
Structure-Antioxidative and Anti-Inflammatory Activity Relationships of Purpurin and Related Anthraquinones in Chemical and Cell Assays


Abstract
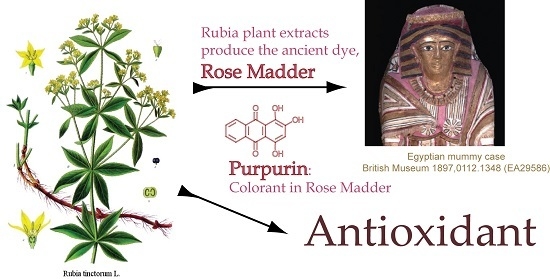
:
1. Introduction
2. Results and Discussion
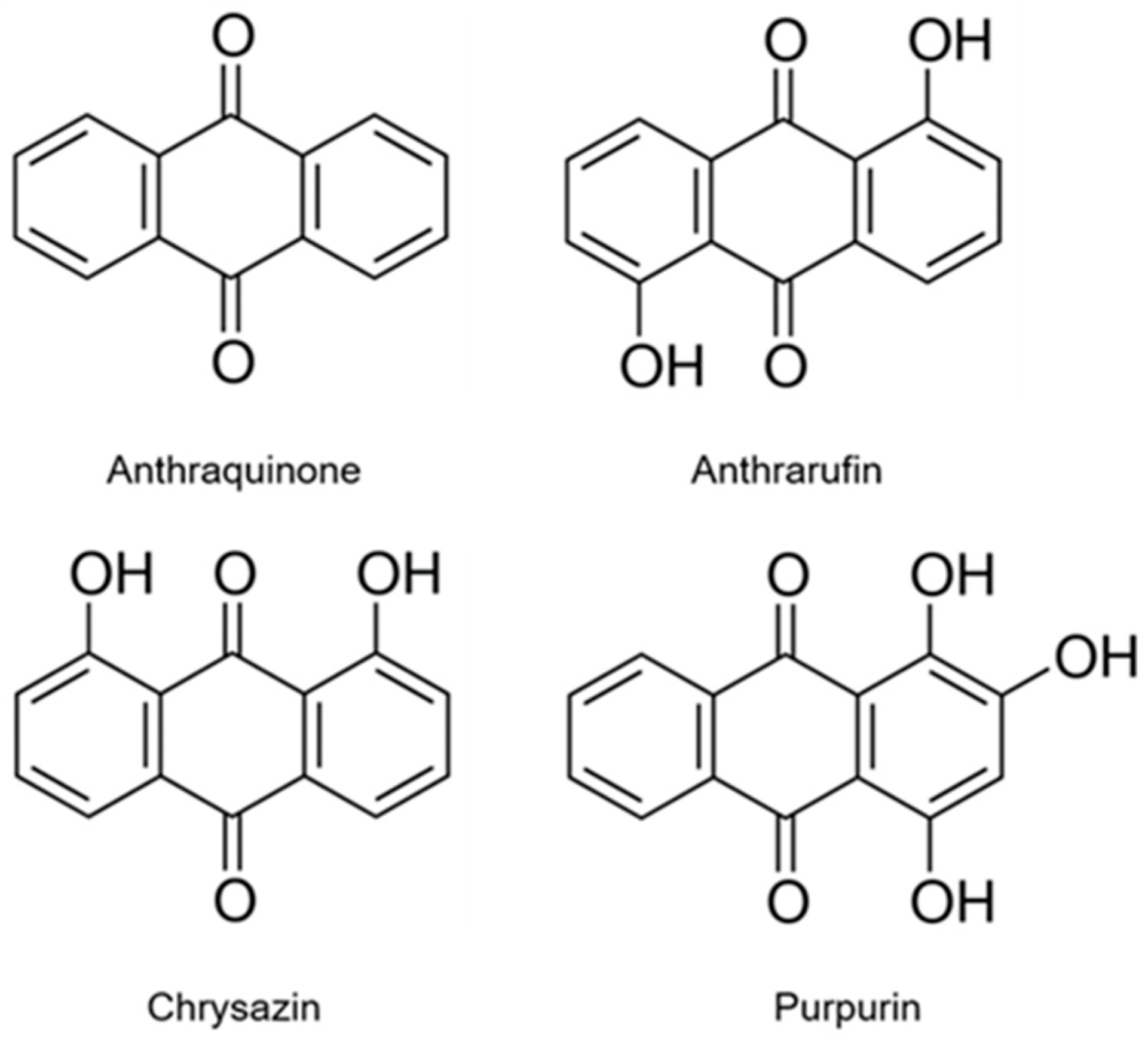
2.1. Antioxidative Chemical Assays
2.1.1. DPPH Radical Scavenging Activity
2.1.2. Inhibition of Lipid Peroxidation
2.1.3. ABTS Radical Cation Scavenging Activity
2.1.4. Hydrogen Peroxide Scavenging Activity
2.1.5. Reducing Power Measured by Ferricyanide Assay
2.2. Antioxidative and Anti-Inflammatory Cell Assays
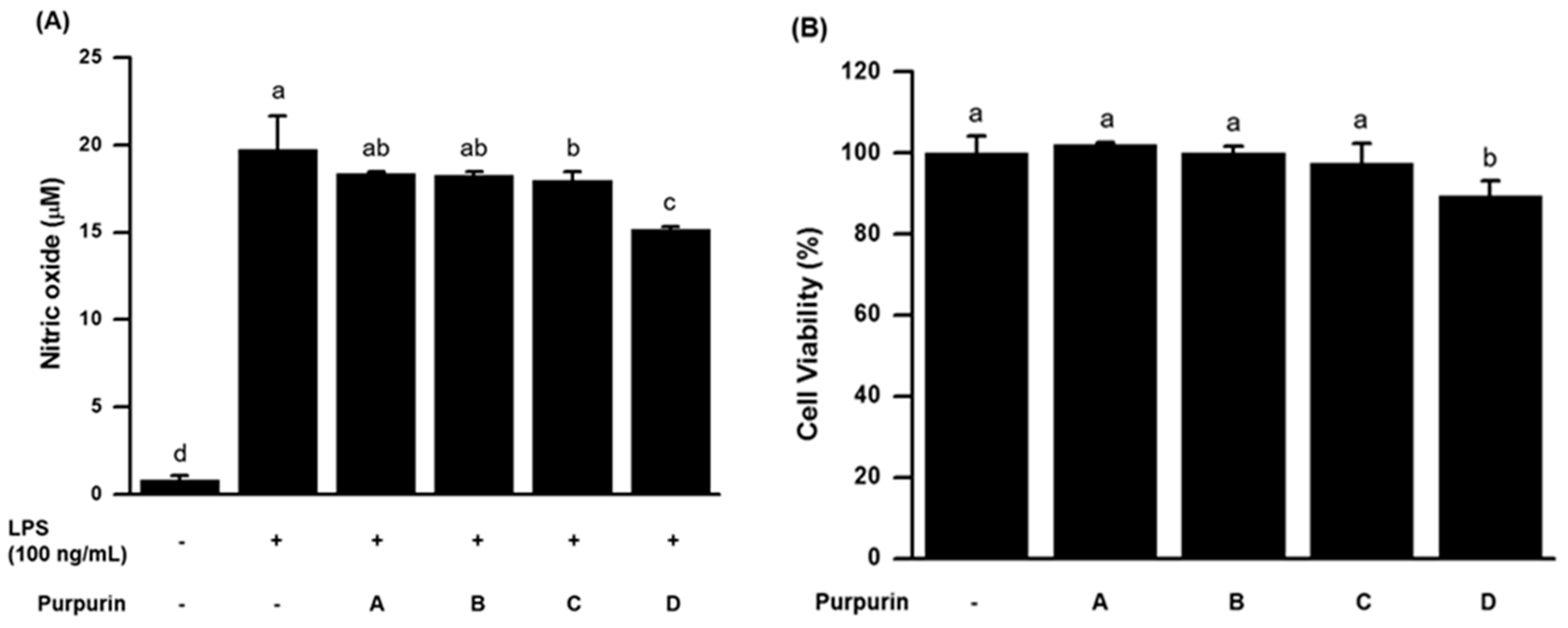
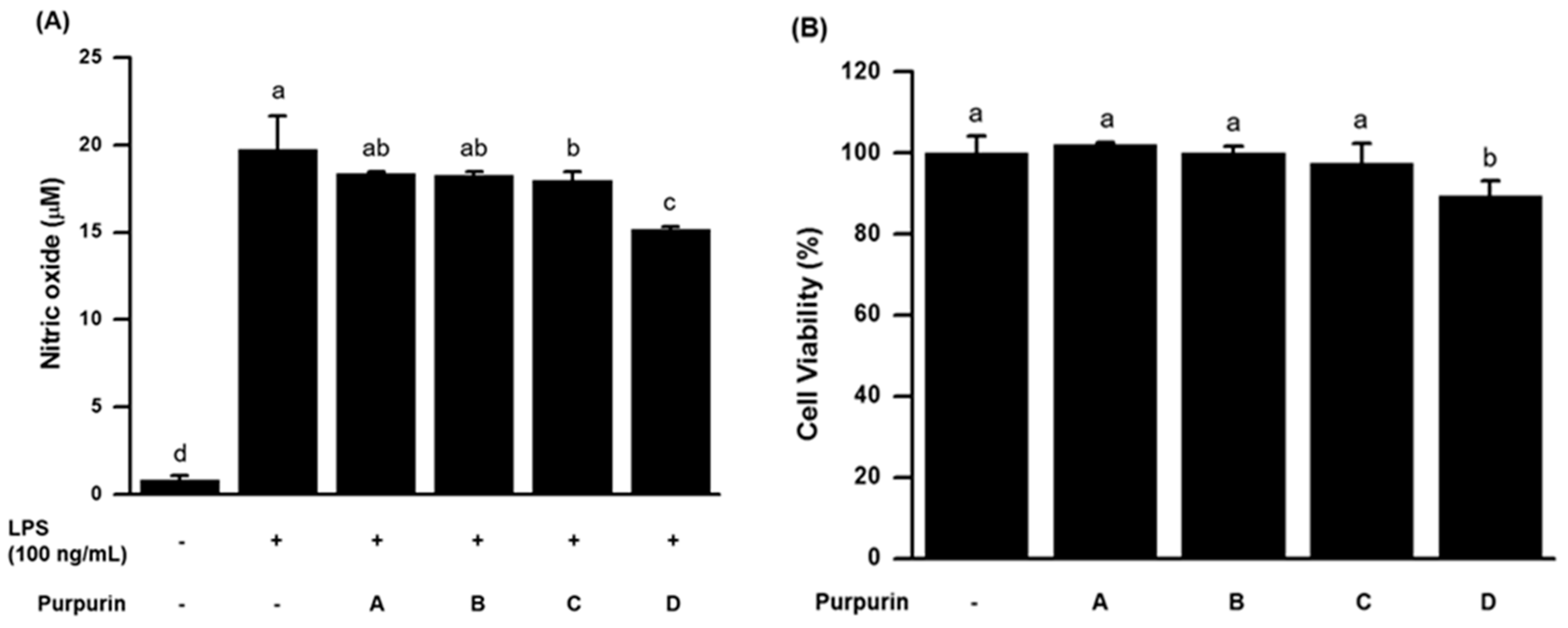
2.2.1. Effects on NO Production and Cell Viability in Murine RAW 264.7 Macrophage Cells
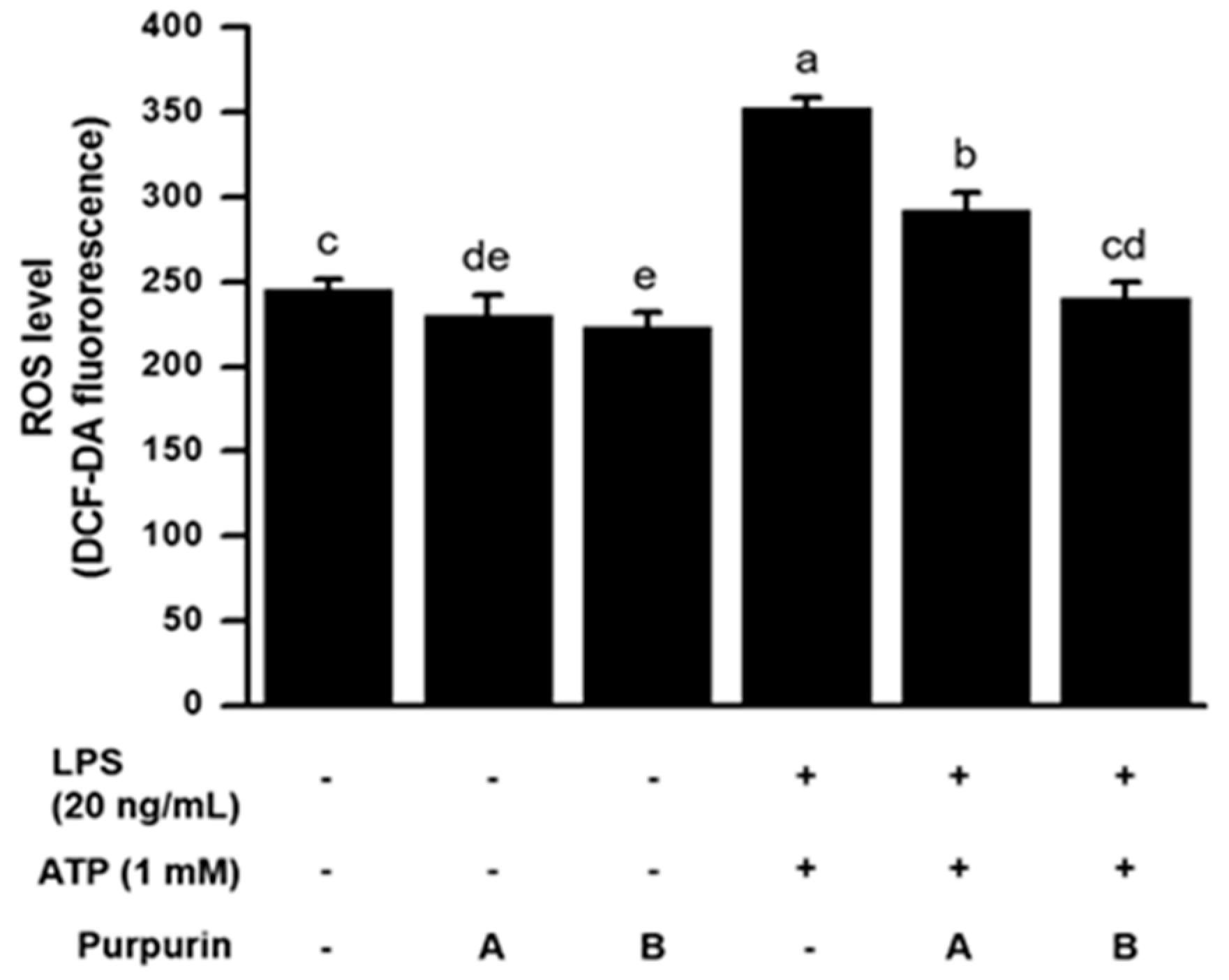
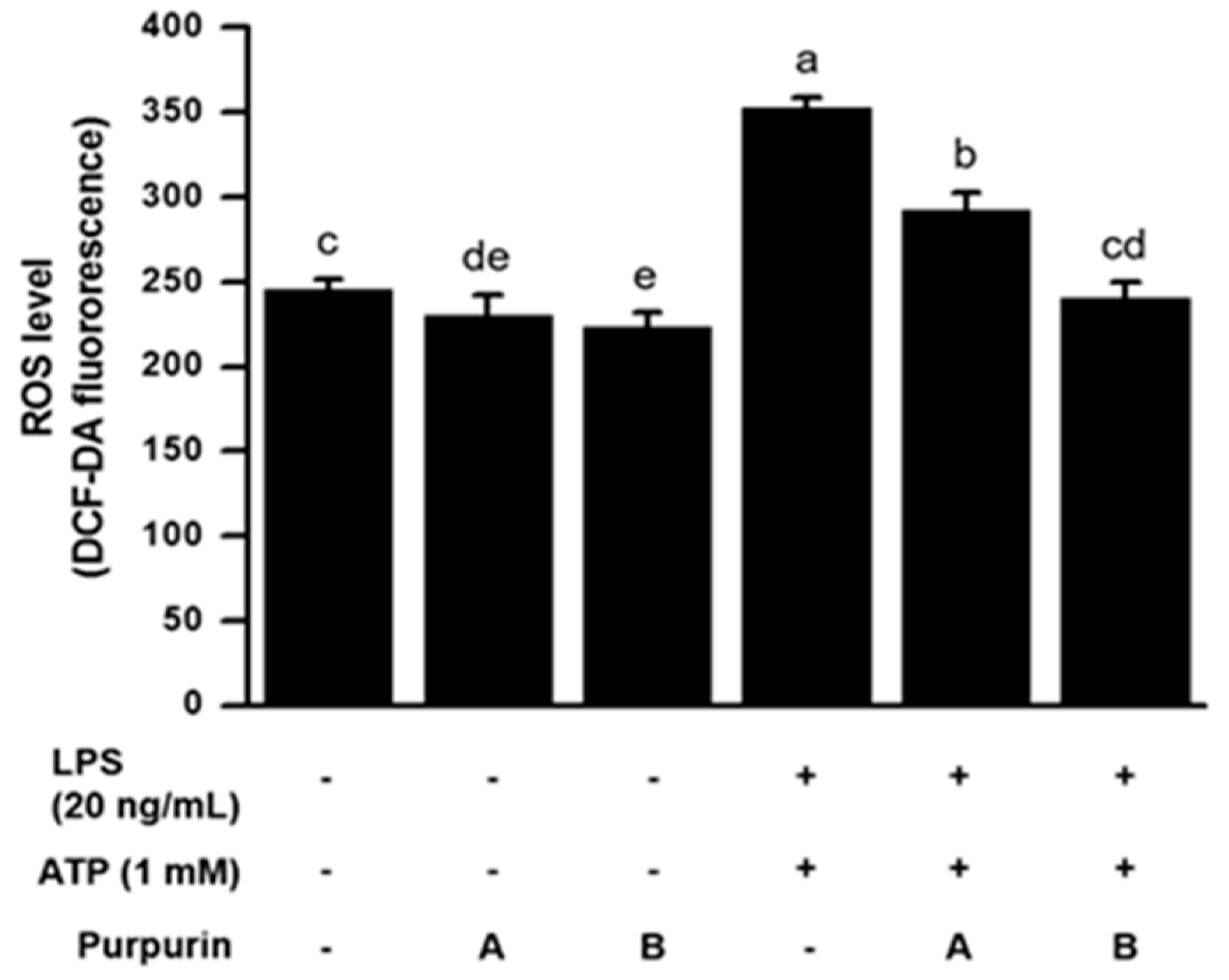
2.2.2. Suppression of Cellular Oxidative Stress
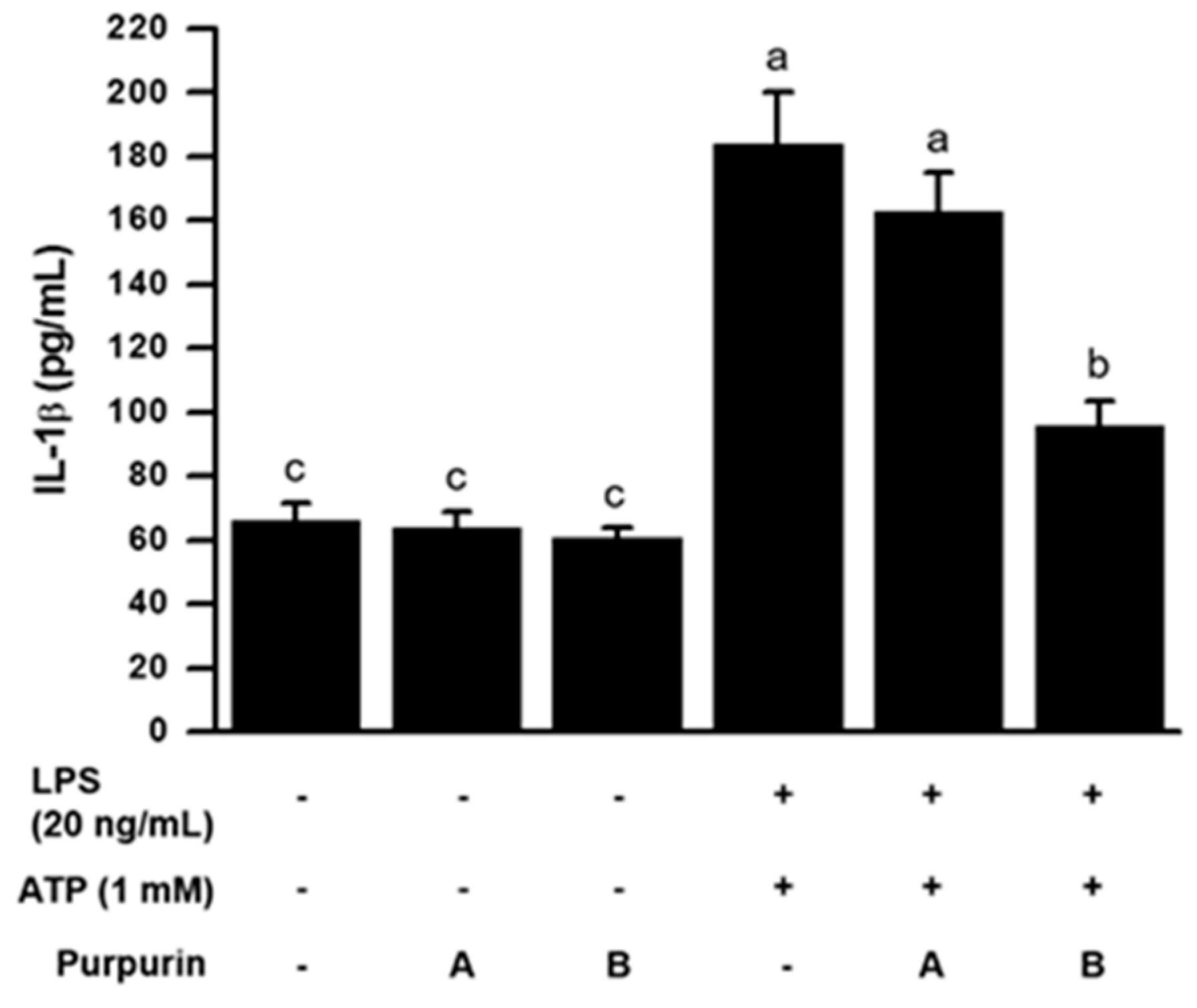
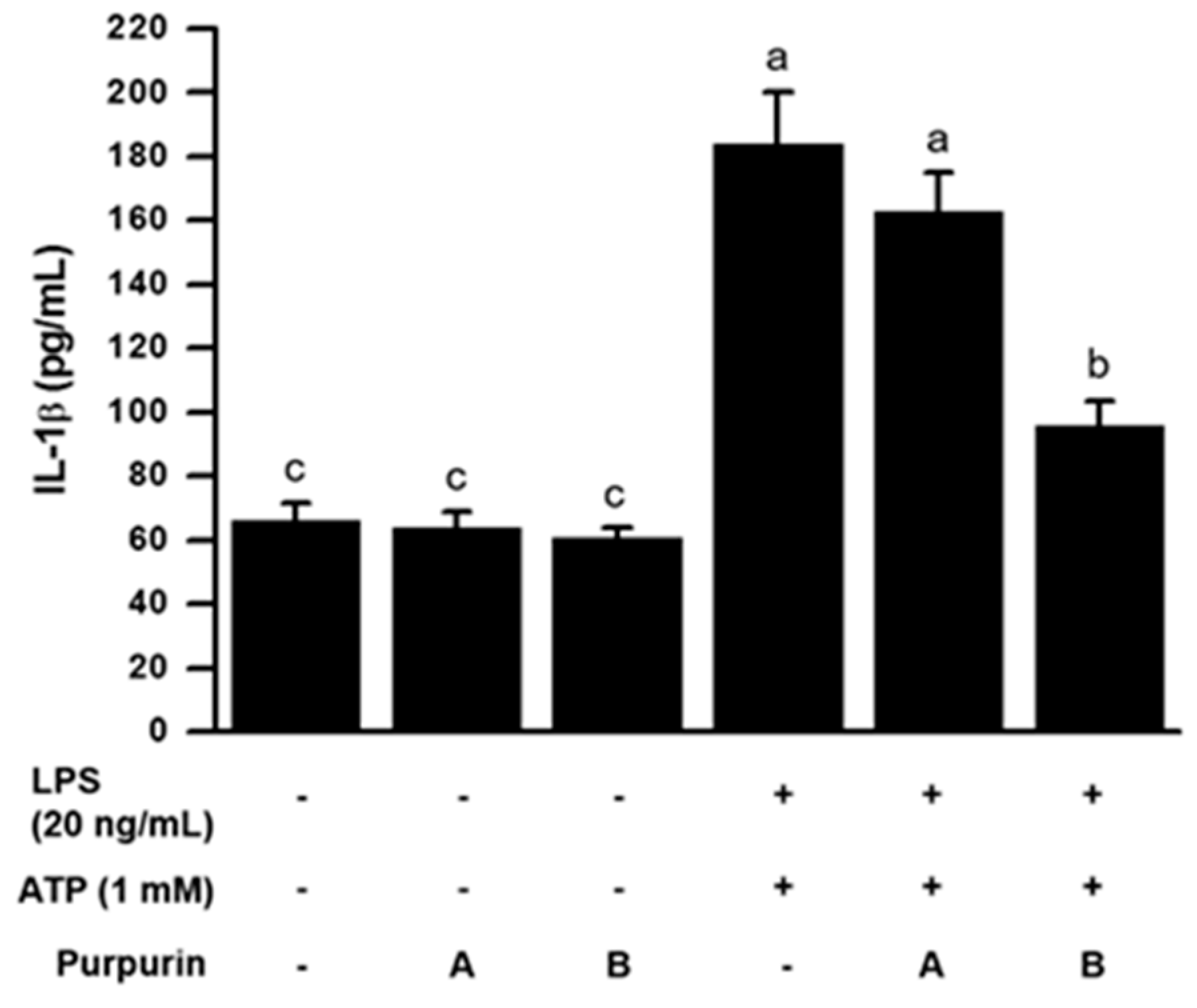
2.2.3. Inhibition of Inflammasome Formation
2.2.4. Structure-Activity Aspects
3. Materials and Methods
3.1. Materials
3.2. Antioxidative Chemical Tests
3.2.1. DPPH Free Radical Scavenging Assay
3.2.2. Inhibition of Linoleic Acid Peroxidation
3.2.3. ABTS Radical Cation Decolorization Assay
3.2.4. Hydrogen Peroxide Scavenging Assay
3.2.5. Ferricyanide Reducing Power Assay
3.2.6. Ferrous Ion Binding Assay
3.3. Cell Assays for Antioxidative and Anti-Inflammatory Activities
3.3.1. Cell Culture
3.3.2. Cell Viability MTT Assay
3.3.3. Nitric Oxide Generation Assay
3.3.4. Cell Treatment for Inflammasome Induction
3.3.5. Quantitation of Cytokine
3.3.6. Intracellular Hydroxyl Radical Scavenging Assay
3.3.7. Western Blot Analysis of Cell Proteins
3.4. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xu, K.; Wang, P.; Wang, L.; Liu, C.; Xu, S.; Cheng, Y.; Wang, Y.; Li, Q.; Lei, H. Quinone derivatives from the genus Rubia and their bioactivities. Chem. Biodivers. 2014, 11, 341–363. [Google Scholar] [CrossRef] [PubMed]
