and C3H, EtO-Phe), 6.91 (t, 3J = 7.7 Hz, 1H, C4H, EtO-Phe), 6.97 (bs, 1H, C3H, Pyr), 7.00–7.05 (t, 3J = 8.7 Hz, 2H, C3/5H, F-Phe), 7.21 (bs, 1H, NH), 7.37–7.41 (m, 2H, C2/6H, F-Phe), 7.49 (d, 3J = 7.7 Hz, 1H, C6H, EtO-Phe), 8.00 (d, 3J = 5.5 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 15.0 (CH3CH2), 16.6 (SCH3), 64.3 (CH2CH3), 106.5 (C3H, Pyr), 111.7 (C3H, EtO-Phe), 112.9 (C5H, Pyr), 116.0 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 120.0 (C6H, EtO-Phe), 120.7 (C5H, EtO-Phe), 122.6 (C4H, EtO-Phe), 129.4 (C1, EtO-Phe), 130.5 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 143.2 (C2, Imdz), 146.8 (C6H, Pyr), 148.7 (COEt), 155.5 (C2, Pyr), 162.5 (d, 1JCF = 249.0 Hz, CF) ppm; MS (ESI, 70 eV) m/z = 421 [MH]+.
N-(3,4-Dimethoxyphenethyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-amine (10d). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and 3,4-dimethoxyphenethylamine (900 µL, 5.31 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10d as yellow crystals. Yield 550 mg (90%); C25H25FN4O2S (Mr 464.56); m.p. 87 °C; 1H-NMR (CDCl3): δ = 2.55 (s, 3H, SCH3), 2.69 (t, 3J = 6.9 Hz, 2H, CH2CH2NH), 3.29 (m, 2H, CH2CH2NH), 3.74 (s, 3H, C3OCH3), 3.75 (s, 3H, C4OCH3), 4.86 (bs, 1H, CH2CH2NH), 6.51 (d, 3J = 5.2 Hz, 1H, C5H, Pyr), 6.57 (d, 4J = 2.0 Hz, 1H, C3H, Pyr), 6.57–6.70 (m, 3H, C2/5/6H, (OCH3)2-Phe), 6.91–6.96 (m, 2H, C3/5H, F-Phe), 7.32–7.36 (m, 2H, C2/6H, F-Phe), 7.74 (d, 3J = 5.8 Hz, 1H, C6H, Pyr), 10.77 (bs, 1H, NH) ppm; 13C-NMR (CDCl3): δ = 16.4 (SCH3), 35.1 (CH2CH2NH), 43.5 (CH2CH2NH), 55.9 (C3OCH3), 55.9 (C4OCH3), 104.1 (C3H, Pyr), 111.0 (C5H, Pyr), 111.4 (C5H, (OCH3)2-Phe), 112.1 (C2H, (OCH3)2-Phe), 115.7 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 120.7 (C6H, (OCH3)2-Phe), 130.3 (d, 3JCF = 8.2 Hz, C2/6H, F-Phe), 131.4 (C4/5, Imdz), 142.9 (C4, Pyr), 146.7 (C6H, Pyr, 147.7 (C4OCH3), 149.0 (C3OCH3), 158.3 (C2, Pyr), 162.6 (d, 1JCF = 248.5 Hz, CF) ppm; MS (ESI, 70 eV) m/z 465 [MH]+.
N-(2-(1H-Indol-3-yl)-ethyl)-4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-amine (10e). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and tryptamine (845 mg, 5.27 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10e as yellowish-brown crystals. Yield 427 mg (73%); C25H22FN5S (Mr 443.54); m.p. 122 °C; 1H-NMR (CDCl3): δ = 2.61 (s, 3H, SCH3), 2.95 (t, 3J = 6.6 Hz, 2H, CH2CH2NH), 3.39 (t, 3J = 6.6 Hz, 2H, CH2CH2NH), 6.52 (d, 3J = 5.7 Hz, 1H, C5H, Pyr), 6.52 (bs, 1H, C2H, Indole), 6.87 (d, 4J = 2.1 Hz, 1H, C3H, Pyr), 6.95–7.01 (m, 2H, C3/5H, F-Phe), 7.08 (t, 3J = 7.8 Hz, 1H, C6H, Indole), 7.16 (t, 3J = 7.9 Hz, 1H, C5H, Indole), 7.30 (d, 3J = 7.9 Hz, 1H, C4H, Indole), 7.35–7.39 (m, 2H, C2/6H, F-Phe), 7.52 (d, 3J = 7.7 Hz, 1H, C7H, Indole), 7.71 (d, 3J = 5.7 Hz, 1H, C6H, Pyr), 8.38 (s, 1H, NH, Indole) ppm; 13C-NMR (CDCl3): δ = 16.4 (SCH3), 25.1 (CH2CH2NH), 42.2 (CH2CH2NH), 104.0 (C3H, Pyr), 110.6 (C5H, Pyr), 111.3 (C4H, Indole), 112.6 (C3, Indole), 115.8 (d, 2JCF = 21.6 Hz, C3/5H, F-Phe), 118.6 (C7H, Indole), 119.4 (C6H, Indole), 122.1 (C5H, Indole), 122.5 (C2H, Indole), 127.2 (C8a, Indole), 130.3 (3JCF = 8.3 Hz, C2/6H, F-Phe), 136.4 (C3a, Indole), 143.3 (C2, Imdz), 145.1 (C6H, Pyr), 158.0 (C2, Pyr) ppm; MS (ESI, 70 eV) m/z 444 [MH]+.
N-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-N′,N′-dimethyl-propan-1,3-diamine (10f). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and N1,N1-dimethylpropane-1,3-diamine (1.00 mL, 7.93 mmol). Purification was achieved by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether, then methanol) and subsequent filtration, to afford 10f as yellow solid. Yield 175 mg (35%); C20H24FN5S (Mr 385.51); m.p. 129 °C; 1H-NMR (CDCl3): δ = 1.61 (t, 3J = 6.8 Hz, 2H, CH2CH2CH2), 2.13 (s, 6H, N(CH3)2), 2.29 (t, 3J = 6.9 Hz, 2H, CH2N(CH3)2), 2.58 (s, 3H, SCH3), 3.11 (t, 3J = 6.7 Hz, 2H, CH2NH), 5.22 (bs, 1H, CH2NH), 6.42–6.46 (m, 2H, C3/5H, Pyr), 6.95–7.01 (m, 2H, C3/5H, F-Phe), 7.35–7.39 (m, 2H, C2/6H, F-Phe), 7.78 (dd, 3J = 5.4 Hz, 5J = 0.6 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 16.5 (SCH3), 26.7 (CH2CH2CH2), 45.2 (N(CH3)2), 57.6 (CH2N(CH3)2), 104.0 (C3H, Pyr), 111.1 (C5H, Pyr), 115.5 (d, 2JCF = 21.5 Hz, C3/5H, F-Phe), 130.2 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 142.7 (C2, Imdz), 147.9 (C6H, Pyr), 159.2 (C2, Pyr), 162.4 (d, 1JCF = 247.7 Hz, CF) ppm; MS (ESI, 70 eV) m/z 386 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-piperazine (10g). Synthesis was performed according to the general procedure from 6 (300 mg, 989 µmol) and piperazine (341 mg, 3.96 mmol). Purification was achieved by flash chromatography (bas. Al2O3, 100% methanol) to afford 10g as yellow solid. Yield 63.0 mg (17%); C19H20FN5S (Mr 369.46); m.p. 146 °C; 1H-NMR (DMSO-d6): δ = 2.60 (bs, 3H, SCH3), 2.73 (t, 3J = 4.5 Hz, 4H, C2/6H2, Piperazine), 3.31 (t, 3J = 4.6 Hz, 4H, C3/5H2, Piperazine), 6.56 (d, 3J = 5.1 Hz, 1H, C5H, Pyr), 6.80 (bs, 1H, C3H, Pyr), 7.20–7.26 (m, 2H, C3/5H, F-Phe), 7.47–7.51 (m, 2H, C2/6H, F-Phe), 7.97 (d, 3J = 5.1 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 45.3 (C2/6H2, Piperazine), 45.8 (C3/5H2, Piperazine), 103.8 (C3H, Pyr), 110.6 (C5H, Pyr), 115.4 (d, 2JCF = 21.6 Hz, C3/5H, F-Phe), 130.3 (d, 3JCF = 7.6 Hz, C2/6H, F-Phe), 142.3 (C2, Imdz), 147.6 (C6H, Pyr), 159.6 (C2, Pyr), 161.6 (d, 1JCF = 245.1 Hz, CF) ppm; MS (ESI, 70 eV) m/z 370 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-4-methyl-piperazine (10h). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and N-methylpiperazine (600 µL, 5.39 mmol). Purification was achieved by flash chromatography (RP-18, 20%–90% methanol/H2O) to afford 10h as pale yellow solid. Yield 115 mg (23%); C20H22FN5S (Mr 383.49); m.p. 111 °C; 1H-NMR (CDCl3): δ = 2.28 (s, 3H, CH3), 2.45 (t, 3J = 4.8 Hz, 4H, C3/5H2, Piperazine), 2.57 (s, 3H, SCH3), 3.40 (t, 3J = 4.8 Hz, 4H, C2/6H2, Piperazine), 6.54 (dd, 3J = 5.3 Hz, 4J = 1.1 Hz, 1H, C5H, Pyr), 6.73 (bs, 1H, C3H, Pyr), 6.91–6.97 (m, 2H, C3/5H, F-Phe), 7.32–7.37 (m, 2H, C2/6H, F-Phe), 7.94 (d, 3J = 5.3 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 16.5 (SCH3), 44.7 (C2/6H2, Piperazine), 45.6 (CH3), 54.4 (C3/5H2, Piperazine), 104.9 (C3H, Pyr), 111.9 (C5H, Pyr), 115.7 (d, 2JCF = 21.6 Hz, C3/5H, F-Phe), 128.2 (d, 4JCF = 4.1 Hz, CC1, F-Phe), 130.2 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 142.8 (C2, Imdz), 147.9 (C6H, Pyr), 159.6 (C2, Pyr), 162.5 (d, 1JCF = 248.3 Hz, CF) ppm; MS (ESI, 70 eV) m/z 384 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-4-phenyl-piperazine (10i). Synthesis was performed according to the general procedure from 6 (400 mg, 1.32 mmol) and N-phenylpiperazine (800 µL, 5.23 mmol). The combined organic phases were extracted with 2 M aq. HCl solution, the aq. layer was neutralized with 2 M aq. KOH solution, and the pale yellow precipitate was collected by filtration, and purified by flash chromatography (SiO2, 20%–90% ethyl acetate/petrol ether) to afford 10i as yellow crystals. Yield 291 mg (50%); C20H22FN5S (Mr 445.56); m.p. 101 °C; 1H- NMR (CDCl3): δ = 2.64 (s, 3H, SCH3), 3.19 (t, 3J = 5.1 Hz, 4H, C2/6H2, Piperazine), 3.58 (t, 3J = 5.1 Hz, 4H, C3/5H2, Piperazine), 6.64 (dd, 3J = 5.4 Hz, 4J = 1.2 Hz, 1H, C5H, Pyr), 6.93–6.85 (m, 4H, C3H, Pyr and C2/4/6H, Phe), 6.99–7.05 (m, 2H, C3/5H, F-Phe), 7.26 (t, 3J = 8.0 Hz, 2H, C3/5H, Phe), 7.39–7.44 (m, 2H, C2/6H, F-Phe), 8.00 (d, 3J = 5.5 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 16.5 (SCH3), 45.4 (C2/6H2, Piperazine), 49.0 (C3/5H2, Piperazine), 105.1 (C3H, Pyr), 111.6 (C5H, Pyr), 115.8 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 116.3 (C2/6H, Phe), 120.2 (C4H, Phe), 129.2 (C3/5H, Phe), 130.3 (d, 3JCF = 8.1 Hz, C2/6H, F-Phe), 143.0 (C2, Imdz), 146.9 (C6H, Pyr), 151.1 (C1, Phe), 159.0 (C2, Pyr), 162.6 (d, 1JCF = 248.5 Hz, CF) ppm; MS (ESI, 70 eV) m/z 446 [MH]+.
3.2.3. Synthesis of N-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-phenyl-propanamides 11a–e
The appropriate methoxy-substituted 3-phenylpropionic acid and CDI (1.1 equiv) were stirred in 3–4 mL anhyd. DMF until formation of CO2 was undetectable. 7 was added to the reaction and the mixture was heated at 110 °C under a nitrogen atmosphere for 12 h. The reaction was cooled to rt and ethyl acetate was added. The resulting mixture was washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (SiO2 and RP-18, eluent and mixing ratio given for each compound, respectively) to afford the particular compound.
3-(2,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)propanamide (11a). Synthesis was performed according to the general procedure from 3-(2,4-dimethoxyphenyl)propionic acid (560 mg, 2.66 mmol) and 7 (400 mg, 1.33 mmol). Purification was achieved by flash chromatography (SiO2, 30%–50% ethyl acetate/petrol ether and RP-18, 30%–80% methanol/H2O) to afford 11a as beige solid. Yield 177 mg (27%); C26H25FN4O3S (Mr 492.57); m.p. 90 °C; 1H-NMR (DMSO-d6): δ = 2.57 (t, 3J = 7.4 Hz, 2H, COCH2CH2), 2.62 (s, 3H, SCH3), 2.75 (t, 3J = 7.5 Hz, 2H, COCH2CH2), 3.72 (s, 3H, C2OH3), 3.77 (s, 3H, C4OCH3), 6.42 (dd, 3J = 8.3 Hz, 4J = 2.4 Hz, 1H, C5H, (OCH3)2-Phe), 6.51 (d, 4J = 2.4 Hz, 1H, C3H, (OCH3)2-Phe), 6.99 (dd, 3J = 5.3 Hz, 1H, C5H, Pyr), 7.03 (d, 3J = 8.3 Hz, 1H, C6H, (OCH3)2-Phe), 7.28 (bs, 2H, C3/5H, F-Phe), 7.46–7.51 (m, 2H, C2/6H, F-Phe), 8.12 (bs, 1H, C6H, Pyr), 8.32 (bs, 1H, C3H, Pyr), 10.29 (bs, 1H, CONH), 12.72 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 24.7 (COCH2CH2), 36.3 (COCH2CH2), 55.1 (C2OCH3), 55.3 (C4OCH3), 98.3 (C3H, (OCH3)2-Phe), 104.3 (C5H, (OCH3)2-Phe), 110.6 (C3H, Pyr), 115.8 (d, 2JCF = 24.1 Hz, C3/5H, F-Phe), 116.4 (C5H, Pyr), 120.9 (C1, (OCH3)2-Phe), 126.7 (C1, F-Phe), 129.8 (C6H, (OCH3)2-Phe), 130.7 (C5, Imdz and C2/6H, F-Phe), 134.5 (C4, Imdz), 143.7 (C2, Imdz), 147.6 (C6H, Pyr), 148.0 (C4, Pyr), 152.5 (C2, Pyr), 157.9 (C4, (OCH3)2-Phe), 159.0 (C2, (OCH3)2-Phe), 161.9 (d, 1JCF = 246.7 Hz, CF), 171.4 (CO) ppm; IR (ATR): ν = 2935, 1670, 1609, 1547, 1505, 1414, 1289, 1262, 1221, 1207, 1153, 1121, 1036, 835 cm−1; MS (ESI, 70 eV) m/z 493 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C26H25FN4O3S, 492.1631; found, 492.1631.
3-(2,5-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (11b). Synthesis was performed according to the general procedure from 3-(2,5-dimethoxyphenyl)propionic acid (560 mg, 2.66 mmol) and 7 (400 mg, 1.33 mmol). Purification was achieved by flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether and RP-18, 20%–100% methanol/H2O) to afford 11b as colorless solid. Yield 255 mg (39%); C26H25FN4O3S (Mr 492.57); m.p. 91 °C; 1H-NMR (DMSO-d6): δ = 2.59–2.64 (m, 5H, SCH3 and COCH2CH2), 2.80 (t, 3J = 7.5 Hz, 2H, COCH2CH2), 3.66 (s, 3H, C5OCH3), 3.73 (s, 3H, C2OCH3), 6.72 (dd, 3J = 8.8 Hz, 4J = 3.1 Hz, 1H, C4H, (OCH3)2-Phe), 6.78 (d, 4J = 3.0 Hz, 1H, C6H, (OCH3)2-Phe), 6.86 (d, 3J = 8.8 Hz, 1H, C3H, (OCH3)2-Phe), 6.99 (dd, 3J = 5.2 Hz, 4J = 1.6 Hz, 1H, C5H, Pyr), 7.26 (bs, 2H, C3/5H, F-Phe), 7.46–7.51 (m, 2H, C2/6H, F-Phe), 8.13 (bs, 1H, C6H, Pyr), 8.32 (bs, 1H, C3H, Pyr), 10.34 (bs, 1H, CONH), 12.72 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 25.3 (COCH2CH2), 36.0 (COCH2CH2), 55.2 (C5OCH3), 55.8 (C2OCH3), 110.6 (C3H, Pyr), 111.2 (C4H, (OCH3)2-Phe), 111.5 (C3H, (OCH3)2-Phe), 115.6 (C3/5H, F-Phe), 115.9 (C6H, (OCH3)2-Phe), 116.5 (C5H, Pyr), 126.7 (C1, F-Phe), 130.0 (C1, (OCH3)2-Phe), 130.6 (C5, Imdz and C2/6H, F-Phe), 134.5 (C4, Imdz), 142.3 (C2, Imdz), 147.7 (C4 and C6H, Pyr), 151.2 (C2, (OCH3)2-Phe), 152.5 (C2, Pyr), 153.0 (C5, (OCH3)2-Phe), 161.9 (d, 1JCF = 247.3, CF), 171.3 (CO) ppm; MS (ESI, 70 eV) m/z 494 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C26H25FN4O3S, 492.1631; found, 492.1631. The compound was demonstrated to have the desired structure by small molecule X-ray structure determination (Figure 6). For further details see CCDC and Supporting Information.
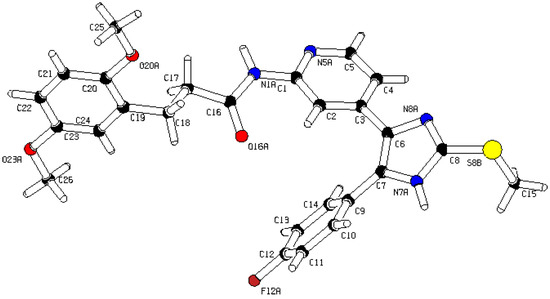
Figure 6.
