
Design, Synthesis and Evaluation of Naphthalimide Derivatives as Potential Anticancer Agents for Hepatocellular Carcinoma

 ,
,
Abstract
:
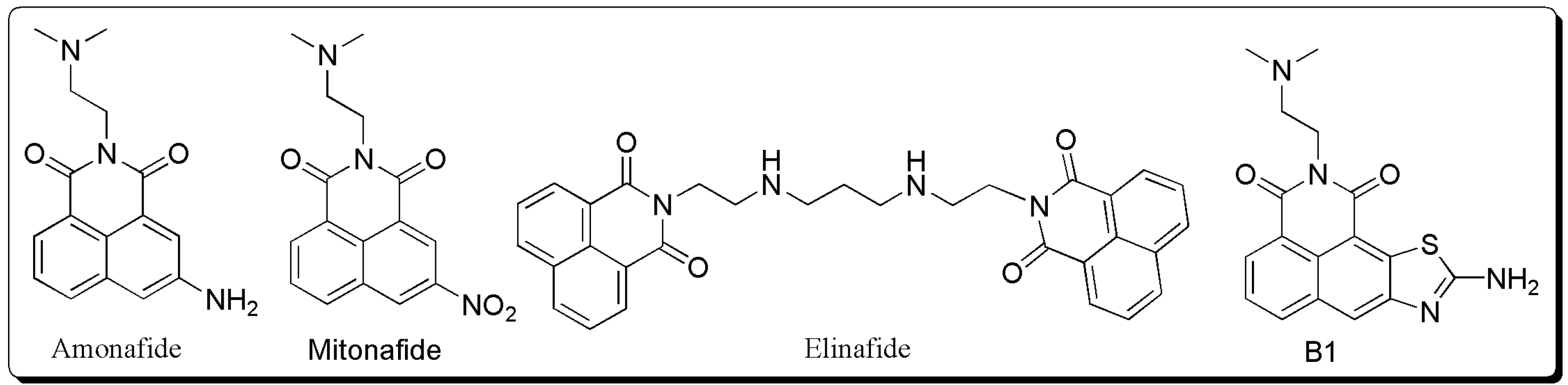
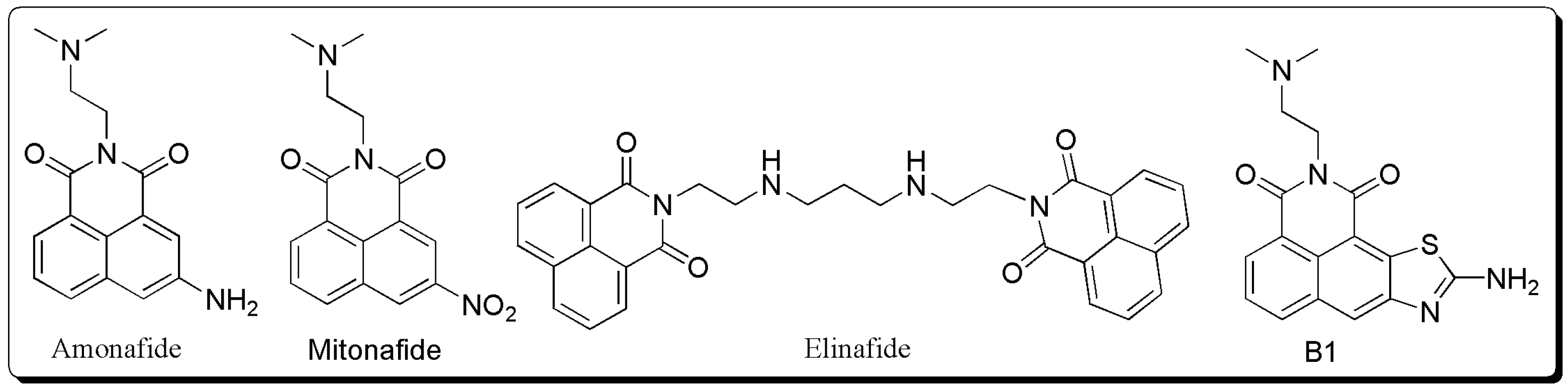
1. Introduction
2. Results and Discussion
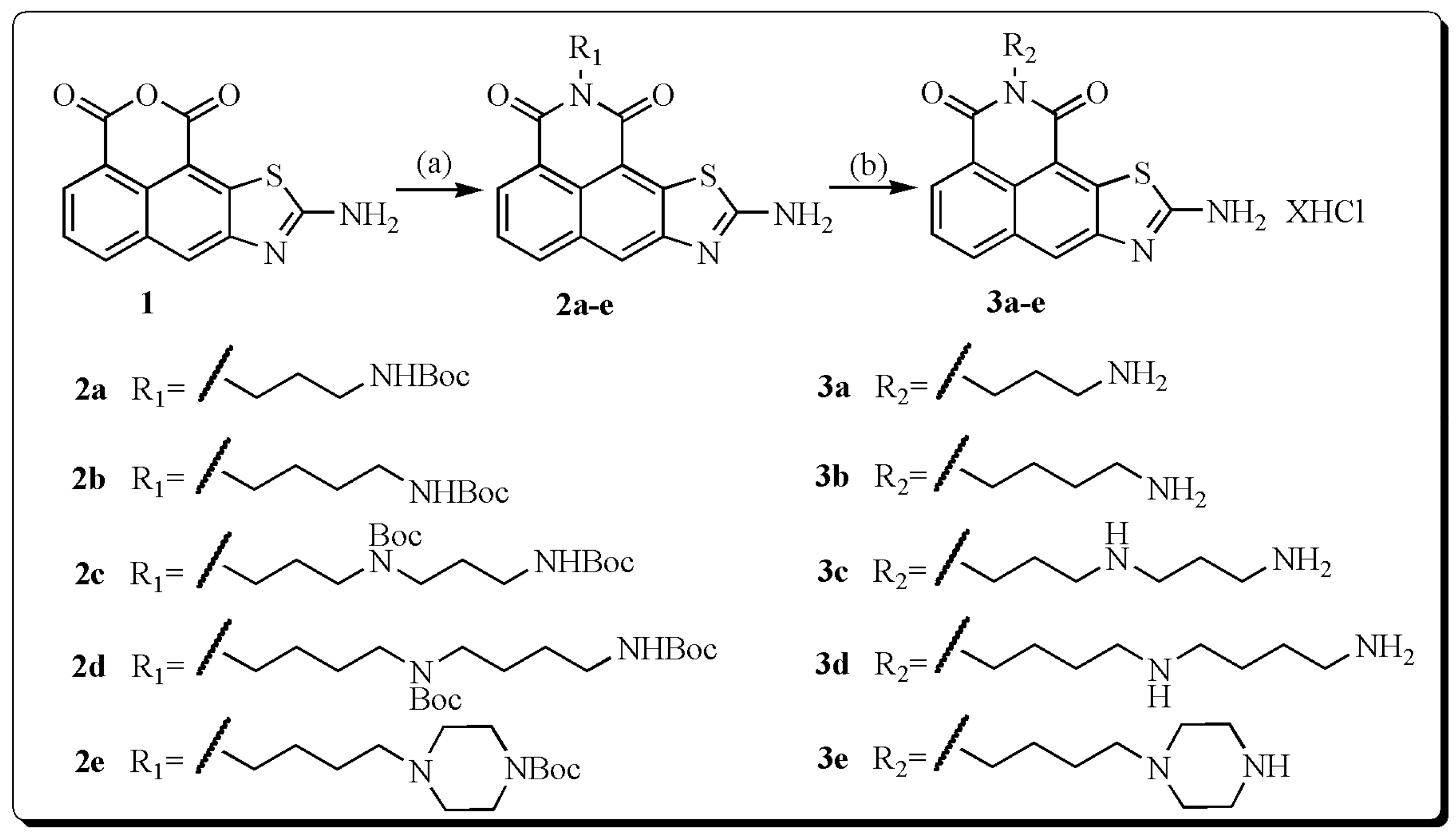
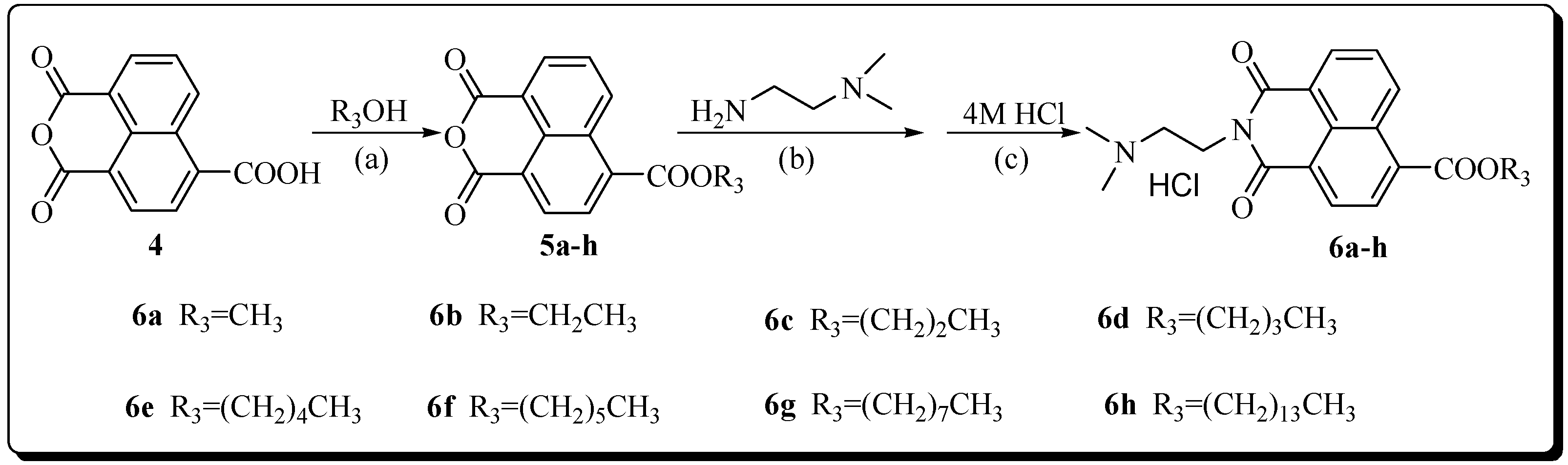
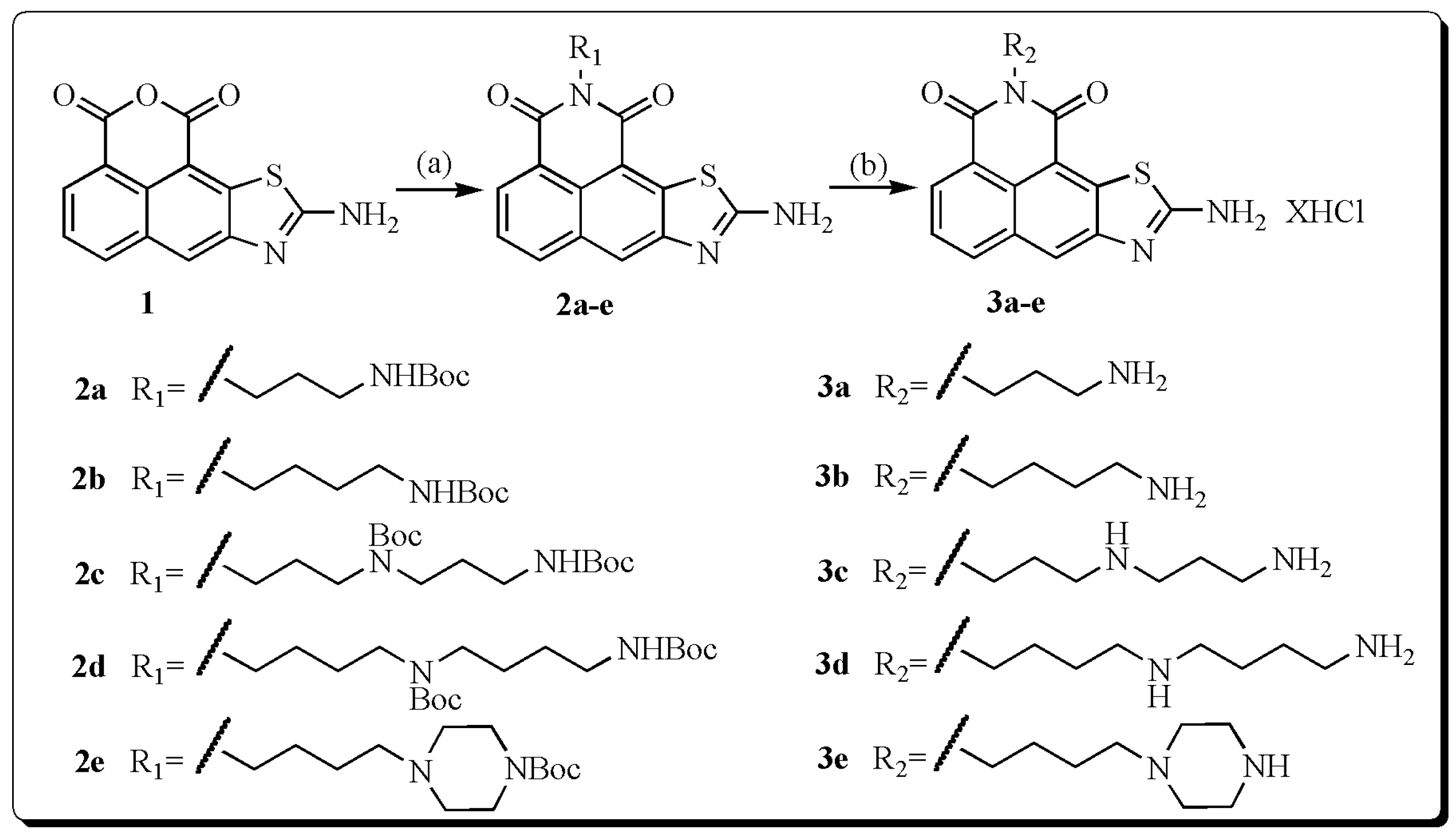
2.1. Synthesis
2.2. Biological Evaluation
2.2.1. Antitumor Activity In Vitro
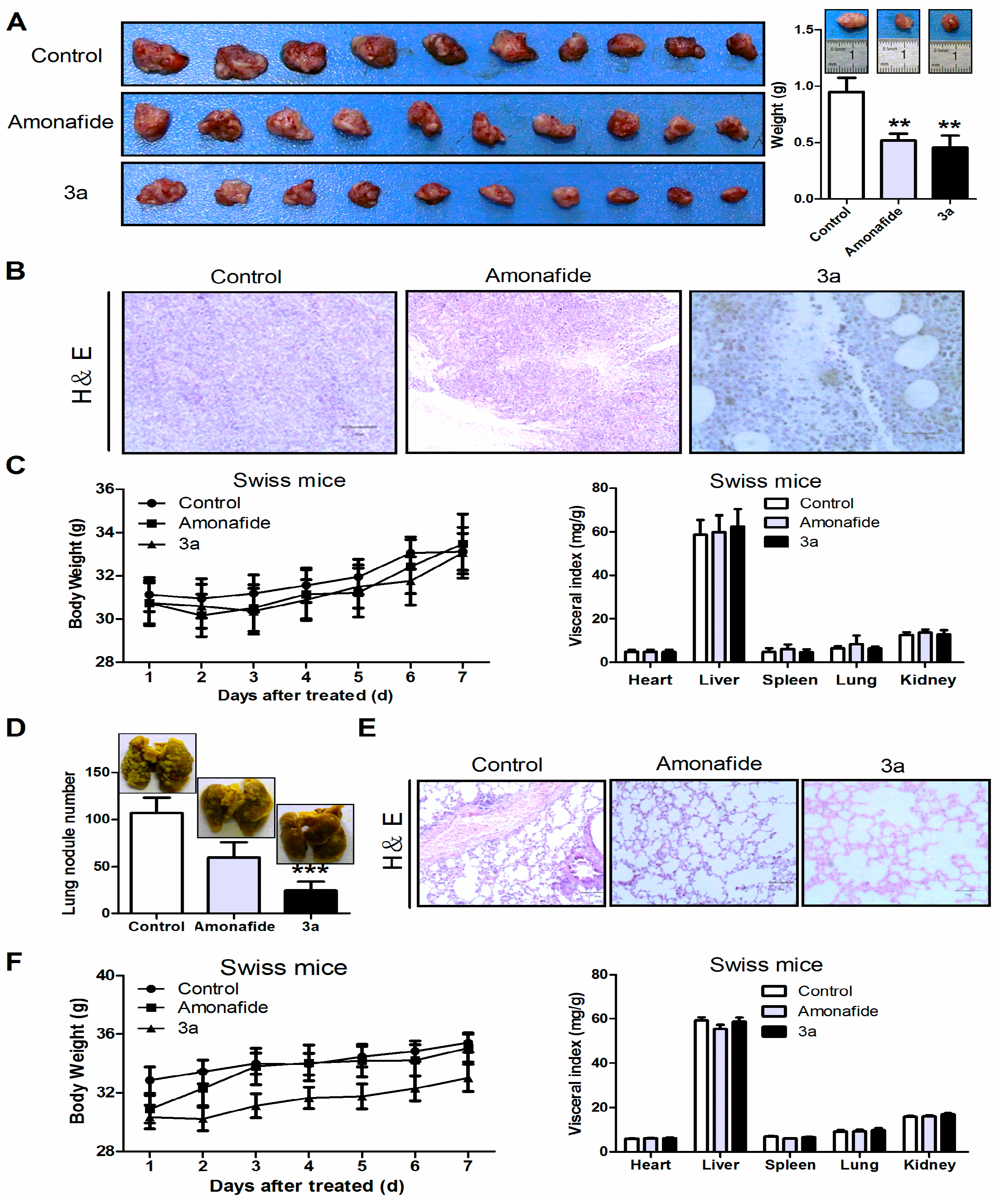
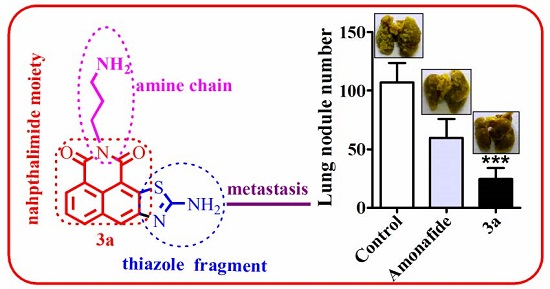
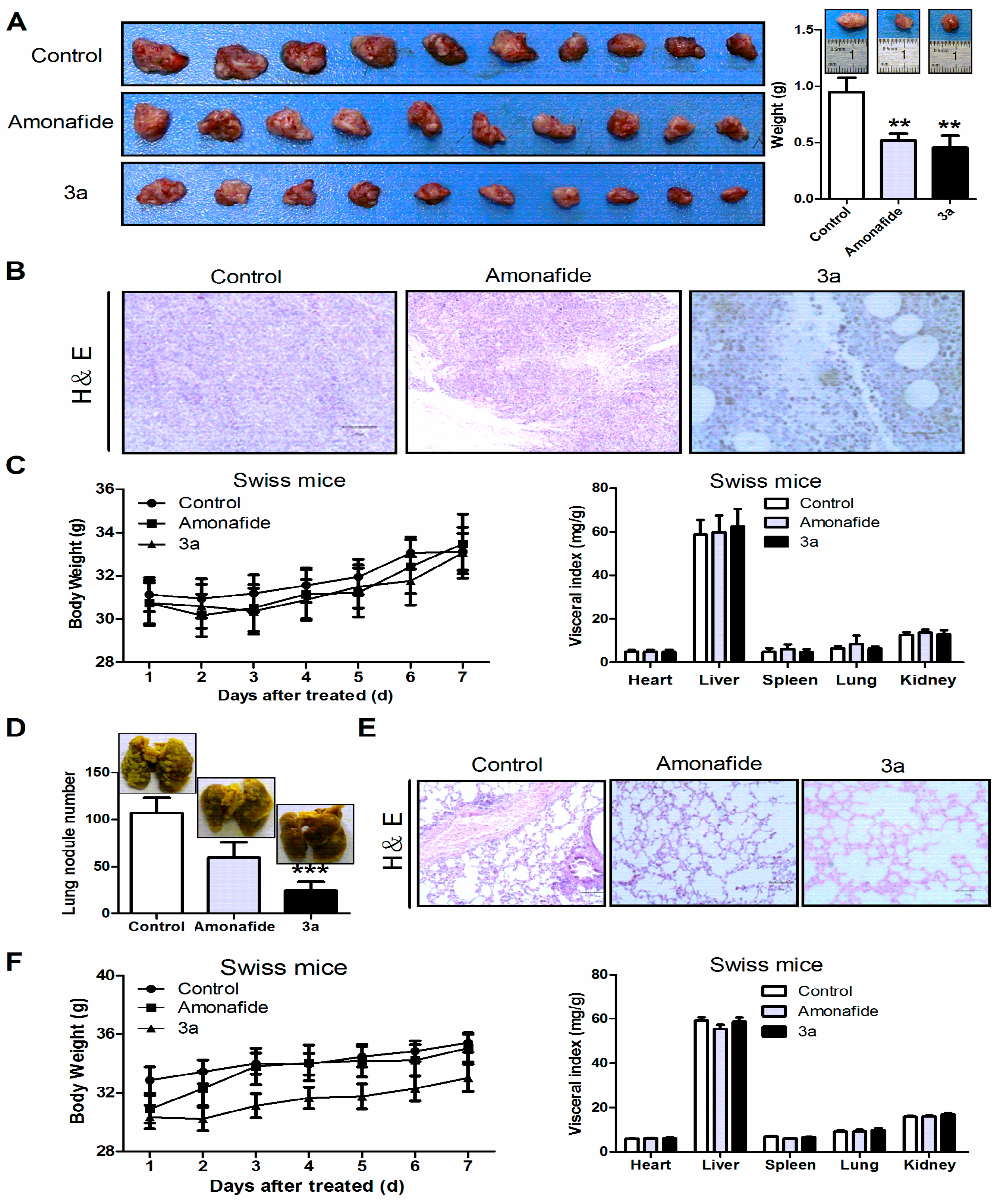
2.2.2. Anti-Tumor Activity In Vivo
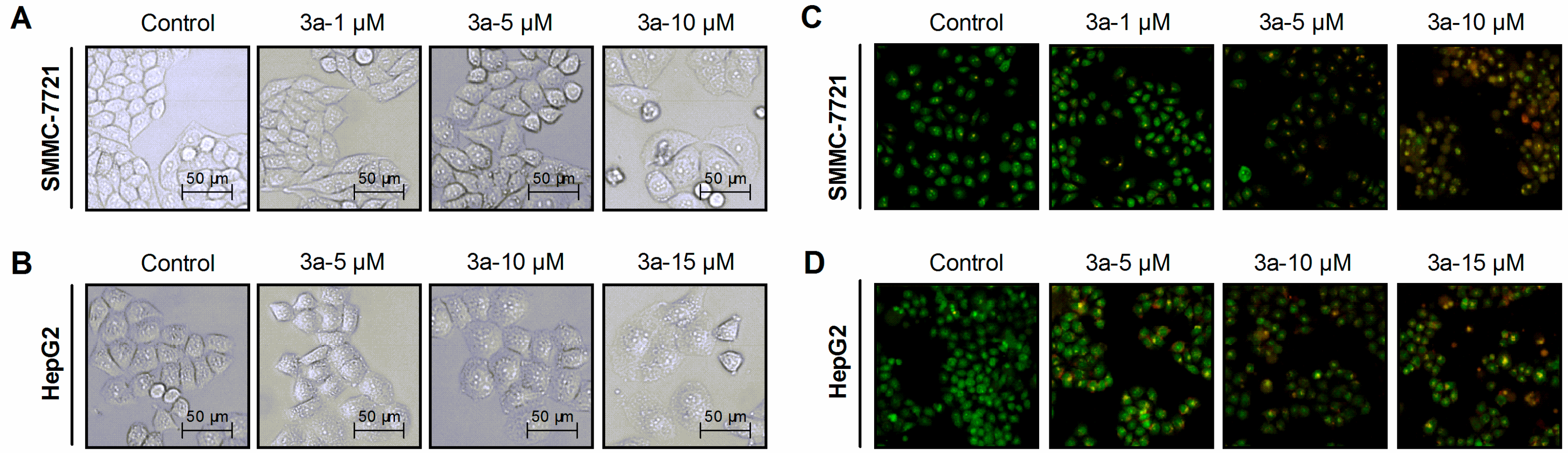
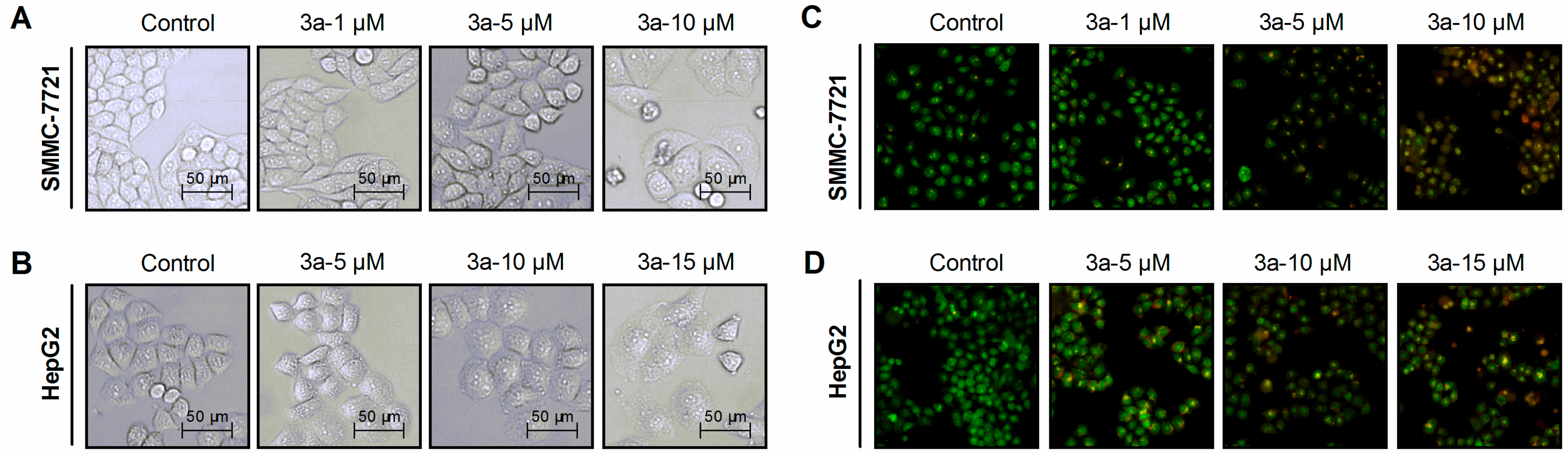
2.2.3. 3a-Induced Cell Morphology Changes and Apoptosis
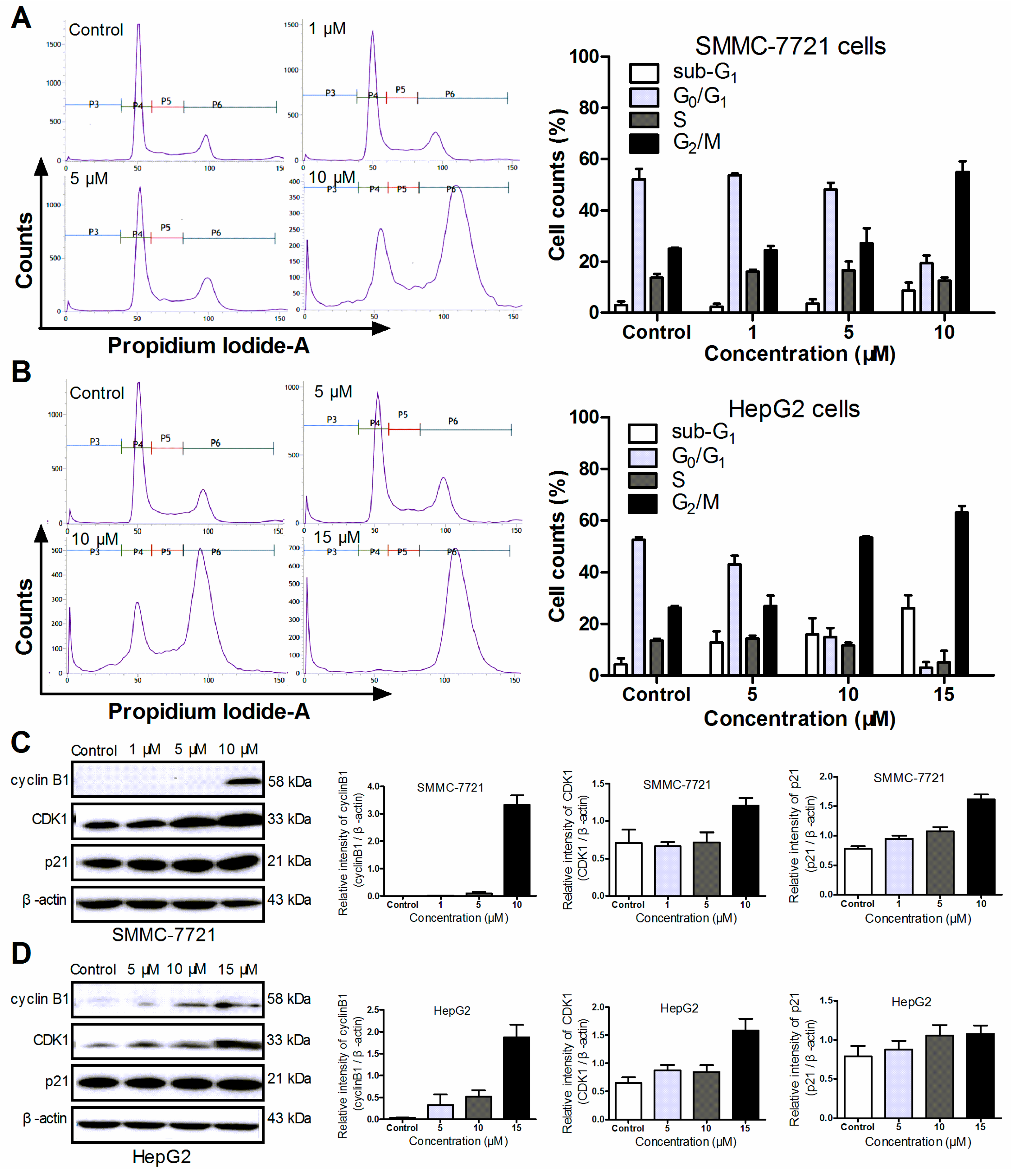
2.2.4. 3a-Induced G2/M Phase Arrest
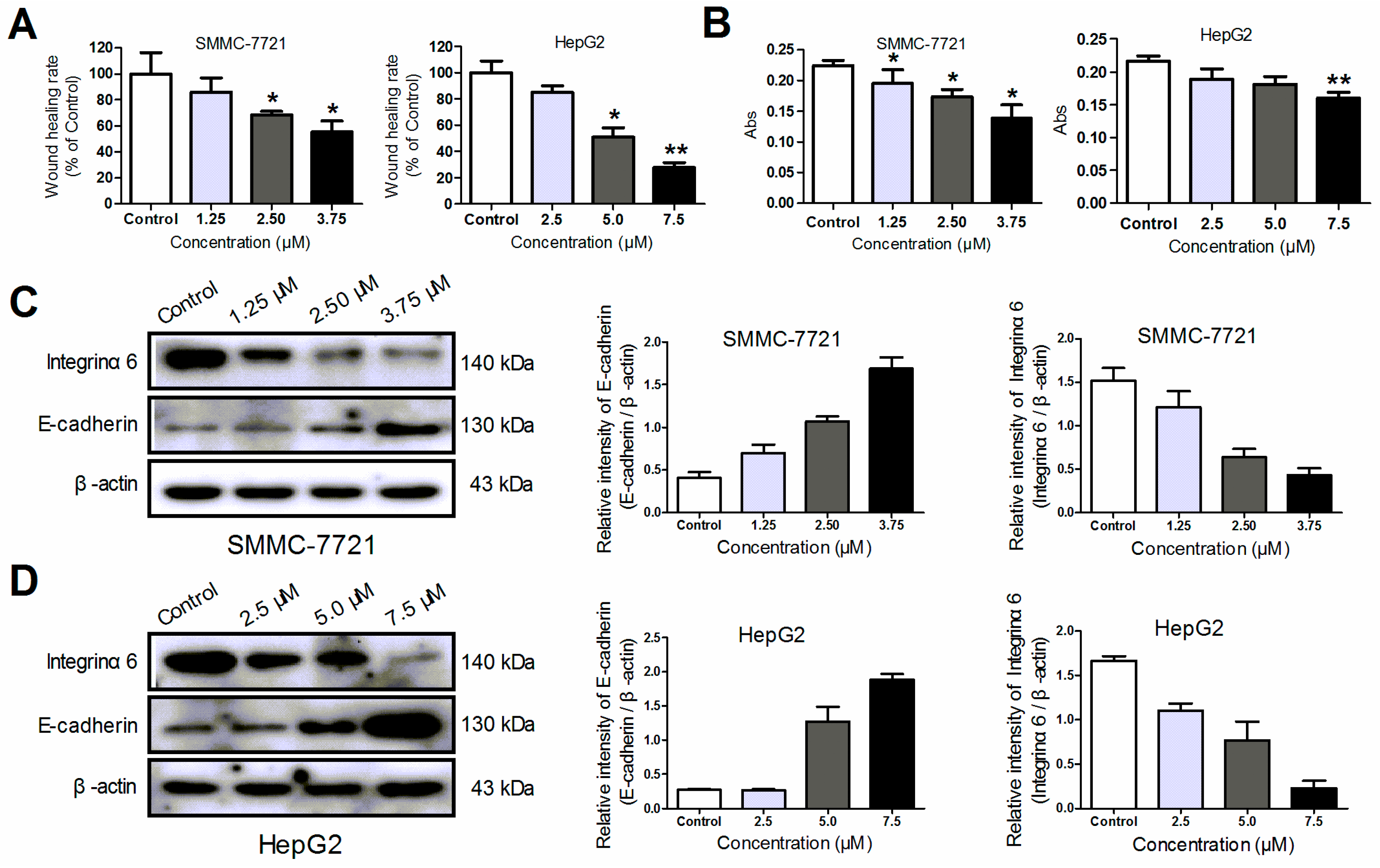
2.2.5. 3a-Induced Inhibition of Migration and Invasion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Compounds 3a–e
3.3. General Procedure for the Synthesis of Compounds 6a–h
3.4. Materials and Cell Lines
3.5. Cytotoxicity against Cancer Cell Lines
3.6. Cell Morphology Observation
3.7. Cellular Apoptotic Evaluation
3.8. Cell Cycle Analysis
3.9. Migration Assay In Vitro
3.10. Transwell Invasion Assay
3.11. Western Blotting
3.12. Evaluation of Antitumor Effects In Vivo
3.13. Systemic Toxicity and Histopathological Evaluation
3.14. Data Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- NSCLC Meta-Analysis Collaborative Group. Preoperative chemotherapy for non-small-cell lung cancer: A systematic review and meta-analysis of individual participant data. Lancet 2014, 383, 1561–1571. [Google Scholar]
