
Betulinic Acid-Mediated Apoptosis in Human Prostate Cancer Cells Involves p53 and Nuclear Factor-Kappa B (NF-κB) Pathways



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
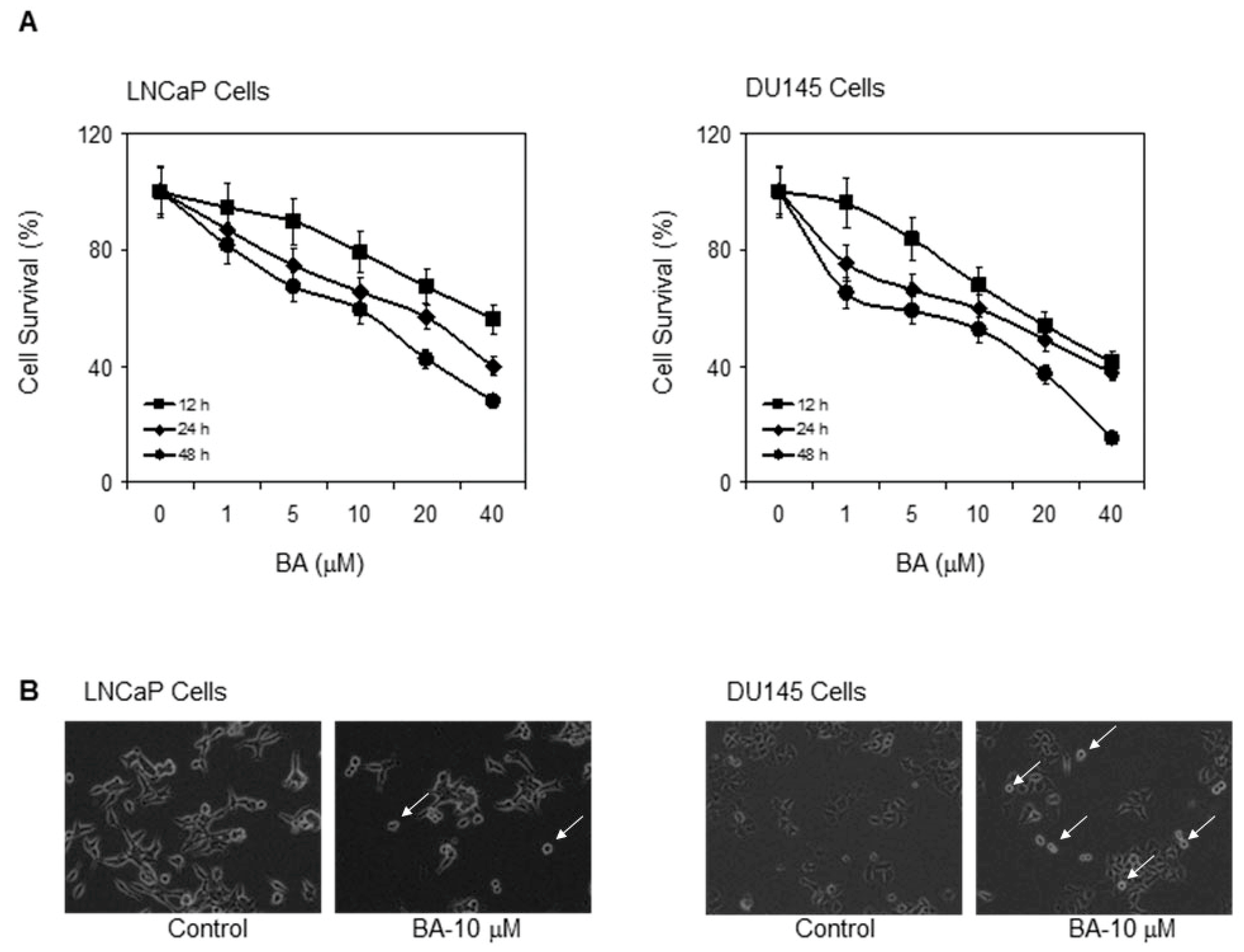
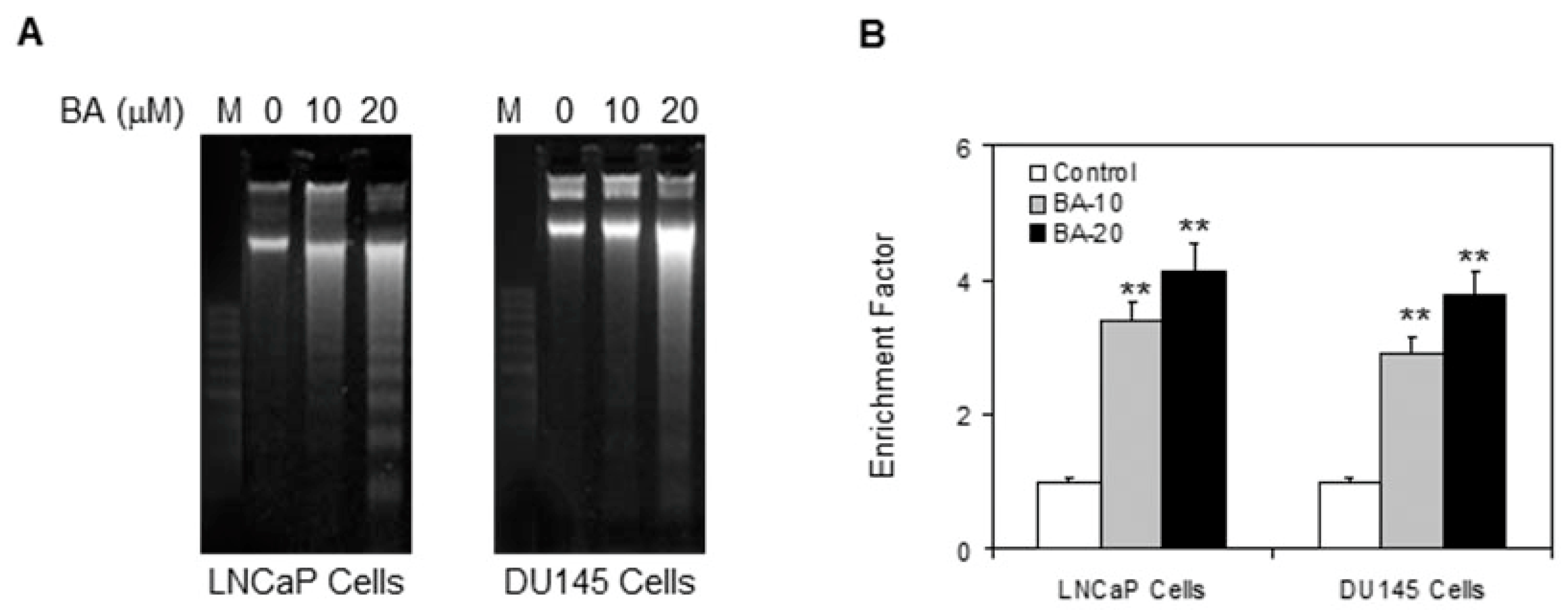
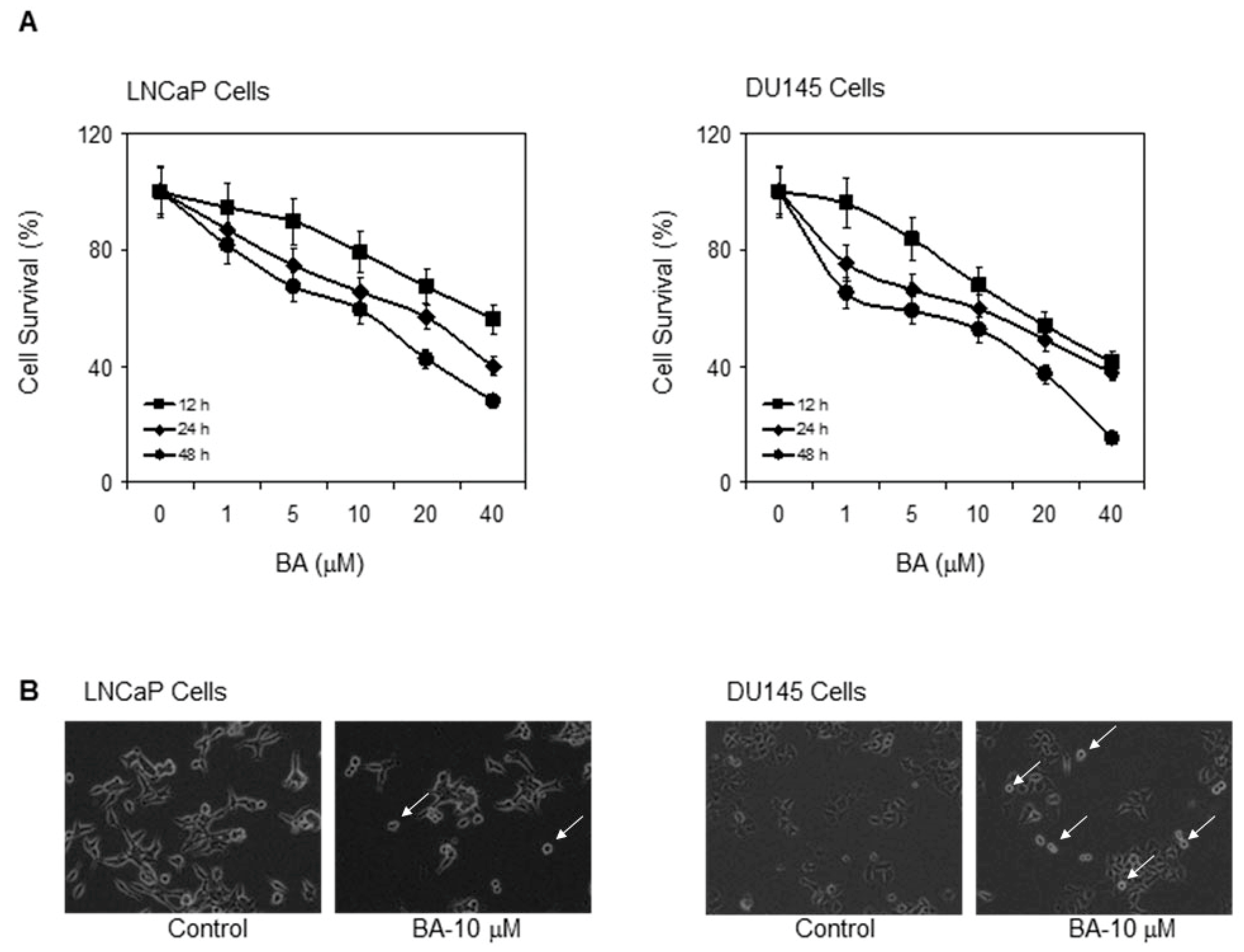
2.1. Cytotoxic Effect of BA in Prostate Cancer Cells
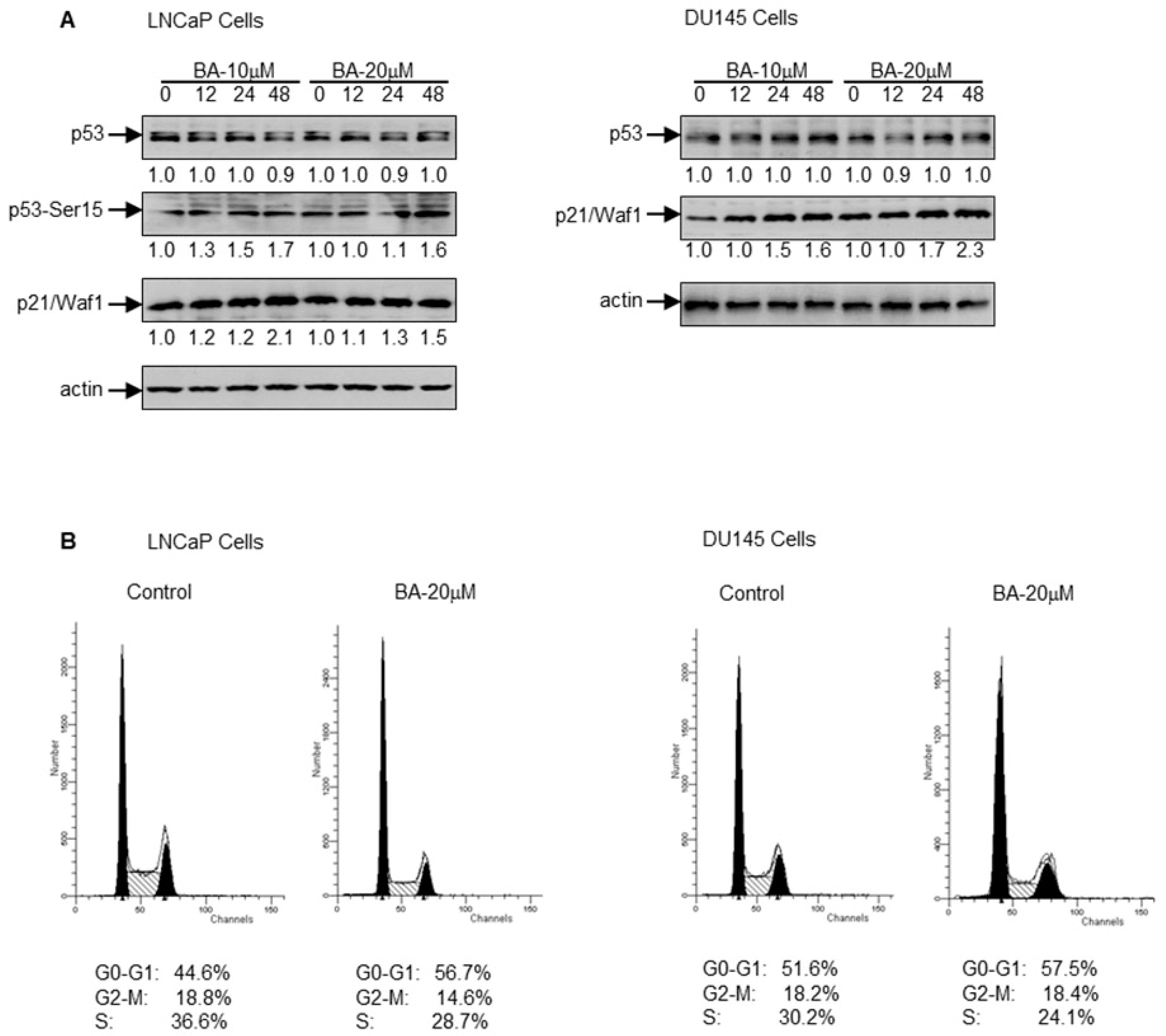
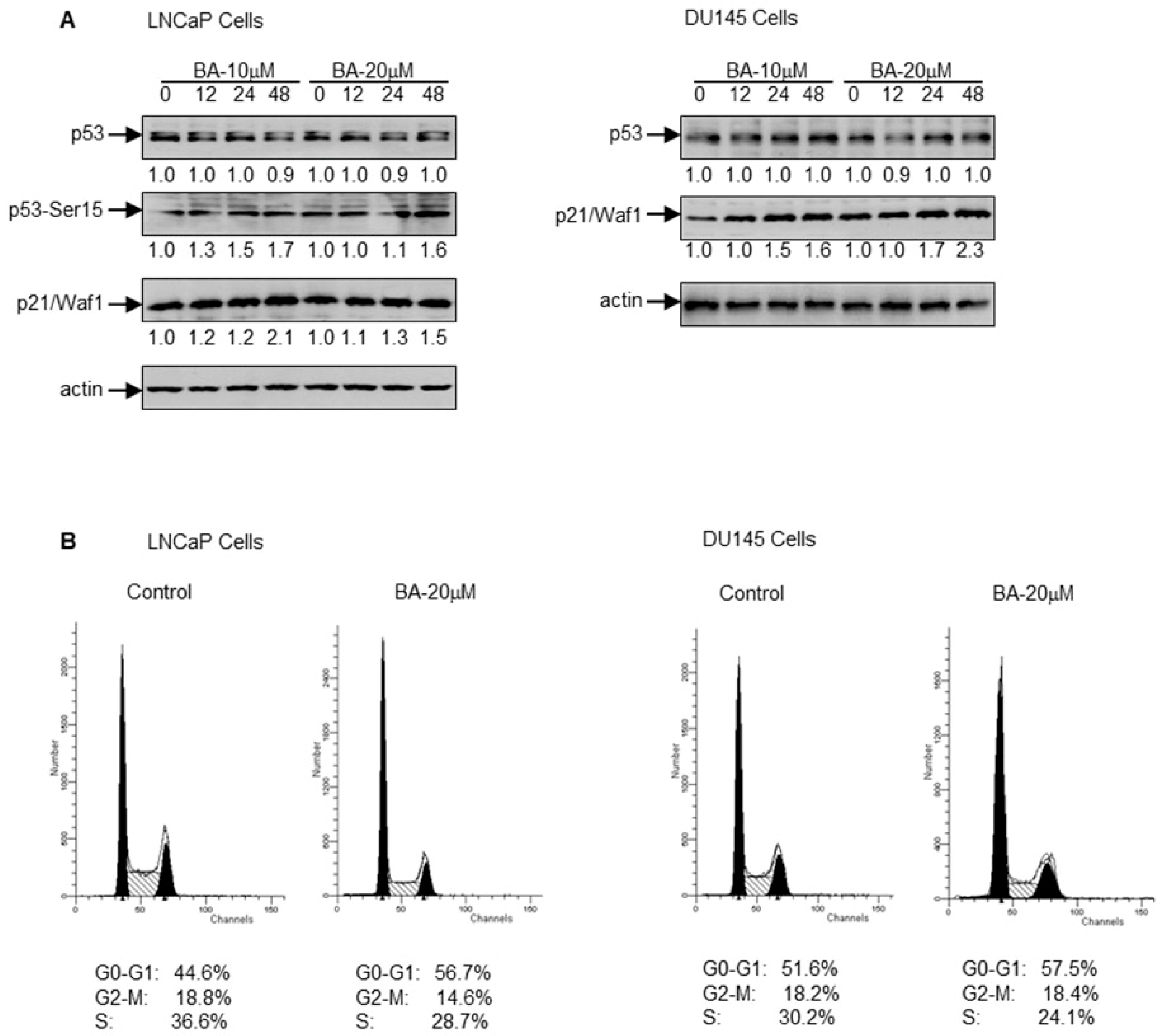
2.2. BA Induces p21/Waf1 in a p53-Dependent and Independent Manner to Cause G1 Cell Cycle Arrest in Prostate Cancer Cells
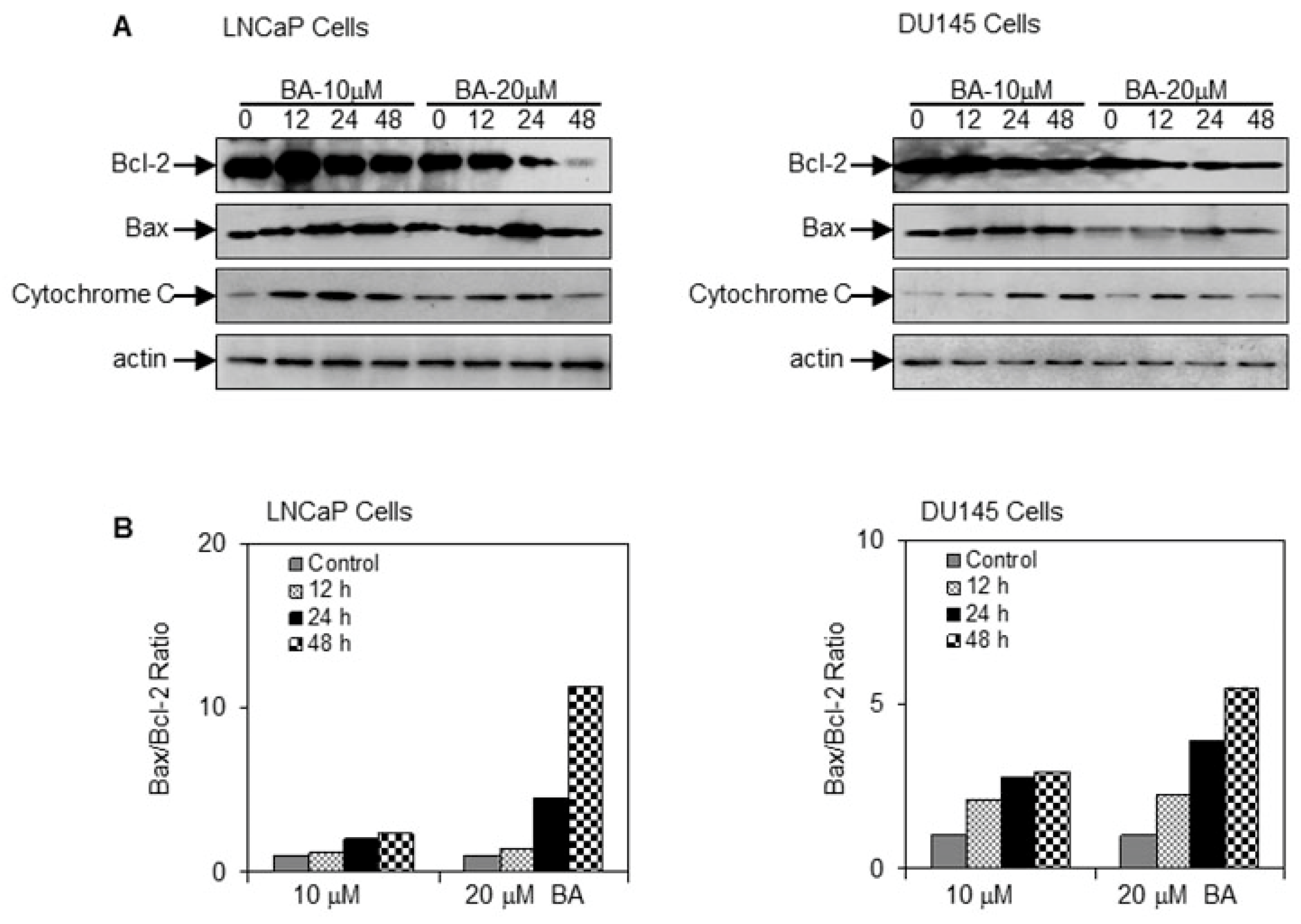
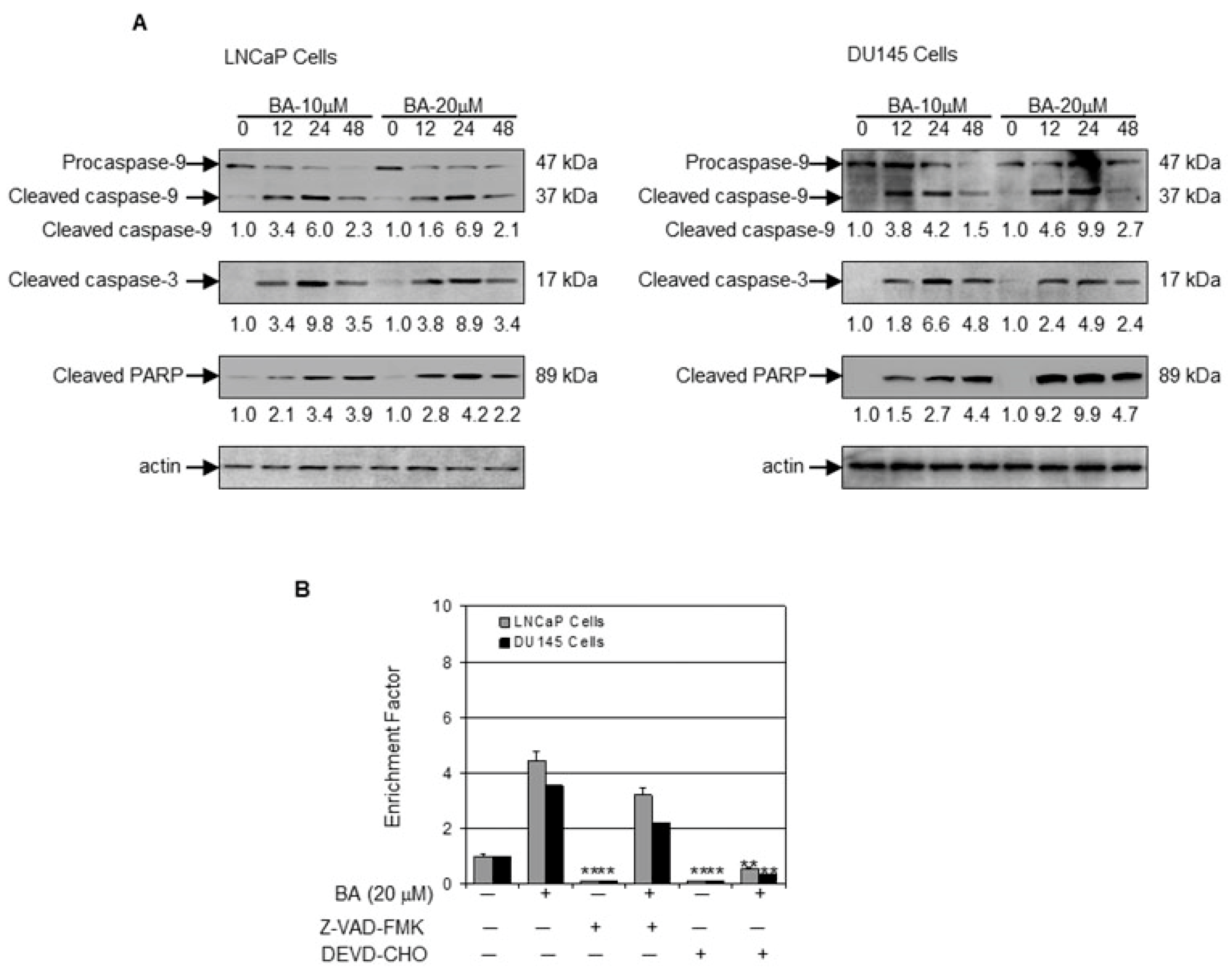
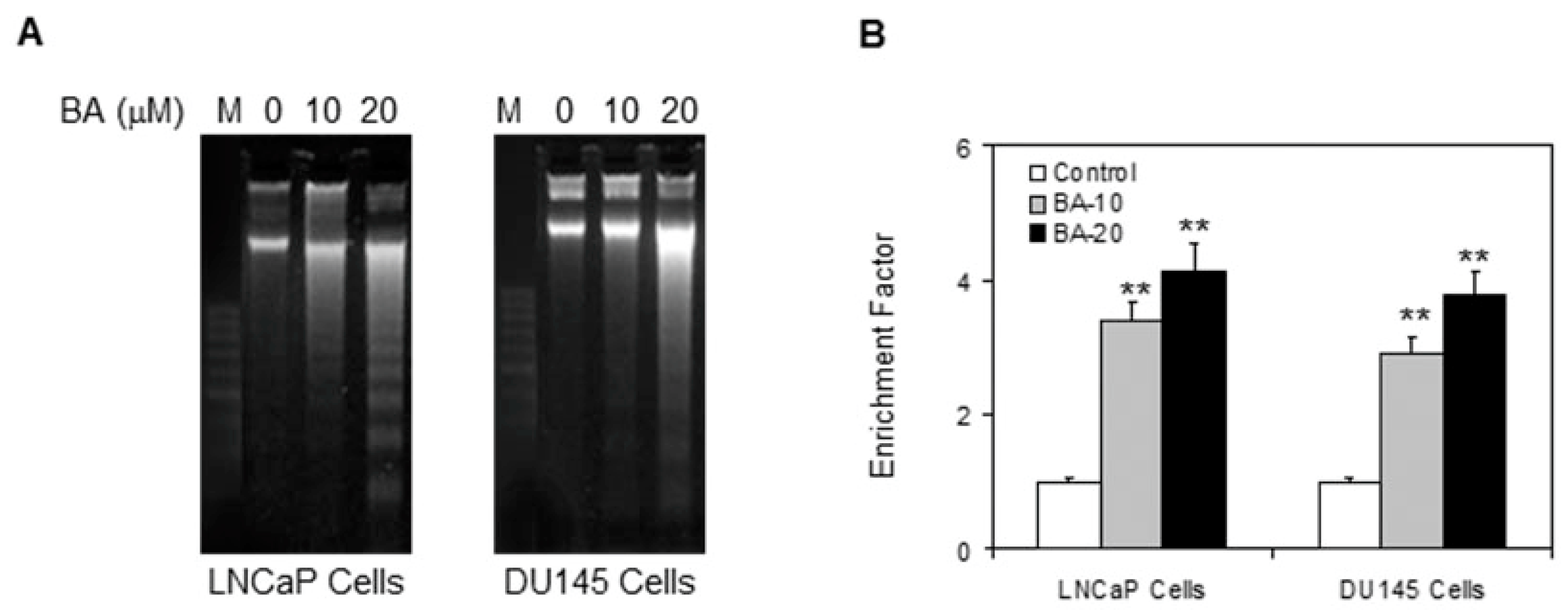
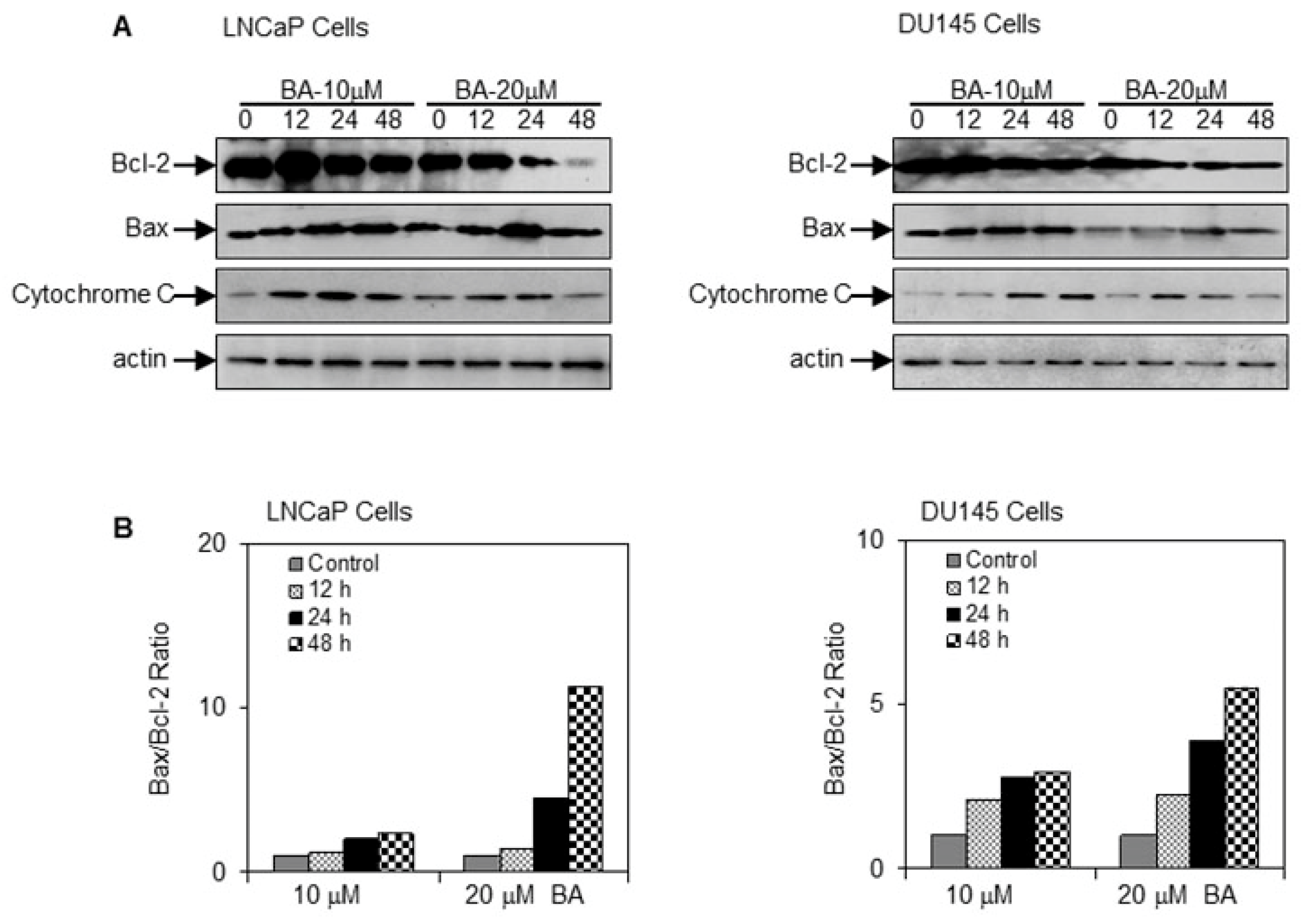
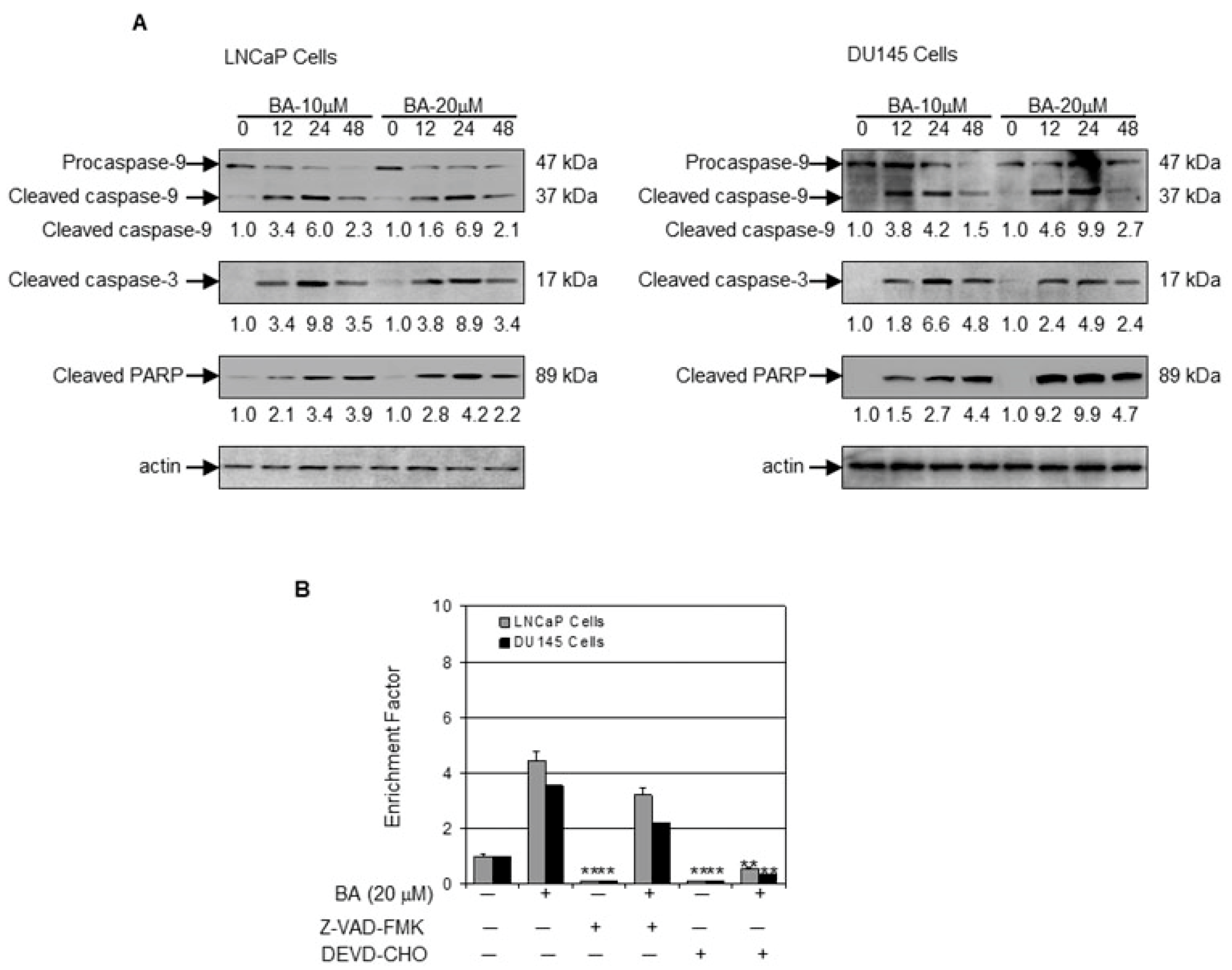
2.3. BA Induces Apoptosis by Altering Bax/Bcl-2 Ratio and Causing Cyctochrome C Release in Prostate Cancer Cells
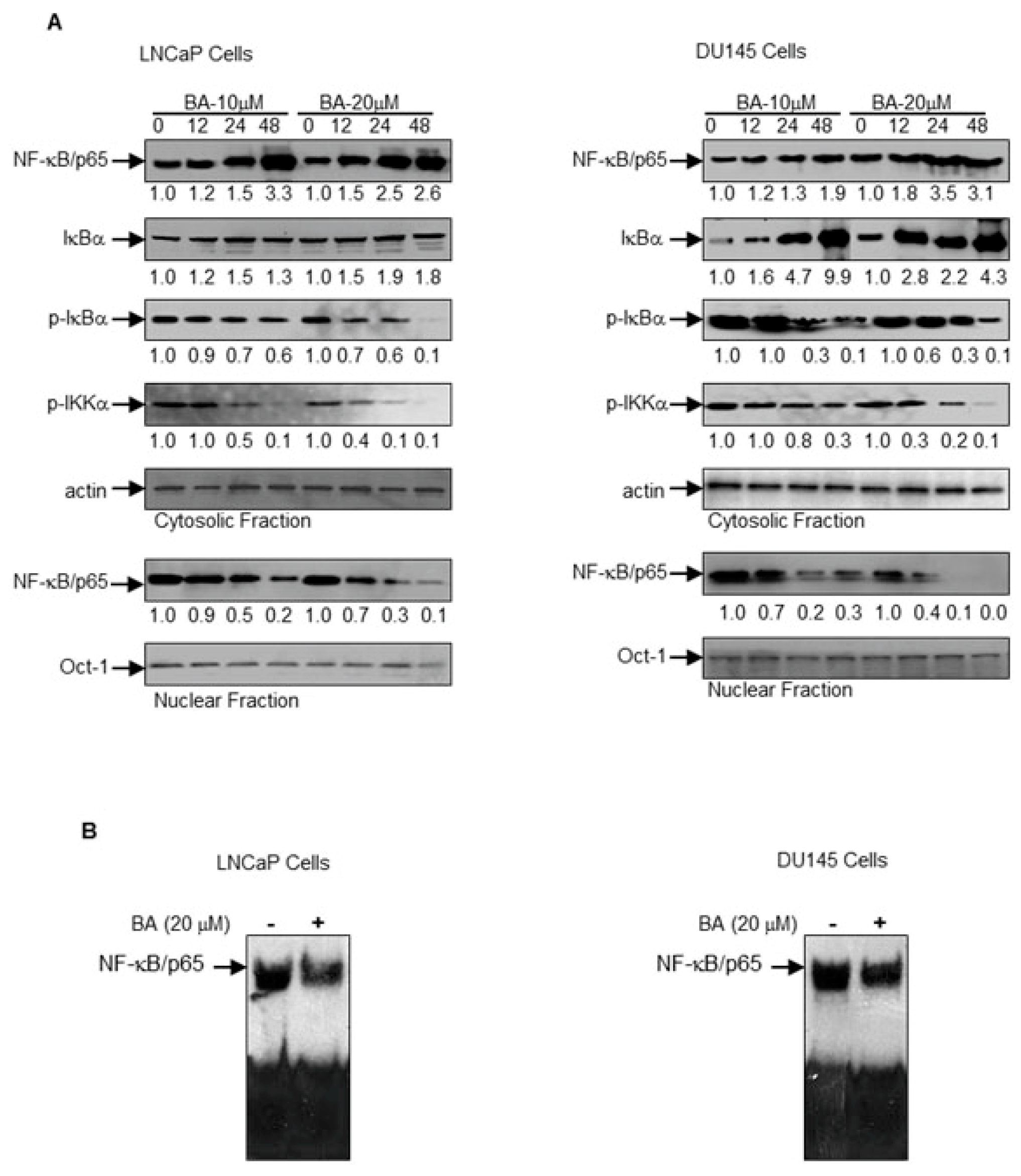
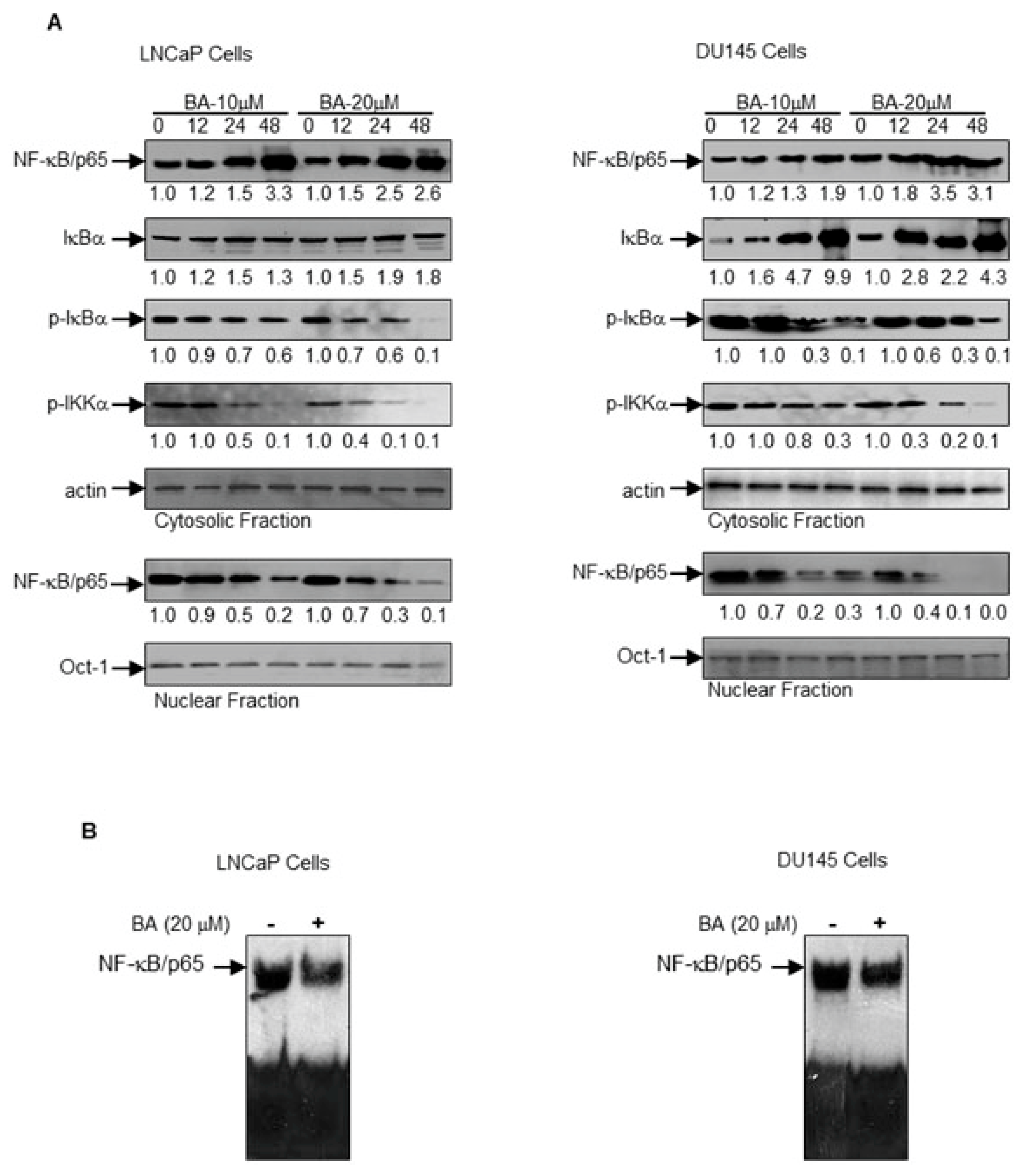
2.4. BA-Mediated Inhibition of NF-κB Pathway in Prostate Cancer Cells
3. Discussion
4. Material and Methods
4.1. Cells and Reagents
4.2. Cell Culture
4.3. Cell Survival Assay
4.4. Cell Microscopy
4.5. DNA Fragmentation Assay
4.6. Cell Cycle Analysis
4.7. Western Blotting
4.8. Apoptosis by ELISA
4.9. Electrophoretic Mobility Shift Assay
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| BA | Betulinic acid |
