
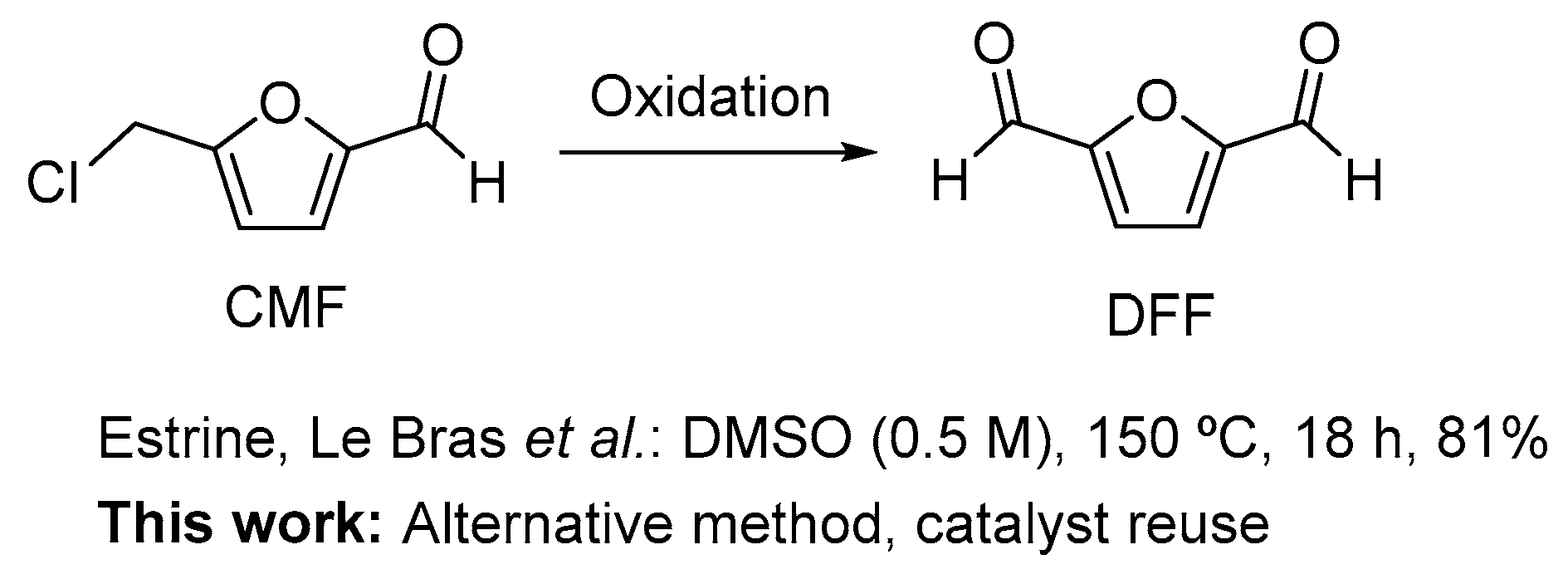
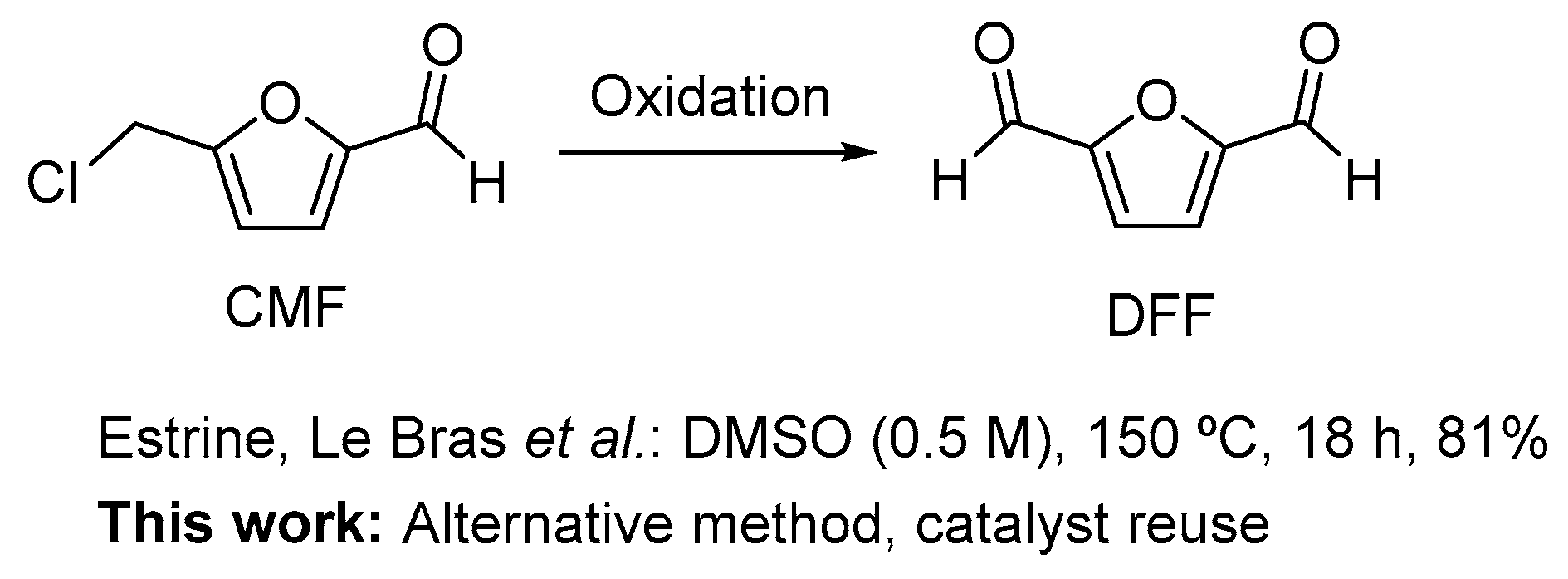
Oxidation of 5-Chloromethylfurfural (CMF) to 2,5-Diformylfuran (DFF)

 ,
,
Abstract
:1. Introduction
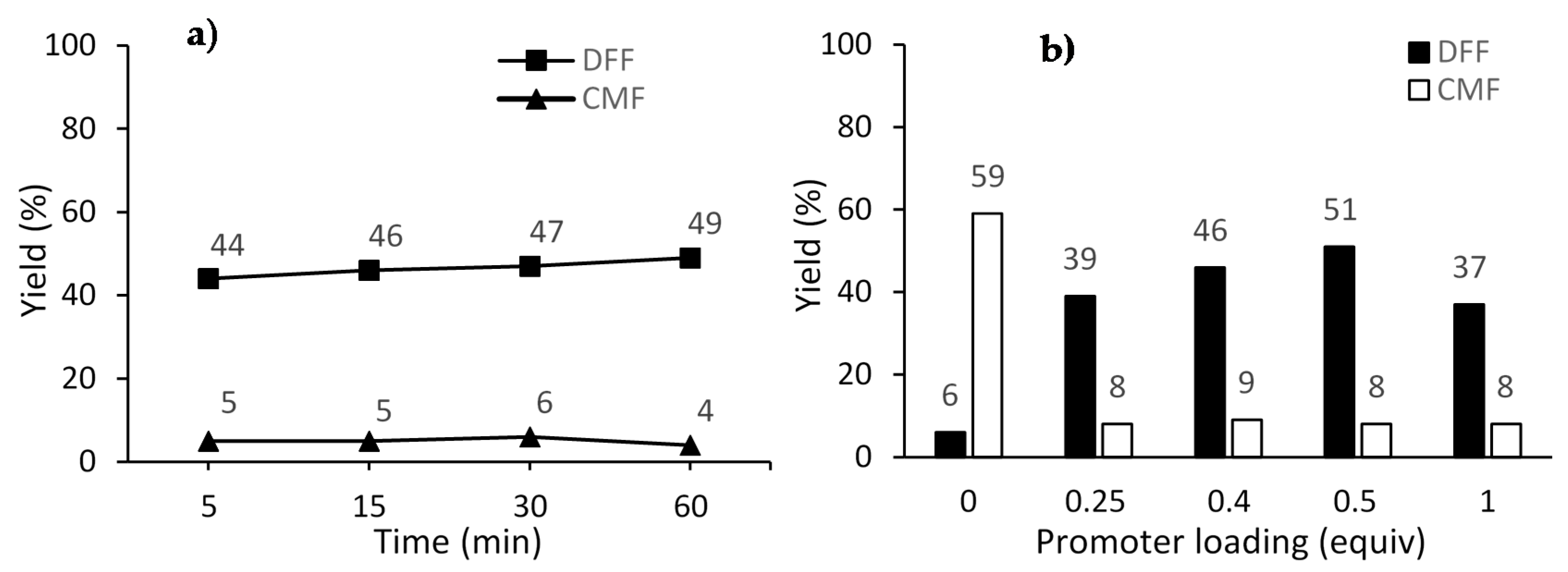
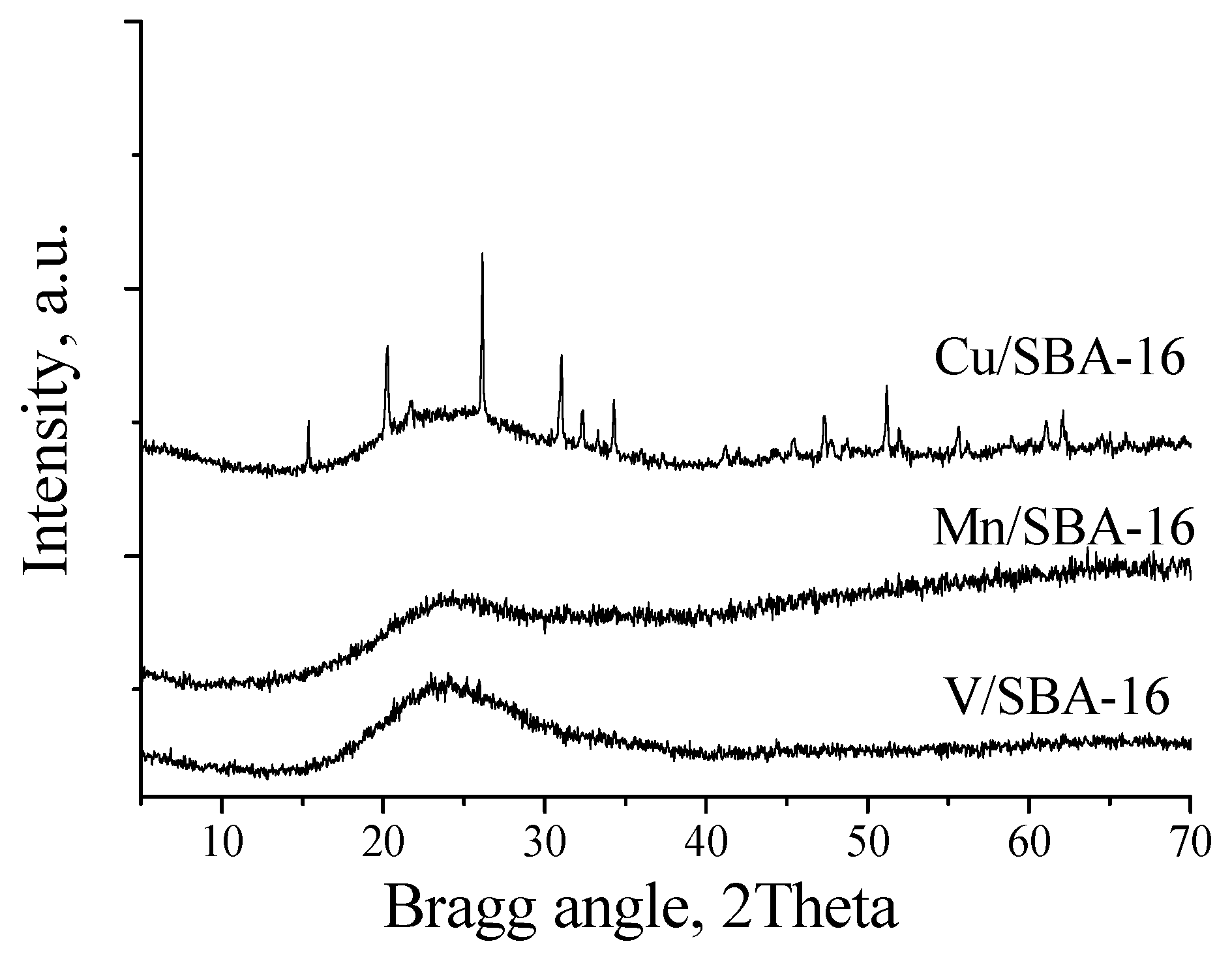
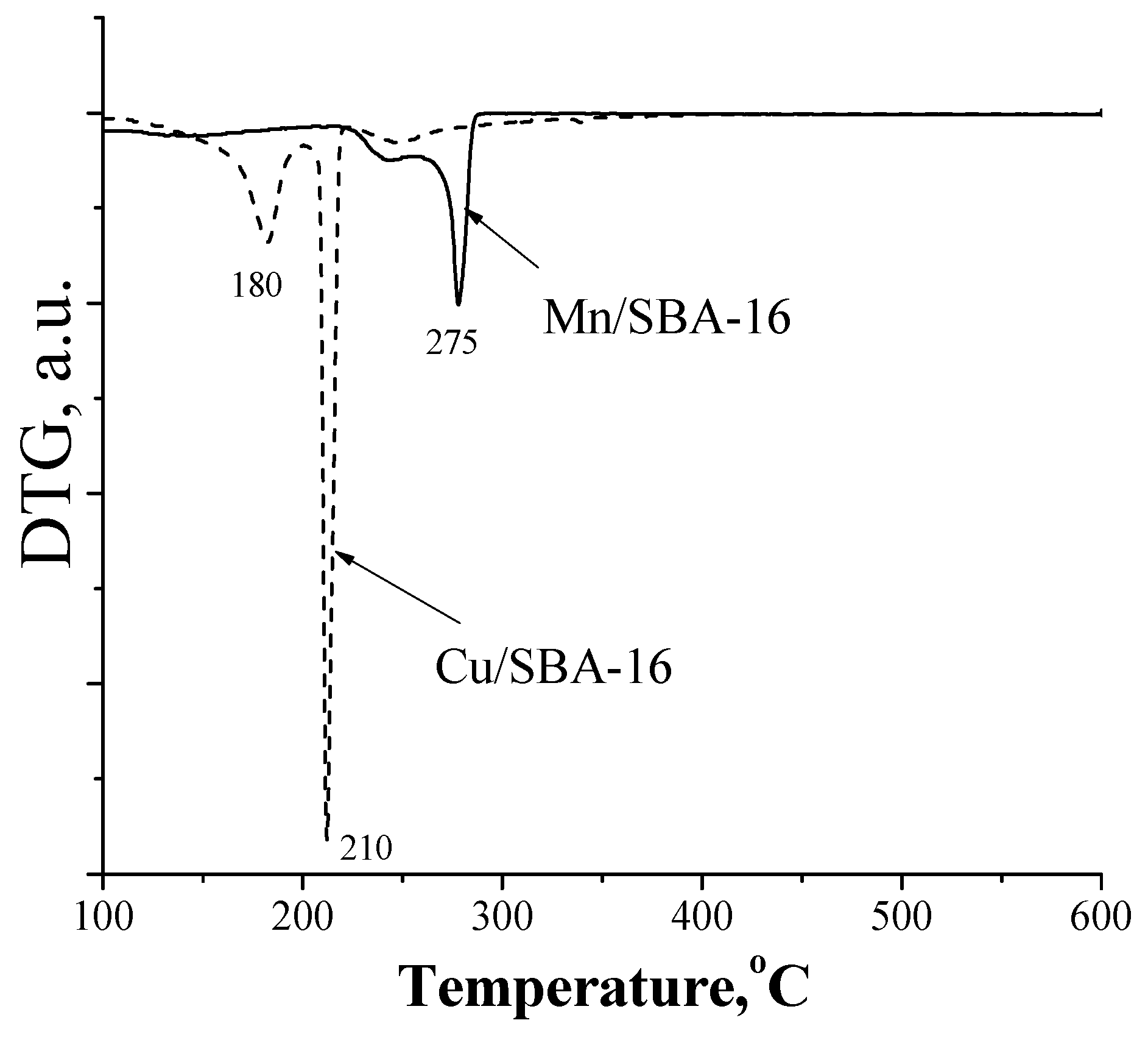
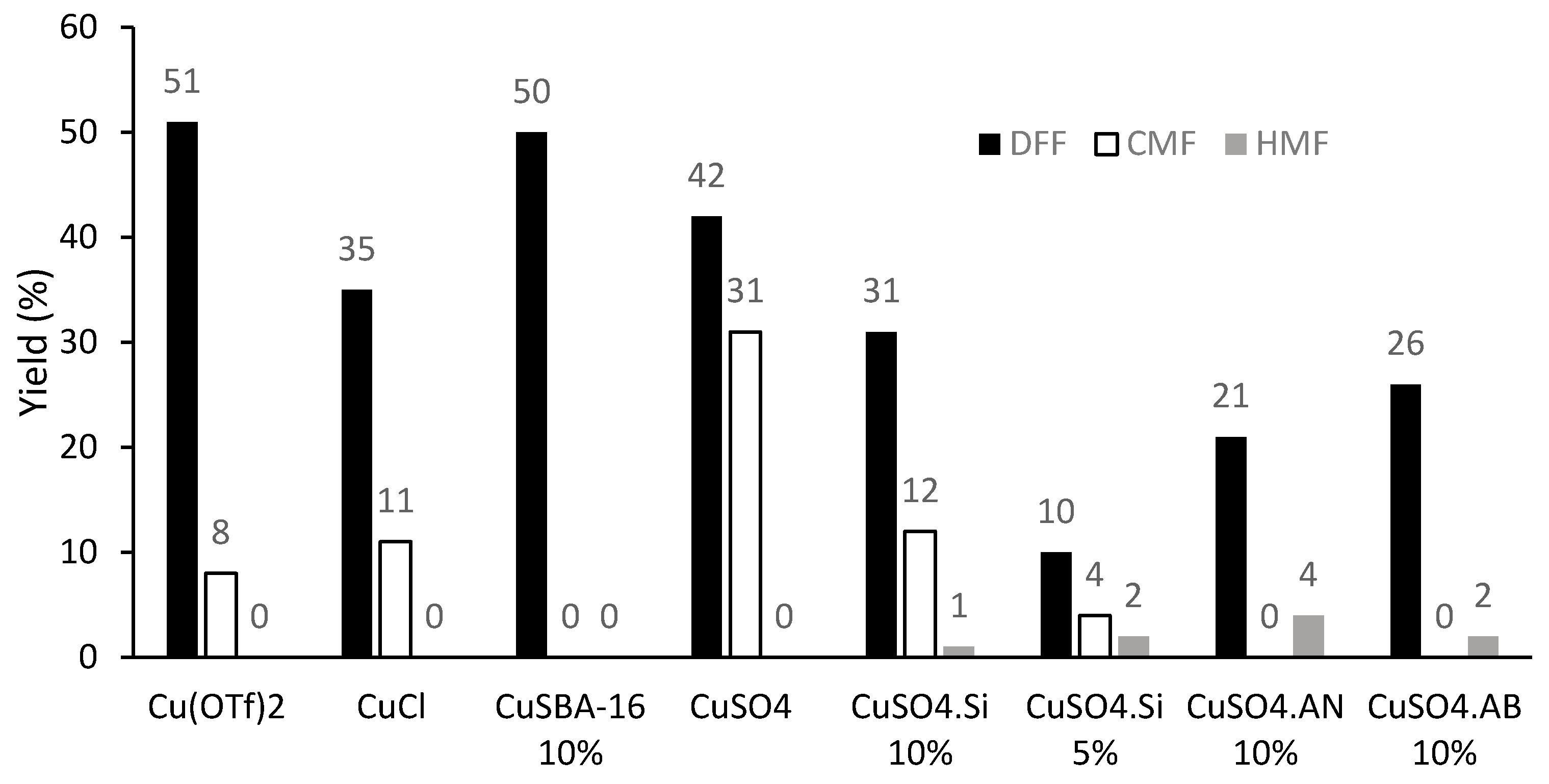
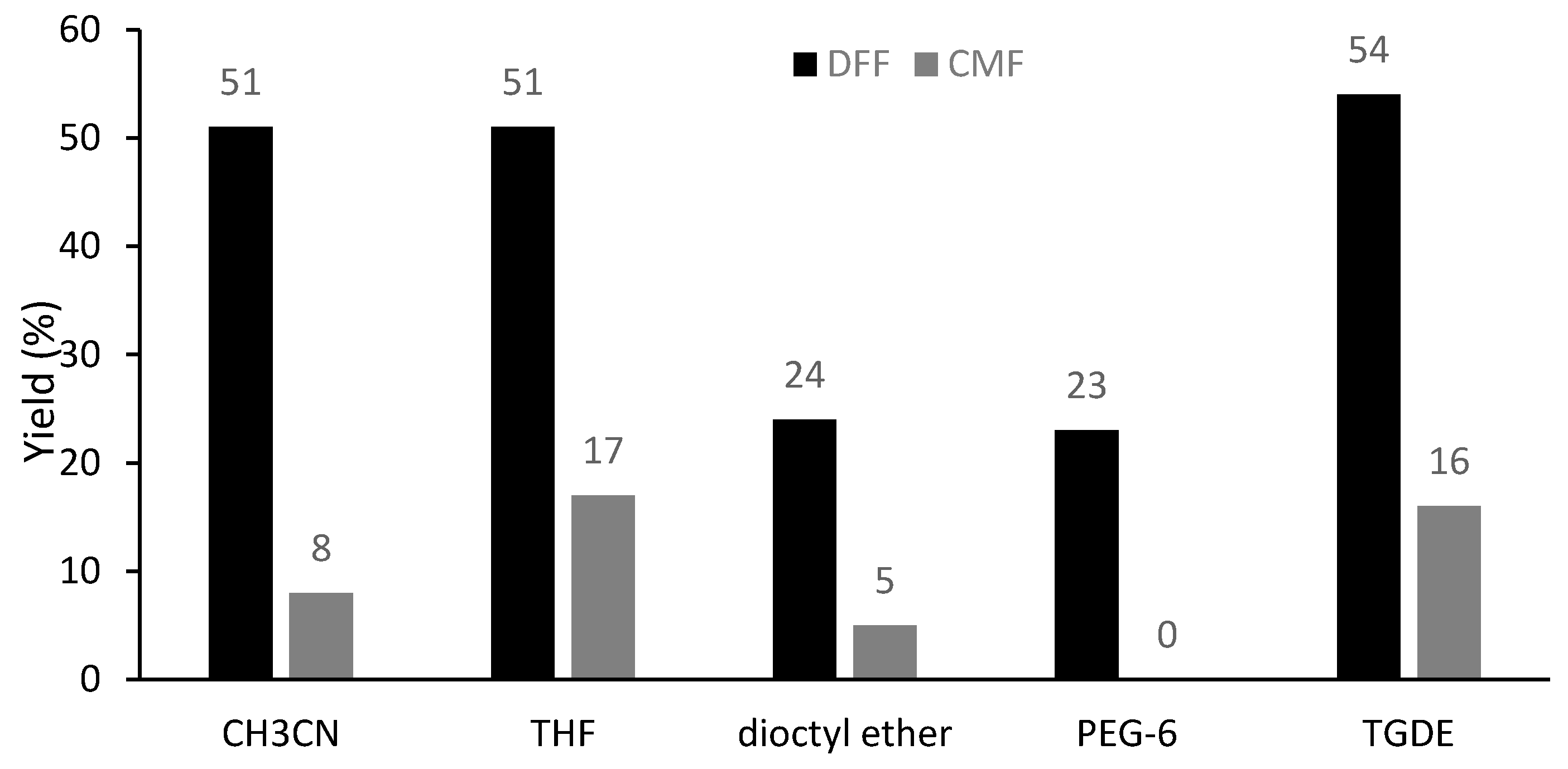
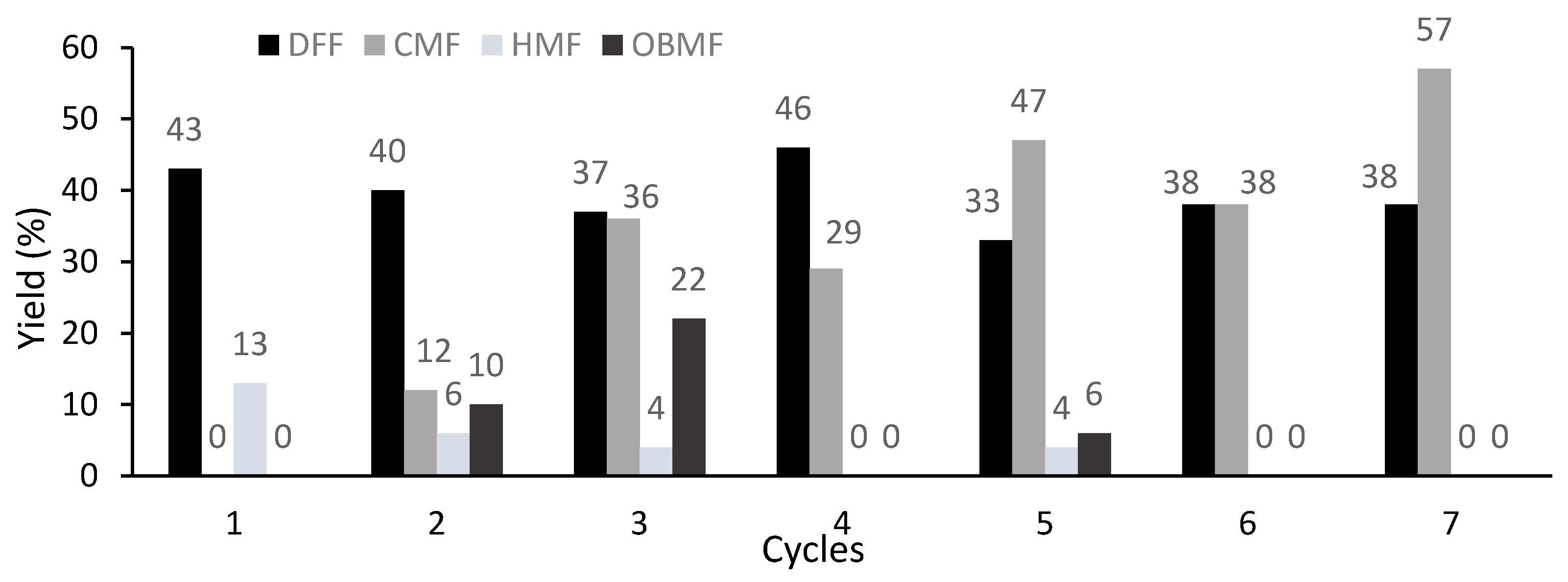
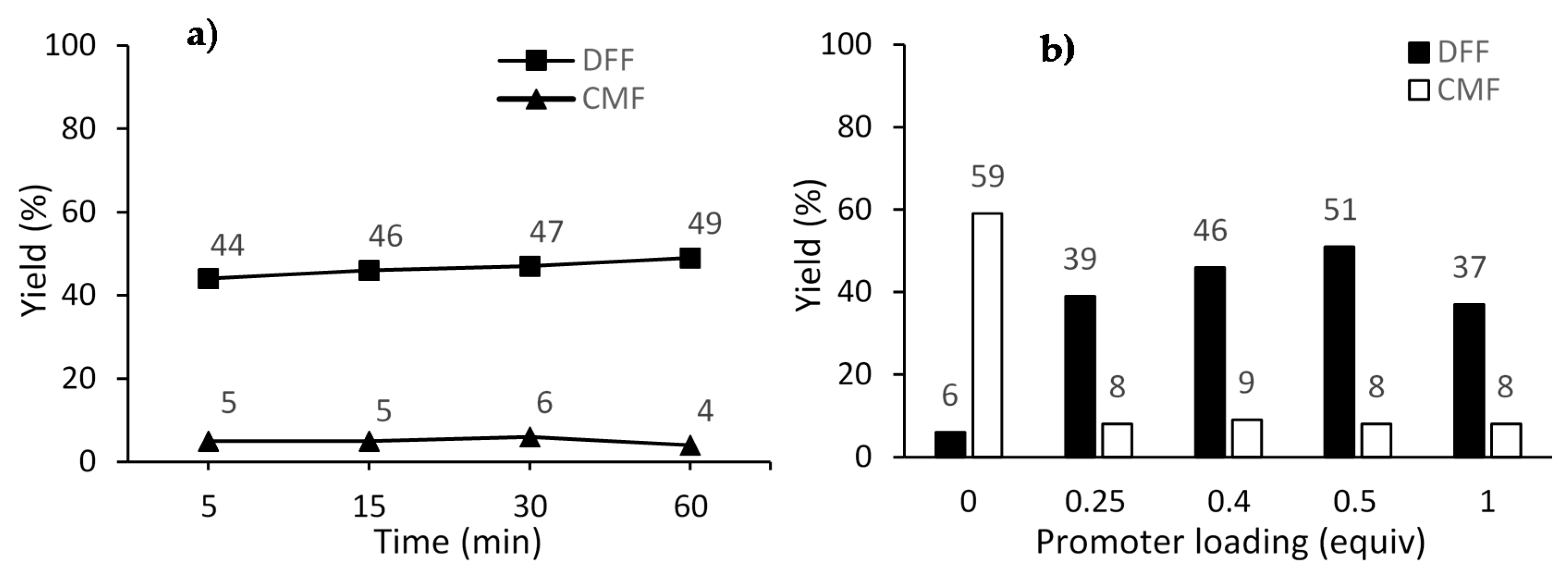
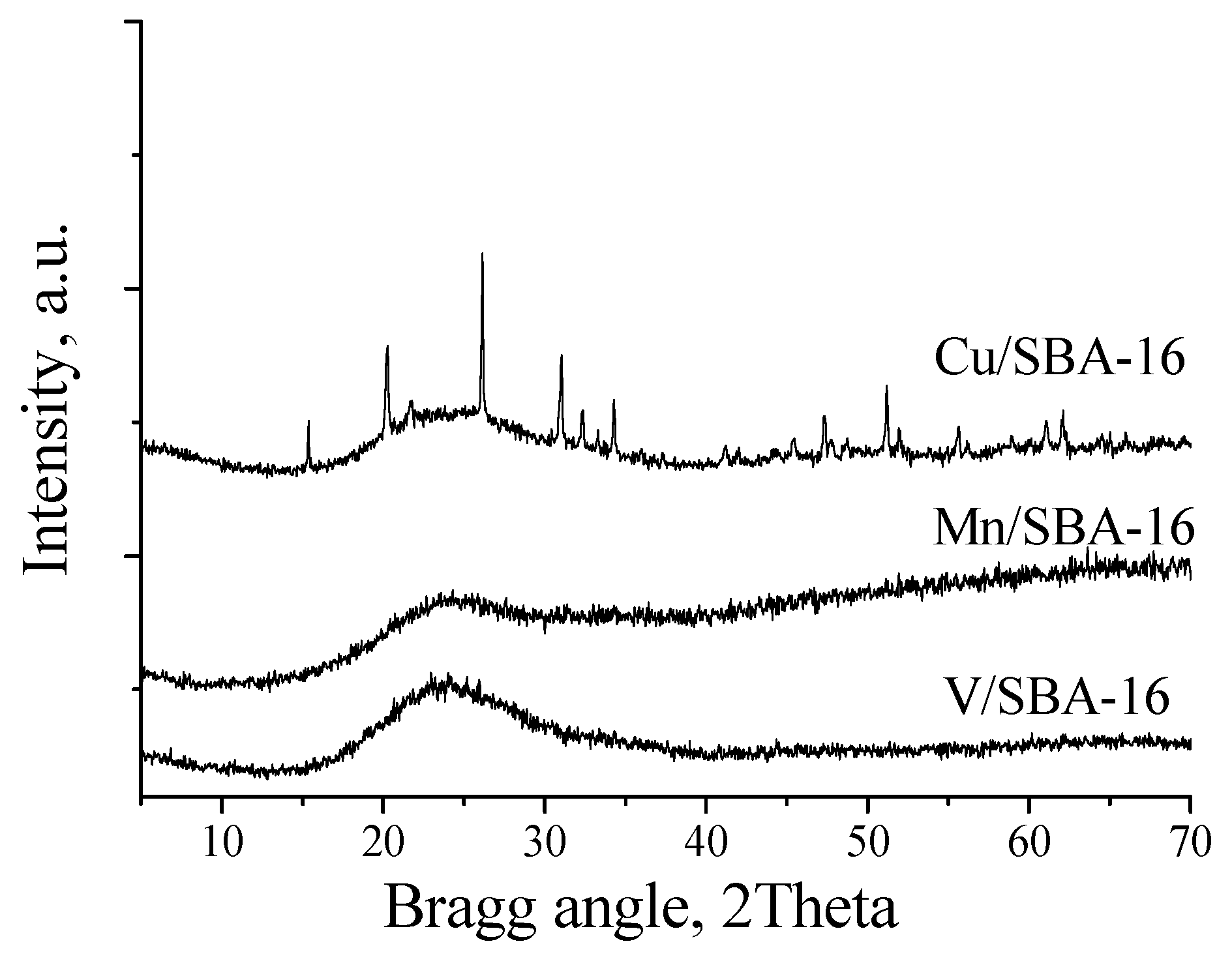
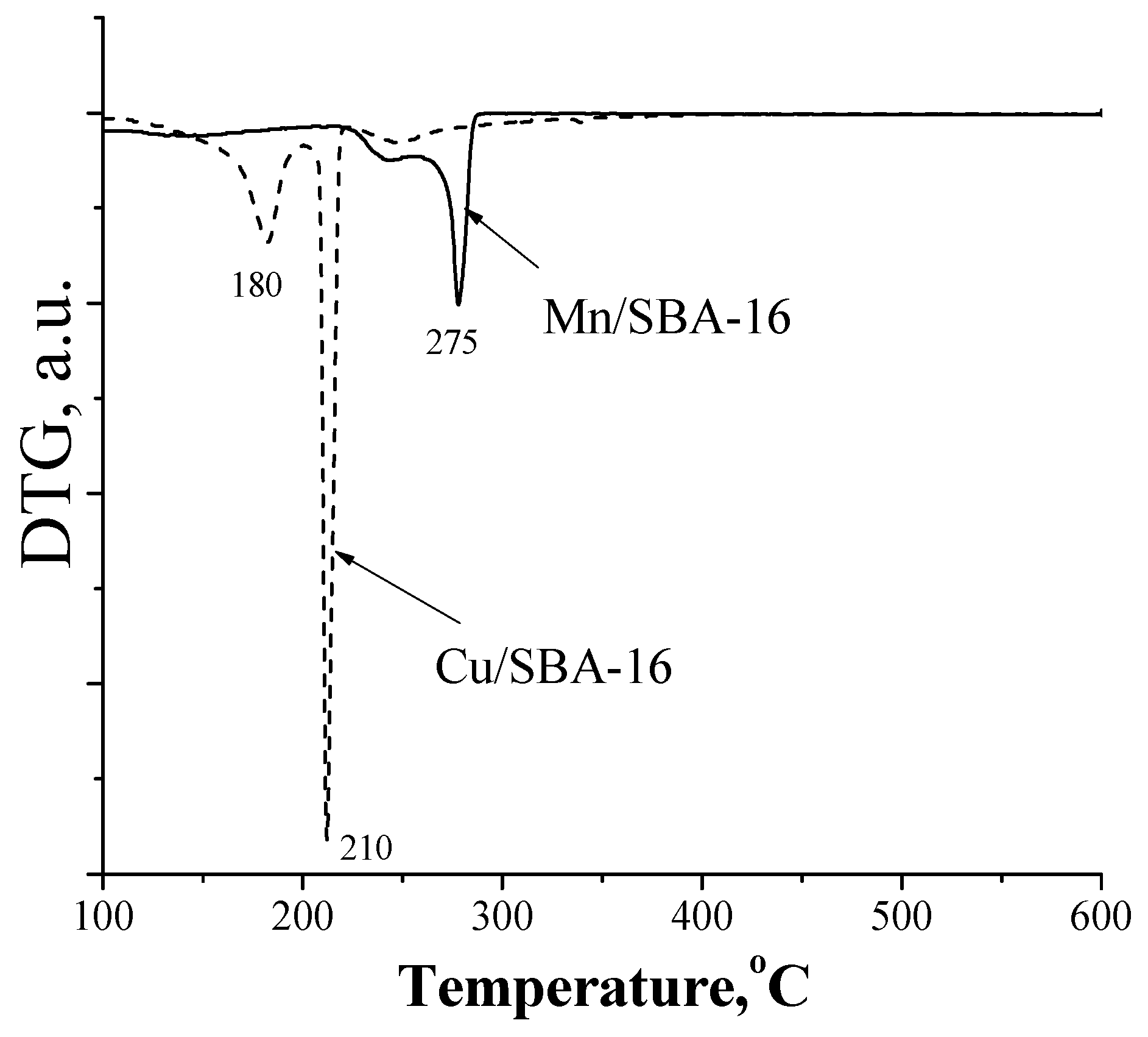
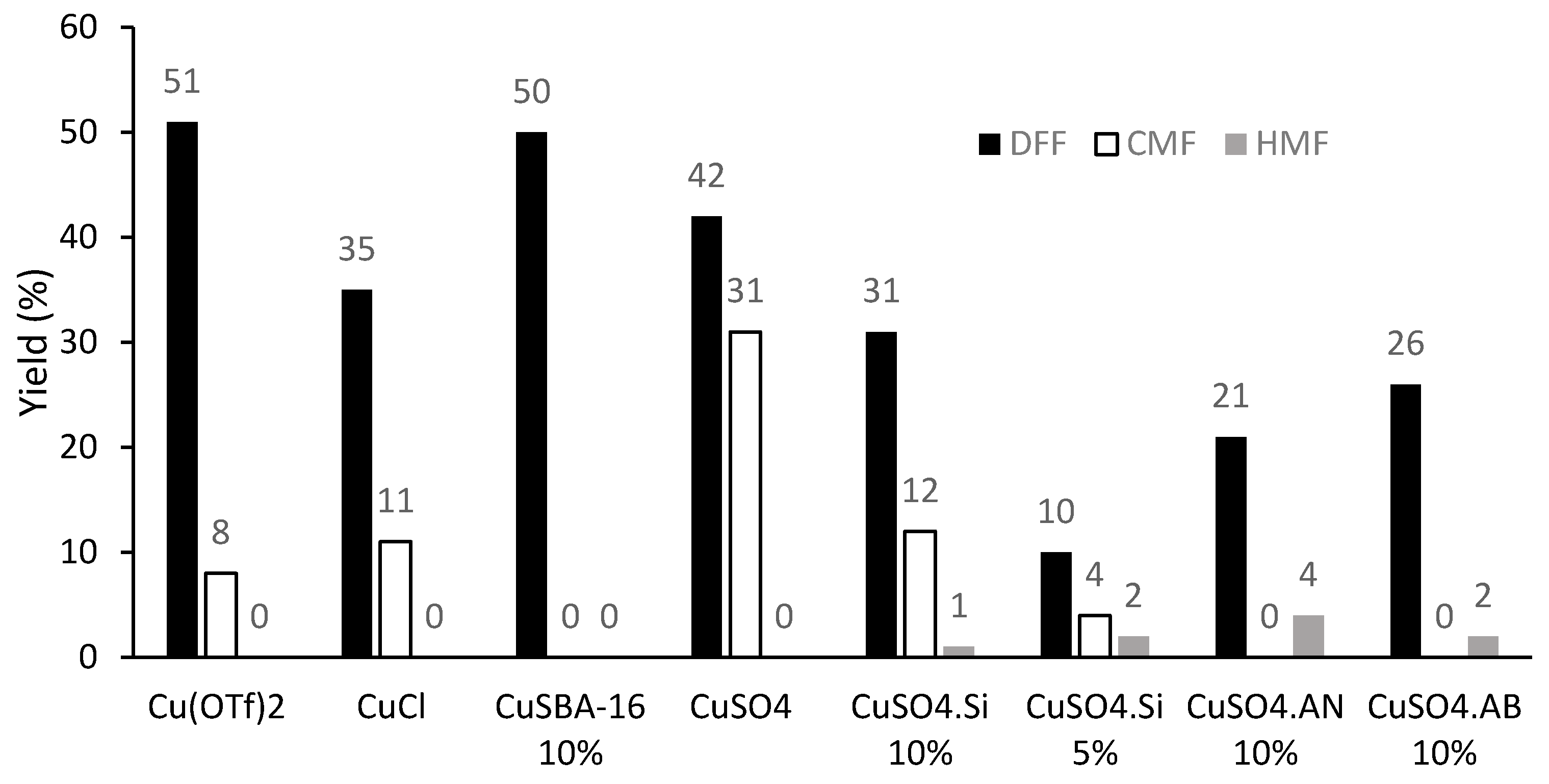
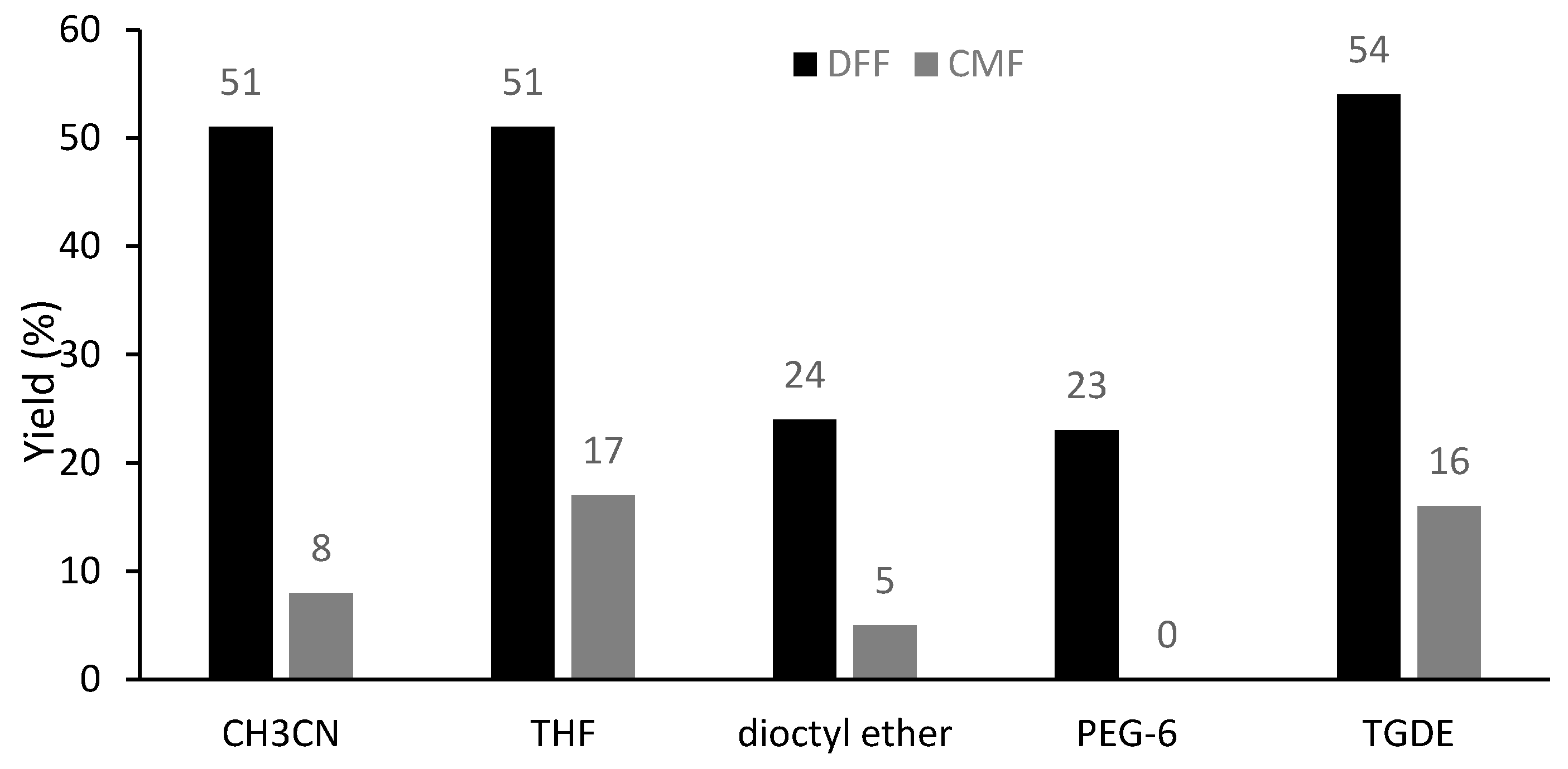
2. Results and Discussion
3. Materials and Methods
3.1. General Materials and Methods
3.2. Preparation of Heterogeneous Catalysts
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Corma, A.; Iborra, S.; Velty, A. Chemical Routes for the Transformation of Biomass into Chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Vispute, T.P.; Zhang, H.; Sanna, A.; Xiao, R.; Huber, G.W. Renewable Chemical Commodity Feedstocks from Integrated Catalytic Processing of Pyrolysis Oils. Science 2010, 330, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, A.; Nishimura, S.; Ebitani, K. Catalytic Transformations of Biomass-Derived Materials into Value-Added Chemicals. Catal. Surv. Asia 2012, 16, 164–182. [Google Scholar] [CrossRef]
- Christensen, C.H.; Rass-Hansen, J.; Marsden, C.C.; Taarning, E.; Egeblad, K. The Renewable Chemicals Industry. ChemSusChem 2008, 1, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; De, S.; Saha, B. A Brief Summary of the Synthesis of Polyester Building-Block Chemicals and Biofuels from 5-Hydroxymethylfurfural. ChemPlusChem 2012, 77, 259–272. [Google Scholar] [CrossRef]
- Gandini, A. The irruption of polymers from renewable resources on the scene of macromolecular science and technology. Green Chem. 2011, 13, 1061–1083. [Google Scholar] [CrossRef]
- Lewkowski, J. Synthesis, chemistry and applications of 5-hydroxymethyl-furfural and its derivatives. ARKIVOC 2001, 17–54. [Google Scholar] [CrossRef]
- Rosatella, A.A.; Simeonov, S.P.; Frade, R.F.M.; Afonso, C.A.M. 5-Hydroxymethylfurfural (HMF) as a building block platform: Biological properties, synthesis and synthetic applications. Green Chem. 2011, 13, 754–793. [Google Scholar] [CrossRef]
