
Ginkgolic Acid C 17:1, Derived from Ginkgo biloba Leaves, Suppresses Constitutive and Inducible STAT3 Activation through Induction of PTEN and SHP-1 Tyrosine Phosphatase


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
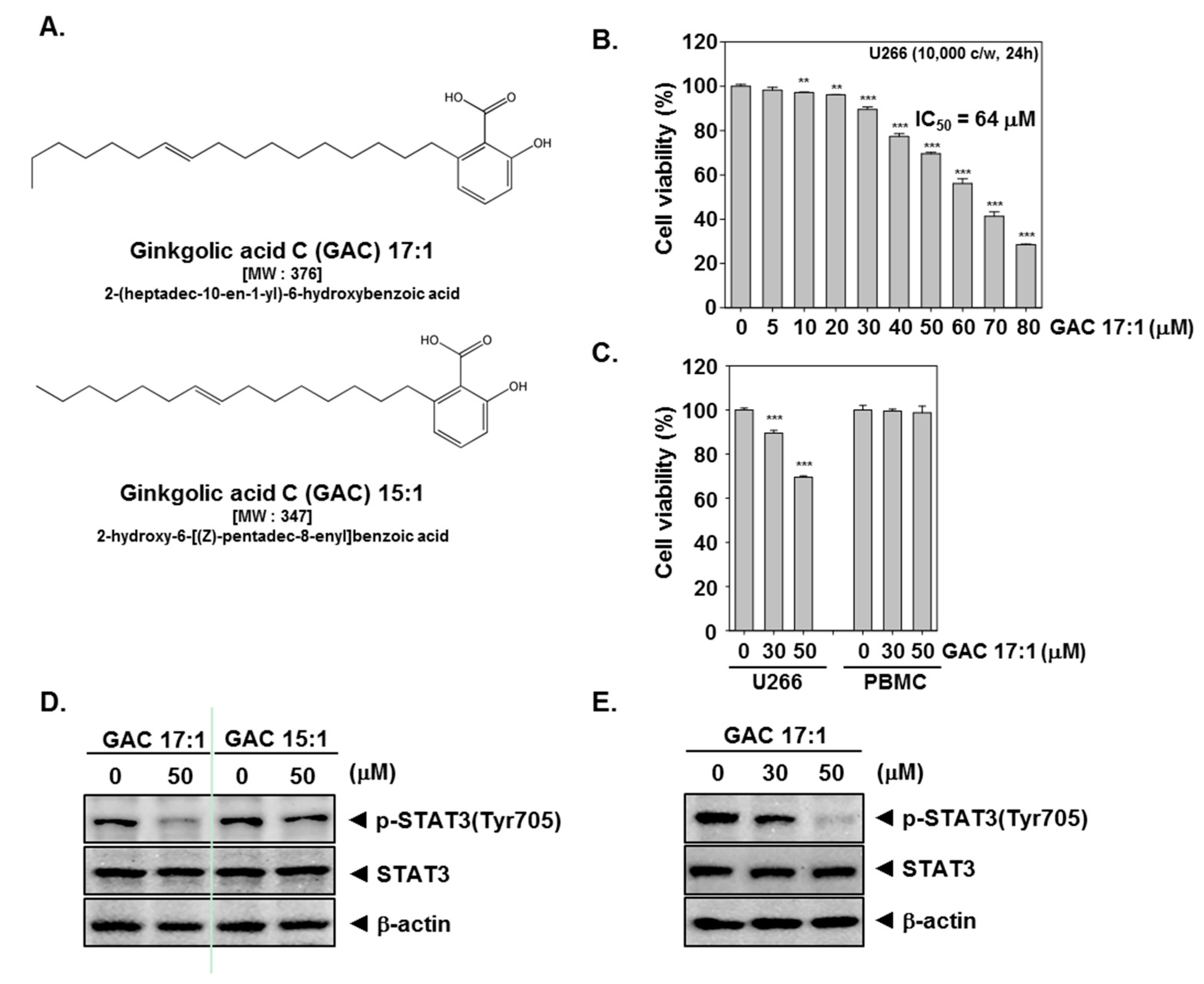
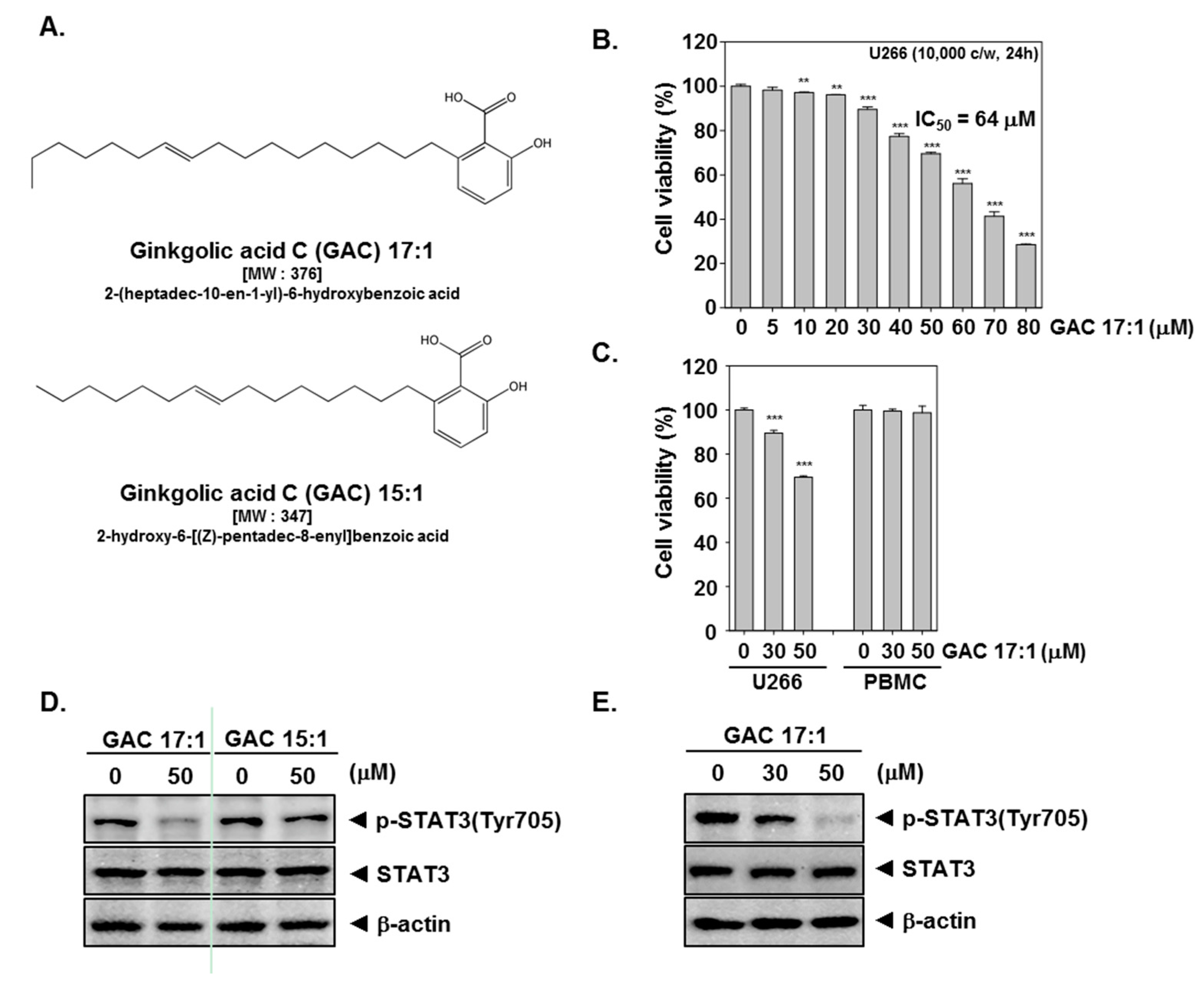
2.1. GAC 17:1 Selectively Exerted Cytotoxic Effects against Multiple Myeloma Cells
2.2. GAC 17:1 Suppressed Constitutive STAT3 Phosphorylation in U266 Cells
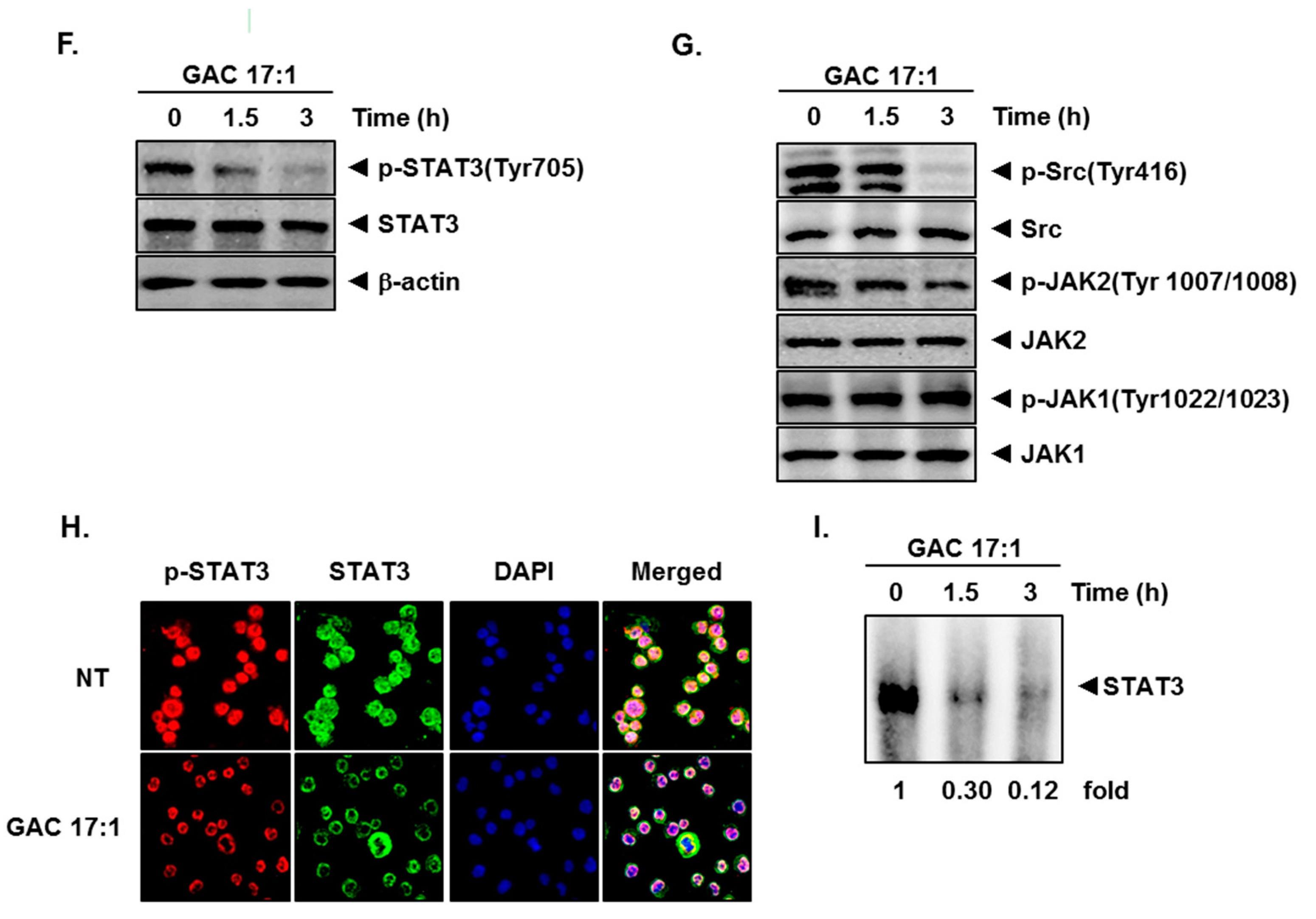
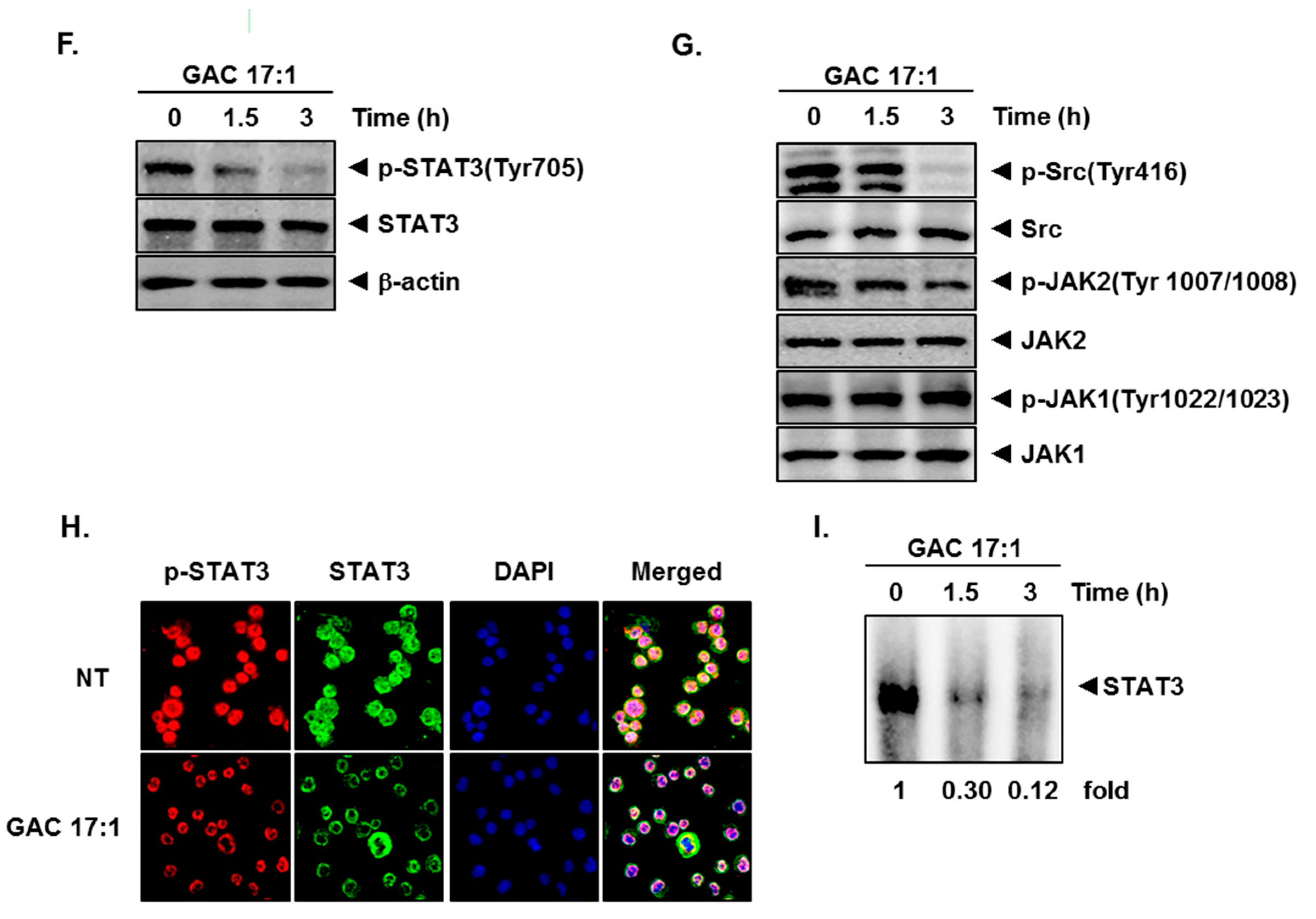
2.3. GAC 17:1 Attenuated Phosphorylation of Upstream Kinases in U266 Cells
2.4. GAC 17:1 Repressed Nuclear Translocation of STAT3 in U266 Cells
2.5. GAC 17:1 Inhibited Binding of STAT3 to the DNA
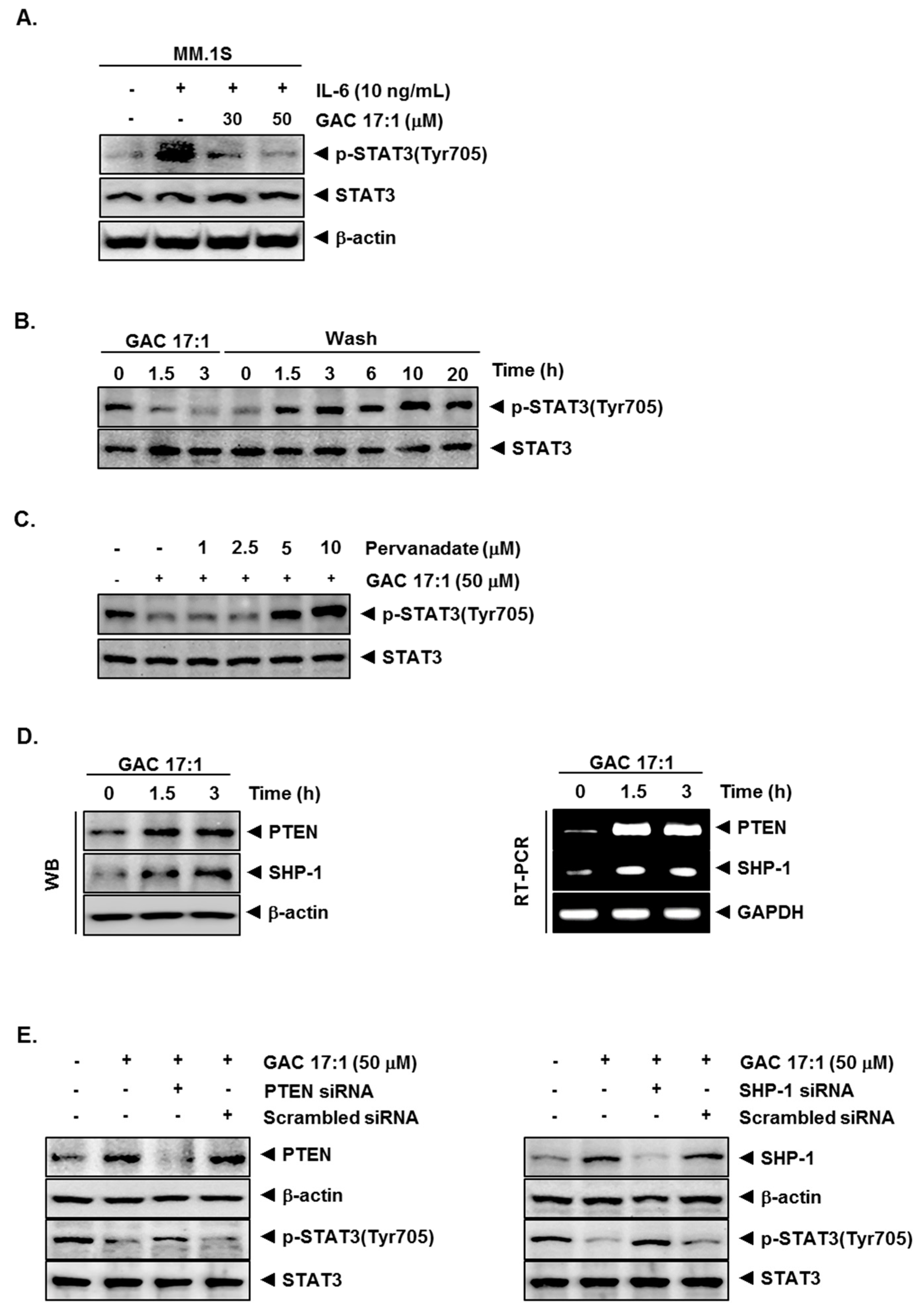
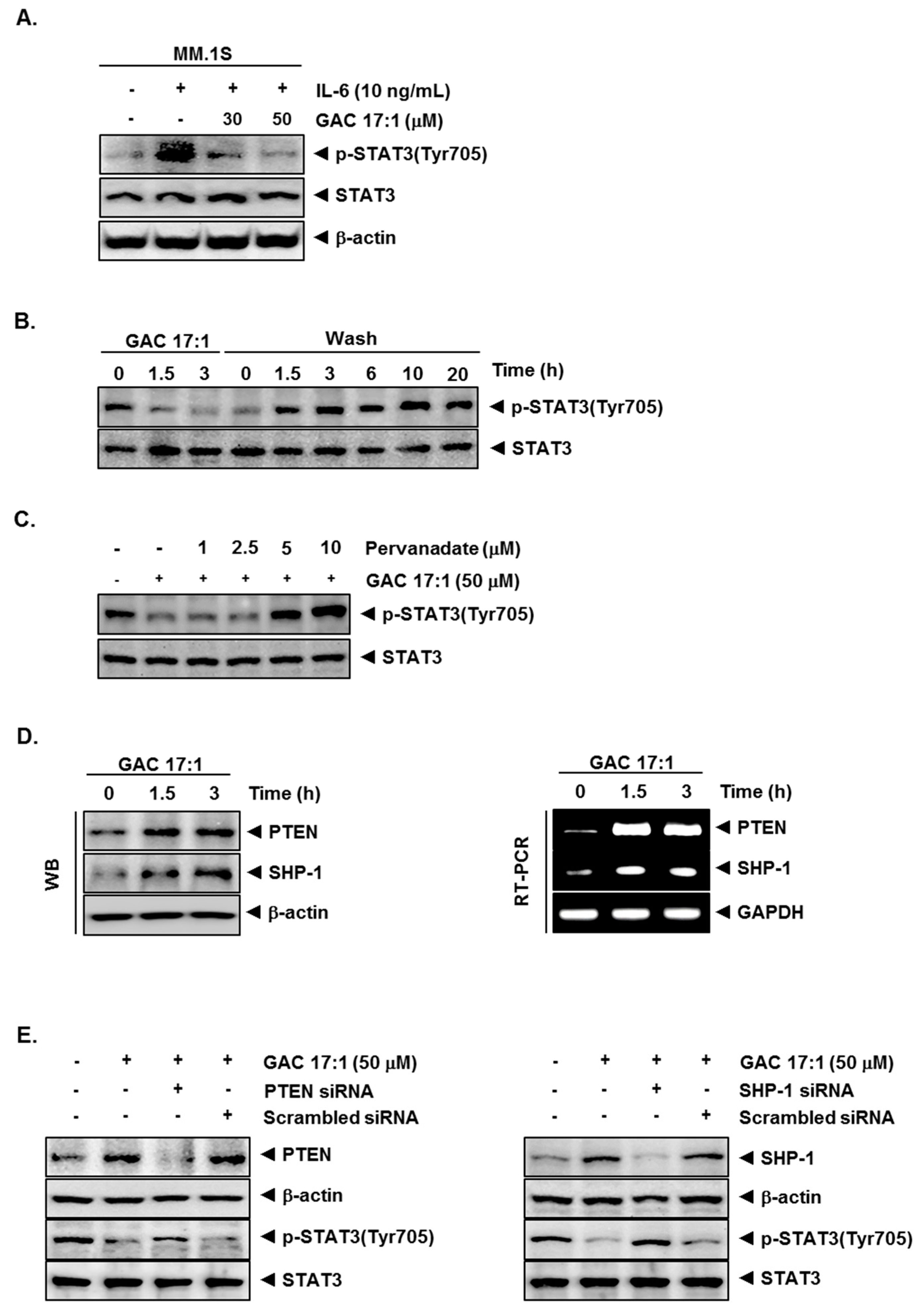
2.6. GAC 17:1 Decreased IL-6-Induced Phosphorylation of STAT3 in U266 Cells
2.7. Inhibition of STAT3 Phosphorylation by GAC 17:1 Is Reversible
2.8. Tyrosine Phosphatase Inhibitor Blocked the Suppression of STAT3 Phosphorylation by GAC 17:1
2.9. GAC 17:1 Induced the Expression of PTEN and SHP-1 in Protein and mRNA Level
2.10. Silencing of SHP-1 and PTEN Reversed the Effect of GAC 17:1 on STAT3 Activation
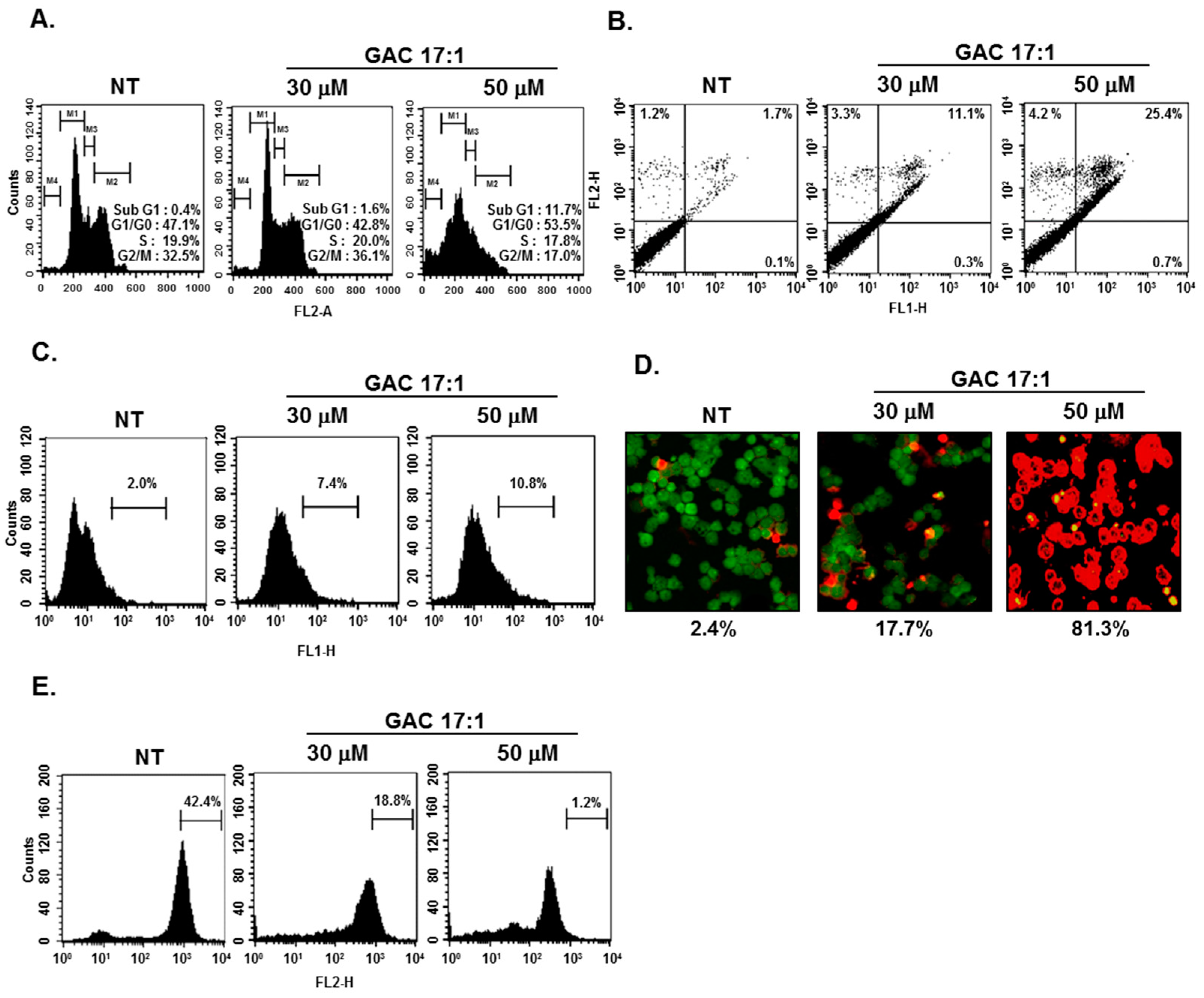
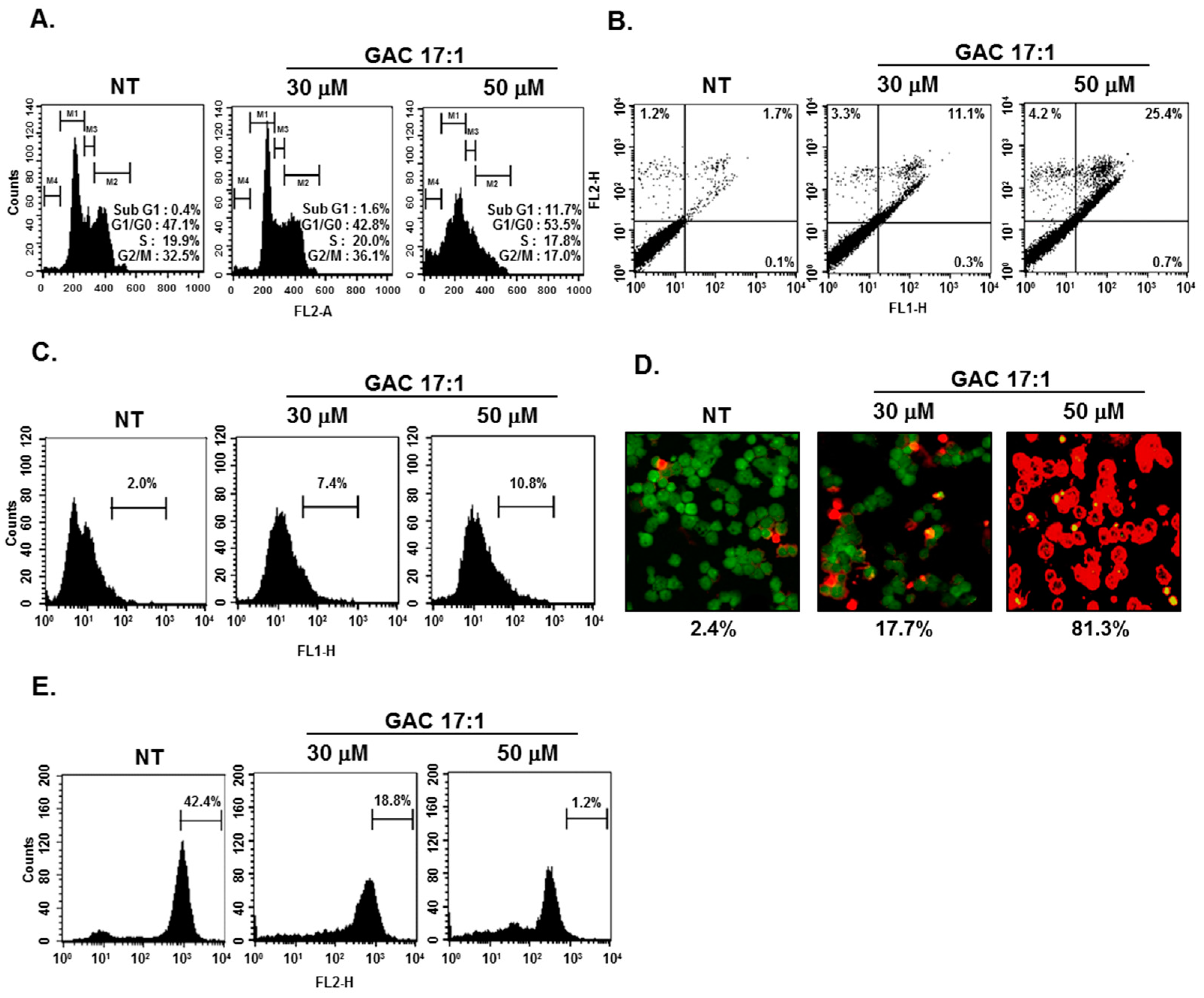
2.11. GAC 17:1 Caused the Accumulation of the Cells in the Sub-G1 Phase of the Cell Cycle, and Increased Annexin V Positive Cells
2.12. GAC 17:1 Elicited Apoptosis and Caused Loss of Mitochondrial Membrane Potential in U266 Cells
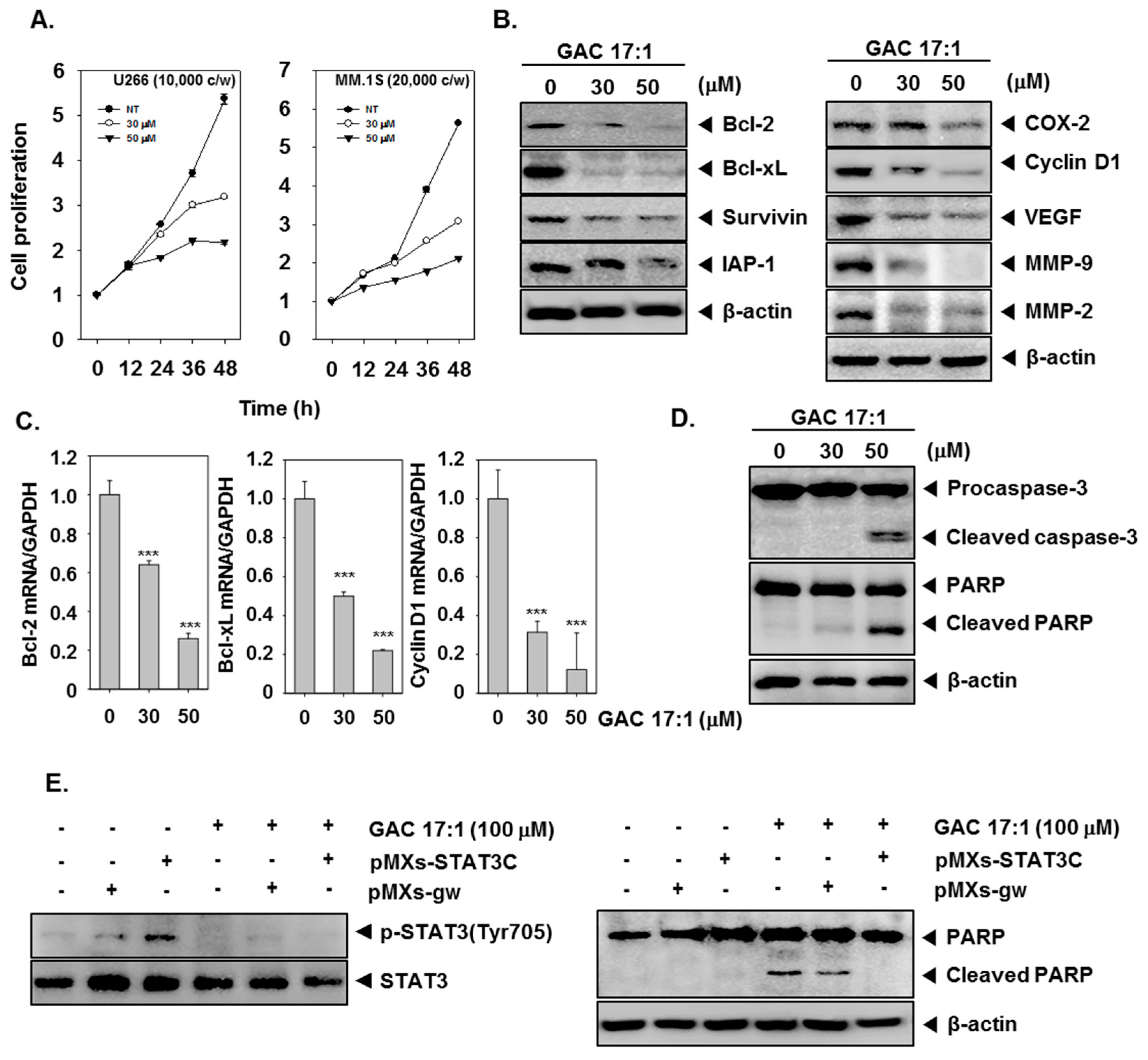
2.13. GAC 17:1 Suppressed the Proliferation of Multiple Myeloma Cells
2.14. GAC 17:1 Down-Regulated the Expression of STAT3-Regulated Gene Products
2.15. GAC 17:1 Induced the Cleavage of Caspase-3 and PARP
2.16. GAC 17:1 Inhibited pMXs-STAT3C-Induced Phosphorylation of STAT3 in MEF Cells