Small molecule X-ray crystal structure determination of compound 11b.
3-(2,3-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (11c). Synthesis was performed according to the general procedure from 3-(2,3-dimethoxyphenyl)propionic acid (700 mg, 3.33 mmol) and 7 (500 mg, 1.67 mmol). Purification was achieved by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 11c as colorless solid. Yield 515 mg (63%); C26H25FN4O3S (Mr 492.57); 1H-NMR (DMSO-d6): δ = 2.62 (s, 3H, SCH3), 2.59–2.66 (m, 2H, COCH2CH2), 2.84 (t, 3J = 7.7 Hz, 2H, COCH2CH2), 3.73 (s, 3H, C2OCH3), 3.78 (s, 3H, C3OCH3), 6.79 (dd, 3J = 7.5 Hz, 4J = 1.6 Hz, 1H, C6H, (OCH3)2-Phe), 6.88 (dd, 3J = 8.1 Hz, 4J = 1.2 Hz, 1H, C4H, (OCH3)2-Phe), 6.97 (t, 3J = 7.8 Hz, 1H, C5H, (OCH3)2-Phe), 7.01 (dd, 3J = 5.3 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.26–7.32 (m, 2H, C3/5H, F-Phe), 7.45–7.51 (m, 2H, C2/6H, F-Phe), 8.11 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 8.34 (bs, 1H, C3H, Pyr), 10.35 (s, 1H, CONH), 12.70 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 24.8 (COCH2CH2), 36.8 (COCH2CH2), 55.3 (C3OCH3), 60.0 (C2OCH3), 110.6 (C3H, Pyr), 110.9 (C4H, (OCH3)2-Phe), 115.8 (d, 2JCF = 21.8 Hz, C3/5H, F-Phe), 116.4 (C5H, Pyr), 121.4 (C6H, (OCH3)2-Phe), 123.7 (C5H, (OCH3)2-Phe), 126.6 (d, 4JCF = 3.1 Hz, C1, F-Phe), 130.1 (C5, Imdz), 130.7 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 134.4 (C1, (OCH3)2-Phe), 134.5 (C4, Imdz), 142.2 (C2, Imdz), 143.8 (C4, Pyr), 146.6 (C2OCH3), 147.6 (C6H, Pyr), 152.4 (C3OCH3), 152.5 (C2, Pyr), 162.0 (d, 1JCF = 244.7 Hz, CF), 171.1 (CO) ppm; MS (ESI, 70 eV) m/z 493 [MH]+.
3-(3,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (11d). Synthesis was performed according to the general procedure from 3-(3,4-dimethoxyphenyl)propionic acid (700 mg, 3.33 mmol) and 7 (500 mg, 1.67 mmol). Purification was achieved by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 11d as colorless solid. Yield 468 mg (57%); 1H-NMR (DMSO-d6): δ = 2.62 (s, 3H, SCH3), 2.62–2.67 (m, 2H, COCH2CH2), 2.81 (t, 3J = 7.4 Hz, 2H, COCH2CH2), 3.70 (s, 3H, C4OCH3), 3.72 (s, 3H, C3OCH3), 6.73 (dd, 3J = 8.2 Hz, 4J = 1.9 Hz, 1H, C6H, (OCH3)2-Phe), 6.84 (d, 3J = 8.3 Hz, 1H, C5H, (OCH3)2-Phe), 6.85 (bs, 1H, C2H, (OCH3)2-Phe), 7.00 (dd, 3J = 5.2 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.26–7.31 (m, 2H, C3/5H, F-Phe), 7.46–7.50 (m, 2H, C2/6H, F-Phe), 8.10–8.34 (m, 2H, C3/6H Pyr), 10.33 (s, 1H, CONH), 12.71 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 30.4 (COCH2CH2), 38.0 (COCH2CH2), 55.4 (C4OCH3), 55.5 (C3OCH3), 110.6 (C3H, Pyr), 111.9 (C5H, (OCH3)2-Phe), 112.3 (C2H, (OCH3)2-Phe), 115.8 (d, 2JCF = 22.5 Hz, C3/5H, F-Phe), 116.4 (C5H, Pyr), 120.0 (C6H, (OCH3)2-Phe), 130.6 (C2/6H, F-Phe), 133.5 (C1, (OCH3)2-Phe), 147.1 (C4OCH3), 147.6 (C6H, Pyr), 148.6 (C3OCH3), 152.5 (C2, Pyr), 165.8 (d, 1JCF = 242.6 Hz, CF), 171.2 (CO) ppm; MS (ESI, 70 eV) m/z 493 [MH]+.
3-(3,4,5-Trimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (11e). Synthesis was performed according to the general procedure from 3-(3,4,5-dimethoxyphenyl)propionic acid (800 mg, 3.33 mmol) and 7 (500 mg, 1.67 mmol). Purification was achieved by flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 11e as pale yellowish solid. Yield 422 mg (49%); C27H27FN4O4S (Mr 522.60); 1H-NMR (DMSO-d6): δ = 2.62 (s, 3H, SCH3), 2.62–2.69 (m, 2H, COCH2CH2), 2.80–2.85 (m, 2H, COCH2CH2), 3.61 (s, 3H, C4OCH3), 3.74 (bs, 6H, C3/5OCH3), 6.56 (s, 2H, C2/6H, (OCH3)3-Phe), 7.01 (dd, 3J = 5.3 Hz, 4J = 1.6 Hz, 1H, C5H, Pyr), 7.26–7.32 (m, 2H, C3/5H, F-Phe), 7.46–7.51 (m, 2H, C2/6H, F-Phe), 8.11 (dd, 3J = 5.3 Hz, 5J = 0.5 Hz, 1H, C6H, Pyr), 8.36 (bs, 1H, C3H, Pyr), 10.36 (s, 1H, CONH), 12.70 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 31.3 (COCH2CH2), 37.9 (COCH2CH2), 55.7 (C3/5OCH3), 59.9 (C4OCH3), 105.5 (C2/6H, (OCH3)3-Phe), 110.6 (C3H, Pyr), 115.8 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 116.4 (C5H, Pyr), 126.6 (d, 4JCF = 3.3 Hz, C1, F-Phe), 130.1 (C5, Imdz), 130.7 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 134.5 (C4, Imdz), 135.7 (C4OCH3), 136.8 (C1, (OCH3)3-Phe), 142.2 (C2, Imdz), 143.8 (C4, Pyr), 147.6 (C6H, Pyr), 152.5 (C2, Pyr), 152.7 (C3/5OCH3), 162.0 (d, 1JCF = 245.4 Hz, CF), 171.2 (CO) ppm; MS (ESI, 70 eV) m/z 523 [MH]+.
3.2.4. Synthesis of 4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-carbamides 12a–g
A solution of 7 (1.0 equiv), the appropriate isocyanate (1.1 equiv) and DIPEA (1.2 equiv) in 5 mL anhyd. DMF was stirred under a nitrogen atmosphere at rt for 12 h. The solvent was removed under reduced pressure, the residue resuspended in ethyl acetate, and washed with 0.1 M aq. HCl, sat. aq. NaHCO3 solution, and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and concentrated under reduced pressure. The crude product was purified by flash chromatography (SiO2 and RP-18, eluent and mixing ratio given for each compound, respectively) to afford the particular compound.
1-(2,4-Dimethoxyphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)carbamide (12a). Synthesis was performed according to the general procedure from 2,4-dimethoxyphenyl isocyanate (262 mg, 1.47 mmol). Purification was achieved by flash chromatography (SiO2, 0%–10% methanol/DCM and RP-18, 50%–100% methanol/H2O) to afford 12a as colorless solid. Yield 135 mg (21%); C24H22FN5O3S (Mr 479.53); m.p. 214 °C; 1H-NMR (DMSO-d6): δ = 2.63 (s, 3H, SCH3), 3.74 (s, 3H, C4OCH3), 3.88 (s, 3H, C2OCH3), 6.48 (dd, 3J = 8.9 Hz, 4J = 2.7 Hz, 1H, C5H, (OCH3)2-Phe), 6.62 (d, 4J = 2.7 Hz, 1H, C3H, (OCH3)2-Phe), 6.92 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.30–7.51 (m, 5H, C3/5H, F-Phe and C3H, Pyr and C2/6H, F-Phe), 8.03 (d, 3J = 8.9 Hz, 1H, C6H, (OCH3)2-Phe), 8.12 (bs, 1H, C6H, Pyr), 9.64 (bs, 1H, NH), 10.89–11.26 (m, 1H, NH), 12.74 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 55.3 (C4OCH3), 56.0 (C2OCH3), 98.8 (C3H, (OCH3)2-Phe), 104.1 (C5H, (OCH3)2-Phe), 108.6 (C3H, Pyr), 114.6 (C5H, Pyr), 115.7 (C2/6H, F-Phe), 119.7 (C6H, (OCH3)2-Phe), 121.9 (C1, (OCH3)2-Phe), 130.7 (C3/5H, F-Phe), 134.2 (C4, Pyr), 142.3 (C2, Imdz), 146.1 (C6H, Pyr), 149.4 (C2, (OCH3)2-Phe), 152.2 (CO), 153.6 (C2, Pyr), 155.2 (C4, (OCH3)2-Phe), 161.8 (d, 1JCF = 241.6 Hz, CF) ppm; MS (ESI, 70 eV) m/z 480 [MH]+.
1-(2,5-Dimethoxyphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)carbamide (12b). Synthesis was performed according to the general procedure from 2,5-dimethoxyphenyl isocyanate (300 mg, 1.67 mmol). Purification was achieved by flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether) to afford 12b as pale yellowish solid. Yield 79.9 mg (15%); C24H22FN5O3S (Mr 479.53); m.p. 184 °C; 1H-NMR (DMSO-d6): δ = 2.63 (s, 3H, SCH3), 3.70 (s, 3H, C5OCH3), 3.84 (s, 3H, C2OCH3), 6.52 (dd, 3J = 8.8 Hz, 4J = 2.9 Hz, 1H, C4H, (OCH3)2-Phe), 6.91–6.96 (m, 1H, C3H, (OCH3)2-Phe), 6.95 (dd, 3J = 5.5 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.28–7.38 (m, 2H, C3/5H, F-Phe), 7.47–7.51 (m, 3H, C2/6H, F-Phe and C3H, Pyr), 7.92 (d, 4J = 2.9 Hz, 1H, C6H, (OCH3)2-Phe), 8.13 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 9.76 (bs, 1H, NH), 11.52 (vbs, 1H, NH), 12.74 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 55.3 (C5OCH3), 55.6 (C2OCH3), 105.6 (C6H, (OCH3)2-Phe), 105.8 (C4H, (OCH3)2-Phe), 108.5 (C3H, Pyr), 111.7 (C3H, (OCH3)2-Phe), 114.7 (C5H, Pyr), 115.9 (d, 2JCF = 22.0 Hz, C3/5H, F-Phe), 126.5 (d, 4JCF = 3.3 Hz, C1, F-Phe), 129.6 (C1, (OCH3)2-Phe), 130.4 (C5, Imdz), 130.8 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 134.1 (C4, Imdz), 142.3 (C2, Imdz), 142.4 (C2OCH3), 144.2 (C4, Pyr), 146.1 (C6H, Pyr), 152.2 (CO), 153.4 (C5OCH3), 153.4 (C2, Pyr), 162.1 (d, 1JCF = 245.9 Hz, CF) ppm; MS (ESI, 70 eV) m/z 480 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(4-methoxy-phenyl)carbamide (12c). Synthesis was performed according to the general procedure from 4-methoxyphenyl isocyanate (165 µL, 1.27 mmol). Purification was achieved by flash chromatography (SiO2, 50%–100% ethyl acetate/petrol ether and RP-18, 60%–100% methanol/H2O) to afford 12c as pale yellow solid. Yield 137 mg (26%); C23H20FN5O2S (Mr 449.50); m.p. 201 °C; 1H-NMR (DMSO-d6): δ = 2.62 (s, 3H, SCH3), 3.72 (s, 3H, OCH3), 6.86–6.90 (m, 2H, C3/5H, H3CO-Phe), 6.92 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.30 (bs, 2H, C3/5H, F-Phe), 7.39–7.44 (m, 2H, C2/6H, H3CO-Phe), 7.47–7.51 (m, 2H, C2/6H, F-Phe), 7.58 (bs, 1H, C3H, Pyr), 8.11 (bs, 1H, C6H, Pyr), 9.40 (bs, 1H, NH), 10.35–10.71 (m, 1H, NH), 12.74 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 55.2 (OCH3), 105.3 (C4, Pyr), 108.7 (C3H, Pyr), 114.0 (C3/5H, H3CO-Phe), 114.6 (C5H, Pyr), 115.8 (d, 2JCF = 22.0 Hz, C3/5H, F-Phe), 120.6 (C2/6H, H3CO-Phe), 130.6–130.8 (C2/6H, F-Phe), 132.0 (C1, H3CO-Phe), 142.3 (C2, Imdz), 146.4 (C6H, Pyr), 152.3 (CO), 153.5 (C2, Pyr), 154.9 (C4, H3CO-Phe), 162.1 (d, 1JCF = 252.6 Hz, CF) ppm; MS (ESI, 70 eV) m/z 450 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(m-tolyl)-carbamide (12d). Synthesis was performed according to the general procedure from 3-methylphenyl isocyanate (284 µL, 2.20 mmol). Purification was achieved by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 60%–80% methanol/H2O) to afford 12d as colorless solid. Yield 241 mg (28%); C23H20FN5OS (Mr 433.51); m.p. 211 °C; 1H-NMR (DMSO-d6): δ = 2.29 (s, 3H, CH3), 2.63 (s, 3H, SCH3), 6.83 (d, 3J = 7.4 Hz, 1H, C4H, Tol), 6.95 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.15–7.35 (m, 2H, C3/5H, F-Phe), 7.17 (t, 3J = 7.6 Hz, 1H, C5H, Tol), 7.31 (d, 3J = 8.0 Hz, 1H, C6H, Tol), 7.35 (s, 1H, C2H, Tol), 7.47–7.52 (m, 2H, C2/6H, F-Phe), 7.61 (bs, 1H, C3H, Pyr), 8.13 (bs, 1H, C6H, Pyr), 9.44 (s, 1H, NH), 10.42–10.78 (m, 1H, NH), 12.74 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 21.2 (CH3), 108.7 (C3H, Pyr), 114.8 (C5H, Pyr), 115.8 (d, 2JCF = 22.3 Hz, C3/5H, F-Phe), 116.0 (C6H, Tol), 119.3 (C2H, Tol), 123.2 (C4H, Tol), 126.6 (d, 4JCF = 2.6 Hz, C1, F-Phe), 128.7 (C5H, Tol), 129.7 (C5, Imdz), 130.8 (C2/6H, F-Phe), 134.2 (C4, Imdz), 138.1 (C3, Tol), 139.0 (C1, Tol), 142.4 (C2, Imdz), 144.3 (C4, Pyr), 146.4 (C6H, Pyr), 152.1 (CO), 153.4 (C2, Pyr), 162.0 (d, 1JCF = 243.0 Hz, CF) ppm; MS (ESI, 70 eV) m/z 434 [MH]+.