| DMSO | dimethyl sulfoxide |
| EMSA | Electrophoretic; mobility shift assay |
| IκBα | I-kappa-B-alpha |
| IKK | IκB kinase |
| NF-κB | Nuclear Factor-kappa B |
| PARP | poly(ADP)ribose polymerase |
| RAID | Rapid Access to Intervention in Development program |
| Sp | Specificity Protein |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts and Figures 2016. Available online: http://www.cancer.org/Cancer/ProstateCancer/index (accessed on 15 December 2016).
- Sfanos, K.S.; de Marzo, A.M. Prostate cancer and inflammation the evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Stark, T.; Livas, L.; Kyprianou, N. Inflammation in prostate cancer progression and therapeutic targeting. Transl. Androl. Urol. 2015, 4, 455–463. [Google Scholar] [PubMed]
- Lucia, M.S.; Torkko, K.C. Inflammation as a target for prostate cancer chemoprevention: Pathological and laboratory rationale. J. Urol. 2004, 171, S30–S34. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.J.; Lho, Y.; Connelly, L.; Wang, Y.; Yu, X.; Saint Jean, L.; Case, T.C.; Ellwood-Yen, K.; Sawyers, C.L.; Bhowmick, N.A.; et al. The nuclear Factor-Kappa-B pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 2008, 68, 6762–6769. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Li, J.; Yadav, S.S.; Tewari, A.K. Recent insights into NF-κB signaling pathways and the link between inflammation and prostate cancer. BJU Int. 2014, 114, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; MacLennan, G.T.; Fu, P.; Patel, J.; Marengo, S.R.; Resnick, M.I.; Gupta, S. Nuclear factor-kappaB/p65 (Rel A) is constitutively activated in human prostate adenocarcinoma and correlates with disease progression. Neoplasia 2004, 6, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, C.; Li, L.; Shanmugam, R.; Bhat-Nakshatri, P.; Jayaprakasan, V.; Baldridge, L.A. Nuclear factor-kappaB is constitutively activated in prostate cancer in vitro and is overexpressed in prostatic intraepithelial neoplasia and adenocarcinoma of the prostate. Clin. Cancer Res. 2004, 10, 5501–5507. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Maclennan, G.T.; Marengo, S.R.; Resnick, M.I.; Gupta, S. Constitutive activation of PI3K-Akt and NF-kappaB during prostate cancer progression in autochthonous transgenic mouse model. Prostate 2005, 64, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Sawyers, C.L. NF-kappa B activates prostate-specific antigen expression and is upregulated in androgen-independent prostate cancer. Mol. Cell. Biol. 2002, 22, 2862–2870. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Bookstein, R.; MacGrogan, D.; Hilsenbeck, S.G.; Sharkey, F.; Allred, D.C. p53 is mutated in a subset of advanced-stage prostate cancers. Cancer Res. 1993, 53, 3369–3373. [Google Scholar] [PubMed]