- Duffy, A.; Greten, T. Developing better treatments in hepatocellular carcinoma. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Brider, T.; Redko, B.; Grynszpan, F.; Gellerman, G. Three overlooked chemical approaches toward 3-naphthalimide amonafide N-derivatives. Tetrahedron Lett. 2014, 55, 6675–6679. [Google Scholar] [CrossRef]
- Banerjee, S.; Veale, E.B.; Phelan, C.M.; Murphy, S.A.; Tocci, G.M.; Gillespie, L.J.; Frimannsson, D.O.; Kelly, J.M.; Gunnlaugsson, T. Recent advances in the development of 1,8-naphthalimide based DNA targeting binders, anticancer and fluorescent cellular imaging agents. Chem. Soc. Rev. 2013, 42, 1601–1618. [Google Scholar] [CrossRef] [PubMed]
- Seliga, R.; Pilatova, M.; Sarissky, M.; Viglasky, V.; Walko, M.; Mojzis, J. Novel naphthalimide polyamine derivatives as potential antitumor agents. Mol. Biol. Rep. 2013, 40, 4129–4137. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Bolla, N.R.; Srikanth, P.S.; Srivastava, A.K. Naphthalimide derivatives with therapeutic characteristics: A patent review. Expert Opin. Ther. Pat. 2013, 23, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Gellerman, G. Recent Developments in the Synthesis and Applications of Anticancer Amonafide Derivatives. A Mini Review. Lett. Drug Des. Dis. 2016, 13, 47–63. [Google Scholar] [CrossRef]
- Lv, M.; Xu, H. Overview of naphthalimide analogs as anticancer agents. Curr. Med. Chem. 2009, 16, 4797–4813. [Google Scholar] [CrossRef] [PubMed]
- Ingrassia, L.; Lefranc, F.; Kiss, R.; Mijatovic, T. Naphthalimides and azonafides as promising anti-cancer agents. Curr. Med. Chem. 2009, 16, 1192–1213. [Google Scholar] [CrossRef] [PubMed]