2.17. Activation of STAT3 Abolished the Apoptotic Effect of GAC 17:1 in MEF Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Isolation of Human Peripheral Blood Mononuclear Cells (PBMCs)
4.4. Western Blot Analysis
4.5. Immunocytochemistry for STAT3 Localization
4.6. EMSA for STAT3-DNA Binding
4.7. RNA Analysis and Reverse Transcription Polymerase Chain Reaction
4.8. Electroporation-Mediated Transfection in U266 and MEF Cells
4.9. Cell Cycle Analysis
4.10. Annexin V assay
4.11. TdT-Mediated dUTP Nick end Labeling (TUNEL) Assay
4.12. Mitochondrial Membrane Potential
4.13. Live/Dead Assay
4.14. MTT Assay
4.15. Real-Time Quantitative PCR
4.16. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.C.; Carrasco, R.D. Pathogenesis of myeloma. Annu. Rev. Pathol. 2011, 6, 249–274. [Google Scholar] [CrossRef] [PubMed]
- Tarkun, P.; Atalay, F.; Atesoglu, E.B.; Mehtap, O.; Simsek, M.; Terzi, E.; Geduk, A.; Balli, F.; Batman, A.; Baydemir, C.; et al. Treatment of patients with multiple myeloma over 65 years: More tolerability or better response? Eur. J. Haematol. 2015, 94, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Manu, K.A.; Shanmugam, M.K.; Ong, T.H.; Subramaniam, A.; Siveen, K.S.; Perumal, E.; Samy, R.P.; Bist, P.; Lim, L.H.; Kumar, A.P.; et al. Emodin suppresses migration and invasion through the modulation of CXCR4 expression in an orthotopic model of human hepatocellular carcinoma. PLoS ONE 2013, 8, e57015. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Bharti, A.C.; Donato, N.; Aggarwal, B.B. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J. Immunol. 2003, 171, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Crowe, P.J.; Goldstein, D.; Yang, J.L. STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers (review). Int. J. Oncol. 2012, 41, 1181–1191. [Google Scholar] [PubMed]