- Teong, S.P.; Yi, G.; Zhang, Y. Hydroxymethylfurfural production from bioresources: Past, present and future. Green Chem. 2014, 16, 2015–2026. [Google Scholar] [CrossRef]
- Mukherjee, A.; Dumont, M.-J.; Raghavan, V. Review: Sustainable production of hydroxymethylfurfural and levulinic acid: Challenges and opportunities. Biomass Bioenergy 2015, 72, 143–183. [Google Scholar] [CrossRef]
- van Putten, R.-J.; van der Waal, J.C.; de Jong, E.; Rasrendra, C.B.; Heeres, H.J.; de Vries, J.G. Hydroxymethylfurfural, A Versatile Platform Chemical Made from Renewable Resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef] [PubMed]
- Mascal, M.; Nikitin, E.B. Direct, High-Yield Conversion of Cellulose into Biofuel. Angew. Chem. Int. Ed. 2008, 47, 7924–7926. [Google Scholar] [CrossRef] [PubMed]
- Mascal, M.; Nikitin, E.B. Dramatic Advancements in the Saccharide to 5-(Chloromethyl)furfural Conversion Reaction. ChemSusChem 2009, 2, 859–861. [Google Scholar] [CrossRef] [PubMed]
- Mascal, M. 5-(Chloromethyl)furfural is the New HMF: Functionally Equiv.alent But More Practical in Terms of its Production From Biomass. ChemSusChem 2015, 8, 3391–3395. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.S.; Sádaba, I.; García-Suárez, E.J.; Riisager, A. Cu catalyzed oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran and 2,5-furandicarboxylic acid under benign reaction conditions. Appl. Catal. A 2013, 456, 44–50. [Google Scholar] [CrossRef]
- Kompanets, M.O.; Kushch, O.V.; Litvinov, Y.E.; Pliekhov, O.L.; Novikova, K.V.; Novokhatko, A.O.; Shendrik, A.N.; Vasilyev, A.V.; Opeida, I.O. Oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran with molecular oxygen in the presence of N-hydroxyphthalimide. Catal. Commun. 2014, 57, 60–63. [Google Scholar] [CrossRef]
- Le, N.-T.; Lakshmanan, P.; Cho, K.; Han, Y.; Kim, H. Selective oxidation of 5-hydroxymethyl-2-furfural into 2,5-diformylfuran over VO2+ and Cu2+ ions immobilized on sulfonated carbon catalysts. Appl. Catal. A 2013, 464–465, 305–312. [Google Scholar] [CrossRef]
- Saha, B.; Gupta, D.; Abu-Omar, M.M.; Modak, A.; Bhaumik, A. Porphyrin-based porous organic polymer-supported iron(III) catalyst for efficient aerobic oxidation of 5-hydroxymethyl-furfural into 2,5-furandicarboxylic acid. J. Catal. 2013, 299, 316–320. [Google Scholar] [CrossRef]