1-(3-Chloro-4-methylphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-carbamide (12e). Synthesis was performed according to the general procedure from 3-chloro-4-methylyphenyl isocyanate (205 µL, 1.47 mmol). Purification was achieved by flash chromatography (SiO2, 0%–10% methanol/DCM and RP-18, 70%–100% methanol/H2O and SiO2, 20%–100% ethyl acetate/petrol ether) to afford 12e as colorless solid. Yield 210 mg (34%); C23H19ClFN5OS (Mr 467.95); m.p. 210 °C; 1H-NMR (DMSO-d6): δ = 2.27 (s, 3H, CH3), 2.63 (s, 3H, SCH3), 6.96 (dd, 3J = 5.5 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.26 (m, 2H, C5/6H, Cl-Tol), 7.28–7.34 (m, 2H, C3/5H, F-Phe), 7.46–7.50 (m, 2H, C2/6H, F-Phe), 7.59 (bs, 1H, C3H, Pyr), 7.77 (s, 1H, C2H, Cl-Tol), 8.12 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 9.51 (s, 1H, NH), 10.99 (s, 1H, NH), 12.73 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 18.8 (CH3), 108.7 (C3H, Pyr), 114.8 (C5H, Pyr), 115.9 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 117.6 (C6H, Cl-Tol), 118.7 (C2H, Cl-Tol), 126.5 (d, 4JCF = 3.1 Hz, C1, F-Phe), 128.9 (C4, Cl-Tol), 130.4 (C5, Imdz), 130.8 (d, 3JCF = 8.3 Hz, C2/6H, F-Phe), 131.2 (C5H, Cl-Tol), 133.2 (C3, Cl-Tol), 134.1 (C4, Imdz), 138.2 (C1, Cl-Tol), 142.3 (C2, Imdz), 144.3 (C4, Pyr), 146.4 (C6H, Pyr), 152.2 (CO), 153.2 (C2, Pyr), 162.1 (d, 1JCF = 245.5 Hz, CF) ppm; MS (ESI, 70 eV) m/z 468 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(3-(trifluoromethyl)phenyl)-carbamide (12f). Synthesis was performed according to the general procedure from 3-(trifluoromethyl)phenyl isocyanate (300 µL, 2.20 mmol). Purification was achieved by flash chromatography (SiO2, 40% methanol/DCM) and subsequent filtration to afford 12f as colorless solid. Yield 255 mg (26%); C23H17FN5OS (Mr 487.48); m.p. 234 °C; 1H-NMR (DMSO-d6): δ = 2.63 (s, 3H, SCH3), 6.99 (dd, 3J = 5.6 Hz, 4J = 1.2 Hz, 1H, C5H, Pyr), 7.28–7.37 (m, 3H, C3/5H, F-Phe and C4H, F3C-Phe), 7.46–7.66 (m, 5H, C3H, Pyr and C2/6H, F-Phe and C5/6H, F3C-Phe), 8.08 (bs, 1H, C2H, F3C-Phe), 8.14 (d, 3J = 5.5 Hz, 1H, C6H, Pyr), 9.56 (s, 1H, NH), 11.12 (s, 1H, NH), 12.74 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 108.7 (C3H, Pyr), 114.7 (C2H, F3C-Phe), 115.0 (C5H, Pyr), 115.9 (d, 2JCF = 21.8 Hz, C3/5H, F-Phe), 118.7 (C4H, F3C-Phe), 122.5 (C6H, F3C-Phe), 124.2 (q, 1JCF = 272.1 Hz, CF3, F3C-Phe), 126.5 (d, 4JCF = 2.9 Hz, C1, F-Phe), 129.6 (q, 2JCF = 31.3 Hz, C3, F3C-Phe), 130.0 (C5H, F3C-Phe), 130.5 (C5, Imdz), 130.8 (d, 3JCF = 8.3 Hz, C2/6H, F-Phe), 134.0 (C4, Imdz), 139.9 (C1, F3C-Phe), 142.3 (C2, Imdz), 144.3 (C4, Pyr), 146.6 (C6H, Pyr), 152.2 (CO), 153.0 (C2, Pyr), 162.0 (d, 1JCF = 245.2 Hz, CF) ppm; MS (ESI, 70 eV) m/z 488 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C23H17FN5OS, 487.1090; found 487.1090.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(naphthalen-1-yl)carbamide (12g). Synthesis was performed according to the general procedure from naphthyl isocyanate (211 µL, 1.47 mmol). Purification was achieved by flash chromatography (SiO2, 0%–10% methanol/DCM and SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 12g as pale yellow solid. Yield 257 mg (41%); C26H20FN5OS (Mr 469.54); m.p. 249 °C; 1H-NMR (DMSO-d6): δ = 2.64 (s, 3H, SCH3), 7.01 (dd, 3J = 5.5 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.30–7.36 (m, 2H, C3/5H, F-Phe), 7.42–7.59 (m, 5H, C3H, Pyr and C2/6H, F-Phe and C3/6H, Naph), 7.63–7.68 (m, 2H, C4/7H, Naph), 7.95 (d, 3J = 8.1 Hz, 1H, C5H, Naph), 8.16–8.22 (m, 2H, C2/8H, Naph), 8.27 (d, 3J = 5.5 Hz, 1H, C6H, Pyr), 9.87 (s, 1H, NH), 11.86 (bs, 1H, NH), 12.77 (s, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 15.1 (SCH3), 108.7 (C3H, Pyr), 114.7 (C5H, Pyr), 115.9 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 116.7 (C2H, Naph), 120.9 (C8H, Naph), 123.0 (C4H, Naph), 125.4 (C1, Naph), 126.0 (C3/6H, Naph), 126.3 (C7H, Naph), 126.5 (d, 4JCF = 3.2 Hz, C1, F-Phe), 128.6 (C5H, Naph), 130.6 (C5, Imdz), 130.8 (d, 3JCF = 8.3 Hz, C2/6H, F-Phe), 133.7 (C4a, Naph), 134.0 (C8a, Naph), 134.2 (C4, Imdz), 142.4 (C2, Imdz), 144.5 (C4, Pyr), 146.1 (C6H, Pyr), 152.6 (CO), 153.5 (C2, Pyr), 162.1 (d, 1JCF = 246.2 Hz, CF) ppm; MS (ESI, 70 eV) m/z 471 [MH]+.
3.2.5. Synthesis of N-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-1-methyl-4-phenyl-1H-pyrrole-2-carboxamides 13, 14a–b, 15a–b, 16a–b
Ethyl 4-bromo-1-methyl-1H-pyrrole-2-carboxylate (13). NaH (240 mg 60% dispersion in mineral oil, 6.05 mmol) was added in one portion to a solution of 4-bromo-1H-pyrrole-2-carboxylate (1.17 g, 5.34 mmol) in 15 mL anhyd. DMF at 0 °C under a nitrogen atmosphere. The suspension was stirred for 20 min at the same temp. before methyl iodide (400 µL, 6.43 mmol) was carefully added and stirring continued at 0 °C for 15 min and another 2.5 h at r.t. The reaction was quenched with H2O and extracted with ethyl acetate. The combined organic phases were washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. Purification was achieved by flash chromatography (SiO2, 2%–10% ethyl acetate/petrol ether) to afford 13 as clear colorless oil that crystallized on standing as colorless needles. Yield 1.22 g (98%); C8H10BrNO2 (Mr 232.08); m.p. 43 °C; 1H-NMR (DMSO-d6): δ = 1.26 (t, 3J = 7.1 Hz, 3H, COOCH2CH3), 3.83 (d, J = 0.2 Hz, 3H, CH3), 4.21 (q, 3J = 7.1 Hz, 2H, COOCH2CH3), 6.84 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.28 (d, 4J = 1.7 Hz, 1H, C5H, Pyrrole) ppm; 13C-NMR (DMSO-d6): δ = 14.2 (COOCH2CH3), 36.6 (CH3), 59.8 (COOCH2CH3), 93.8 (CBr), 118.1 (C3H, Pyrrole), 122.7 (C2, Pyrrole), 129.5 (C5H, Pyrrole), 159.5 (COOCH2CH3) ppm.
Ethyl 4-(2,4-dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylate (14a). 27 mL 2 M aq. Na2CO3 solution were added to a stirred solution of 13 (1.22 g, 5.26 mmol), 2,4-dimethoxyphenyl boronic acid (2.89 g, 15.9 mmol), and Pd(PPh3)4 (306 mg, 265 µmol) in 80 mL DMF. Stirring continued for 4 h under reflux and 12 h at r.t. H2O was added and the mixture was extracted with ethyl acetate. The combined organic phases were washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (SiO2, 5%–10% ethyl acetate/petrol ether and RP-18, 50%–70% methanol/H2O) to afford 14a as colorless solid. Yield 1.02 g (67%); C16H19NO4 (Mr 289.33); 1H-NMR (DMSO-d6): δ = 1.29 (t, 3J = 7.1 Hz, 3H, COOCH2CH3), 3.77 (s, 3H, C4OCH3), 3.84 (s, 3H, C2OCH3), 3.87 (s, 3H, CH3), 4.23 (q, 3J = 7.1 Hz, 2H, COOCH2CH3), 6.53 (dd, 3J = 8.5 Hz, 4J = 2.5 Hz, 1H, C5H, (OCH3)2-Phe), 6.60 (d, 4J = 2.5 Hz, 1H, C3H, (OCH3)2-Phe), 7.17 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.44 (d, 3J = 8.5 Hz, 1H, C6H, (OCH3)2-Phe), 7.46 (dd, 4J = 2.0 Hz, 5J = 0.4 Hz, 1H, C5H, Pyrrole) ppm; 13C-NMR (DMSO-d6): δ = 14.4 (COOCH2CH3), 36.4 (CH3), 55.2 (C4OCH3), 55.4 (C2OCH3), 59.4 (CH2), 98.8 (C3H, (OCH3)2-Phe), 105.2 (C5H, (OCH3)2-Phe), 115.4 (C3H, Pyrrole), 115.5 (C1, (OCH3)2-Phe), 119.0 (C4, Pyrrole), 121.3 (C2, Pyrrole), 127.7 (C6H, (OCH3)2-Phe), 129.0 (C5H, Pyrrole), 156.7 (C4OCH3), 158.74 (C2OCH3), 160.5 (COOCH2CH3) ppm; MS (ESI, 70 eV) m/z 290 [MH]+.
Ethyl 4-(2,5-dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylate (14b). Synthesis was performed according to the procedure described for 14a starting from 13 (1.11 g, 4.78 mmol), 2,5-dimethoxyphenyl boronic acid (2.65 g, 14.6 mmol), Pd(PPh3)4 (289 mg, 250 µmol) in 80 mL DMF, and 25 mL 2 M aq. Na2CO3 solution. Purification was achieved by flash chromatography (SiO2, 2%–10% ethyl acetate/petrol ether and RP-18, 65% methanol/H2O) to afford 14b as colorless solid. Yield 984 mg (71%); C16H19NO4 (Mr 289.33); 1H-NMR (DMSO-d6): δ = 1.30 (t, 3J = 7.1 Hz, 3H, COOCH2CH3), 3.74 (s, 3H, C5OCH3), 3.79 (s, 3H, C2OCH3), 3.89 (s, 3H, CH3), 4.24 (q, 3J = 7.1 Hz, 2H, COOCH2CH3), 6.74 (dd, 3J = 8.9 Hz, 4J = 3.1 Hz, 1H, C4H, (OCH3)2-Phe), 6.96 (d, 3J = 8.9 Hz, 1H, C3H, (OCH3)2-Phe), 7.09 (d, 4J = 3.1 Hz, 1H, C6H, (OCH3)2-Phe), 7.28 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.60 (dd, 4J = 2.0 Hz, 5J = 0.4 Hz, 1H, C5H, Pyrrole) ppm; 13C-NMR (DMSO-d6): δ = 14.3 (COOCH2CH3), 36.5 (CH3), 55.4 (C5OCH3), 55.9 (C2OCH3), 59.4 (COOCH2CH3), 111.7 (C4H, (OCH3)2-Phe), 112.5 (C6H, (OCH3)2-Phe), 112.9 (C3H, (OCH3)2-Phe), 116.1 (C3H, Pyrrole), 118.9 (C4, Pyrrole), 121.6 (C1, Pyrrole), 123.4 (C1, (OCH3)2-Phe), 130.0 (C5H, Pyrrole), 150.0 (C2OCH3), 153.4 (C5OCH3), 160.5 (COOCH2CH3) ppm; MS (ESI, 70 eV) m/z 290 [MH]+.
4-(2,4-Dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylic acid (15a). 7 mL 4 M aq. NaOH were added to a solution of 14a (990 mg, 3.42 mmol) in 18 mL THF and 9 mL methanol and the mixture was stirred at 50 °C for 5 h and then 12 h at r.t. H2O was added to the reaction, the pH was adjusted to 3 using 1 M aq. HCl, and the mixture was extracted with ethyl acetate. The combined organic phases were washed with H2O and sat. aq. NaCl solution, the organic phase was dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure to afford 15a as brown solid. Yield 894 mg (quant.); C14H15NO4 (Mr 261.28); m.p. 164 °C; 1H-NMR (DMSO-d6): δ = 3.76 (s, 3H, C4OCH3), 3.83 (s, 3H, C2OCH3), 3.86 (s, 3H, CH3), 6.53 (dd, 3J = 8.5 Hz, 4J = 2.4 Hz, 1H, C5H, (OCH3)2-Phe), 6.59 (d, 4J = 2.4 Hz, 1H, C3H, (OCH3)2-Phe), 7.14 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.41 (d, 4J = 2.0 Hz, 1H, C5H, Pyrrole), 7.42 (d, 3J = 8.4 Hz, 1H, C6H, (OCH3)2-Phe), 12.17 (bs, 1H, COOH) ppm; 13C-NMR (DMSO-d6): δ = 36.3 (CH3), 55.2 (C4OCH3), 55.4 (C2OCH3), 98.8 (C3H, (OCH3)2-Phe), 105.2 (C5H, (OCH3)2-Phe), 115.7 (C3H, Pyrrole), 115.7 (C1, (OCH3)2-Phe), 118.8 (C4, Pyrrole), 122.0 (C2, Pyrrole), 127.6 (C6H, (OCH3)2-Phe), 128.6 (C5H, Pyrrole), 156.7 (C2OCH3), 158.7 (C4OCH3), 162.1 (COOH) ppm; MS (ESI, 70 eV) m/z 262 [MH]+.
4-(2,5-Dimethoxyphenyl)-1-methyl-1H-pyrrole-2-carboxylic acid (15b). Synthesis was performed according to the procedure described for 15a starting from 14b (980 mg, 3.39 mmol) in 19 mL THF and 10 mL methanol, and 7 mL 4 M aq. NaOH to afford 15b as brown solid. Yield 885 mg (quant.); C14H15NO4 (Mr 261.28); m.p. 149 °C; 1H-NMR (DMSO-d6): δ = 3.74 (s, 3H, C2OCH3), 3.79 (s, 3H, C5OCH3), 3.88 (CH3), 6.72 (dd, 3J = 8.9 Hz, 4J = 3.1 Hz, 1H, C4H, (OCH3)2-Phe), 6.95 (d, 3J = 8.9 Hz, 1H, C3H, (OCH3)2-Phe), 7.08 (d, 4J = 3.1 Hz, 1H, C6H, (OCH3)2-Phe), 7.25 (d, 4J = 2.0 Hz, 1H, C3H, Pyrrole), 7.55 (d, 4J = 2.0 Hz, 1H, C5H, Pyrrole), 12.15 (bs, 1H, COOH) ppm; 13C-NMR (DMSO-d6): δ = 36.4 (CH3), 55.4 (C2OCH3), 55.9 (C5OCH3), 111.5 (C4H, (OCH3)2-Phe), 112.5 (C6H, (OCH3)2-Phe), 112.9 (C3H, (OCH3)2-Phe), 116.2 (C3H, Pyrrole), 118.7 (C4, Pyrrole), 122.4 (C2, Pyrrole), 123.6 (C1, (OCH3)2-Phe), 129.6 (C5H, Pyrrole), 150.1 (C5OCH3), 153.4 (C2OCH3), 162.1 (COOH) ppm; MS (ESI, 70 eV) m/z 262 [MH]+.
4-(2,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorphenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-1-methyl-1H-pyrrole-2-carboxamide (16a). A solution of 15a (1.01 g, 4.07 mmol), PyBOP (2.54 g, 4.88 mmol), and DIPEA (2.15 mL, 12.3 mmol) was stirred in 14 mL anhyd. DMF under a nitrogen atmosphere for 30 min at r.t. 7 (1.60 g, 5.31 mmol) was added in one portion and the mixture was stirred for 12 h at 110 °C. The reaction was quenched with H2O and extracted with ethyl acetate. The combined organic phases were washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 16a as beige solid. Yield 303 mg (24%); C29H26FN5O3S (Mr 543.62); m.p. 236 °C; 1H-NMR (DMSO-d6): δ = 2.64 (s, 3H, SCH3), 3.78 (s, 3H, C2OCH3), 3.86 (s, 3H, C4OCH3), 3.90 (s, 3H, CH3), 6.57 (dd, 3J = 8.5 Hz, 4J = 2.3 Hz, 1H, C5H, (OCH3)2-Phe), 6.61 (d, 4J = 2.3 Hz, 1H, C3H, (OCH3)2-Phe), 7.06 (dd, 3J = 5.3 Hz, 4J = 1.2 Hz, 1H, C5H, Pyr), 7.28–7.34 (m, 2H, C3/5H, F-Phe), 7.41 (d, 4J = 1.3 Hz, 1H, C3H, Pyrrole), 7.46 (d, 3J = 8.4 Hz, 1H, C6H, (OCH3)2-Phe), 7.49–7.54 (m, 2H, C2/6H, F-Phe), 7.66 (d, 4J = 1.4 Hz, 1H, C5H, Pyrrole), 8.18 (d, 3J = 5.2 Hz, 1H, C6H, Pyr), 8.38 (bs, 1H, C3H, Pyr), 10.10 (bs, 1H, CONH), 12.73 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.3 (SCH3), 36.5 (CH3), 55.2 (C2OCH3), 55.5 (C4OCH3), 98.8 (C3H, (OCH3)2-Phe), 105.2 (C5H, (OCH3)2-Phe), 111.3 (C3H, Pyr), 113.2 (C5H, Pyrrole), 115.9 (d, 2JCF = 21.9 Hz, C3/5H, F-Phe), 115.9 (C1, (OCH3)2-Phe), 116.3 (C5H, Pyr), 118.5 (C4, Pyrrole), 124.1 (C2, Pyrrole), 126.7 (d, 4JCF = 3.5 Hz, C1, F-Phe), 127.5 (C6H, (OCH3)2-Phe), 128.5 (C3H, Pyrrole), 130.8 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 134.5 (C5, Imdz), 142.1 (C2, Imdz), 143.6 (C4, Imdz), 147.6 (C6H, Pyr), 152.7 (C2, Pyr), 156.7 (C4OCH3), 158.6 (C2OCH3), 159.8 (CONH), 162.0 (d, 1JCF = 245.6 Hz, CF) ppm; MS (ESI, 70 eV) m/z 544 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C29H26FN5O3S, 543.1740; found, 543.1740.
4-(2,5-Dimethoxyphenyl)-N-(4-(5-(4-fluorphenyl)-2-(methylthio)-1H-imidazol-4-yl)-pyridin-2-yl)-1-methyl-1H-pyrrole-2-carboxamide (16b). Synthesis was performed according to the procedure described for 16a starting from 15b (604 mg, 2.31 mmol), PyBOP (1.45 g, 2.79 mmol), DIPEA (1.20 mL, 6.87 mmol), and 7 (904 mg, 3.01 mmol) in 12 mL anhyd. DMF. The crude product was purified by flash chromatography (SiO2, 30%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) to afford 16b as pale yellowish solid. Yield 255 mg (20%); m.p. 127 °C; 1H-NMR (DMSO-d6): δ = 2.64 (s, 3H, SCH3), 3.76 (s, 3H, C5OCH3), 3.81 (s, 3H, C2OCH3), 3.92 (s, 3H, CH3), 6.73 (dd, 3J = 8.9 Hz, 4J = 3.1 Hz, 1H, C4H, (OCH3)2-Phe), 6.96 (d, 3J = 9.0 Hz, 1H, C3H, (OCH3)2-Phe), 7.06 (dd, 3J = 5.3 Hz, 4J = 1.6 Hz, 1H, C5H, Pyr), 7.17 (d, 4J = 3.1 Hz, 1H, C6H, (OCH3)2-Phe), 7.24–7.30 (m, 2H, C3/5H, F-Phe), 7.50–7.55 (m, 2H, C2/6H, F-Phe), 7.56 (d, 4J = 1.7 Hz, 1H, C5H, Pyrrole), 7.80 (d, 4J = 1.8 Hz, 1H, C3H, Pyrrole), 8.22 (d, 3J = 5.2 Hz, 1H, C6H, Pyr), 8.35 (bs, 1H, C3H, Pyr), 10.15 (bs, 1H, CONH), 12.77 (vbs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 15.2 (SCH3), 36.7 (CH3), 55.4 (C5OCH3), 55.9 (C2OCH3), 111.3 (C4H, (OCH3)2-Phe and C3H, Pyr), 112.5 (C6H, (OCH3)2-Phe), 112.8 (C3H, (OCH3)2-Phe), 113.6 (C3H, Pyrrole), 115.7 (d, 2JCF = 21.5 Hz, C3/5H, F-Phe), 116.5 (C5H, Pyr), 118.3 (C4, Pyrrole), 123.8 (C1, (OCH3)2-Phe), 124.3 (C2, Pyrrole), 129.7 (C5H, Pyrrole), 130.42 (C2/6H, F-Phe), 142.5 (C2, Imdz and C4, Pyr), 147.7 (C6H, Pyr), 150.1 (C2OCH3), 152.7 (C1, Pyr), 153.4 (C5OCH3), 159.8 (CONH), 161.9 (d, 1JCF = 247.7 Hz, CF) ppm; MS (ESI, 70 eV) m/z 544 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C29H26FN5O3S, 543.1740; found, 543.1740.