- Zengin, G.; Locatelli, M.; Ceylan, R.; Aktumsek, A. Anthraquinone profile, antioxidant and enzyme inhibitory effect of root extracts of eight Asphodeline taxa from Turkey: Can Asphodeline roots be considered as a new source of natural compounds? J. Enzym. Inhib. Med. Chem. 2016, 31, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Usai, M.; Marchetti, M. Anthraquinone distribution in the hypogeal apparatus of Rubia peregrina L. growing wild in Sardinia. Nat. Prod. Res. 2010, 24, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.R.; Thomson, R.H. Naturally occurring quinones. Part XV. Biogenesis of the anthraquinones in Rubia tinctorum L. (Madder). J. Chem. Soc. C 1968, 2437–2441. [Google Scholar] [CrossRef]
- Cuoco, G.; Mathe, C.; Archier, P.; El Maataoui, M.; Vieillescazes, C. Cytohistological and phytochemical study of madder root extracts obtained by ultrasonic and classical extractions. Phytochem. Anal. 2009, 20, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Derksen, G.C.H.; Van Beek, T.A. Rubia tinctorum L. Stud. Nat. Prod. Chem. 2002, 26, 629–684. [Google Scholar]
- Zengin, G.; Degirmenci, N.S.; Alpsoy, L.; Aktumsek, A. Evaluation of antioxidant, enzyme inhibition, and cytotoxic activity of three anthraquinones (alizarin, purpurin, and quinizarin). Hum. Exp. Toxicol. 2016, 35, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, E.; Fujita, K.; Kamataki, T.; Arimoto-Kobayashi, S.; Okamoto, K.; Negishi, T. Inhibition of human cytochrome P450 1B1, 1A1 and 1A2 by antigenotoxic compounds, purpurin and alizarin. Mutat. Res. 2002, 508, 147–156. [Google Scholar] [CrossRef]
- Takahashi, E.; Marczylo, T.H.; Watanabe, T.; Nagai, S.; Hayatsu, H.; Negishi, T. Preventive effects of anthraquinone food pigments on the DNA damage induced by carcinogens in Drosophila. Mutat. Res. Fundam. Mol. Mech. Mutag. 2001, 480–481, 139–145. [Google Scholar] [CrossRef]
- Friedman, M.; Zhu, L.; Feinstein, Y.; Ravishankar, S. Carvacrol facilitates heat-induced inactivation of Escherichia coli O157:H7 and inhibits formation of heterocyclic amines in grilled ground beef patties. J. Agric. Food Chem. 2009, 57, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Marczylo, T.; Sugiyama, C.; Hayatsu, H. Protection against Trp-P-2 DNA adduct formation in C57bl6 mice by purpurin is accompanied by induction of cytochrome P450. J. Agric. Food Chem. 2003, 51, 3334–3337. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, E.; Arimoto, S.; Okamoto, K.; Negishi, T. Enhancement of phase II enzyme activity by purpurin resulting in the suppression of MeIQx-DNA-adduct formation in mice. Mutat. Res. 2007, 626, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Malterud, K.E.; Farbrot, T.L.; Huse, A.E.; Sund, R.B. Antioxidant and radical scavenging effects of anthraquinones and anthrones. Pharmacology 1993, 47 (Suppl. 1), 77–85. [Google Scholar] [CrossRef] [PubMed]