- Aggarwal, B.B.; Sethi, G.; Ahn, K.S.; Sandur, S.K.; Pandey, M.K.; Kunnumakkara, A.B.; Sung, B.; Ichikawa, H. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: Modern target but ancient solution. Ann. N. Y. Acad. Sci. 2006, 1091, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R.; Hurt, E.M.; Farrar, W.L. The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 2005, 41, 2502–2512. [Google Scholar] [CrossRef] [PubMed]
- Mali, S.B. Review of STAT3 (Signal Transducers and Activators of Transcription) in head and neck cancer. Oral Oncol. 2015, 51, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Steinberg, B.M. PTEN is a negative regulator of STAT3 activation in human papillomavirus-infected cells. J. Gen. Virol. 2002, 83, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Amin, H.M.; Franko, B.; Frantz, C.; Shi, X.; Lai, R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood 2006, 108, 2796–2803. [Google Scholar] [CrossRef] [PubMed]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Ahn, K.S.; Kim, C.; Siveen, K.S.; Ong, T.H.; Shanmugam, M.K.; Li, F.; Shi, J.; Kumar, A.P.; Wang, L.Z.; et al. Ascochlorin, an isoprenoid antibiotic inhibits growth and invasion of hepatocellular carcinoma by targeting STAT3 signaling cascade through the induction of PIAS3. Mol. Oncol. 2015, 9, 818–833. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Chatterjee, S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Wong, K.F.; Kumar, A.P.; Senapati, P.; Behera, A.K.; Hui, K.M.; et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer 2014, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, A.; Shanmugam, M.K.; Ong, T.H.; Li, F.; Perumal, E.; Chen, L.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br. J. Pharmacol. 2013, 170, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Hay, H.S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P.; et al. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-kappaB and STAT3 regulated gene products in multiple myeloma cells. Br. J. Pharmacol. 2011, 164, 1506–1521. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Manu, K.A.; Shanmugam, M.K.; Loo, S.Y.; Kumar, A.P.; Sethi, G. gamma-Tocotrienol is a novel inhibitor of constitutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma: Potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. Br. J. Pharmacol. 2011, 163, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Ahlemeyer, B.; Krieglstein, J. Pharmacological studies supporting the therapeutic use of Ginkgo biloba extract for Alzheimer’s disease. Pharmacopsychiatry 2003, 36 (Suppl. 1), S8–S14. [Google Scholar] [PubMed]
- Kuo, I.; Chen, J.; Chang, T.K. Effect of Ginkgo biloba extract on rat hepatic microsomal CYP1A activity: Role of ginkgolides, bilobalide, and flavonols. Can. J. Physiol. Pharmacol. 2004, 82, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Han, Y. Ginkgo terpene component has an anti-inflammatory effect on Candida albicans-caused arthritic inflammation. Int. Immunopharmacol. 2005, 5, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, S.; Park, Y. Multifaceted therapeutic benefits of Ginkgo biloba L.: Chemistry, efficacy, safety, and uses. J. Food Sci. 2008, 73, R14–R19. [Google Scholar] [CrossRef] [PubMed]
- Mahady, G.B. Ginkgo biloba for the prevention and treatment of cardiovascular disease: A review of the literature. J. Cardiovasc. Nurs. 2002, 16, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.M.; Wang, Y.F.; Li, Y.Y.; Ma, H.L. Thermal stability of ginkgolic acids from Ginkgo biloba and the effects of ginkgol C17:1 on the apoptosis and migration of SMMC7721 cells. Fitoterapia 2014, 98, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yao, Q.Q.; Xu, S.Y.; Hu, H.H.; Shen, Q.; Tian, Y.; Pan, L.Y.; Zhou, H.; Jiang, H.D.; Lu, C.; et al. Cyclosporin A affects the bioavailability of ginkgolic acids via inhibition of P-gp and BCRP. Eur. J. Pharm. Biopharm. 2014, 88, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Duan, W.; Han, S.; Lei, J.; Xu, Q.; Chen, X.; Jiang, Z.; Nan, L.; Li, J.; Chen, K.; et al. Ginkgolic acid suppresses the develol.pment of pancreatic cancer by inhibiting pathways driving lipogenesis. Oncotarget 2015, 6, 20993–21003. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Hwang, I.H.; Hong, C.E.; Lyu, S.Y.; Na, M. Inhibition of fatty acid synthase by ginkgolic acids from the leaves of Ginkgo biloba and their cytotoxic activity. J. Enzym. Inhib. Med. Chem. 2013, 28, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, X.; Du, W.; Feng, Y.; Kong, X.; Li, Y.; Xiao, L.; Zhang, P. Antitumor effects of ginkgolic acid in human cancer cell occur via cell cycle arrest and decrease the Bcl-2/Bax ratio to induce apoptosis. Chemotherapy 2010, 56, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.C.; Du, W.; Wen, Z.; Li, J.Y.; Zhang, P. [Effects of natural plant ginkgolic acids on the apoptosis of human Hep-2 cancer cells]. Sichuan Da Xue Xue Bao Yi Xue Ban 2009, 40, 459–461. [Google Scholar] [PubMed]
- Fukuda, I.; Ito, A.; Hirai, G.; Nishimura, S.; Kawasaki, H.; Saitoh, H.; Kimura, K.; Sodeoka, M.; Yoshida, M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009, 16, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Hirohama, M.; Kumar, A.; Fukuda, I.; Matsuoka, S.; Igarashi, Y.; Saitoh, H.; Takagi, M.; Shin-ya, K.; Honda, K.; Kondoh, Y.; et al. Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS Chem. Biol. 2013, 8, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Won, C.; Lee, Y.H.; Choi, J.S.; Noh, K.H.; Han, S.; Lee, H.; Lee, C.S.; Lee, D.S.; Ye, S.K.; et al. Sophoraflavanone G induces apoptosis of human cancer cells by targeting upstream signals of STATs. Biochem. Pharmacol. 2013, 86, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef] [PubMed]
- Kamran, M.Z.; Patil, P.; Gude, R.P. Role of STAT3 in cancer metastasis and translational advances. BioMed Res. Int. 2013, 2013, 421821. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.L. On the role of tyrosine phosphatases as negative regulators of STAT signaling in breast cancers: new findings and future perspectives. Breast Cancer Res. 2013, 15, 312. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Tolentino, J.H.; Hazlehurst, L.A. Role of STAT3 in Transformation and Drug Resistance in CML. Front. Oncol. 2012, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.Y.; Kim, H.J.; Kim, S.M.; Park, K.R.; Park, S.Y.; Kim, S.W.; Nam, D.; Jang, H.J.; Lee, S.G.; Ahn, K.S.; et al. Embelin suppresses STAT3 signaling, proliferation, and survival of multiple myeloma via the protein tyrosine phosphatase PTEN. Cancer Lett. 2011, 308, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Park, K.R.; Yun, H.M.; Quang, T.H.; Oh, H.; Lee, D.S.; Auh, Q.S.; Kim, E.C. 4-Methoxydalbergione suppresses growth and induces apoptosis in human osteosarcoma cells in vitro and in vivo xenograft model through down-regulation of the JAK2/STAT3 pathway. Oncotarget 2016, 7, 6960–6971. [Google Scholar] [PubMed]
- Hadisaputri, Y.E.; Miyazaki, T.; Suzuki, S.; Kubo, N.; Zuhrotun, A.; Yokobori, T.; Abdulah, R.; Yazawa, S.; Kuwano, H. Molecular characterization of antitumor effects of the rhizome extract from Curcuma zedoaria on human esophageal carcinoma cells. Int. J. Oncol. 2015, 47, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Kim, C.; Lee, J.H.; Nam, D.; Lee, J.; Lee, S.G.; Chung, W.S.; Jang, H.J.; Kim, S.H.; Ahn, K.S. Cinobufagin exerts anti-proliferative and pro-apoptotic effects through the modulation ROS-mediated MAPKs signaling pathway. Immunopharmacol. Immunotoxicol. 2015, 37, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Vogel, T.P.; Milner, J.D.; Cooper, M.A. The Ying and Yang of STAT3 in Human Disease. J. Clin. Immunol. 2015, 35, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.M.; Putoczki, T.L.; Ernst, M. STAT3-Activating Cytokines: A Therapeutic Opportunity for Inflammatory Bowel Disease? J. Interferon Cytokine Res. 2015, 35, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Haghikia, A.; Ricke-Hoch, M.; Stapel, B.; Gorst, I.; Hilfiker-Kleiner, D. STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc. Res. 2014, 102, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Sabri, N.; Li, J.; Li, W.X. Role of STAT3 in lung cancer. JAKSTAT 2014, 3, e999503. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, O.; Nepstad, I.; Hauge, M.; Hatfield, K.J.; Reikvam, H. STAT3 as a possible therapeutic target in human malignancies: Lessons from acute myeloid leukemia. Expert Rev. Hematol. 2015, 8, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Harada, D.; Takigawa, N.; Kiura, K. The Role of STAT3 in Non-Small Cell Lung Cancer. Cancers (Basel) 2014, 6, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ren, Y.; Liu, A.; Han, L.; Zhang, K.; Li, S.; Li, P.; Li, P.; Kang, C.; Wang, X.; Zhang, L. STAT3 inhibitor WP1066 attenuates miRNA-21 to suppress human oral squamous cell carcinoma growth in vitro and in vivo. Oncol. Rep. 2014, 31, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.C.; Shiau, C.W.; Tai, W.T.; Hung, M.H.; Chu, P.Y.; Hsieh, F.S.; Lin, H.; Yu, H.C.; Chen, K.F. SHP-1 is a negative regulator of epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene 2015, 34, 5252–5263. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Chen, G.; Zhang, J.; Li, Z.; Lu, W.; Liu, M.; Pang, X. Small molecule 1′-acetoxychavicol acetate suppresses breast tumor metastasis by regulating the SHP-1/STAT3/MMPs signaling pathway. Breast Cancer Res. Treat. 2014, 148, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, H.A.; Mat Jusoh, S.A.; Hassan, R.; Johan, M.F. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer 2015, 15, 869. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Lee, J.H.; Ko, J.H.; Lee, H.; Nam, D.