3.2.6. Synthesis of Sulfoxides 10j–k, 11f–g, 12h–m, 16c
The sulfide (1.0 equiv) was dissolved in THF and H2O was added (approx. 3:1). The mixture was stirred at 0 °C for 10 min before an ice-cold aq. solution of potassium peroxomonosulfate (Oxone®, 0.6 equiv) was added and stirring continued for 0.5–2 h at the same temp. After completion of the reaction sat. aq. NaHCO3 solution, H2O, and ethyl acetate were added and the phases were separated. The organic layer was washed with H2O and sat. aq. NaCl solution, dried over anhyd. Na2SO4, and the solvent was removed under reduced pressure. Purification of the crude products was achieved by crystallization from ethyl acetate or flash chromatography (SiO2 and RP-18, eluent and mixing ratio given for each compound) to afford the appropriate compound.
N-(2-Ethoxyphenyl)-4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-amine (10j). Synthesis was performed according to the general procedure for sulfoxidation starting from 10c (300 mg, 713 µmol) in 5 mL THF and 2 mL H2O. Flash chromatography (RP-18, 20%–90% methanol/H2O) afforded 10j as voluminous yellow solid. Yield 255 mg (82%); C23H21FN4O2S (Mr 436.51); m.p. 152 °C; 1H-NMR (CDCl3): δ = 1.42 (t, 3J = 7.0 Hz, 3H, CH3), 3.03 (s, 3H, SCH3), 4.07 (q, 3J = 7.0 Hz, 2H, CH2), 6.94–6.78 (m, 4H, C4–6H, EtO-Phe and C5H, Pyr), 7.03 (dd, 4J = 1.4 Hz, 5J = 0.7 Hz, 1H, C3H, Pyr), 7.07–7.13 (m, 3H, C3/5H, F-Phe and NH), 7.46–7.50 (m, 2H, C2/6H, F-Phe), 7.62 (dd, 3J = 7.8 Hz, 4J = 1.5 Hz, 1H, C3H, EtO-Phe), 8.14 (dd, 3J = 5.3 Hz, 5J = 0.7 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 14.9 (CH3), 40.8 (SCH3), 64.1 (CH2), 106.9 (C3H, Pyr), 111.5 (C3H, EtO-Phe), 113.3 (C5H, Pyr), 116.0 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 118.2 (C6H, EtO-Phe), 120.6 (C5H, EtO-Phe), 121.9 (C4H, EtO-Phe), 129.9 (C1, EtO-Phe), 130.6 (d, 3JCF = 8.2 Hz, C2/6H, F-Phe), 146.7 (C2, Imdz), 148.2 (C2, EtO-Phe), 148.5 (C6H, Pyr), 156.0 (C2, Pyr), 162.9 (d, 1JCF = 248.8 Hz, CF) ppm; MS (ESI, 70 eV) m/z 437 [MH]+.
N-(3,4-Dimethoxyphenethyl)-4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-amine (10k). Synthesis was performed according to the general procedure for sulfoxidation starting from 10d (500 mg, 1.08 mmol) in 5 mL H2O in 9 mL THF and 3 mL H2O. Flash chromatography (RP-18, 20%–90% methanol/H2O) afforded 10d as pale yellow solid. Yield 208 mg (39%); C25H25FN4O3S (Mr 480.56); m.p. 183 °C; 1H-NMR (CDCl3): δ = 2.74 (t, 3J = 6.8 Hz, 1H, CH2CH2NH), 2.97 (s, 3H, SCH3), 3.36 (m, 2H, CH2CH2NH), 3.76 (s, 6H, 2 OCH3), 5.00 (bs, 1H, CH2CH2NH), 6.49 (bs, 1H, C3H, Pyr), 6.52 (dd, 3J = 5.4 Hz, 4J = 1.3 Hz, 1H, C5H, Pyr), 6.60–6.63 (m, 2H, C2/6H, (OCH3)2-Phe), 6.71 (d, 3J = 8.7 Hz, 1H, C5H, (OCH3)2-Phe), 6.96–7.02 (m, 2H, C3/5H, F-Phe), 7.36–7.41 (m, 2H, C2/6H, F-Phe), 7.80 (d, 3J = 5.5 Hz, 1H, C6H, Pyr) ppm; 13C-NMR (CDCl3): δ = 35.1 (CH2CH2NH), 40.7 (SCH3), 43.3 (CH2CH2NH), 55.9 (C3OCH3), 55.9 (C4OCH3), 104.6 (C3H, Pyr), 111.2 (C5H, Pyr), 111.4 (C5H, (OCH3)2-Phe), 112.1 (C2H, (OCH3)2-Phe), 115.9 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 120.7 (C6H, (OCH3)2-Phe), 127.2 (C1, F-Phe), 130.5 (d, 3JCF = 8.2 Hz, C2/6H, F-Phe), 131.5 (C1, (OCH3)2-Phe), 135.3 (C4/5, Imdz), 141.6 (C4, Pyr), 146.8 (C2, Imdz), 147.4 (C6H, Pyr), 147.7 (C4OCH3), 149.0 (C3OCH3), 158.6 (C2, Pyr), 162.9 (d, 1JCF = 249.2 Hz, CF) ppm; MS (ESI, 70 eV) m/z 481 [MH]+.
3-(2,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorphenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-yl)-propanamide (11f). Synthesis was performed according to the general procedure for sulfoxidation starting from 11a (100 mg, 203 µmol) in 2 mL THF and 0.6 mL H2O. Flash chromatography (SiO2, 40%–100% ethyl acetate/petrol ether) afforded 11f as colorless solid. Yield 96.1 mg (93%); C26H25FN4O4S (Mr 508.57); m.p. 209 °C; 1H-NMR (DMSO-d6): δ = 2.58 (t, 3J = 7.5 Hz, 2H, CH2CH2CO), 2.75 (t, 3J = 7.4 Hz, 2H, CH2CH2CO), 3.08 (s, 3H, SCH3), 3.72 (s, 3H, C2OCH3), 3.77 (s, 3H, C4OCH3), 6.42 (dd, 3J = 8.3 Hz, 4J = 2.4 Hz, 1H, C5H, (OCH3)2-Phe), 6.51 (d, 4J = 2.4 Hz, 1H, C3H, (OCH3)2-Phe), 7.01–7.05 (m, 2H, C5H, Pyr and C6H, (OCH3)2-Phe), 7.26–7.32 (m, 2H, C3/5H, F-Phe), 7.51–7.55 (m, 2H, C2/6H, F-Phe), 8.19 (d, 3J = 5.2 Hz, 1H, C6H, Pyr), 8.34 (bs, 1H, C3H, Pyr), 10.40 (s, 1H, CONH), 13.89 (bs, NH) ppm; 13C-NMR (DMSO-d6): δ = 24.7 (CH2CH2CO), 36.3 (CH2CH2CO), 39.1 (SCH3), 55.1 (C2OCH3), 55.3 (C4OCH3), 98.3 (C3H, (OCH3)2-Phe), 104.3 (C5H, (OCH3)2-Phe), 111.1 (C3H, Pyr), 115.8 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 117.0 (C5H, Pyr), 120.9 (C1, (OCH3)2-Phe), 127.3 (C1, F-Phe), 129.8 (C6H, (OCH3)2-Phe), 130.8 (d, 3JCF = 8.2 Hz, C2/6H, F-Phe), 133.4 (C4, Imdz), 133.9 (C5, Imdz), 142.2 (C4, Pyr), 147.9 (C6H, Pyr), 148.9 (C2, Imdz), 152.6 (C2, Pyr), 157.9 (C4OCH3), 159.0 (C2OCH3), 162.1 (d, 1JCF = 245.5 Hz, CF), 171.54 (CO) ppm; MS (ESI, 70 eV) m/z 509 [MH]+; HRMS (EI, 70 eV) m/z [M]+ calcd. for C26H25FN4O4S, 508.1581; found, 508.1581.
3-(2,5-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-yl)propanamide (11g). Synthesis was performed according to the general procedure for sulfoxidation starting from 11b (100 mg, 203 µmol) in 2 mL THF and 0.6 mL H2O. Crystallization from ethyl acetate afforded 11g as colorless solid. Yield 26.6 mg (26%); m.p. 204 °C; 1H-NMR (DMSO-d6): δ = 2.62 (t, 3J = 7.7 Hz, 2H, CH2CH2CO), 2.80 (t, 3J = 7.5 Hz, 2H, CH2CH2CO), 3.09 (s, 3H, SCH3), 3.66 (s, 3H, C5OCH3), 3.72 (s, 3H, C2OCH3), 6.72 (dd, 3J = 8.8 Hz, 4J = 3.1 Hz, 1H, C4H, (OCH3)2-Phe), 6.77 (d, 4J = 3.0 Hz, 1H, C6H, (OCH3)2-Phe), 6.86 (d, 3J = 8.8 Hz, 1H, C3H, (OCH3)2-Phe), 7.02 (dd, 3J = 5.2 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.25–7.31 (m, 2H, C3/5H, F-Phe), 7.51–7.55 (m, 2H, C2/6H, F-Phe), 8.19 (d, 3J = 5.0 Hz, 1H, C6H, Pyr), 8.33 (s, 1H, C3H, Pyr), 10.45 (s, 1H, CONH), 14.00 (bs, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 25.3 (CH2CH2CO), 36.0 (CH2CH2CO), 39.0 (SCH3), 55.2 (C5OCH3), 55.8 (C2OCH3), 111.2 (C4H, (OCH3)2-Phe), 111.3 (C3H, Pyr), 111.5 (C3H, (OCH3)2-Phe), 115.8 (d, 2JCF = 21.3 Hz, C3/5H, F-Phe), 115.9 (C6H, (OCH3)2-Phe), 126.5 (C1, F-Phe), 130.8 (d, 3JCF = 8.3 Hz, C2/6H, F-Phe), 142.1 (C4, Pyr), 148.0 (C6H, Pyr), 148.8 (C2, Imdz), 151.2 (C2OCH3), 152.58 (C2, Pyr), 153.0 (C5OCH3), 162.1 (d, 1JCF = 245.6 Hz, CF), 171.42 (CO) ppm; MS (ESI, 70 eV) m/z 509 [MH]+.
1-(2,4-Dimethoxyphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-yl)-carbamide (12h). Synthesis was performed according to the general procedure for sulfoxidation starting from 12a (85.0 mg, 177 µmol) in 3.4 mL THF and 1 mL H2O. Flash chromatography (SiO2, 35%–100% ethyl acetate/petrol ether and RP-18, 55%–100% methanol/H2O) afforded 12h as colorless solid. Yield 18.9 mg (22%); C24H22FN5O4S (Mr 495.53); m.p. 227 °C; 1H-NMR (DMSO-d6): δ = 3.08 (s, 3H, SCH3), 3.74 (s, 3H, C4OCH3), 3.88 (s, 3H, C2OCH3), 6.48 (dd, 3J = 8.9 Hz, 4J = 2.7 Hz, 1H, C5H, (OCH3)2-Phe), 6.62 (d, 4J = 2.7 Hz, 1H, C3H, (OCH3)2-Phe), 6.95 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.27–7.33 (m, 2H, C3/5H, F-Phe), 7.46 (bs, 1H, C3H, Pyr), 7.51–7.56 (m, 2H, C2/6H, F-Phe), 8.02 (d, 3J = 8.9 Hz, 1H, C6H, (OCH3)2-Phe), 8.19 (d, 3J = 5.3 Hz, 1H, C6H, Pyr), 9.70 (s, 1H, NH), 11.07 (bs, 1H, NH), 13.87 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 39.1 (SCH3), 55.3 (C2OCH3), 56.0 (C4OCH3), 98.8 (C3H, (OCH3)2-Phe), 104.1 (C5H, (OCH3)2-Phe), 109.2 (C3H, Pyr), 115.1 (C5H, Pyr), 115.8 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 119.8 (C6H, (OCH3)2-Phe), 121.8 (C1, (OCH3)2-Phe), 127.2 (C1, F-Phe), 130.8 (d, 3JCF = 8.4 Hz, C2/6H, F-Phe), 132.7 (C4, Imdz), 134.1 (C5, Imdz), 142.8 (C4, Pyr), 146.5 (C6H, Pyr), 149.1 (C2, Imdz), 149.4 (C2OCH3), 152.2 (CO), 153.6 (C2, Pyr), 155.2 (C4OCH3), 162.14 (d, 1JCF = 244.6 Hz, CF) ppm; MS (ESI, 70 eV) m/z 496 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-yl)-3-(4-meth-oxyphenyl)carbamide (12i). Synthesis was performed according to the general procedure for sulfoxidation starting from 12c (100 mg, 225 µmol) in 2 mL THF and 0.6 mL H2O. Flash chromatography (RP-18, 50%–70% methanol/H2O) afforded 12i as colorless solid. Yield 62.6 mg (61%); C23H20FN5O3S (Mr 465.50); m.p. 233 °C; 1H-NMR (DMSO-d6): δ = 3.09 (s, 3H, SCH3), 3.73 (s, 3H, C4OCH3), 6.89 (d, 3J = 9.1 Hz, 2H, C3/5H, H3CO-Phe), 6.95 (dd, 3J = 5.4 Hz, 4J = 1.5 Hz, 1H, C5H, Pyr), 7.28–7.34 (m, 2H, C3/5H, F-Phe), 7.41 (d, 3J = 9.1 Hz, 2H, C2/6H, H3CO-Phe), 7.51–7.56 (m, 2H, C2/6H, F-Phe), 7.65 (bs, 1H, C3H, Pyr), 8.18 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 9.43 (s, 1H, NH), 10.47 (bs, 1H, NH), 13.92 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 39.1 (SCH3), 55.2 (C4OCH3), 109.4 (C3H, Pyr), 114.0 (C3/5H, H3CO-Phe), 115.2 (C5H, Pyr), 115.9 (d, 2JCF = 21.7 Hz, C3/5H, F-Phe), 120.6 (C2/6H, H3CO-Phe), 127.5 (C1, F-Phe), 130.9 (d, 3JCF = 8.7 Hz, C2/6H, F-Phe), 132.0 (C1, H3CO-Phe), 146.9 (C6H, Pyr), 148.8 (C2, Imdz), 152.2 (CO), 153.5 (C2, Pyr), 154.9 (C4OCH3), 162.2 (d, 1JCF = 246.7 Hz, CF) ppm; MS (ESI, 70 eV) m/z 466 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-yl)-3-(m-tolyl)-carbamide (12j). Synthesis was performed according to the general procedure for sulfoxidation starting from 12d (100 mg, 231 µmol) in 2 mL THF and 0.6 mL H2O. Flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether) afforded 12j as colorless solid. Yield 74.8 mg (72%); C23H20FN5O2S (Mr 449.50); m.p. 235 °C; 1H-NMR (DMSO-d6): δ = 2.29 (s, 3H, CH3), 3.09 (s, 3H, SCH3), 6.83 (d, 3J = 7.4 Hz, 1H, C4H, Tol), 6.97 (dd, 3J = 5.4 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.18 (t, 3J = 7.7 Hz, 1H, C5H, Tol), 7.29–7.35 (m, 4H, C3/5H, F-Phe and C2/6H, Tol), 7.52–7.57 (m, 2H, C2/6H, F-Phe), 7.69 (s, 1H, C3H, Pyr), 8.19 (d, 3J = 5.1 Hz, 1H, C6H, Pyr), 9.48 (s, 1H, NH), 10.54 (bs, 1H, NH), 13.92 (bs, 1H, NH, Imdz) ppm; 13C-NMR (75 MHz, DMSO-d6): δ = 21.2 (CH3), 39.1 (SCH3), 109.3 (C3H, Pyr), 115.3 (C5H, Pyr), 115.9 (d, 2JCF = 21.5 Hz, C3/5H, F-Phe), 116.0 (C6H, Tol), 119.3 (C2H, Tol), 123.2 (C4H, Tol), 128.7 (C5H, Tol), 130.9 (C2/6H, F-Phe), 138.1 (C3, Tol), 138.9 (C1, Tol), 143.1 (C4, Pyr), 147.0 (C6H, Pyr), 148.7 (C2, Imdz), 152.1 (CO), 153.4 (C2, Pyr), 162.2 (d, 1JCF = 244.0 Hz, CF) ppm; MS (ESI, 70 eV) m/z 450 [MH]+.