- Nie, J.; Xie, J.; Liu, H. Efficient aerobic oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran on supported Ru catalysts. J. Catal. 2013, 301, 83–91. [Google Scholar] [CrossRef]
- Jia, X.; Ma, J.; Wang, M.; Du, Z.; Lu, F.; Wang, F.; Xu, J. Promoted role of Cu(NO3)2 on aerobic oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran over VOSO4. Appl. Catal. A 2014, 482, 231–236. [Google Scholar] [CrossRef]
- Yadav, G.D.; Sharma, R.V. Biomass derived chemicals: Environmentally benign process for oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran by using nano-fibrous Ag-OMS-2-catalyst. Appl. Catal. B Environ. 2014, 147, 293–301. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, Z.; Tang, D.; Ren, Y.; Lv, K.; Liu, B. Iron Oxide Encapsulated by Ruthenium Hydroxyapatite as Heterogeneous Catalyst for the Synthesis of 2,5-Diformylfuran. ChemSusChem 2014, 7, 3496–3504. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, B.; Huang, K.; Zhang, Z. Aerobic Oxidation of Biomass-Derived 5-(Hydroxymethyl)furfural into 2,5-Diformylfuran Catalyzed by the Trimetallic Mixed Oxide (Co–Ce–Ru). Ind. Eng. Chem. Res. 2014, 53, 1313–1319. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Liu, B.; Li, J. Environmentally Friendly Oxidation of Biomass Derived 5-Hydroxymethylfurfural into 2,5-Diformylfuran Catalyzed by Magnetic Separation of Ruthenium Catalyst. Ind. Eng. Chem. Res. 2014, 53, 5820–5827. [Google Scholar] [CrossRef]
- Artz, J.; Palkovits, R. Base-Free Aqueous-Phase Oxidation of 5-Hydroxymethylfurfural over Ruthenium Catalysts Supported on Covalent Triazine Frameworks. ChemSusChem 2015, 8, 3832–3838. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Geng, L.; Guo, Y.; Jia, R.; Liu, X.; Zhang, Y.; Wang, Y. Base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over a Pt/C-O-Mg catalyst. Green Chem. 2016, 18, 1597–1604. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, J. Bicomponent Assembly of VO2 and Polyaniline-Functionalized Carbon Nanotubes for the Selective Oxidation of Biomass-Based 5-Hydroxymethylfurfural to 2,5-Diformylfuran. ChemPlusChem 2015, 80, 1760–1768. [Google Scholar] [CrossRef]
- Neațu, F.; Marin, R.S.; Florea, M.; Petrea, N.; Pavel, O.D.; Pârvulescu, V.I. Selective oxidation of 5-hydroxymethyl furfural over non-precious metal heterogeneous catalysts. Appl. Catal. B Environ. 2016, 180, 751–757. [Google Scholar] [CrossRef]