- Braña, M.F.; Cacho, M.; Garcia, M.A.; de Pascual-Teresa, B.; Ramos, A.; Dominguez, M.T.; Pozuelo, J.M.; Abradelo, C.; Rey-Stolle, M.F.; Yuste, M.; et al. New analogues of amonafide and elinafide, containing aromatic heterocycles: Synthesis, antitumor activity, molecular modeling, and DNA binding properties. J. Med. Chem. 2004, 47, 1391–1399. [Google Scholar]
- Li, Z.; Yang, Q.; Qian, X. Novel thiazonaphthalimides as efficient antitumor and DNA photocleaving agents: Effects of intercalation, side chains, and substituent groups. Bioorg. Med. Chem. 2005, 13, 4864–4870. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, Q.; Qian, X. Novel 2-aminothiazonaphthalimides as visible light activatable photonucleases: Effects of intercalation, heterocyclic-fused area and side chains. Bioorg. Med. Chem. Lett. 2005, 15, 1769–1772. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, Z.; Yang, Q. Highly efficient antitumor agents of heterocycles containing sulfur atom: Linear and angular thiazonaphthalimides against human lung cancer cell in vitro. Bioorg. Med. Chem. 2007, 15, 6846–6851. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xu, K.; Xu, Y.; Liu, J.; Qian, X. B1-induced caspase-independent apoptosis in MCF-7 cells is mediated by down-regulation of Bcl-2 via p53 binding to P2 promoter TATA box. Toxicol. Appl. Pharmacol. 2011, 256, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Xu, Y.; Xu, K.; Liu, J.; Qian, X. B1, a novel amonafide analogue, overcomes the resistance conferred by Bcl-2 in human promyelocytic leukemia HL60 Cells. Mol. Cancer Res. 2010, 8, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zhang, X.B.; Zhao, J.; Xie, S.Q.; Wang, C.J. Nonhematotoxic naphthalene diimide modified by polyamine: Synthesis and biological evaluation. J. Med. Chem. 2012, 55, 3502–3512. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.Y.; Xie, S.Q.; Du, Y.W.; Ma, Y.F.; Zhao, J.; Gao, W.Y.; Wang, C.J. Synthesis, cytotoxicity and apoptosis of naphthalimide polyamine conjugates as antitumor agents. Eur. J. Med. Chem. 2009, 44, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Delcros, J.G.; Biggerstaff, J.; Phanstiel, O. Molecular requirements for targeting the polyamine transport system. Synthesis and biological evaluation of polyamine-anthracene conjugates. J. Med. Chem. 2003, 46, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.H.; Yao, R.; Tian, H. Synthesis of novel electro-transporting emitting compounds. Dyes Pigment. 2002, 54, 147–154. [Google Scholar] [CrossRef]
- Chen, M.; Wu, Y.Y.; Luo, Y.; He, M.Q.; Xie, J.M.; Li, H.M.; Yuan, X.H. One-pot synthesis of 5-acetylacenaphthene using heteropoly acid catalysts. React. Kinet. Mech. Catal. 2011, 102, 103–111. [Google Scholar] [CrossRef]
- Stacey, D.W.; Hitomi, M.; Kanovsky, M.; Gan, L.; Johnson, E.M. Cell cycle arrest and morphological alterations following microinjection of NIH3T3 cells with Pur alpha. Oncogene 1999, 18, 4254–4261. [Google Scholar] [CrossRef] [PubMed]
- Brandhagen, B.N.; Tieszen, C.R.; Ulmer, T.M.; Tracy, M.S.; Goyeneche, A.A.; Telleria, C.M. Cytostasis and morphological changes induced by mifepristone in human metastatic cancer cells involve cytoskeletal filamentous actin reorganization and impairment of cell adhesion dynamics. BMC Cancer 2013, 13, 1–15. [Google Scholar] [CrossRef]