- Yen, G.-C.; Duh, P.-D.; Chuang, D.-Y. Antioxidant activity of anthraquinones and anthrone. Food Chem. 2000, 70, 437–441. [Google Scholar] [CrossRef]
- Baghiani, A.; Charef, N.; Djarmouni, M.; Saadeh, H.A.; Arrar, L.; Mubarak, M.S. Free radical scanvenging and antioxidant effects of some anthraquinone derivatives. Med. Chem. 2011, 7, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Chandel, M.; Kumar, S.; Kaur, S. Amelioration of oxidative stress by anthraquinones in various in vitro assays. Asian Pac. J. Trop. Dis. 2012, 2, S692–S698. [Google Scholar] [CrossRef]
- Marković, Z.; Jeremić, S.; Dimitrić Marković, J.; Stanojević Pirković, M.; Amić, D. Influence of structural characteristics of substituents on the antioxidant activity of some anthraquinone derivatives. Comput. Theor. Chem. 2016, 1077, 25–31. [Google Scholar] [CrossRef]
- Özbakır Işın, D. Theoretical study on the investigation of antioxidant properties of some hydroxyanthraquinones. Mol. Phys. 2016, 114, 3578–3588. [Google Scholar] [CrossRef]
- Damodaran, S.; Parkin, K.L.; Fennema, O.R. Fennema’s Food Chemistry, 4th ed.; CRC Press: Boca Raton, FL, USA, 2007; p. 571. [Google Scholar]
- Oyaizu, M. Studies on products of the browning reaction. Antioxidative activities of browning reaction products prepared from glucosamine. Eiyogaku Zasshi 1986, 44, 307–315. [Google Scholar] [CrossRef]
- Lopes, G.K.B.; Schulman, H.M.; Hermes-Lima, M. Polyphenol tannic acid inhibits hydroxyl radical formation from the Fenton reaction by complexing ferrous ions. Biochim. Biophys. Acta 1999, 1472, 142–152. [Google Scholar] [CrossRef]
- Hsieh, T.J.; Liu, T.Z.; Chia, Y.C.; Chern, C.L.; Lu, F.J.; Chuang, M.; Mau, S.Y.; Chen, S.H.; Syu, Y.H.; Chen, C.H. Protective effect of methyl gallate from Toona sinensis (Meliaceae) against hydrogen peroxide-induced oxidative stress and DNA damage in DDCK cells. Food Chem. Toxicol. 2004, 42, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Buttery, L.D.K.; Springall, D.R.; Chester, A.H.; Evans, T.J.; Standfield, N.; Parums, D.V.; Yacoub, M.H.; Polak, J.M. Inducible nitric oxide synthase is present within human atherosclerotic lesions and promotes the formation and activity of peroxynitrite. Lab. Investig. 1996, 75, 77–85. [Google Scholar] [PubMed]