- Mittal, N.; Nisola, G.M.; Malihan, L.B.; Seo, J.G.; Kim, H.; Lee, S.-P.; Chung, W.-J. One-pot synthesis of 2,5-diformylfuran from fructose using a magnetic bi-functional catalyst. RSC Adv. 2016, 6, 25678–25688. [Google Scholar] [CrossRef]
- Li, S.; Su, K.; Li, Z.; Cheng, B. Selective oxidation of 5-hydroxymethylfurfural with H2O2 catalyzed by a molybdenum complex. Green Chem. 2016, 18, 2122–2128. [Google Scholar] [CrossRef]
- Lv, G.; Wang, H.; Yang, Y.; Li, X.; Deng, T.; Chen, C.; Zhu, Y.; Hou, X. Aerobic selective oxidation of 5-hydroxymethyl-furfural over nitrogen-doped graphene materials with 2,2,6,6-tetramethylpiperidin-oxyl as co-catalyst. Catal. Sci. Tech. 2016, 6, 2377–2386. [Google Scholar] [CrossRef]
- Neaţu, F.; Petrea, N.; Petre, R.; Somoghi, V.; Florea, M.; Parvulescu, V.I. Oxidation of 5-hydroxymethyl furfural to 2,5-diformylfuran in aqueous media over heterogeneous manganese based catalysts. Catal. Today 2016, 278, 66–73. [Google Scholar] [CrossRef]
- Liao, L.; Liu, Y.; Li, Z.; Zhuang, J.; Zhou, Y.; Chen, S. Catalytic aerobic oxidation of 5-hydroxymethylfurfural into 2,5-diformylfuran over VO2+ and Cu2+ immobilized on amino-functionalized core-shell magnetic Fe3O4@SiO2. RSC Adv. 2016, 6, 94976–94988. [Google Scholar] [CrossRef]
- Baruah, D.; Hussain, F.L.; Suri, M.; Saikia, U.P.; Sengupta, P.; Dutta, D.K.; Konwar, D. Bi(NO3)3·5H2O and cellulose mediated Cu-NPs—A highly efficient and novel catalytic system for aerobic oxidation of alcohols to carbonyls and synthesis of DFF from HMF. Catal. Commun. 2016, 77, 9–12. [Google Scholar] [CrossRef]
- Ghosh, K.; Molla, R.A.; Iqubal, M.A.; Islam, S.S.; Islam, S.M. Ruthenium nanoparticles supported on N-containing mesoporous polymer catalyzed aerobic oxidation of biomass-derived 5-hydroxymethylfurfural (HMF) to 2,5-diformylfuran (DFF). Appl. Catal. A 2016, 520, 44–52. [Google Scholar] [CrossRef]
- Wang, F.; Jiang, L.; Wang, J.; Zhang, Z. Catalytic Conversion of Fructose and 5-Hydroxymethylfurfural into 2,5-Diformylfuran over SBA-15 Supported Ruthenium Catalysts. Energy Fuels 2016, 30, 5885–5892. [Google Scholar] [CrossRef]
- Zhang, W.; Xie, J.; Hou, W.; Liu, Y.; Zhou, Y.; Wang, J. One-Pot Template-Free Synthesis of Cu–MOR Zeolite toward Efficient Catalyst Support for Aerobic Oxidation of 5-Hydroxymethylfurfural under Ambient Pressure. ACS Appl. Mater. Interfaces 2016, 8, 23122–23132. [Google Scholar] [CrossRef] [PubMed]