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Lee, J.; Kim, S.H.; Shim, B.S.; et al. Ginkgetin Blocks Constitutive STAT3 Activation and Induces Apoptosis through Induction of SHP-1 and PTEN Tyrosine Phosphatases. Phytother Res. 2016, 30, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Lee, J.H.; Sethi, G.; Kim, C.; Baek, S.H.; Nam, D.; Chung, W.S.; Kim, S.H.; Shim, B.S.; Ahn, K.S. Bergamottin, a natural furanocoumarin obtained from grapefruit juice induces chemosensitization and apoptosis through the inhibition of STAT3 signaling pathway in tumor cells. Cancer Lett. 2014, 354, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, S.H.; Lee, J.H.; Kim, C.; Ko, J.-H.; Ryu, S.-H.; Lee, S.-G.; Yang, W.M.; Um, J.-Y.; Chinnathambi, A.; Alharbi, S.A.; et al. Ginkgolic Acid C 17:1, Derived from Ginkgo biloba Leaves, Suppresses Constitutive and Inducible STAT3 Activation through Induction of PTEN and SHP-1 Tyrosine Phosphatase. Molecules 2017, 22, 276. https://doi.org/10.3390/molecules22020276
Baek SH, Lee JH, Kim C, Ko J-H, Ryu S-H, Lee S-G, Yang WM, Um J-Y, Chinnathambi A, Alharbi SA, et al. Ginkgolic Acid C 17:1, Derived from Ginkgo biloba Leaves, Suppresses Constitutive and Inducible STAT3 Activation through Induction of PTEN and SHP-1 Tyrosine Phosphatase. Molecules. 2017; 22(2):276. https://doi.org/10.3390/molecules22020276
Chicago/Turabian StyleBaek, Seung Ho, Jong Hyun Lee, Chulwon Kim, Jeong-Hyeon Ko, Seung-Hee Ryu, Seok-Geun Lee, Woong Mo Yang, Jae-Young Um, Arunachalam Chinnathambi, Sulaiman Ali Alharbi, and et al. 2017. "Ginkgolic Acid C 17:1, Derived from Ginkgo biloba Leaves, Suppresses Constitutive and Inducible STAT3 Activation through Induction of PTEN and SHP-1 Tyrosine Phosphatase" Molecules 22, no. 2: 276. https://doi.org/10.3390/molecules22020276
APA StyleBaek, S. H., Lee, J. H., Kim, C., Ko, J.-H., Ryu, S.-H., Lee, S.-G., Yang, W. M., Um, J.-Y., Chinnathambi, A., Alharbi, S. A., Sethi, G., & Ahn, K. S. (2017). Ginkgolic Acid C 17:1, Derived from Ginkgo biloba Leaves, Suppresses Constitutive and Inducible STAT3 Activation through Induction of PTEN and SHP-1 Tyrosine Phosphatase. Molecules, 22(2), 276. https://doi.org/10.3390/molecules22020276