1-(3-Chloro-4-methylphenyl)-3-(4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-yl)-carbamide (12k). Synthesis was performed according to the general procedure for sulfoxidation starting from 12e (100 mg, 214 µmol) in 4 mL THF and 1 mL H2O. Flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether and RP-18, 50%–100% methanol/H2O) afforded 12k as beige solid. Yield 78.0 mg (75%); C23H19ClFN5O2S (Mr 483.95); 1H-NMR (DMSO-d6): δ = 2.27 (s, 3H, CH3), 3.09 (s, 3H, SCH3), 6.99 (dd, 3J = 5.4 Hz, 4J = 1.3 Hz, 1H, C5H, Pyr), 7.26 (s, 1H, C5H, Cl-Tol), 7.26 (s, 1H, C6H, Cl-Tol), 7.29–7.35 (m, 2H, C3/5H, F-Phe), 7.52–7.56 (m, 2H, C2/6H, F-Phe), 7.65 (bs, 1H, C3H, Pyr), 7.76 (bs, 1H, C2H, Cl-Tol), 9.55 (s, 1H, NH), 10.77 (bs, 1H, NH), 13.93 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 18.8 (CH3), 39.1 (SCH3), 109.2 (C3H, Pyr), 115.4 (C5H, Pyr), 115.9 (d, 2JCF = 21.5 Hz, C3/5H, F-Phe), 117.5 (C6H, Cl-Tol), 118.7 (C2H, Cl-Tol), 129.0 (C4, Cl-Tol), 131.1 (C2/6H, F-Phe), 131.2 (C5, Imdz and C5H, Cl-Tol), 133.2 (C3, Cl-Tol), 134.6 (C4, Imdz), 138.2 (C1, Cl-Tol), 147.0 (C6H, Pyr), 148.7 (C2, Imdz), 152.1 (CO), 153.2 (C2, Pyr), 162.3 (d, 1JCF = 249.9 Hz, CF) ppm; MS (ESI, 70 eV) m/z 484 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)pyridin-2-yl)-3-(3-(trifluormethyl)phenyl)carbamide (12l). Synthesis was performed according to the general procedure for sulfoxidation starting from 12f (20.0 mg, 41.0 µmol) in 0.4 mL THF and 0.1 mL water. Flash chromatography (SiO2, 40%–100% ethyl acetate/petrol ether) afforded 12l as a colorless solid. Yield 15.1 mg (73%); m.p. 235 °C; 1H-NMR (DMSO-d6): δ = 3.10 (s, 3H, SCH3), 7.02 (dd, 3J = 5.4 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.30–7.37 (m, 2H, C3/5H, F-Phe), 7.36 (d, 3J = 7.7 Hz, 1H, C4H, F3C-Phe), 7.51–7.57 (m, 3H, C2/6H, F-Phe and C5H, F3C-Phe), 7.65 (d, 3J = 8.3 Hz, 1H, C6H, F3C-Phe), 7.71 (s, 1H, C2H, F3C-Phe), 8.08 (bs, 1H, C3H, Pyr), 8.22 (bs, 1H, C6H, Pyr), 9.59 (s, 1H, NH), 10.90 (bs, 1H, NH), 13.93 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 39.1 (SCH3), 109.3 (C2H, F3C-Phe), 114.7 (q, 3JCF = 2.5 Hz, C3H, Pyr), 115.5 (C5H, Pyr), 115.9 (d, 3JCF = 21.9 Hz, C3/5H, F-Phe), 118.7 (q, 3JCF = 2.1 Hz, C4H, F3C-Phe), 122.5 (C6H, F3C-Phe), 124.2 (q, 1JCF = 272.2 Hz, CF3), 125.9 (d, 4JCF = 3.2 Hz, C1, F-Phe), 129.6 (q, 2JCF = 31.4 Hz, C3, F3C-Phe), 130.0 (C5H, F3C-Phe), 130.8 (C5, Imdz), 131.1 (d, 4JCF = 11.4 Hz, C2/6H, F-Phe), 134.5 (C4, Imdz), 139.9 (C1, F3C-Phe), 143.4 (C4, Pyr), 147.1 (C6H, Pyr), 148.7 (C2, Imdz), 152.2 (CO), 162.4 (d, 1JCF = 242.12 Hz, CF) ppm; MS (ESI, 70 eV) m/z 504 [MH]+.
1-(4-(5-(4-Fluorophenyl)-2-(methylthio)-1H-imidazol-4-yl)pyridin-2-yl)-3-(naphthalen-1-yl)carbamide (12m). Synthesis was performed according to the general procedure for sulfoxidation starting from 12g (51.0 mg, 109 µmol) in 5 mL THF and 1.5 mL H2O. Flash chromatography (SiO2, 20%–100% ethyl acetate/petrol ether) afforded 12m as colorless solid. Yield 37.0 mg (70%); C26H20FN5O2S (Mr 485.54); m.p. 244 °C; 1H-NMR (DMSO-d6): δ = 3.10 (s, 3H, SCH3), 7.04 (dd, 3J = 5.4 Hz, 4J = 1.4 Hz, 1H, C5H, Pyr), 7.31–7.36 (m, 2H, C3/5H, F-Phe), 7.49 (t, 3J = 7.9 Hz, 1H, C3H, Naph), 7.54–7.59 (m, 4H, C3H, Pyr and C2/6H, F-Phe and C6H, Naph), 7.63–7.68 (m, 2H, C4/7H, Naph), 7.96 (dd, 3J = 8.1 Hz, 4J = 0.8 Hz, 1H, C5H, Naph), 8.16–8.20 (m, 2H, C2/8H, Naph), 8.34 (d, 3J = 5.4 Hz, 1H, C6H, Pyr), 8.93 (s, 1H, NH), 11.58 (bs, 1H, NH), 13.95 (bs, 1H, NH, Imdz) ppm; 13C-NMR (DMSO-d6): δ = 39.1 (SCH3), 109.4 (C3H, Pyr), 115.3 (C5H, Pyr), 115.9 (d, 2JCF = 21.8 Hz, C3/5H, F-Phe), 116.9 (C2H, Naph), 120.9 (C8H, Naph), 123.1 (C4H, Naph), 125.5 (C1, Naph), 126.0 (C6H, Naph), 126.0 (C3H, Naph), 126.3 (C7H, Naph), 128.6 (C5H, Naph), 130.7 (C5, Imdz), 130.9 (C2/6H, F-Phe), 133.7 (C4a, Naph), 134.1 (C8a, Naph), 143.5 (C4, Pyr), 146.7 (C6H, Pyr), 148.9 (C2, Imdz), 152.6 (CO), 153.6 (C2, Pyr), 162.3 (d, 1JCF = 246.9 Hz, CF) ppm; MS (ESI, 70 eV) m/z 486 [MH]+.
4-(2,4-Dimethoxyphenyl)-N-(4-(5-(4-fluorophenyl)-2-(methylsulfinyl)-1H-imidazol-4-yl)-pyridin-2-yl)-1-methyl-1H-pyrrol-2-carboxamide (16c). Synthesis was performed according to the general procedure for sulfoxidation starting from 16a (196 mg, 368 µmol) in 4 mL THF and 1 mL H2O. Flash chromatography (SiO2, 40%–100% ethyl acetate/petrol ether) afforded 16c as yellow solid. Yield 182 mg (88%); C29H26FN5O4S (Mr 559.62); m.p. 136 °C; 1H-NMR (DMSO-d6): δ = 3.11 (s, 3H, SCH3), 3.78 (s, 3H, C2OCH3), 3.86 (s, 3H, C4OCH3), 3.90 (s, 3H, CH3), 6.57 (dd, 3J = 8.5 Hz, 4J = 2.4 Hz, 1H, C5H, (OCH3)2-Phe), 6.61 (d, 4J = 2.3 Hz, 1H, C3H, (OCH3)2-Phe), 7.07 (d, 3J = 4.8 Hz, 1H, C5H, Pyr), 7.23–7.38 (m, 2H, C3/5H, F-Phe), 7.42 (bs, 1H, C3H, Pyrrole), 7.46 (d, 3J = 8.4 Hz, 1H, C6H, (OCH3)2-Phe), 7.54–7.59 (m, 2H, C2/6H, F-Phe), 7.68 (bs, 1H, C5H, Pyrrole), 8.22 (d, 3J = 4.5 Hz, 1H, C6H, Pyr), 8.42 (s, 1H, C3H, Pyr), 10.17 (s, 1H, CONH), 13.89 (s, 1H, NH) ppm; 13C-NMR (DMSO-d6): δ = 36.6 (CH3), 39.2 (SCH3), 55.2 (C2OCH3), 55.4 (C4OCH3), 98.8 (C3H, (OCH3)2-Phe), 105.2 (C5H, (OCH3)2-Phe), 111.8 (C3H, Pyr), 113.3 (C5H, Pyrrole), 115.9 (C1, (OCH3)2-Phe), 116.0 (d, 2JCF = 22.4 Hz, C3/5H, F-Phe), 116.1 (C5H, Pyr), 118.5 (C4, Pyrrole), 124.0 (C2, Pyrrole), 125.8 (d, 4JCF = 3.3 Hz, C1, F-Phe), 127.5 (C6H, (OCH3)2-Phe), 128.6 (C3H, Pyrrole), 131.3 (d, 3JCF = 8.3 Hz, C2/6H. F-Phe), 131.9 (C4, Imdz), 135.1 (C5, Imdz), 143.0 (C4, Pyr), 147.8 (C6H, Pyr), 148.5 (C2, Imdz), 152.9 (C2, Pyr), 156.7 (C4OCH3), 158.6 (C2OCH3), 159.9 (CO), 162.4 (d, 1JCF = 244.3 Hz, CF) ppm; MS (ESI, 70 eV) m/z 560 [MH]+.
3.3. Kinase Assays and IC50 Determination
In vitro kinase assays using 2 μCi 32P-γ-ATP per reaction as co-factor were carried out in kinase buffer containing 25 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 100 µM EDTA, and 10 µM ATP. Potential inhibitor compounds were used in a dilution series ranging from 10 μM to 5 nM final reaction concentration which was prepared by serial dilution in DMSO. Recombinant human CK1δ transcription variant 1 (expressed and purified as GST fusion protein as described earlier [60]) and human GST-CK1ε (Invitrogen) were used as sources of enzyme while α-casein (C6780; Sigma-Aldrich) was used as substrate. Kinase reactions were incubated for 30 min at 30 °C. Subsequently, reactions were separated by SDS-PAGE and phosphorylated protein bands were visualized on dried gels by autoradiography. The phosphorylated substrate protein bands were excised and phosphorylation was quantified by Cherenkov counting. Dose-response analyses and calculation of IC50 values were carried out using GraphPad Prism 6 statistical software (San Diego, CA, USA).
3.4. Cell Culture
3.4.1. Cell Lines
The human pancreatic cancer cell lines Colo357 [61], Panc-1 [62], and MiaPaCa-2 [63] were grown in Dulbecco’s modified Eagle’s medium (DMEM). Panc89 [64] was grown in DMEM:RPMI-1640 medium in ratio 1:1. The human colon adenocarcinoma cell line HT-29 [65] was grown in McCoy’s 5A medium. All media were supplemented with 10% fetal calf serum (FCS), 100 units·mL−1 penicillin, 100 µg∙mL−1 streptomycin and 2 mM glutamine. All cells were grown at 37 °C in a humidified 5% carbon dioxide atmosphere.
3.4.2. Cell Assays and EC50 Determination
In order to determine the cytotoxic effects of investigated inhibitors conventional MTT assay were used. 1 × 104 cells∙well−1 were seeded in 96-well cell culture plates and cultivated for 24 h. Cell lines were exposed to increasing concentrations of the indicated inhibitor compounds, with untreated and DMSO-treated cells serving as control. After an incubation period of 48 h 10 µL of MTT solution (5 mg∙mL−1 in PBS) were added and cells were incubated for 4 h. MTT-containing medium was carefully removed and 100 µL acidic isopropanol (0.04 N HCl in isopropanol) per well were added. For dissolution of formazan crystals plates were shaken for 30 min on an orbital shaker in the dark. Finally, dissolved crystals were measured spectrophotometrically at 570 nM. All experiments were performed in triplicate with four technical replicates per assay. Results were normalized considering the mean optical density value of control wells as 100%. GraphPad Prism 6 (La Jolla, CA, USA) software was used to calculate EC50 values.
3.5. X-ray Crystallography
3.5.1. Protein Expression, Purification, and Crystallisation of CK1δ
BL21 (DE3) TaKaRa 2 cells (Clontech) were transformed with the plasmid pET28a-tCK1δ which contains a codon-optimized construct of CK1δ spanning residues 1–294 in a pET28a vector (NdeI and XhoI restriction sites) and streaked out on LB agar plates containing 50 μg∙mL−1 kanamycin and 20 µg∙mL−1 chloramphenicol as selective antibiotics. A pre-culture was prepared by inoculating cells from a single colony in LB medium supplemented with selective antibiotics and 4 mg∙mL−1 l-arabinose and overnight cultivation at 37 °C. Expression cultures were prepared by diluting a fresh pre-culture with LB medium (plus selective antibiotics and 4 mg∙mL−1 l-arabinose) to an optical density of 0.1 at 600 nm (OD600). Expression cultures were cultivated at 37 °C until an OD600 of 0.6 was reached, then cultivation temperature was reduced to 20 °C and tCK1δ expression was induced by adding 0.5 mM IPTG (isopropyl-β-d-1-thiogalactopyranoside). 16 h after IPTG addition, cells were harvested by centrifugation (4000× g, 20 min 4 °C), washed with TBS (20 mM Tris pH 7.5, 300 mM NaCl), and stored at −80 °C until purification.
Cells were thawed and resuspended (4 mL∙g−1 wet cell pellet) in TBS plus 0.5 mM TCEP and subsequently lysed on ice by sonication (Vibra-Cell VCX500, Sonics, Newtown, CT, USA). The resultant lysate was clarified by ultracentrifugation (165,000× g at 4 °C for 30 min) and supplemented with 5 mM imidazole pH 7.5 before loading it on 2 mL TALON® (Clontech Takara Bio Europe SAS, Saint-Germain-en-Laye, France) resin. After a wash step with TBS containing 10 mM imidazole, tCK1δ was eluted with TBS and 120 mM imidazole, concentrated, and applied to a Superdex S200 16/600 size exclusion chromatography column using TBS as chromatography buffer. Fractions containing monomeric tCK1δ were pooled and concentrated to 10 mg/mL and stored at −80 °C.
For co-crystallization of tCK1δ with 16b, protein stock solution (10 mg∙mL−1) was mixed 30:1 with 10 mM 16b (solubilized in DMSO) and incubated for 30 min at room temperature. Sitting drop crystallization trials were set up at 4 °C with drop ratios of 3 µL protein/inhibitor solution to 2 µL precipitant solution. Crystals appeared after three to four days in drops containing 0.1 M Hepes pH 7.0, 0.7 M NaH2PO4, and 0.7 M KH2PO4. For data collection, these crystals were cryo-protected by swiping them though reservoir solution supplemented with sucrose (60% saturation) and 0.3 mM 16b and subsequently flash frozen.
Diffraction data was collected at beamLine X06DA at the Swiss Light Source, Paul-Scherrer-Institute, Villigen, Switzerland and processed using XDS [66]. The structure was solved by molecular replacement using the program PHASER [67] with a truncated crystal structure of CK1δ (pdb 4TWC [25]) as search model. Between iterative cycles of refinement using phenix.refine [68] missing loops as well as 16b were manually built with Coot [69]. Restrains of 16b were calculated using phenix.elbow [70].
3.5.2. Protein Expression, Purification, and Crystallization of p38α MAPK
The expression and purification of inactive, non-phosphorylated p38α wt MAPK was done as previously reported [71]. Briefly, an N-terminal His6-p38α wt construct was transformed into E. coli BL21 (DE3) and expressed overnight at 18 °C. The protein was purified by Ni2+-NTA-affinity chromatography, followed by anion exchange and size exclusion chromatography after removal of the His-tag by proteolytic cleavage. The pure protein was subsequently concentrated to 10–30 mg·mL−1, aliquoted, flash frozen in liquid N2, and stored at −80 °C.
11b was co-crystallized with p38α wt using conditions similar to those as described previously [72]. Briefly, protein-ligand complexes were prepared by mixing 20 µL p38α wt (10 mg·mL−1,) with 0.2 µl compound (50 mM in DMSO) that were subsequently incubated for 60 min on ice. The samples were centrifuged at 13,000 rpm for 10 min to remove excess ligand. Crystals were grown in 24-well crystallisation plates (EasyXtal Tool, Qiagen, Hilden, Germany) using the hanging drop vapor diffusion method and by mixing 1.5 µL protein-ligand solution with 0.5 µL reservoir (100 mM MES pH 5.6–6.2, 20%–30% PEG4000 and 50 mM β-gluco-d-pyranoside). The crystals were protected using 25% PEG400 before they were flash frozen in liquid N2. Diffraction data of the p38α-ligand complexes were collected at the PX II beam line of the Swiss Light Source (Paul-Scherrer-Institute, Villigen, Switzerland) using wavelengths close to 1 Å. The datasets were integrated with XDS and scaled using XSCALE [73]. The complex structures were solved by molecular replacement with PHASER [67]. using the published p38α structure (pdb 4DLI [74]) as template. Molecules in the asymmetric unit were manually modified using the program COOT [75]. The final refinement was performed with REFMAC [76]. Inhibitor topology files were generated using the Dundee PRODRG server [77]. Refined structures were validated by Ramachandran plot analysis with RAMPAGE [78]. Data collection, structure refinement statistics, and the Ramachandran plot results are shown in Supplementary Table S6.
4. Conclusions
Deriving from hit compounds 1 and 2 we report on design and synthesis of novel sets of highly potent and specific ATP-competitive inhibitors of CK1δ thereby confirming modeled binding modes by X-ray analysis in CK1δ and also in comparison to p38α. Especially 11b (CK1δ IC50 ≤ 3–4 nM, CK1ε IC50 = 25 nM, p38α IC50 = 10 nM) has been identified as promising agent with IC50 values in the single-digit nanomolar range and is therefore among the most potent inhibitors of CK1δ reported so far. Interestingly, X-ray analysis of ligand-protein complexes demonstrated a binding mode for 11b deviating from typical type I within the active site of p38α, thereby stabilizing the kinase in an intermediate conformation between DFG-in and DFG-out (DFG-in-between). CK1δ, however, is addressed by conventional type I inhibition in a DFG-in conformation as confirmed by co-crystallization with 16b. In addition, at 100 nM 11b is rather selective for CK1δ/ε hitting only six off-targets from a panel of 321 protein kinases, among them p38α. Compound 11b further exhibited single-digit micromolar efficacy in MTT viability assays in pancreatic Colo357 and Panc89 carcinoma cell lines. Protein kinase CK1δ, however, executes diverse physiological and pathophysiological functions and specific inhibitors might therefore strongly depend on tissue and cellular background [2,19,28,79]. Consequently, additional experiments using cellular systems as well as optimization of physicochemical properties might prove beneficial. Nevertheless, the implied hit structure has to be considered inept in order to achieve complete CK1 isoform selectivity regarding CK1δ and CK1ε, although moderate discrimination of CK1α and significant selectivity against CK1γ1-3 have been successfully achieved. Based on the current results, additional optimization cycles might therefore focus on decreased fitting into the active site of p38α. Reducing the acrylamide moiety of 1 proved beneficial as the obtained propionic amide 11b does not show a Michael acceptor moiety. All synthesized inhibitors were stable in DMSO solution at room temperature over a period of 72 h. In fact, the compound is among the most potent and stable inhibitors of CK1δ published to date with good selectivity regarding a screen comprising 321 protein kinases and suitable efficacy in different human cancer cell lines. Fortunately, sulfoxidation, which is believed to be the predominant metabolic pathway of this inhibitor class [44], did not abrogate activity. Co-crystallization of compounds with CK1δ and related p38α confirmed our postulated binding modes within these kinases. Taken together, small molecule kinase inhibitor 11b can be suggested for the use as a pharmacological tool to further investigate the significance of CK1δ in signaling pathways, especially linked to proliferative and neurodegenerative diseases.