- Laugel, C.; Estrine, B.; Le Bras, J.; Hoffmann, N.; Marinkovic, S.; Muzart, J. NaBr/DMSO-Induced Synthesis of 2,5-Diformylfuran from Fructose or 5-(Hydroxymethyl)furfural. ChemCatChem 2014, 6, 1195–1198. [Google Scholar] [CrossRef]
- Liu, R.; Chen, J.; Chen, L.; Guo, Y.; Zhong, J. One-Step Approach to 2,5-Diformylfuran from Fructose by Using a Bifunctional and Recyclable Acidic Polyoxometalate Catalyst. ChemPlusChem 2014, 79, 1448–1454. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, L.; Tang, J.; Liu, M.; Cheng, R.; Hu, C. One-pot, One-step Synthesis of 2,5-Diformylfuran from Carbohydrates over Mo-Containing Keggin Heteropolyacids. ChemSusChem 2014, 7, 3541–3547. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, Z. Polyaniline-Grafted VO(acac)2: An Effective Catalyst for the Synthesis of 2,5-Diformylfuran from 5-Hydroxymethylfurfural and Fructose. ChemCatChem 2015, 7, 1470–1477. [Google Scholar] [CrossRef]
- Mittal, N.; Nisola, G.M.; Malihan, L.B.; Seo, J.G.; Lee, S.-P.; Chung, W.-J. Metal-free mild oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran. Korean J. Chem. Eng. 2014, 31, 1362–1367. [Google Scholar] [CrossRef]
- Mittal, N.; Nisola, G.M.; Seo, J.G.; Lee, S.-P.; Chung, W.-J. Organic radical functionalized SBA-15 as a heterogeneous catalyst for facile oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran. J. Mol. Catal. A Chem. 2015, 404–405, 106–114. [Google Scholar] [CrossRef]
- Lv, G.; Wang, H.; Yang, Y.; Deng, T.; Chen, C.; Zhu, Y.; Hou, X. Graphene Oxide: A Convenient Metal-Free Carbocatalyst for Facilitating Aerobic Oxidation of 5-Hydroxymethylfurfural into 2, 5-Diformylfuran. ACS Catal. 2015, 5, 5636–5646. [Google Scholar] [CrossRef]
- Qin, Y.-Z.; Li, Y.-M.; Zong, M.-H.; Wu, H.; Li, N. Enzyme-catalyzed selective oxidation of 5-hydroxymethylfurfural (HMF) and separation of HMF and 2,5-diformylfuran using deep eutectic solvents. Green Chem. 2015, 17, 3718–3722. [Google Scholar] [CrossRef]
- Dutta, S.; Wu, L.; Mascal, M. Production of 5-(chloromethyl)furan-2-carbonyl chloride and furan-2,5-dicarbonyl chloride from biomass-derived 5-(chloromethyl)furfural (CMF). Green Chem. 2015, 17, 3737–3739. [Google Scholar] [CrossRef]