- Ross, R.; Reske-Kunz, A.B. The role of NO in contact hypersensitivity. Int. Immunopharmacol. 2001, 1, 1469–1478. [Google Scholar] [CrossRef]
- Carey, R.M.; Chen, B.; Adappa, N.D.; Palmer, J.N.; Kennedy, D.W.; Lee, R.J.; Cohen, N.A. Human upper airway epithelium produces nitric oxide in response to Staphylococcus epidermidis. Int. Forum Allergy Rhinol. 2016, 6, 1238–1244. [Google Scholar] [CrossRef]
- Jin, C.; Flavell, R.A. Molecular mechanism of NLRP3 inflammasome activation. J. Clin. Immunol. 2010, 30, 628–631. [Google Scholar] [PubMed]
- Zhao, D.; Wu, Y.; Zhuang, J.; Xu, C.; Zhang, F. Activation of NLRP1 and NLRP3 inflammasomes contributed to cyclic stretch-induced pyroptosis and release of IL-1β in human periodontal ligament cells. Oncotarget 2016, 7, 68292–68302. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yan, Z.; Schwartz, D.E.; Yu, J.; Malik, A.B.; Hu, G. Activation of NLRP3 inflammasome in alveolar macrophages contributes to mechanical stretch-induced lung inflammation and injury. J. Immunol. 2013, 190, 3590–3599. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.P.; Park, S.O.; Lee, S.J.; Nam, S.H.; Friedman, M. A polysaccharide isolated from the liquid culture of Lentinus edodes (Shiitake) mushroom mycelia containing black rice bran protects mice against salmonellosis through up-regulation of the Th1 immune reaction. J. Agric. Food Chem. 2014, 62, 2384–2391. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Chemistry, biochemistry, and dietary role of potato polyphenols. A review. J. Agric. Food Chem. 1997, 45, 1523–1540. [Google Scholar] [CrossRef]
- Gao, M.; Yang, J.; Wang, Z.; Yang, B.; Kuang, H.; Liu, L.; Wang, L.; Yang, C. Simultaneous determination of purpurin, munjistin and mollugin in rat plasma by ultra high performance liquid chromatography-tandem mass spectrometry: Application to a pharmacokinetic study after oral administration of Rubia cordifolia L. extract. Molecules 2016, 21, 717. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Abraham, T.E. Studies on the antioxidant activities of cinnamon (Cinnamomum verum) bark extracts, through various in vitro models. Food Chem. 2006, 94, 520–528. [Google Scholar] [CrossRef]