Supplementary Materials
Supplementary materials are available online.
Acknowledgments
We gratefully acknowledge the help of Dieter Schollmeyer, University of Mainz, Institute for Organic Chemistry, Germany, for X-ray analysis of compound 11b. Chiara Ianes is financially supported by a grant of the medical faculty of Ulm University (“Bausteinprogramm”) awarded to Joachim Bischof. We acknowledge financial support by Land Schleswig-Holstein within the funding program Open Access Publikationsfonds.
Author Contributions
J.H., L.W., C.I., M.K., M.B., D.R., C.P., E.B., A.L., U.B., U.K., J.B. and C.P. conceived and designed the experiments; J.H. and L.W. performed synthesis; C.I. and M.K. performed the biological evaluation in vitro; M.B., C.P. and E.B. performed crystallization experiments. J.H. and C.I. analyzed the data; J.H. wrote the paper.
Conflicts of Interest
The authors declare no competing financial interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| ATP | adenosine triphosphate |
| B-Raf | rapidly accelerated fibrosarcoma B |
| Boc | tert-butyloxycarbonyl |
| CDI | N,N’-carbonyldiimidazole |
| CK1 | protein kinase CK1, formerly known as casein kinase 1 |
| DCM | dichloromethane |
| DFG-in/out | sequence motif of aspartic acid (D), phenylalanine (F), and glycine (G) within the activation loop of the kinase domain |
| DIPEA | N,N-diisopropylethylamine |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMF | N,N-dimethylformamide |
| DMSO | dimethyl sulfoxide |
| EDTA | ethylenediaminetetraacetic acid |
| FASPS | familial advanced sleep phase syndrome |
| FCS | fetal calf serum |
| GST | glutathione S-transferase |
| HPI | hydrophobic pocket I |
| HRII | hydrophobic region II |
| JNK | c-Jun N-terminal kinase |
| LB | lysogeny broth |
| LCK | lymphocyte-specific protein tyrosine kinase |
| MAPK | mitogen-activated protein kinase |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NaHMDS | sodium bis(trimethylsilyl)amide |
| NBS | N-bromosuccinimide |
| NTA | nitrilotriacetic acid |
| PBS | phosphate-buffered saline |
| Pd/C | palladium on activated charcoal |
| pdb | Research Collaboratory for Structural Bioinformatics (RCSB) protein data bank |
| PEG | polyethylene glycol |
| PPh3 | triphenylphosphine |
| PyBOP | (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate |
| rt | room temperature |
| RIPK | receptor-interacting Ser/Thr-protein kinase |
| RPMI | Roswell Park Memorial Institute medium |
| SAR | structure-activity relationship |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| TBS | Tris-buffered saline |
| TCEP | tris(2-carboxyethyl)phosphine |
| THF | tetrahydrofuran |
| Tris | tris(hydroxymethyl)aminomethane |
| TTBK | tau tubulin kinase |
| VRK | vaccinia-related kinase |
References
- Venerando, A.; Ruzzene, M.; Pinna, L.A. Casein kinase: The triple meaning of a misnomer. Biochem. J. 2014, 460, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Kruger, M.; Richter, J.; Xu, P.; Garcia-Reyes, B.; Peifer, C.; Halekotte, J.; Bakulev, V.; Bischof, J. The CK1 Family: Contribution to Cellular Stress Response and Its Role in Carcinogenesis. Front. Oncol. 2014, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.D.; Anderson, R.A. Casein Kinase I: Spatial Organization and Positioning of a Multifunctional Protein Kinase Family. Cell. Signal. 1998, 10, 699–711. [Google Scholar] [CrossRef]
- Knippschild, U.; Wolff, S.; Giamas, G.; Brockschmidt, C.; Wittau, M.; Wurl, P.U.; Eismann, T.; Stoter, M. The role of the casein kinase 1 (CK1) family in different signaling pathways linked to cancer development. Onkologie 2005, 28, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Gocht, A.; Wolff, S.; Huber, N.; Lohler, J.; Stoter, M. The casein kinase 1 family: Participation in multiple cellular processes in eukaryotes. Cell. Signal. 2005, 17, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yin, H.; Kuret, J. Casein kinase 1 δ phosphorylates tau and disrupts its binding to microtubules. J. Biol. Chem. 2004, 279, 15938–15945. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, N.; Smiley, J.F.; DeMaggio, A.J.; Hoekstra, M.F.; Cochran, E.J.; Binder, L.I.; Kuret, J. A New Molecular Link between the Fibrillar and Granulovacuolar Lesions of Alzheimer’s Disease. Am. J. Pathol. 1999, 155, 1163–1172. [Google Scholar] [CrossRef]
- Yasojima, K.; Kuret, J.; DeMaggio, A.J.; McGeer, E.; McGeer, P.L. Casein kinase 1 δ mRNA is upregulated in Alzheimer disease brain. Brain Res. 2000, 865, 116–120. [Google Scholar] [CrossRef]
- Salado, I.G.; Redondo, M.; Bello, M.L.; Perez, C.; Liachko, N.F.; Kraemer, B.C.; Miguel, L.; Lecourtois, M.; Gil, C.; Martinez, A.; et al. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J. Med. Chem. 2014, 57, 2755–2772. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Padiath, Q.S.; Shapiro, R.E.; Jones, C.R.; Wu, S.C.; Saigoh, N.; Saigoh, K.; Ptacek, L.J.; Fu, Y.-H. Functional consequences of a CKIδ mutation causing familial advanced sleep phase syndrome. Nature 2005, 434, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Tsai, I.-C.; Woolf, M.; Neklason, D.W.; Branford, W.W.; Yost, H.J.; Burt, R.W.; Virshup, D.M. Disease-associated casein kinase I δ mutation may promote adenomatous polyps formation via a Wnt/β-catenin independent mechanism. Int. J. Cancer 2007, 120, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Ullah, K.; Xu, P.; Alscher, V.; Blatz, A.; Peifer, C.; Halekotte, J.; Leban, J.; Vitt, D.; Holzmann, K.; et al. Effects of altered expression and activity levels of CK1δ and varepsilon on tumor growth and survival of colorectal cancer patients. Int. J. Cancer 2015, 136, 2799–2810. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rajendran, V.; Sethumadhavan, R.; Purohit, R. Relationship between a point mutation S97C in CK1δ protein and its affect on ATP-binding affinity. J. Biomol. Struct. Dyn. 2013, 32, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Schittek, B.; Sinnberg, T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol. Cancer 2014, 13, 231. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Rudeck, S.; Kretz, A.-L.; Kramer, K.; Just, S.; Henne-Bruns, D.; Hillenbrand, A.; Leithäuser, F.; Lemke, J.; Knippschild, U. Decreased CK1δ expression predicts prolonged survival in colorectal cancer patients. Tumor Biol. 2016, 37, 8731–8739. [Google Scholar] [CrossRef] [PubMed]