- Wyllie, A.H.; Kerr, J.F.; Currie, A.R. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [PubMed]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar] [PubMed]
- Buja, L.M.; Eigenbrodt, M.L.; Eigenbrodt, E.H. Apoptosis and necrosis. Basic types and mechanisms of cell death. Arch. Pathol. Lab. Med. 1993, 117, 1208–1214. [Google Scholar] [PubMed]
- Xie, S.Q.; Li, Q.; Zhang, Y.H.; Wang, J.H.; Mei, Z.H.; Zhao, J.; Wang, C.J. NPC-16, a novel naphthalimide-polyamine conjugate, induced apoptosis and autophagy in human hepatoma HepG2 cells and Bel-7402 cells. Apoptosis 2011, 16, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liang, X.; Zhang, H.; Xie, H.; Liu, J.; Xu, Y.; Zhu, W.; Wang, Y.; Wang, X.; Tan, S.; et al. A new class of naphthalimide-based antitumor agents that inhibit topoisomerase II and induce lysosomal membrane permeabilization and apoptosis. J. Med. Chem. 2010, 53, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.; Quinta-Costa, M.; Leite, P.S.; Guimarães, J.E. Critical evaluation of techniques to detect and measure cell death—Study in a model of UV radiation of the leukaemic cell line HL60. Anal. Cell Pathol. 1999, 19, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Stephens, P.A.; Middleton, F.K.; Curtin, N.J. Targeting the S and G2 checkpoint to treat cancer. Drug Discov. Today 2012, 17, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Shin, Y.J.; Riew, T.R.; Lee, M.Y. The indolinone MAZ51 induces cell rounding and G2/M cell cycle arrest in glioma cells without the inhibition of VEGFR-3 phosphorylation: Involvement of the RhoA and Akt/GSK3beta signaling pathways. PLoS ONE 2014, 9, e109055. [Google Scholar] [CrossRef] [PubMed]