- Mitsuda, H.; Yasumoto, K.; Iwami, K. Antioxidative action of indole compounds during the autoxidation of linoleic acid. Eiyo To Shokuryo 1966, 19, 210–214. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Fernando, C.D.; Soysa, P. Optimized enzymatic colorimetric assay for determination of hydrogen peroxide (H2O2) scavenging activity of plant extracts. MethodsX 2015, 2, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Sroka, Z.; Cisowski, W. Hydrogen peroxide scavenging, antioxidant and anti-radical activity of some phenolic acids. Food Chem. Toxicol. 2003, 41, 753–758. [Google Scholar] [CrossRef]
- Dinis, T.C.; Maderia, V.M.; Almeida, L.M. Action of phenolic derivatives (acetaminophen, salicylate, and 5-aminosalicylate) as inhibitors of membrane lipid peroxidation and as peroxyl radical scavengers. Arch. Biochem. Biophys. 1994, 315, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Murakami, A.; Gao, G.; Kim, O.K.; Omura, M.; Yano, M.; Ito, C.; Furukawa, H.; Jiwajinda, S.; Koshimizu, K.; Ohigashi, H. Identification of coumarins from the fruit of Citrus hystrix DC as inhibitors of nitric oxide generation in mouse macrophage raw 264.7 cells. J. Agric. Food Chem. 1999, 47, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.K.; Chen, P.C.; Ho, C.T.; Lin-Shiau, S.Y. Inhibition of xanthine oxidase and suppression of intracellular reactive oxygen species in HL-60 cells by theaflavin-3,3′-digallate, (−)-epigallocatechin-3-gallate, and propyl gallate. J. Agric. Food Chem. 2000, 48, 2736–2743. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Winkler-Moser, J.K. Antioxidant activity of amino acids in soybean oil at frying temperature: Structural effects and synergism with tocopherols. Food Chem. 2017, 221, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, C.; Barrio, A.; Del Mar Cavia, M.; Alonso-Torre, S. Global antioxidant response of meat. J. Sci. Food Agric. 2016. [Google Scholar] [CrossRef] [PubMed]
- Habib, H.M.; Theuri, S.W.; Kheadr, E.; Mohamed, F.E. DNA and BSA damage inhibitory activities, and anti-acetylcholinesterase, anti-porcine α-amylase and antioxidant properties of Dolichos lablab beans. Food Funct. 2016. [Google Scholar] [CrossRef] [PubMed]