- Brockschmidt, C.; Hirner, H.; Huber, N.; Eismann, T.; Hillenbrand, A.; Giamas, G.; Radunsky, B.; Ammerpohl, O.; Bohm, B.; Henne-Bruns, D.; et al. Anti-apoptotic and growth-stimulatory functions of CK1δ and epsilon in ductal adenocarcinoma of the pancreas are inhibited by IC261 in vitro and in vivo. Gut 2008, 57, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S.; Oltmer, F.; Richter, J.; Bischof, J.; Xu, P.; Burster, T.; Leithäuser, F.; Knippschild, U. CK1δ in lymphoma: Gene expression and mutation analyses and validation of CK1δ kinase activity for therapeutic application. Front. Cell Dev. Biol. 2015, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L.H.; Lafitte, M.; Quereda, V.; Grant, W.; Chen, W.; Bibian, M.; Noguchi, Y.; Fallahi, M.; Yang, C.; Chang, J.C.; et al. Therapeutic targeting of casein kinase 1δ in breast cancer. Sci. Transl. Med. 2015, 7, 318ra202. [Google Scholar] [CrossRef] [PubMed]
- Stöter, M.; Bamberger, A.-M.; Aslan, B.; Kurth, M.; Speidel, D.; Löning, T.; Frank, H.-G.; Kaufmann, P.; Löhler, J.; Henne-Bruns, D.; et al. Inhibition of casein kinase I δ alters mitotic spindle formation and induces apoptosis in trophoblast cells. Oncogene 2005, 24, 7964–7975. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2015, 20, 319–340. [Google Scholar] [CrossRef] [PubMed]
- Mashhoon, N.; DeMaggio, A.J.; Tereshko, V.; Bergmeier, S.C.; Egli, M.; Hoekstra, M.F.; Kuret, J. Crystal Structure of a Conformation-selective Casein Kinase-1 Inhibitor. J. Biol. Chem. 2000, 275, 20052–20060. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.K.; Hung, N.T.; Wang, H.; Tan, P.; Voorhoeve, P.M.; Lee, S.H.; Virshup, D.M. IC261 induces cell cycle arrest and apoptosis of human cancer cells via CK1δ/ɛ and Wnt/β-catenin independent inhibition of mitotic spindle formation. Oncogene 2011, 30, 2558–2569. [Google Scholar] [CrossRef] [PubMed]
- Kruger, M.; Kalbacher, H.; Kastritis, P.L.; Bischof, J.; Barth, H.; Henne-Bruns, D.; Vorgias, C.; Sarno, S.; Pinna, L.A.; Knippschild, U. New potential peptide therapeutics perturbing CK1δ/α-tubulin interaction. Cancer Lett. 2016, 375, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Föhr, K.J.; Knippschild, U.; Herkommer, A.; Fauler, M.; Peifer, C.; Georgieff, M.; Adolph, O. State-dependent block of voltage-gated sodium channels by the casein-kinase 1 inhibitor IC261. Investig. New Drugs 2017. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.; Leban, J.; Zaja, M.; Grothey, A.; Radunsky, B.; Othersen, O.; Strobl, S.; Vitt, D.; Knippschild, U. 2-Benzamido-N-(1H-benzo[d]imidazol-2-yl)thiazole-4-carboxamide derivatives as potent inhibitors of CK1δ/ε. Amino Acids 2012, 43, 1577–1591. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Bischof, J.; Zaja, M.; Kohlhof, H.; Othersen, O.; Vitt, D.; Alscher, V.; Pospiech, I.; Garcia-Reyes, B.; Berg, S.; et al. Difluoro-dioxolo-benzoimidazol-benzamides as potent inhibitors of CK1δ and epsilon with nanomolar inhibitory activity on cancer cell proliferation. J. Med. Chem. 2014, 57, 7933–7946. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Koeberle, S.C.; Laufer, S.A. p38α mitogen-activated protein kinase inhibitors, a patent review (2005–2011). Expert Opin. Ther. Pat. 2011, 21, 1843–1866. [Google Scholar] [CrossRef] [PubMed]
- Peifer, C.; Abadleh, M.; Bischof, J.; Hauser, D.; Schattel, V.; Hirner, H.; Knippschild, U.; Laufer, S. 3,4-Diaryl-isoxazoles and -imidazoles as Potent Dual Inhibitors of p38α Mitogen Activated Protein Kinase and Casein Kinase 1δ. J. Med. Chem. 2009, 52, 7618–7630. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.; Dos Santos, R.; Oliva, G.; Andricopulo, A. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Long, A.; Zhao, H.; Huang, X. Structural Basis for the Interaction between Casein Kinase 1 δ and a Potent and Selective Inhibitor. J. Med. Chem. 2012, 55, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Laufer, S.A.; Hauser, D.R.J.; Domeyer, D.M.; Kinkel, K.; Liedtke, A.J. Design, Synthesis, and Biological Evaluation of Novel Tri- and Tetrasubstituted Imidazoles as Highly Potent and Specific ATP-Mimetic Inhibitors of p38 MAP Kinase: Focus on Optimized Interactions with the Enzyme’s Surface-Exposed Front Region. J. Med. Chem. 2008, 51, 4122–4149. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Canagarajah, B.J.; Boehm, J.C.; Kassisà, S.; Cobb, M.H.; Young, P.R.; Abdel-Meguid, S.; Adams, J.L.; Goldsmith, E.J. Structural basis of inhibitor selectivity in MAP kinases. Structure 1998, 6, 1117–1128. [Google Scholar] [CrossRef]
- Traxler, P.; Furet, P. Strategies toward the Design of Novel and Selective Protein Tyrosine Kinase Inhibitors. Pharmacol. Ther. 1999, 82, 195–206. [Google Scholar] [CrossRef]
- Mente, S.; Arnold, E.; Butler, T.; Chakrapani, S.; Chandrasekaran, R.; Cherry, K.; DiRico, K.; Doran, A.; Fisher, K.; Galatsis, P.; et al. Ligand–Protein Interactions of Selective Casein Kinase 1δ Inhibitors. J. Med. Chem. 2013, 56, 6819–6828. [Google Scholar] [CrossRef] [PubMed]
- Peifer, C.; Kinkel, K.; Abadleh, M.; Schollmeyer, D.; Laufer, S. From five- to six-membered rings: 3,4-diarylquinolinone as lead for novel p38MAP kinase inhibitors. J. Med. Chem. 2007, 50, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.E.; Cubbon, R.M.; Cummings, R.T.; Wicker, L.S.; Frankshun, R.; Cunningham, B.R.; Cameron, P.M.; Meinke, P.T.; Liverton, N.; Weng, Y.; et al. Photochemical Preparation of a Pyridone Containing Tetracycle: A Jak Protein Kinase Inhibitor. Bioorg. Med. Chem. Lett. 2002, 12, 1219–1223. [Google Scholar] [CrossRef]
- Laufer, S.A.; Liedtke, A.J. A concise and optimized four-step approach toward 2-(aryl-)alkylsulfanyl-, 4(5)-aryl-, 5(4)-heteroaryl-substituted imidazoles using alkyl- or arylalkyl thiocyanates. Tetrahedron Lett. 2006, 47, 7199–7203. [Google Scholar] [CrossRef]
- Lal, K.; Ghosh, S.; Salomon, R.G. Hydroxyl-Directed Regioselective Monodemethylation of Polymethoxyarenes. J. Org. Chem. 1987, 52, 1072–1078. [Google Scholar] [CrossRef]
- Kiuchi, M.; Marukawa, K.; Hamada, M.; Sugahara, K. Amine Compound and Pharmaceutical Use Thereof. Patent WO2008153159 A1, 18 December 2008. [Google Scholar]
- Engen, W.; O’Brien, T.E.; Kelly, B.; Do, J.; Rillera, L.; Stapleton, L.K.; Youngren, J.F.; Anderson, M.O. Synthesis of Aryl-Heteroaryl Ureas (AHUs) Based on 4-Aminoquinoline and Their Evaluation Against the Insulin-Like Growth Factor Receptor (IGF-1R). Bioorg. Med. Chem. 2010, 18, 5995–6005. [Google Scholar] [CrossRef] [PubMed]
- Handy, S.T.; Zhang, Y.; Bregman, H. A Modular Synthesis of the Lamellarins: Total Synthesis of Lamellarin G Trimethyl Ether. J. Org. Chem. 2004, 69, 2362–2366. [Google Scholar] [CrossRef] [PubMed]
- Buhler, S.; Goettert, M.; Schollmeyer, D.; Albrecht, W.; Laufer, S.A. Chiral sulfoxides as metabolites of 2-thioimidazole-based p38α mitogen-activated protein kinase inhibitors: Enantioselective synthesis and biological evaluation. J. Med. Chem. 2011, 54, 3283–3297. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, K.; Hauser, D.R.J.; Unger, A.; Albrecht, W.; Laufer, S.A. 2-Acylaminopyridin-4-ylimidazoles as p38 MAP Kinase Inhibitors: Design, Synthesis, and Biological and Metabolic Evaluations. ChemMedChem 2009, 4, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Kornev, A.P.; Taylor, S.S.; Eyck, L.F.; Ten Eyck, L.F. A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 14377–14382. [Google Scholar] [CrossRef] [PubMed]