- Ito, M. Factors controlling cyclin B expression. Plant Mol. Biol. 2000, 43, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A. Mechanisms and regulation of the degradation of cyclin B. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.H.; Chuah, Q.Y.; Yang, P.M.; Chen, C.T.; Chao, J.C.; Lin, M.D.; Chiu, S.J. Securin enhances the anti-cancer effects of 6-methoxy-3-(3′,4′,5′-trimethoxy-benzoyl)-1H-indole (BPR0L075) in human colorectal cancer cells. PLoS ONE 2012, 7, e36006. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Chan, J.Y.; Lin, H.C.; Wang, S.L.; Liu, S.T.; Ho, C.L.; Chang, L.C.; Huang, S.M. Modulation of the cyclin-dependent kinase inhibitor p21(WAF1/Cip1) gene by Zac1 through the antagonistic regulators p53 and histone deacetylase 1 in HeLa Cells. Mol. Cancer Res. 2008, 6, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.S.; Li, J.; Singletary, K.W. Effects of genistein on cell proliferation and cell cycle arrest in nonneoplastic human mammary epithelial cells: Involvement of Cdc2, p21(waf/cip1), p27(kip1), and Cdc25C expression. Biochem. Pharmacol. 2001, 61, 979–989. [Google Scholar] [CrossRef]
- Xie, S.Q.; Li, Q.; Zhang, Y.H.; Li, Z.; Zhao, J.; Wang, C.J. BND-12, a novel nonhaematotoxic naphthalimide derivative, inhibits tumour growth and metastasis of hepatocellular carcinoma. J. Pharm. Pharmacol. 