- Fougère, B.; Boulanger, E.; Nourhashémi, F.; Guyonnet, S.; Cesari, M. Chronic inflammation: Accelerator of biological aging. J. Gerontol. A Biol. Sci. Med. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wallace, G.; Cro, S.; Doré, C.; King, L.; Kluzek, S.; Price, A.; Roemer, F.; Guermazi, A.; Keen, R.; Arden, N. Associations between clinical evidence of inflammation and synovitis in symptomatic knee osteoarthritis: A substudy of the VIDEO trial. Arthritis Care Res. (Hoboken) 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-P.; Lee, S.-J.; Nam, S.-H.; Friedman, M. Elm tree (Ulmus parvifolia) bark bioprocessed with mycelia of Shiitake (Lentinus edodes) mushrooms in liquid culture: Composition and mechanism of protection against allergic asthma in mice. J. Agric. Food Chem. 2016, 64, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Lee, H.S.; Sol, I.S.; Kim, M.N.; Hong, J.Y.; Lee, K.E.; Kim, Y.H.; Kim, K.W.; Sohn, M.H.; Kim, K.E. Sputum pentraxin 3 as a candidate to assess airway inflammation and remodeling in childhood asthma. Medicine (Baltimore) 2016, 95, e5677. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol. Cell Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Jiang, H.; Wu, X.; Fang, J. Therapeutic effects of quercetin on inflammation, obesity, and type 2 diabetes. Mediat. Inflamm. 2016, 2016, 9340637. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M. Mushroom polysaccharides: Chemistry and antiobesity, antidiabetes, anticancer, and antibiotic properties in cells, rodents, and humans. Foods 2016, 5, 80. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
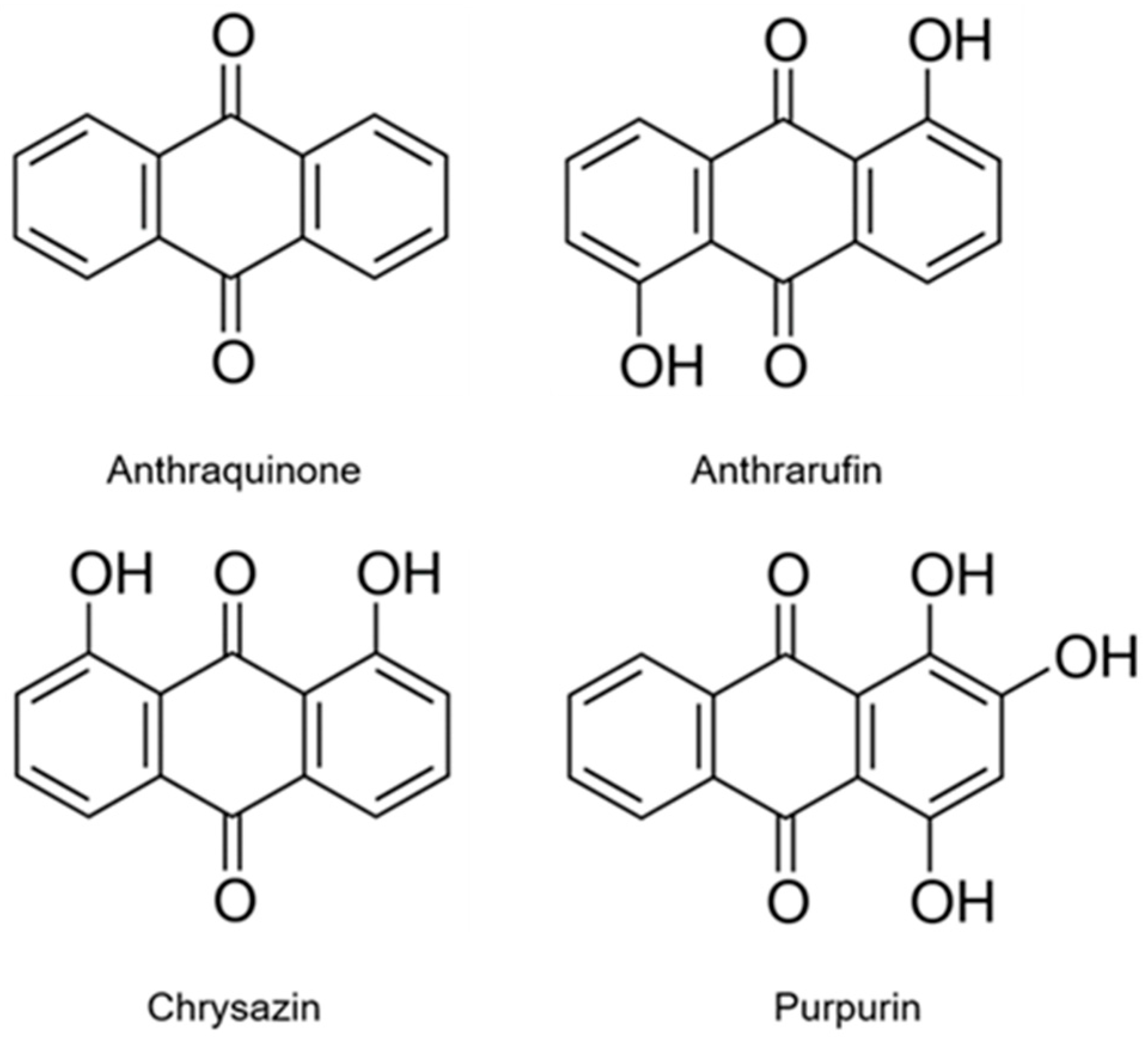


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | DPPH Scavenging Activity (%) | |||||
|---|---|---|---|---|---|---|
| 1 µM | 10 µM | 50 µM | 100 µM | 250 µM | IC50 (µM) | |
| BHA | 1.2 ± 1.9 c | 16.4 ± 2.8 c | 59.93 ± 0.55 b | 65.4 ± 0.40 b | 62.1 ± 3.2 b | 36.5 ± 1.7 b |
| Purpurin | 8.7 ± 1.3 b | 36.8 ± 1.2 a | 78.7 ± 3.1 a | 88.0 ± 1.9 a | 92.5 ± 1.3 a | 20.9 ± 1.4 b |
| Anthrarufin | 8.56 ± 0.67 b | 18.62 ± 0.26 c | 27.5 ± 1.4 c | 35.9 ± 2.3 c | 58.2 ± 1.1 b | 225 ± 19 a |
| Chrysazin | 6.9 ± 2.3 b | 9.1 ± 4.0 d | 12.0 ± 3.1 d | 26.0 ± 7.0 d | 59.0 ± 1.7 b | 215 ± 14 a |