- Heidenberg, H.B.; Bauer, J.J.; McLeod, D.G.; Moul, J.W.; Srivastava, S. The role of the p53 tumor suppressor gene in prostate cancer: A possible biomarker? Urology 1996, 48, 971–979. [Google Scholar] [CrossRef]
- Downing, S.R.; Russell, P.J.; Jackson, P. Alterations of p53 are common in early stage prostate cancer. Can. J. Urol. 2003, 10, 1924–1933. [Google Scholar] [PubMed]
- Effert, P.J.; Neubauer, A.; Walther, P.J.; Liu, E.T. Alterations of the P53 gene are associated with the progression of a human prostate carcinoma. J. Urol. 1992, 147, 789–793. [Google Scholar] [PubMed]
- Buhmeida, A.; Pyrhönen, S.; Laato, M.; Collan, Y. Prognostic factors in prostate cancer. Diagn. Pathol. 2006, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Lehmann, B.D.; Terrian, D.M.; Abrams, S.L.; Steelman, L.S.; McCubrey, J.A. p53 expression controls prostate cancer sensitivity to chemotherapy and the MDM2 inhibitor Nutlin-3. Cell Cycle 2012, 11, 4579–4588. [Google Scholar] [CrossRef] [PubMed]
- Tergaonkar, V. p53 and NF-kappaB fresh breath in the cross talk. Cell Res. 2009, 19, 1313–1315. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G.; Henrich, A.; Greiner, G.; Wolf, V.; Lovas, A.; Wieczorek, M.; Wagner, T.; Reichardt, S.; von Werder, A.; Schmid, R.M.; et al. Crosstalk between stimulated NF-kappaB and the tumor suppressor p53. Oncogene 2010, 29, 2795–2806. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.H.; Suzuki, K.; Downes, M.; Welch, G.L.; de Jesus, P.; Miraglia, L.J.; Orth, A.P.; Chanda, S.K.; Evans, R.M.; Verma, I.M. Tumor suppressor protein p53, is a regulator of NF-kappaB repression by the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 17117–17122. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.A.; Perkins, N.D. Transcriptional cross talk between NF-kappaB and p53. Mol. Cell. Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.M.; Menendez, D.; Bushel, P.R.; Shatz, M.; Kirk, E.L.; Troester, M.A.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. p53 and NF-κB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014, 74, 2182–2192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, J.; Chen, Y. Betulinic acid and the pharmacological effects of tumor suppression (Review). Mol. Med. Rep. 2016, 14, 4489–4495. [Google Scholar] [CrossRef] [PubMed]
- Rabi, T.; Gupta, S. Dietary terpenoids and prostate cancer chemoprevention. Front. Biosci. 2008, 13, 3457–3469. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef]
- Fulda, S.; Jeremias, I.; Debatin, K.M. Cooperation of betulinic acid and TRAIL to induce apoptosis in tumor cells. Oncogene 2004, 23, 7611–7620. [Google Scholar] [CrossRef] [PubMed]
- Selzer, E.; Pimentel, E.; Wacheck, V.; Schlegel, W.; Pehamberger, H.; Jansen, B.; Kodym, R. Effects of betulinic acid alone and in combination with irradiation in human melanoma cells. J. Investig. Dermatol. 2000, 114, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Chintharlapalli, S.; Papineni, S.; Ramaiah, S.K.; Safe, S. Betulinic acid inhibits prostate cancer growth through inhibition of specificity protein transcription factors. Cancer Res. 2007, 67, 2816–2823. [Google Scholar] [CrossRef] [PubMed]
- Rabi, T.; Shukla, S.; Gupta, S. Betulinic acid suppresses constitutive and TNFalpha-induced NF-kappaB activation and induces apoptosis in human prostate carcinoma PC-3 cells. Mol. Carcinog. 2008, 47, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Pisha, E.; Chai, H.; Lee, I.S.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Beecher, C.W.; Fong, H.H.; Kinghorn, A.D.; Brown, D.M. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat. Med. 1995, 1, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Meng, R.D.; El-Deiry, W.S. p53-independent upregulation of KILLER/DR5 TRAIL receptor expression by glucocorticoids and interferon-gamma. Exp. Cell Res. 2001, 262, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Friesen, C.; Los, M.; Scaffidi, C.; Mier, W.; Benedict, M.; Nuñez, G.; Krammer, P.H.; Peter, M.E.; Debatin, K.M. Betulinic acid triggers CD95 (APO-1/Fas)- and p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res. 1997, 57, 4956–4964. [Google Scholar] [PubMed]