2012, 64, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, Q.; Zhang, Y.H.; Tian, Z.Y.; Ma, H.X.; Zhao, J.; Xie, S.Q.; Wang, C.J. Antitumor effects and preliminary systemic toxicity of ANISpm in vivo and in vitro. Anticancer Drugs 2013, 24, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Christofori, G.; Semb, H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends Biochem. Sci. 1999, 24, 73–76. [Google Scholar] [CrossRef]
- Friedrichs, K.; Ruiz, P.; Franke, F.; Gille, I.; Terpe, H.J.; Imhof, B.A. High expression level of alpha 6 integrin in human breast carcinoma is correlated with reduced survival. Cancer Res. 1995, 55, 901–906. [Google Scholar] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J. Transwell® invasion assays. Methods Mol. Biol. 2011, 769, 97–110. [Google Scholar] [PubMed]
- Wang, S.; Jia, L.; Zhou, H.; Shi, W.; Zhang, J. Knockdown of caveolin-1 by siRNA inhibits the transformation of mouse hepatoma H22 cells in vitro and in vivo. Oligonucleotides 2009, 19, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
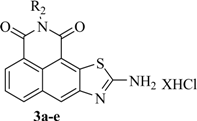 | 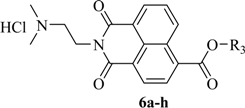 | ||||
|---|---|---|---|---|---|
| Compound | R2 (R3), X= | IC50 (µM) a | |||
| SMMC-7721 | HepG2 | HCT-116 | K562 | ||
| Amonafide | 10.32 ± 1.04 | 11.67 ± 1.45 | 6.86 ± 0.79 | 10.10 ± 1.67 | |
| 3a | -(CH2)3NH2, X = 3 | 6.03 ± 0.84 | 9.33 ± 0.95 | 11.71 ± 1.06 | 27.50 ± 2.31 |
| 3b | -(CH2)4NH2, X = 3 | 21.84 ± 1.85 | 16.48 ± 1.21 | 30.42 ± 2.49 | 42.50 ± 3.87 |
| 3c | -(CH2)3NH(CH2)3NH2, X = 4 | 1.61 ± 0.13 | 4.67 ± 0.31 | 3.81 ± 0.25 | 12.41 ± 1.56 |
| 3d | -(CH2)4NH(CH2)4NH2, X = 4 | 26.86 ± 1.73 | >50 | 15.79 ± 0.98 | >50 |
| 3e | -(CH2)4NH(CH2)4NH, X = 4 | 10.07 ± 0.89 | 19.18 ± 1.84 | 3.18 ± 0.27 | 27.18 ± 2.39 |
| 6a | -CH3, X = 1 | 22.06 ± 1.77 | 22.11 ± 1.69 | 46.48 ± 3.93 | >50 |