| Anthraquinone | 21.0 ± 1.2 a | 29.0 ± 2.1 b | 30.2 ± 1.3 c | 37.33 ± 0.31 c | 54.2 ± 2.4 c | 213 ± 22 a |
| Sample | Inhibition of Lipid Peroxidation (%) | |||||
|---|---|---|---|---|---|---|
| 1 µM | 10 µM | 50 µM | 100 µM | 250 µM | IC50 (µM) | |
| BHA b | 92.2 ± 2.0 a | 95.1 ± 1.4 a | 98.4 ± 2.5 a | 98.9 ± 3.1 a | 98.28 ± 0.64 a | 0.99 ± 0.01 c |
| Purpurin | 42.9 ± 1.9 b | 83.2 ± 1.1 b | 96.0 ± 1.1 a | 98.35 ± 0.65 a | 97.5 ± 1.4 a | 1.27 ± 0.05 c |
| Anthrarufin | 22.12 ± 0.83 d | 34.7 ± 1.5 c | 71.1 ± 2.0 b | 85.01 ± 0.59 b | 88.11 ± 0.82 b | 23.5 ± 1.7 c |
| Chrysazin | 11.9 ± 4.1 e | 17.0 ± 1.7 d | 41.1 ± 1.4 c | 41.54 ± 0.49 d | 50.57 ± 0.29 d | 202 ± 25 a |
| Anthraquinone | 27.0 ± 1.8 c | 33.85 ± 0.55 c | 42.5 ± 3.7 c | 49.7 ± 2.7 c | 54.70 ± 0.21 c | 101 ± 21 b |
| Sample | ABTS Scavenging Activity (%) | |||||
|---|---|---|---|---|---|---|
| 1 µM | 10 µM | 50 µM | 100 µM | 250 µM | IC50 (µM) | |
| BHA | 3.03 ± 0.70 a | 31.49 ± 0.69 a | 98.98 ± 0.04 a | 99.22 ± 0.03 a | 99.07 ± 0.22 a | 17.55 ± 0.13 b |
| Purpurin | 2.49 ± 0.99 a | 21.09 ± 0.27 b | 50.32 ± 0.41 b | 61.21 ± 0.10 b | 76.04 ± 0.30 b | 54.5 ± 0.6 a |
| Anthrarufin | 1.04 ± 0.68 ab | 0.55 ± 0.45 c | 1.4 ± 1.1 c | 1.97 ± 0.94 d | 0.02 ± 0.62 d | N.D. |
| Chrysazin | 1.16 ± 0.65 a,b | 0.62 ± 0.44 c | 0.77 ± 0.40 c | 0.7 ± 1.4 d | 1.75 ± 0.62 d | N.D. |
| Anthraquinone | 0.2 ± 1.9 b | 0.5 ± 1.3 c | 1.8 ± 2.3 c | 16.5 ± 2.1 c | 47.2 ± 2.7 c | N.D. |
| Sample | H2O2 Scavenging Activity (%) | |||||
|---|---|---|---|---|---|---|
| 1 µM | 10 µM | 50 µM | 100 µM | 250 µM | IC50 (µM) | |
| BHA | 9.0 ± 4.6 a | 96.6 ± 1.8 a | 102.2 ± 1.3 a | 103.3 ± 1.4 a | 108.2 ± 1.7 b | 4.81 ± 0.32 b |
| Purpurin | 10.0 ± 3.3 a | 86.8 ± 1.2 b | 98.8 ± 4.2 a | 106.6 ± 2.9 a | 112.0 ± 1.2 a | 5.29 ± 0.25 b |
| Anthrarufin | −1.6 ± 2.2 b | 5.32 ± 0.19 c | 20.88 ± 0.76 b | 28.2 ± 1.2 b | 57.2 ± 2.6 c | 191.1 ± 6.9 a |
| Chrysazin | −2.2 ± 3.5 b | −1.3 ± 1.9 d | 0.3 ± 1.4 c | 3.8 ± 1.6 d | 27.12 ± 0.83 e | N.D. |
| Anthraquinone | 0.2 ± 1.9 b | 0.5 ± 1.3 d | 1.8 ± 2.3 c | 16.5 ± 2.1 c | 47.2 ± 2.7 d | N.D. |
| Sample | Reductive Potential (O.D 700 nm) | ||||
|---|---|---|---|---|---|
| 1 µM | 10 µM | 50 µM | 100 µM | 250 µM | |
| BHA | 0.019 ± 0.001 b | 0.075 ± 0.002 b | 0.324 ± 0.004 b | 0.605 ± 0.003 b | 1.352 ± 0.033 b |