- Strasser, A.; Harris, A.W.; Jacks, T.; Cory, S. DNA damage can induce apoptosis in proliferating lymphoid cells via p53-independent mechanisms inhibitable by Bcl-2. Cell 1994, 79, 329–339. [Google Scholar] [CrossRef]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappaB signaling: Balancing life and death—A new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Aggarwal, B.B. Betulinic acid suppresses carcinogen-induced NF-kappa B activation through inhibition of I kappa B alpha kinase and p65 phosphorylation: Abrogation of cyclooxygenase-2 and matrix metalloprotease-9. J. Immunol. 2003, 171, 3278–3286. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Scaffidi, C.; Susin, S.A.; Krammer, P.H.; Kroemer, G.; Peter, M.E.; Debatin, K.M. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem. 1998, 273, 33942–33948. [Google Scholar] [CrossRef] [PubMed]
- Udeani, G.O.; Zhao, G.M.; Geun Shin, Y.; Cooke, B.P.; Graham, J.; Beecher, C.W.; Kinghorn, A.D.; Pezzuto, J.M. Pharmacokinetics and tissue distribution of betulinic acid in CD-1 mice. Biopharm. Drug Dispos. 1999, 20, 379–383. [Google Scholar] [CrossRef]
- Srivastava, J.K.; Gupta, S. Antiproliferative and apoptotic effects of chamomile extract in various human cancer cells. J. Agric. Food Chem. 2007, 55, 9470–9478. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Gupta, S. Apigenin-induced prostate cancer cell death is initiated by reactive oxygen species and p53 activation. Free Radic. Biol. Med. 2008, 44, 1833–1845. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shankar, E.; Zhang, A.; Franco, D.; Gupta, S. Betulinic Acid-Mediated Apoptosis in Human Prostate Cancer Cells Involves p53 and Nuclear Factor-Kappa B (NF-κB) Pathways. Molecules 2017, 22, 264. https://doi.org/10.3390/molecules22020264
Shankar E, Zhang A, Franco D, Gupta S. Betulinic Acid-Mediated Apoptosis in Human Prostate Cancer Cells Involves p53 and Nuclear Factor-Kappa B (NF-κB) Pathways. Molecules. 2017; 22(2):264. https://doi.org/10.3390/molecules22020264
Chicago/Turabian StyleShankar, Eswar, Ailin Zhang, Daniel Franco, and Sanjay Gupta. 2017. "Betulinic Acid-Mediated Apoptosis in Human Prostate Cancer Cells Involves p53 and Nuclear Factor-Kappa B (NF-κB) Pathways" Molecules 22, no. 2: 264. https://doi.org/10.3390/molecules22020264
APA StyleShankar, E., Zhang, A., Franco, D., & Gupta, S. (2017). Betulinic Acid-Mediated Apoptosis in Human Prostate Cancer Cells Involves p53 and Nuclear Factor-Kappa B (NF-κB) Pathways. Molecules, 22(2), 264. https://doi.org/10.3390/molecules22020264