- Hashemi, M.M.; Beni, Y.A. Copper(I) Chloride/Kieselguhr: A Versatile Catalyst for Oxidation of Alkyl Halides and Alkyl Tosylates to the Carbonyl Compounds. J. Chem. Res. Synop. 1999, 434–435. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Sridhar, M. A bifunctional approach towards the mild oxidation of organic halides: 2-Dimethylamino-N,N-dimethylaniline N-oxide. Tetrahedron Lett. 2000, 41, 5423–5425. [Google Scholar] [CrossRef]
- Badri, R.; Soleymani, M. Selective Oxidation of Benzylic Substrates to Their Corresponding Carbonyl Compounds with 3,6-Bis(Triphenylphosphonium)cyclohexene Peroxodisulfate. Synth. Commun. 2003, 33, 1325–1332. [Google Scholar] [CrossRef]
- Das, S.; Panigrahi, A.K.; Maikap, G.C. NaIO4–DMF: A novel reagent for the oxidation of organic halides to carbonyl compounds. Tetrahedron Lett. 2003, 44, 1375–1377. [Google Scholar] [CrossRef]
- Hu, Y.L.; Liu, Q.F.; Lu, T.T.; Lu, M. Highly efficient oxidation of organic halides to aldehydes and ketones with H5IO6 in ionic liquid [C12mim][FeCl4]. Catal. Commun. 2010, 11, 923–927. [Google Scholar] [CrossRef]
- Liu, Q.; Lu, M.; Yang, F.; Wei, W.; Sun, F.; Yang, Z.; Huang, S. Aerobic Oxidation of Benzylic Halides to Carbonyl Compounds with Molecular Oxygen Catalyzed by TEMPO/KNO2 in Aqueous Media. Synth. Commun. 2010, 40, 1106–1114. [Google Scholar] [CrossRef]
- Khumraksa, B.; Phakhodee, W.; Pattarawarapan, M. Rapid oxidation of organic halides with N-methylmorpholine N-oxide in an ionic liquid under microwave irradiation. Tetrahedron Lett. 2013, 54, 1983–1986. [Google Scholar] [CrossRef]
- Bayat, A.; Shakourian-Fard, M.; Ramezanpour, S.; Hashemi, M.M. A green procedure for direct oxidation of organic halides to aldehydes and ketones catalyzed by a molybdate-based catalyst. New J. Chem. 2015, 39, 3845–3851. [Google Scholar] [CrossRef]
- Carniti, P.; Gervasini, A.; Modica, V.H.; Ravasio, N. Catalytic selective reduction of NO with ethylene over a series of copper catalysts on amorphous silicas. Appl. Catal. B Environ. 2000, 28, 175–185. [Google Scholar] [CrossRef]
- Hu, Y.; Zhi, Z.; Zhao, Q.; Wu, C.; Zhao, P.; Jiang, H.; Jiang, T.; Wang, S. 3D cubic mesoporous silica microsphere as a carrier for poorly soluble drug carvedilol. Microporous Mesoporous Mater. 2012, 147, 94–101. [Google Scholar] [CrossRef]