| Purpurin | 0.021 ± 0.000 a | 0.082 ± 0.001 a | 0.351 ± 0.003 a | 0.665 ± 0.002 a | 1.452 ± 0.018 a |
| Anthrarufin | 0.012 ± 0.000 e | 0.02 ± 0.005 c | 0.016 ± 0.000 d | 0.035 ± 0.001 c | 0.056 ± 0.001 c |
| Chrysazin | 0.015 ± 0.000 c | 0.015 ± 0.000 d | 0.017 ± 0.000 cd | 0.015 ± 0.000 e | 0.017 ± 0.012 d |
| Anthraquinone | 0.014 ± 0.000 d | 0.014 ± 0.002 d | 0.02 ± 0.001 c | 0.024 ± 0.004 d | 0.027 ± 0.001 cd |
| Sample | Metal Ion Chelating Activity (%) | |||||
|---|---|---|---|---|---|---|
| 1 µM | 10 µM | 50 µM | 100 µM | 250 µM | IC50 (µM) | |
| EDTA | 1.60 ± 0.15 a | 1.75 ± 0.13 a | 10.46 ± 0.40 a | 57.02 ± 0.13 a | 99.91 ± 0.05 a | 91.46 ± 0.17 |
| BHA | −0.20 ± 0.50 b | −1.46 ± 0.83 c | −1.58 ± 0.25 e | −2.41 ± 0.30 f | −1.12 ± 0.69 f | N.D. |
| Purpurin | −0.83 ± 0.41 bc | 2.01 ± 0.45 a | 8.17 ± 0.53 b | 12.26 ± 0.91 b | 24.2 ± 1.5 b | N.D. |
| Anthrarufin | −0.20 ± 0.57 b | 0.03 ± 0.39 b | 3.81 ± 0.54 c | 6.9 ± 1.2 c | 12.75 ± 0.73 d | N.D. |
| Chrysazin | −0.92 ± 0.65 bc | −0.80 ± 0.89 bc | 2.41 ± 0.79 d | 3.95 ± 0.39 d | 8.48 ± 0.26 e | N.D. |
| Anthraquinone | −1.55 ± 0.17 c | −0.11 ± 0.09 b | 3.47 ± 0.28 c | 0.8 ± 1.0 e | 15.30 ± 0.89 c | N.D. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, W.; Kim, S.P.; Nam, S.H.; Friedman, M. Structure-Antioxidative and Anti-Inflammatory Activity Relationships of Purpurin and Related Anthraquinones in Chemical and Cell Assays. Molecules 2017, 22, 265. https://doi.org/10.3390/molecules22020265
Nam W, Kim SP, Nam SH, Friedman M. Structure-Antioxidative and Anti-Inflammatory Activity Relationships of Purpurin and Related Anthraquinones in Chemical and Cell Assays. Molecules. 2017; 22(2):265. https://doi.org/10.3390/molecules22020265
Chicago/Turabian StyleNam, Woo, Sung Phil Kim, Seok Hyun Nam, and Mendel Friedman. 2017. "Structure-Antioxidative and Anti-Inflammatory Activity Relationships of Purpurin and Related Anthraquinones in Chemical and Cell Assays" Molecules 22, no. 2: 265. https://doi.org/10.3390/molecules22020265
APA StyleNam, W., Kim, S. P., Nam, S. H., & Friedman, M. (2017). Structure-Antioxidative and Anti-Inflammatory Activity Relationships of Purpurin and Related Anthraquinones in Chemical and Cell Assays. Molecules, 22(2), 265. https://doi.org/10.3390/molecules22020265