- Meharena, H.S.; Chang, P.; Keshwani, M.M.; Oruganty, K.; Nene, A.K.; Kannan, N.; Taylor, S.S.; Kornev, A.P. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 2013, 11, e1001680. [Google Scholar] [CrossRef] [PubMed]
- Bukhtiyarova, M.; Karpusas, M.; Northrop, K.; Namboodiri, H.V.M.; Springman, E.B. Mutagenesis of p38α MAP kinase establishes key roles of Phe169 in function and structural dynamics and reveals a novel DFG-OUT state. Biochemistry 2007, 46, 5687–5696. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.R.; Grutter, C.; Pawar, V.; Aust, B.; Wolf, A.; Rabiller, M.; Wulfert, S.; Robubi, A.; Kluter, S.; Ottmann, C.; et al. High-throughput screening to identify inhibitors which stabilize inactive kinase conformations in p38α. J. Am. Chem. Soc. 2009, 131, 18478–18488. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Wentsch, H.K.; Mayer-Wrangowski, S.C.; Zimmermann, M.; Bauer, S.M.; Storch, K.; Niess, R.; Koeberle, S.C.; Grütter, C.; Boeckler, F.M.; et al. Dibenzosuberones as p38 Mitogen-Activated Protein Kinase Inhibitors with Low ATP Competitiveness and Outstanding Whole Blood Activity. J. Med. Chem. 2013, 56, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D. 25 years of small molecular weight kinase inhibitors: Potentials and limitations. Mol. Pharmacol. 2015, 87, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Laufer, S.; Hauser, D.; Stegmiller, T.; Bracht, C.; Ruff, K.; Schattel, V.; Albrecht, W.; Koch, P. Tri- and tetrasubstituted imidazoles as p38α mitogen-activated protein kinase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 6671–6675. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.L.; Frederickson, M.; Cleasby, A.; Woodhead, S.J.; Carr, M.G.; Woodhead, A.J.; Walker, M.T.; Congreve, M.S.; Devine, L.A.; Tisi, D.; et al. Identification of Novel p38α MAP Kinase Inhibitors Using Fragment-Based Lead Generation. J. Med. Chem. 2005, 48, 414–426. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Bain, J.; Elliott, M.; Cohen, P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004, 5, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A Conformational Search Method for Efficient Generation of Bioactive Conformers. J. Chem. Inf. Model. 2010, 50, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Xiao, Z.; Wittau, M.; Sussner, N.; Stoter, M.; Knippschild, U. Interaction of casein kinase 1 δ (CK1δ) with the light chain LC2 of microtubule associated protein 1A (MAP1A). Biochim. Biophys. Acta 2005, 1745, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.T.; Woods, L.K.; Moore, G.E.; Quinn, L.A.; McGavran, L.; Gordon, S.G. Human cell line (COLO 357) of metastatic pancreatic adenocarcinoma. Int. J. Cancer 1980, 25, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.; Mazzetta, J.; Nelson-Rees, W.; Kaplan, M.; Todaro, G. Establishment of a continuous tumor-cell line (panc-1) from a human carcinoma of the exocrine pancreas. Int. J. Cancer 1975, 15, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Yunis, A.A.; Arimura, G.K.; Russin, D.J. Human pancreatic carcinoma (MIA PaCa-2) in continuous culture: Sensitivity to asparaginase. Int. J. Cancer 1977, 19, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Okabe, T.; Yamaguchi, N.; Ohsawa, N. Establishment and characterization of a carcinoembryonic antigen (CEA)-producing cell line from a human carcinoma of the exocrine pancreas. Cancer 1983, 51, 662–668. [Google Scholar] [CrossRef]
- Fogh, J.; Trempe, G. New Human Tumor Cell Lines. In Human Tumor Cells In Vitro; Fogh, J., Ed.; Springer: Boston, MA, USA, 1975; pp. 115–159. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Read, R.J. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001, 57, 1373–1382. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 352–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, N.W.; Grosse-Kunstleve, R.W.; Adams, P.D. electronic Ligand Builder and Optimization Workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.R.; Getlik, M.; Grutter, C.; Pawar, V.; Wulfert, S.; Rabiller, M.; Rauh, D. Development of a fluorescent-tagged kinase assay system for the detection and characterization of allosteric kinase inhibitors. J. Am. Chem. Soc. 2009, 131, 13286–13296. [Google Scholar] [CrossRef] [PubMed]
- Bukhtiyarova, M.; Northrop, K.; Chai, X.; Casper, D.; Karpusas, M.; Springman, E. Improved expression, purification, and crystallization of p38α MAP kinase. Protein Expr. Purif. 2004, 37, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993, 26, 795–800. [Google Scholar] [CrossRef]
- Getlik, M.; Simard, J.R.; Termathe, M.; Grutter, C.; Rabiller, M.; van Otterlo, W.A.L.; Rauh, D. Fluorophore labeled kinase detects ligands that bind within the MAPK insert of p38α kinase. PLoS ONE 2012, 7, e39713. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Schuttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: Phi, psi and Cβ deviation. Proteins 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Löhler, J.; Hirner, H.; Schmidt, B.; Kramer, K.; Fischer, D.; Thal, D.R.; Leithäuser, F.; Knippschild, U. Immunohistochemical Characterisation of Cell-Type Specific Expression of CK1δ in Various Tissues of Young Adult BALB/c Mice. PLoS ONE 2009, 4, e4174. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1, 3, 4 and 5 are available from the authors. |
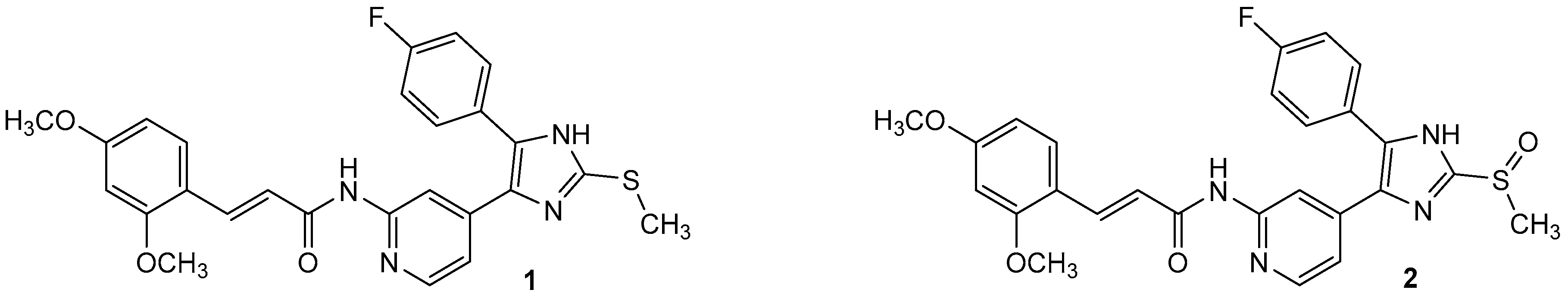
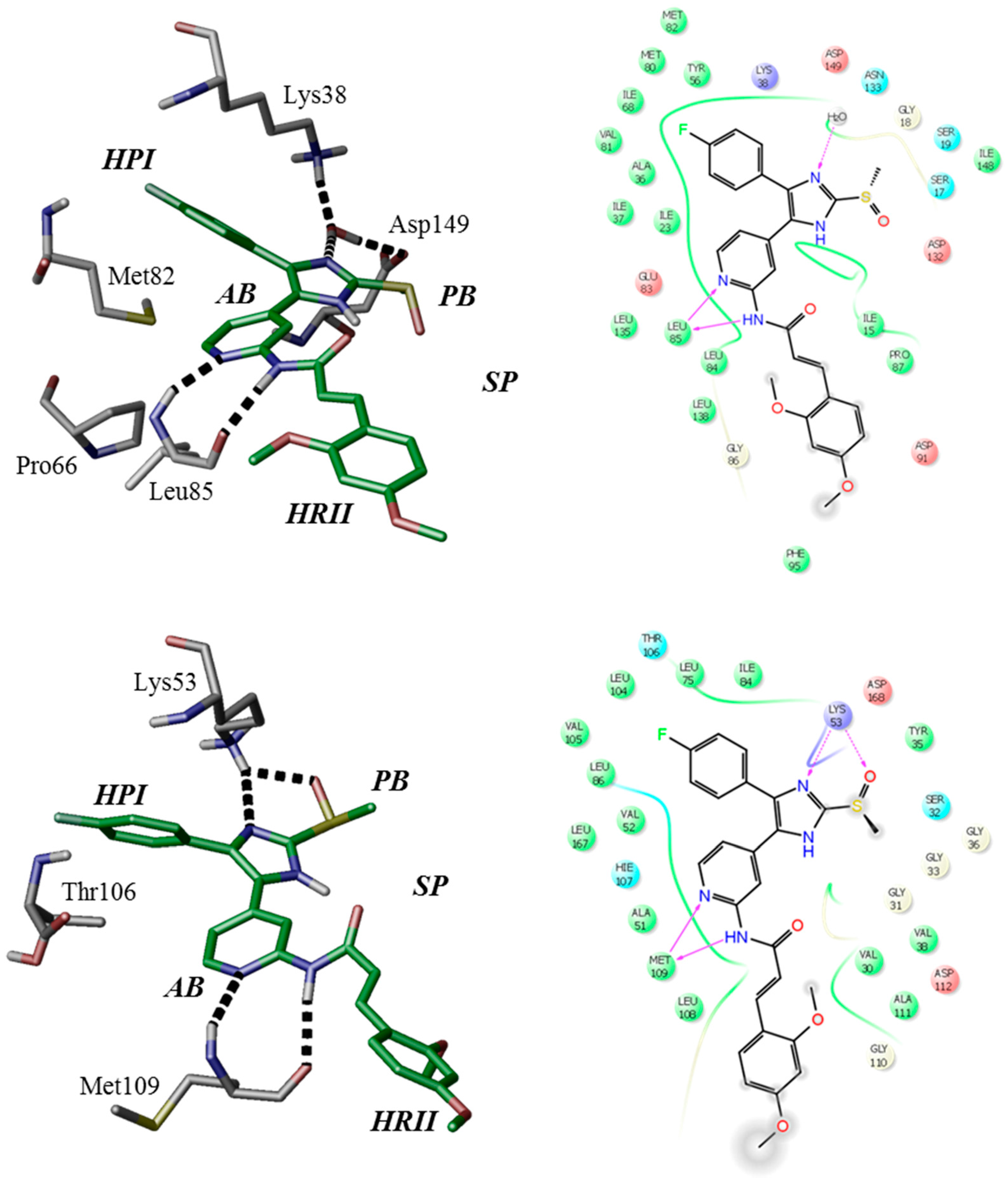
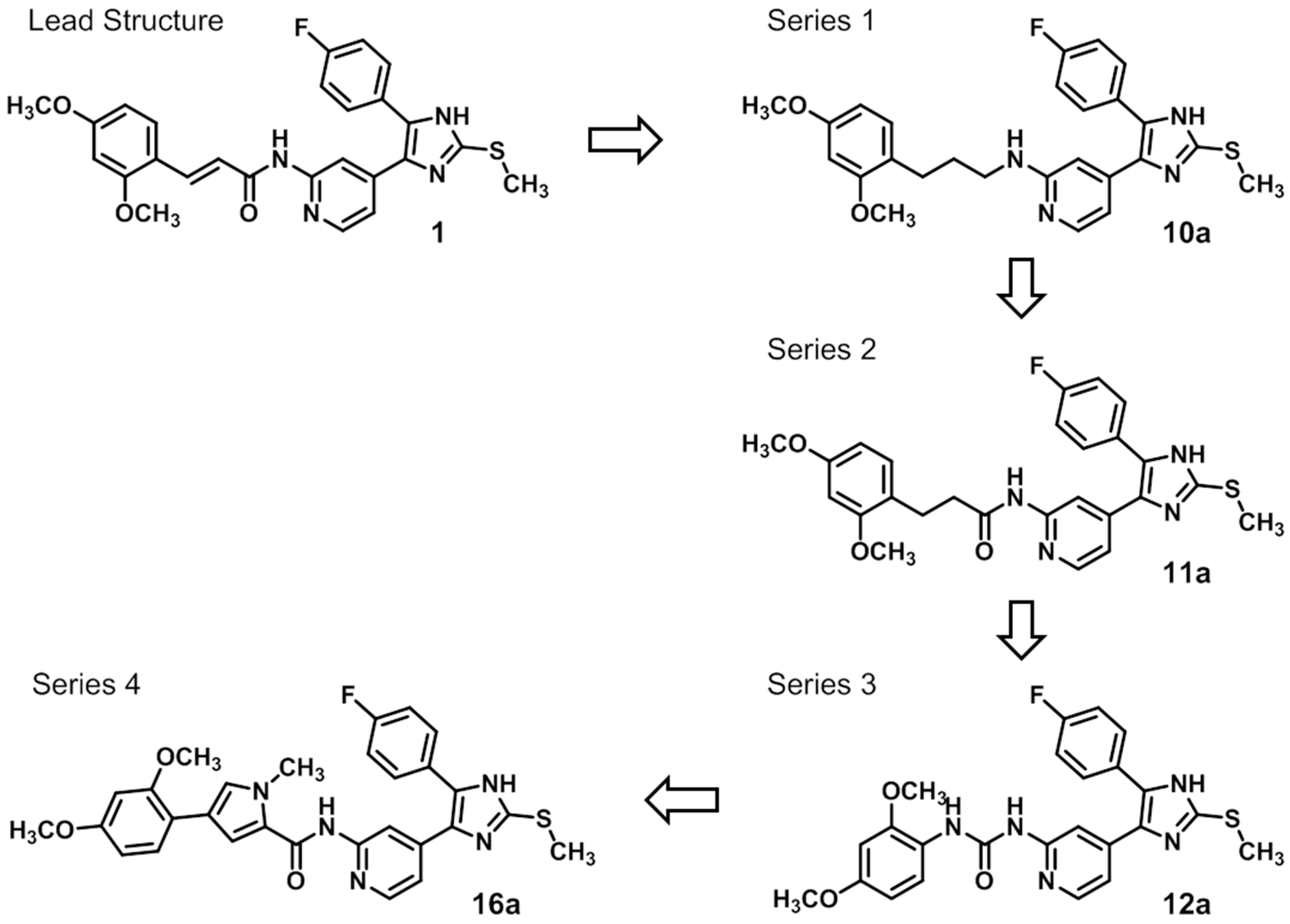
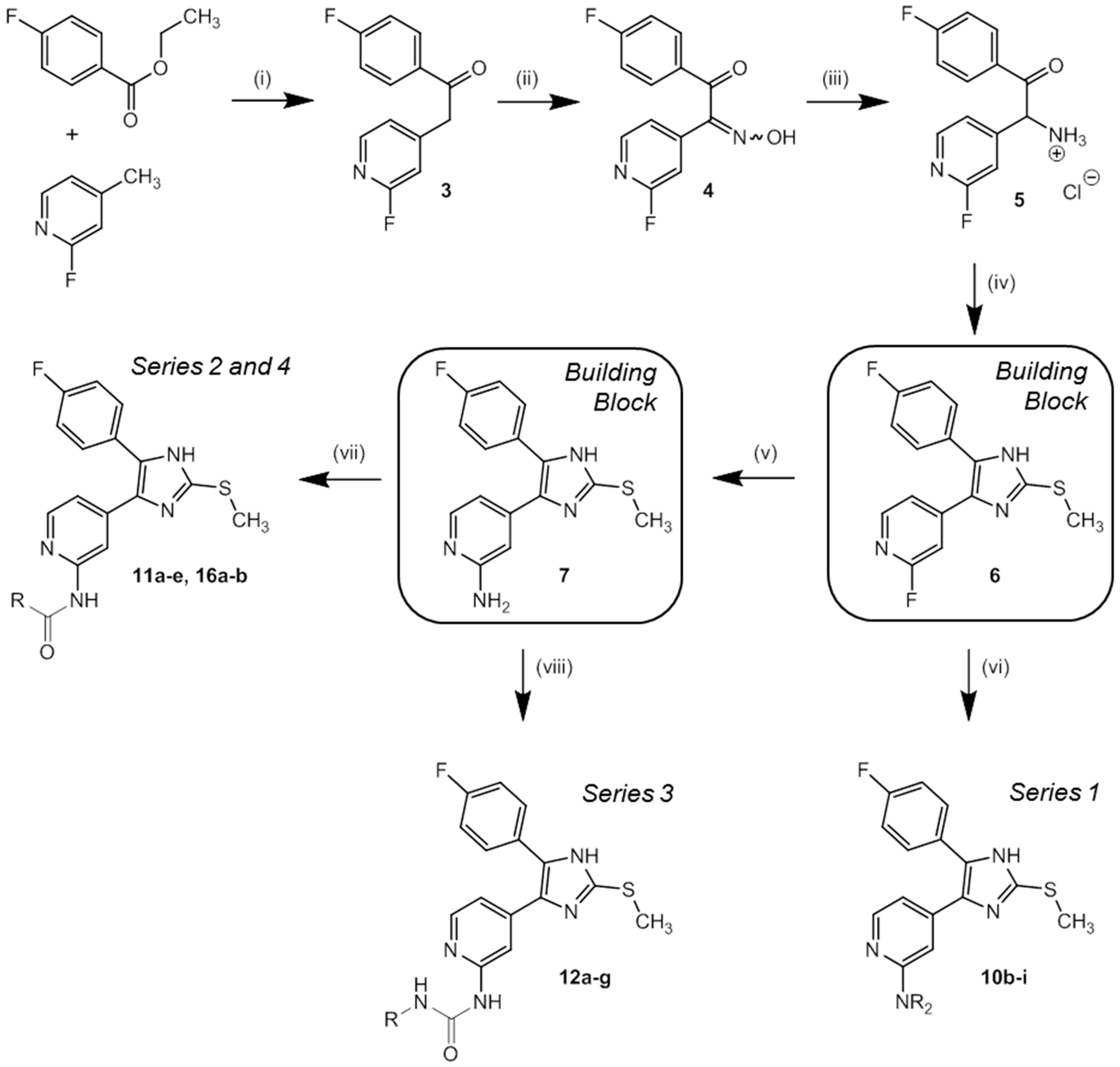
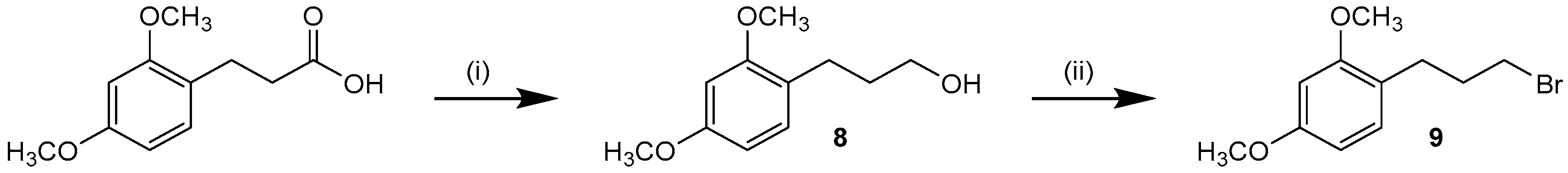
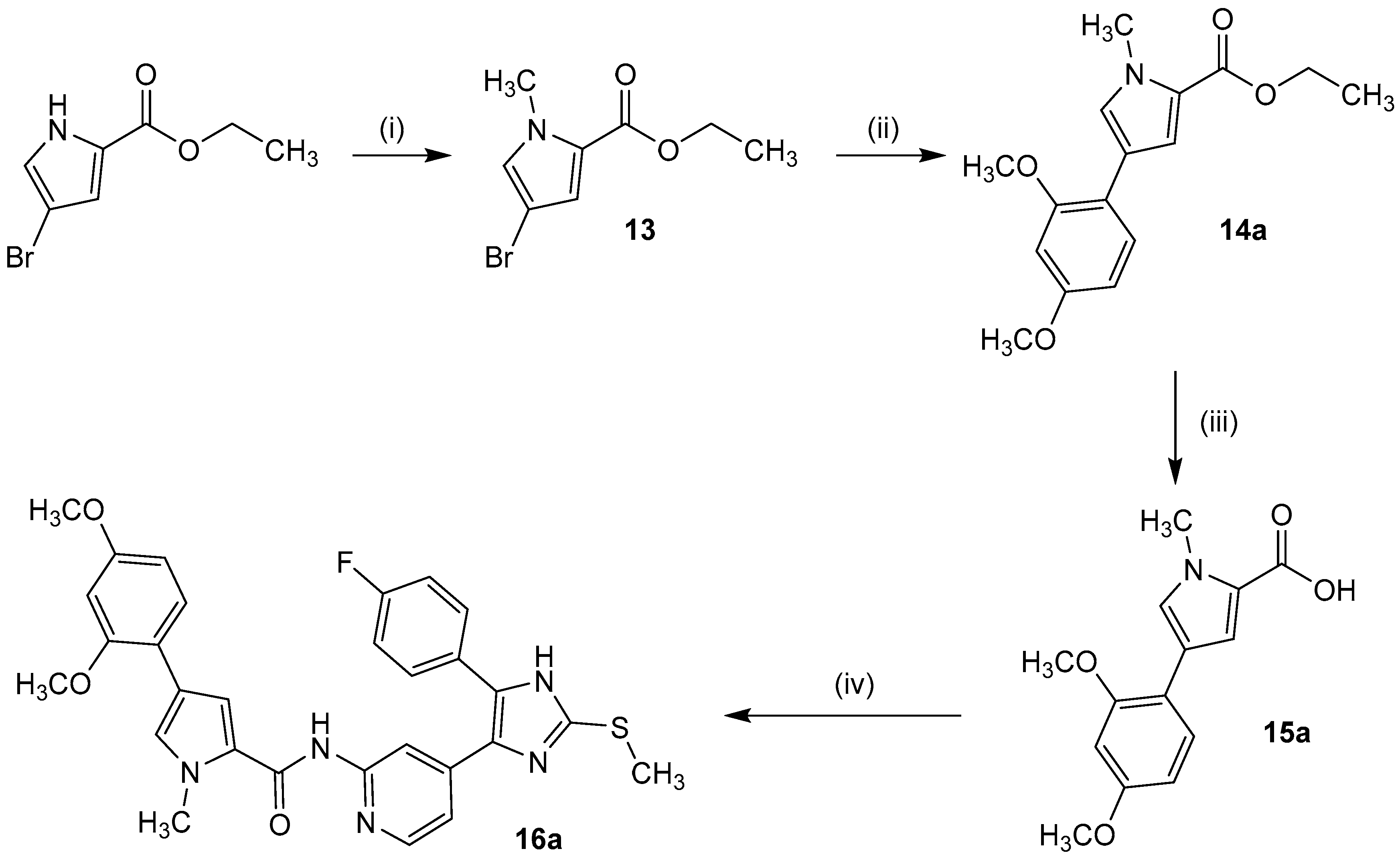
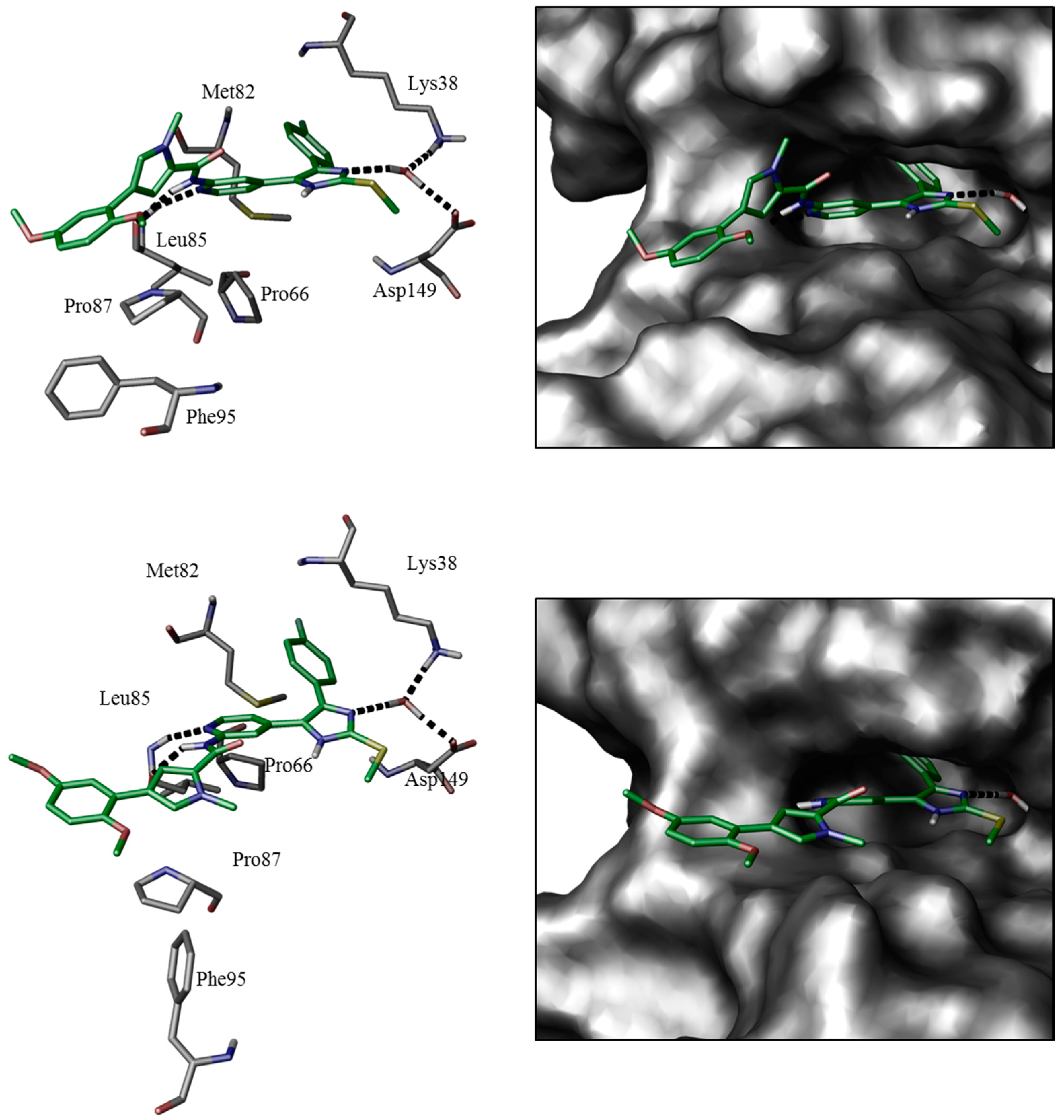
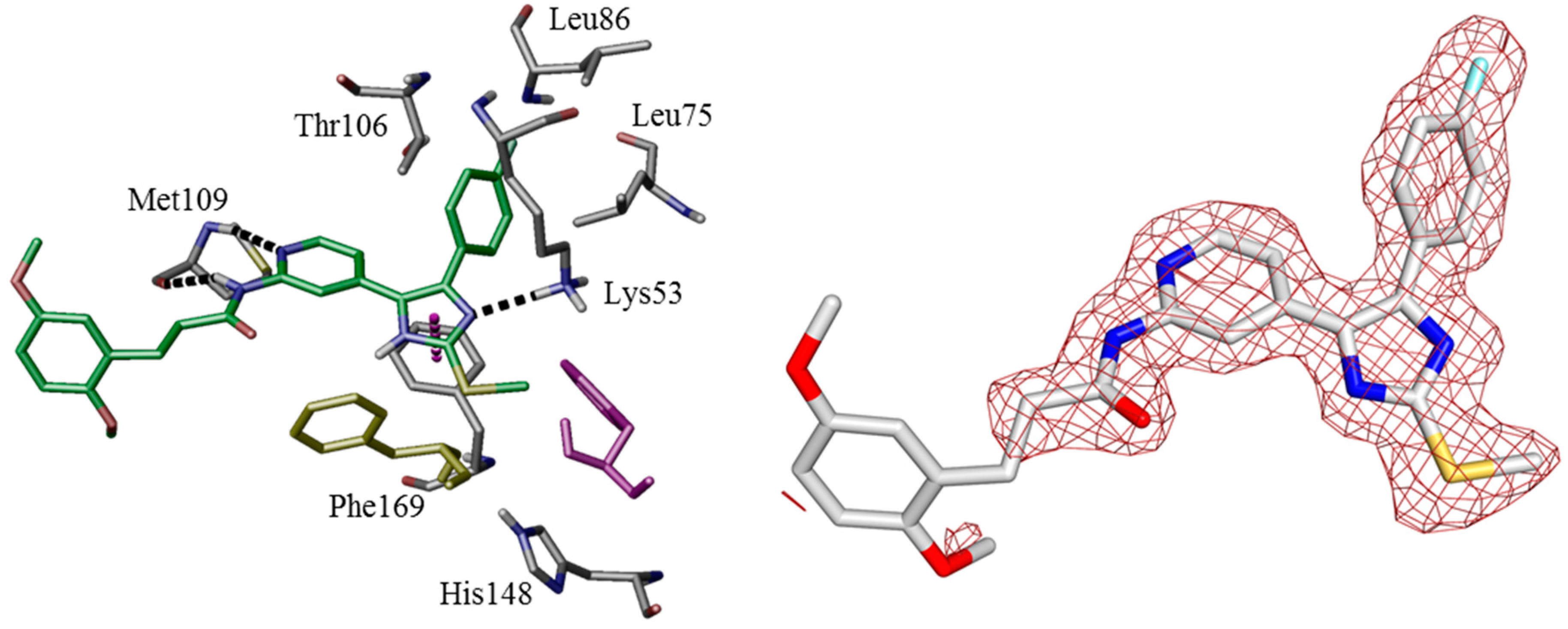
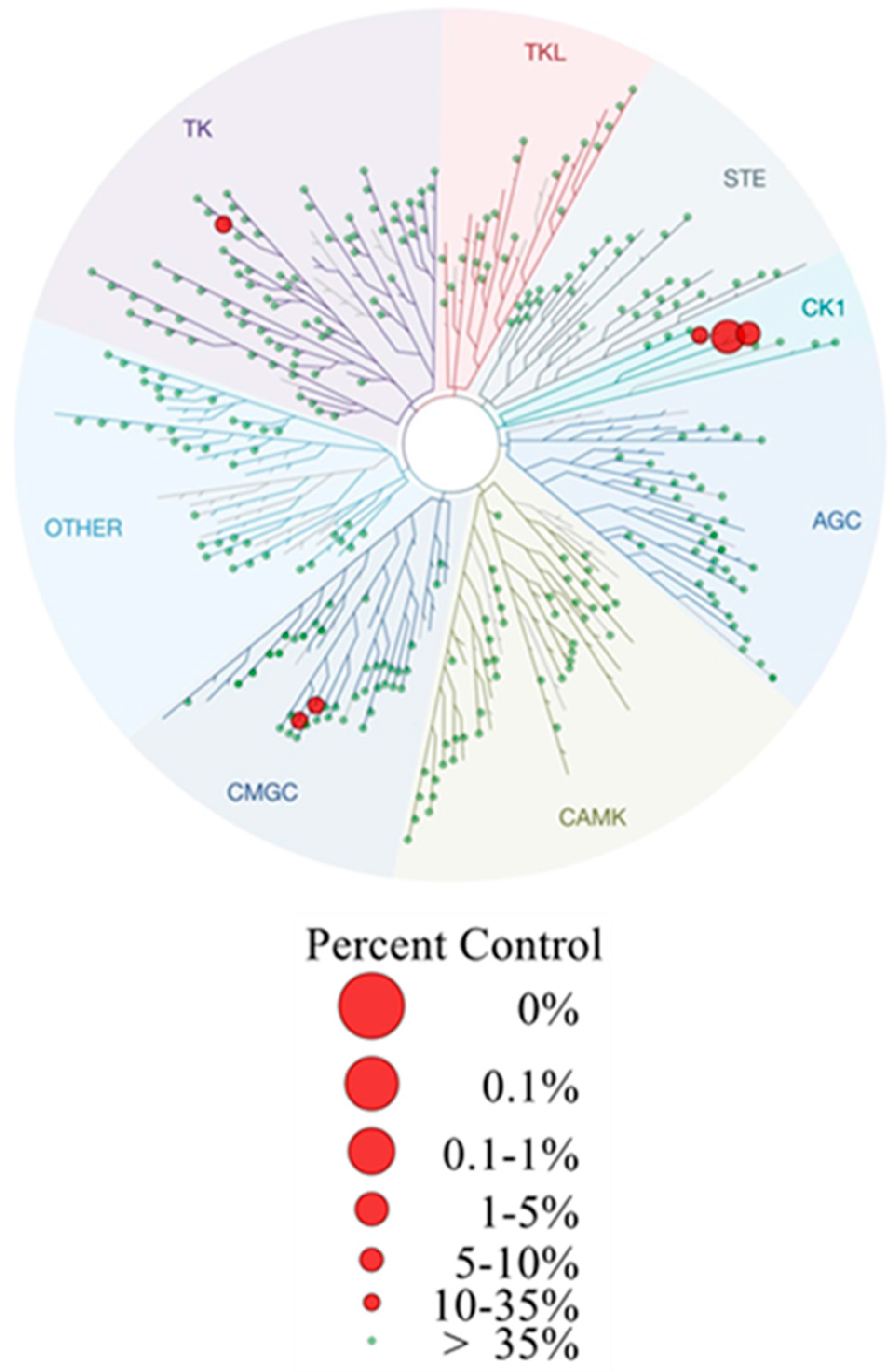
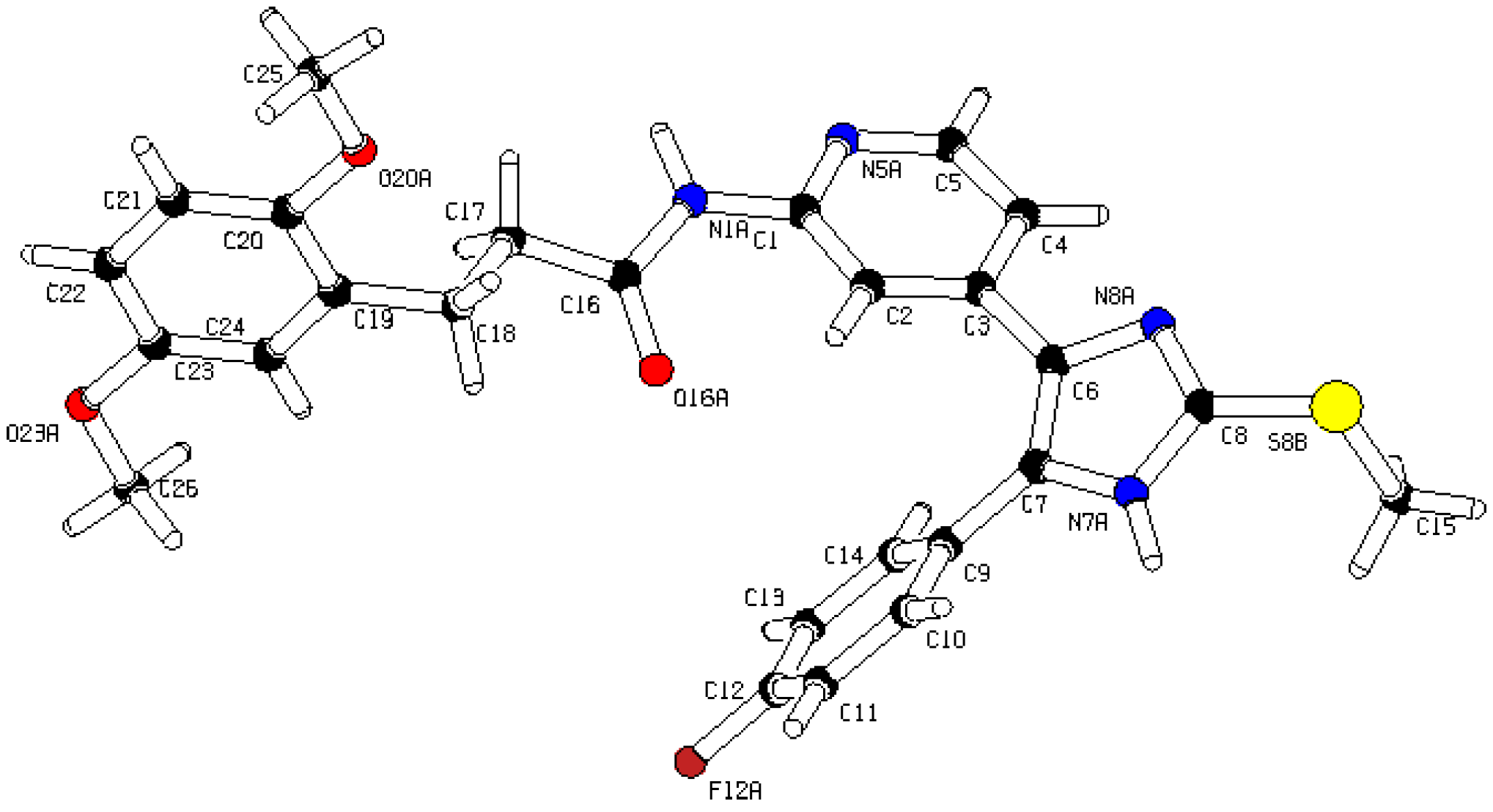
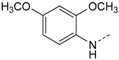
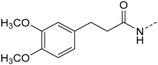
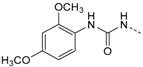
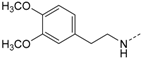
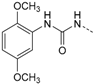
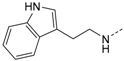
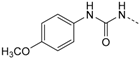
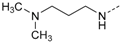
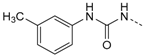
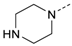
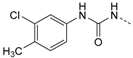
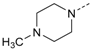
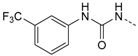
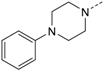
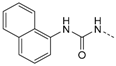
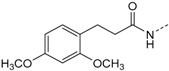
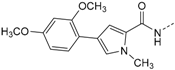
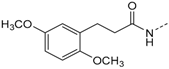
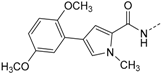
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).