- Simeonov, S.P.; Coelho, J.A.S.; Afonso, C.A.M. An Integrated Approach for the Production and Isolation of 5-Hydroxymethylfurfural from Carbohydrates. ChemSusChem 2012, 5, 1388–1391. [Google Scholar] [CrossRef] [PubMed]
- Svilen, P.; Simeonov, J.A.S.C.; Afonso, C.A.M. Synthesis of 5-(Hydroxymethyl)furfural (HMF). Org. Synth. 2016, 93, 29–36. [Google Scholar]
- Sanda, K.; Rigal, L.; Gaset, A. Synthèse du 5-bromométhyl-et du 5-chlorométhyl-2-furannecarboxaldéhyde. Carbohydr. Res. 1989, 187, 15–23. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds HMF, CMF, DFF and OBMF are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Promoter | Oxidant (equiv.) | T (°C) | Yield (%) | |||
|---|---|---|---|---|---|---|---|---|
| DFF | CMF | HMF | Total | |||||
| 1 | CH3CN | CuCl | 2 | 82 | 9 | 49 | n.d. | 58 |
| 2 | t-BuOH | CuCl | 2 | 82 | 5 | 47 | 48 | 100 |
| 3 | THF | CuCl | 2 | 82 | 6 | n.d. | 96 | 100 |
| 4 | CH3CN | None | 2 | 82 | n.d. | 100 | n.d. | 100 |
| 5 | CH3CN:H2O 1:1 | CuCl | 2 | 82 | n.d. | n.d. | 100 | 100 |
| 6 | CH3CN | CuCl | 4 | 82 | 37 | 3 | 26 | 66 |
| 7 | CH3CN a | CuCl | 4 | 82 | 25 | 3 | 53 | 81 |
| 8 | CH3CN b | CuCl | 4 | 82 | 34 | 14 | n.d. | 48 |
| 9 | CH3CN | CuCl | 8 | 82 | 37 | 8 | 4 | 49 |
| 10 | CH3CN | CuCl | 4 | 60 | 13 | 2 | 45 | 60 |
| 11 | CH3CN | CuCl | 4 | 110 | 19 | n.d. | n.d. | 19 |
| 12 | CH3CN | Cu(OTf)2 | 4 | 82 | 35 | 5 | n.d. | 40 |
| 13 | CH3CN c | Cu(OTf)2 | 4 | 82 | 32 | 2 | 4 | 38 |
| 14 | CH3CN d | CuCl | 4 | 82 | n.d. | 100 | n.d. | 100 |
| Entry | Time (min) | Temp. (°C) | Oxidant (equiv.) | Promoter (equiv.) | Yield (%) | ||
|---|---|---|---|---|---|---|---|
| DFF | CMF | Total | |||||
| 1 | 360 | 82 | 4 | 0.5 | 49 | 24 | 73 |
| 2 | 120 | 100 | 4 | 0.5 | 39 | 4 | 43 |
| 3 | 60 | 120 | 4 | 0.5 | 39 | 7 | 46 |
| 4 | 5 | 140 | 4 | 0.5 | 44 | 5 | 49 |
| 5 | 15 | 140 | 4 | 0.5 | 46 | 5 | 51 |
| 6 | 30 | 140 | 4 | 0.5 | 47 | 6 | 53 |
| 7 | 60 | 140 | 4 | 0.5 | 49 | 4 | 53 |
| 8 | 1 | 160 | 4 | 0.5 | 47 | 18 | 65 |
| 9 | 5 | 160 | 4 | 0.5 | 51 | 8 | 59 |
| 10 | 5 a | 160 | 4 | 0.5 | 50 | 8 | 58 |
| 11 | 5 | 160 | 4 | 0 | 6 | 59 | 65 |
| 12 | 5 | 160 | 4 | 0.25 | 39 | 8 | 47 |
| 13 | 5 | 160 | 4 | 1 | 37 | 8 | 45 |
| 14 | 5 | 160 | 2 | 0.5 | 40 | n.d. | 40 |
| 15 | 5 | 160 | 0 | 0.5 | dark crude | ||
| 16 | 30 | 160 | 4 | 0.5 | 45 | 4 | 49 |
| 17 | 1 | 170 | 4 | 0.5 | 49 | 3 | 52 |
| Samples | SBET (m2/g) | PD a (nm) | Pore Volume (cm3/g) |