| 6b | -CH2CH3, X = 1 | 13.66 ± 1.04 | 25.85 ± 2.48 | 46.76 ± 3.56 | 42.99 ± 2.54 |
| 6c | -n-C3H7, X = 1 | >50 | >50 | >50 | >50 |
| 6d | -n-C4H9, X = 1 | >50 | >50 | >50 | >50 |
| 6e | -n-C5H11, X = 1 | >50 | >50 | >50 | >50 |
| 6f | -n-C6H13, X = 1 | >50 | >50 | >50 | >50 |
| 6g | -n-C8H17, X = 1 | >50 | >50 | >50 | >50 |
| 6h | -n-C14H29, X = 1 | >50 | >50 | >50 | >50 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, C.; Chang, L.; Zhao, Y.; Chang, C.; Xu, X.; He, H.; Wang, Y.; Dai, F.; Xie, S.; Wang, C. Design, Synthesis and Evaluation of Naphthalimide Derivatives as Potential Anticancer Agents for Hepatocellular Carcinoma. Molecules 2017, 22, 342. https://doi.org/10.3390/molecules22020342
Ge C, Chang L, Zhao Y, Chang C, Xu X, He H, Wang Y, Dai F, Xie S, Wang C. Design, Synthesis and Evaluation of Naphthalimide Derivatives as Potential Anticancer Agents for Hepatocellular Carcinoma. Molecules. 2017; 22(2):342. https://doi.org/10.3390/molecules22020342
Chicago/Turabian StyleGe, Chaochao, Liping Chang, Ying Zhao, Congcong Chang, Xiaojuan Xu, Haoying He, Yuxia Wang, Fujun Dai, Songqiang Xie, and Chaojie Wang. 2017. "Design, Synthesis and Evaluation of Naphthalimide Derivatives as Potential Anticancer Agents for Hepatocellular Carcinoma" Molecules 22, no. 2: 342. https://doi.org/10.3390/molecules22020342
APA StyleGe, C., Chang, L., Zhao, Y., Chang, C., Xu, X., He, H., Wang, Y., Dai, F., Xie, S., & Wang, C. (2017). Design, Synthesis and Evaluation of Naphthalimide Derivatives as Potential Anticancer Agents for Hepatocellular Carcinoma. Molecules, 22(2), 342. https://doi.org/10.3390/molecules22020342