|---|---|---|---|
| SBA-16 | 1128 | 7.90 (2.5) | 0.64 |
| CuSBA-16 | 695 | 7.55 (2.5) | 0.50 |
| MnSBA-16 | 630 | 7.86 (2.5) | 0.39 |
| VSBA-16 | 570 | 7.83 (2.5) | 0.37 |
| Consecutive Reactions | Yield (%) | ||||
|---|---|---|---|---|---|
| DFF | CMF | HMF | OBMF | Total | |
| 1st | 43 (20) | 15 (16) | 40 (43) | 0 (20) | 98 (98) |
| 2nd | 22 | 16 | 20 | 36 | 95 |
| 3rd | 11 | 5 | 20 | 56 | 92 |
| 4th | 17 | n.d. | n.d. | 77 | 94 |
| Entry | Catalyst | Flow Rate (mL/min) | Residence Time (min) | Yield (%) | ||||
|---|---|---|---|---|---|---|---|---|
| DFF | CMF | HMF | OBMF | Total | ||||
| 1 | CuSO4·Si 10% a,b | 0.5 | 5.3 | 40 | 16 | 42 | n.d. | 98 |
| 2 | CuSO4·Si 10% dry a,c | 0.5 | 5.3 | 36 | 26 | 36 | n.d. | 98 |
| 3 | CuSO4·Si 10% dry c | 0.5 | 5.3 | 40 | 20 | 25 | 10 | 94 |
| 4 | CuSO4 d | 0.5 | 5.3 | 17 | n.d. | 71 | n.d. | 88 |
| 5 | CuSO4·Si 5% c | 0.5 | 5.3 | 40 | 10 | 32 | n.d. | 82 |
| 6 | Cu2O/silica (1:1) e | 0.5 | 5.3 | 37 | n.d. | n.d. | n.d. | 37 |
| 7 | Cu-SBA16 a,f | 0.5 | 5.3 | 24 | n.d. | 58 | n.d. | 82 |
| 8 | Cu/SiO2 (0.5 mol equiv.) c | 0.5 | 5.3 | 29 | n.d. | 49 | n.d. | 70 |
| 9 | Cu/SiO2 (0.5 mol equiv.) c | 1.0 | 2.7 | 42 | n.d. | 13 | 18 | 73 |
| 10 | Cu/SiO2 (1 mol equiv.) c | 1.0 | 2.7 | 54 | n.d. | 19 | n.d. | 72 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vicente, A.I.; Coelho, J.A.S.; Simeonov, S.P.; Lazarova, H.I.; Popova, M.D.; Afonso, C.A.M. Oxidation of 5-Chloromethylfurfural (CMF) to 2,5-Diformylfuran (DFF). Molecules 2017, 22, 329. https://doi.org/10.3390/molecules22020329
Vicente AI, Coelho JAS, Simeonov SP, Lazarova HI, Popova MD, Afonso CAM. Oxidation of 5-Chloromethylfurfural (CMF) to 2,5-Diformylfuran (DFF). Molecules. 2017; 22(2):329. https://doi.org/10.3390/molecules22020329
Chicago/Turabian StyleVicente, Ana I., Jaime A. S. Coelho, Svilen P. Simeonov, Hristina I. Lazarova, Margarita D. Popova, and Carlos A. M. Afonso. 2017. "Oxidation of 5-Chloromethylfurfural (CMF) to 2,5-Diformylfuran (DFF)" Molecules 22, no. 2: 329. https://doi.org/10.3390/molecules22020329
APA StyleVicente, A. I., Coelho, J. A. S., Simeonov, S. P., Lazarova, H. I., Popova, M. D., & Afonso, C. A. M. (2017). Oxidation of 5-Chloromethylfurfural (CMF) to 2,5-Diformylfuran (DFF). Molecules, 22(2), 329. https://doi.org/10.3390/molecules22020329