
Metal Fluorides, Metal Chlorides and Halogenated Metal Oxides as Lewis Acidic Heterogeneous Catalysts. Providing Some Context for Nanostructured Metal Fluorides

Abstract
:1. Introduction
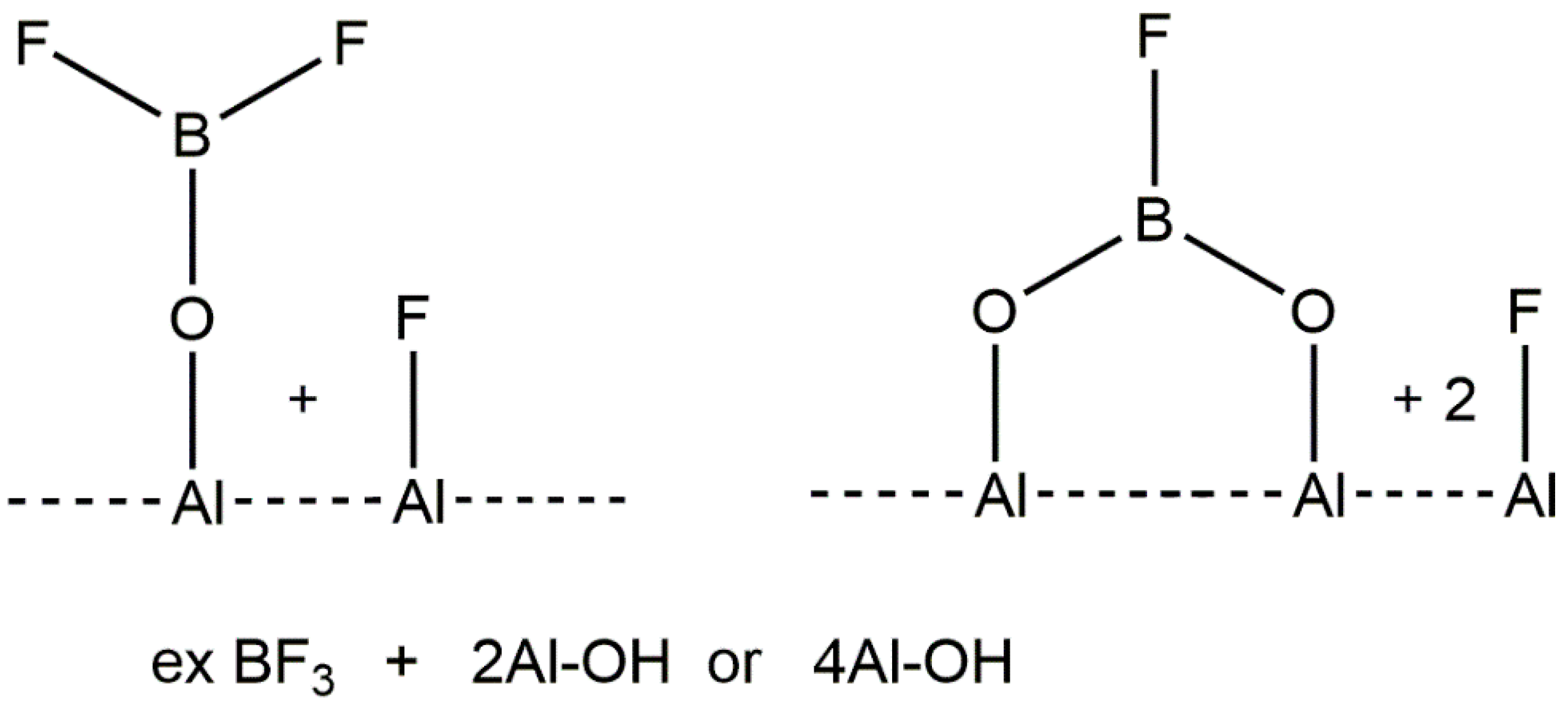
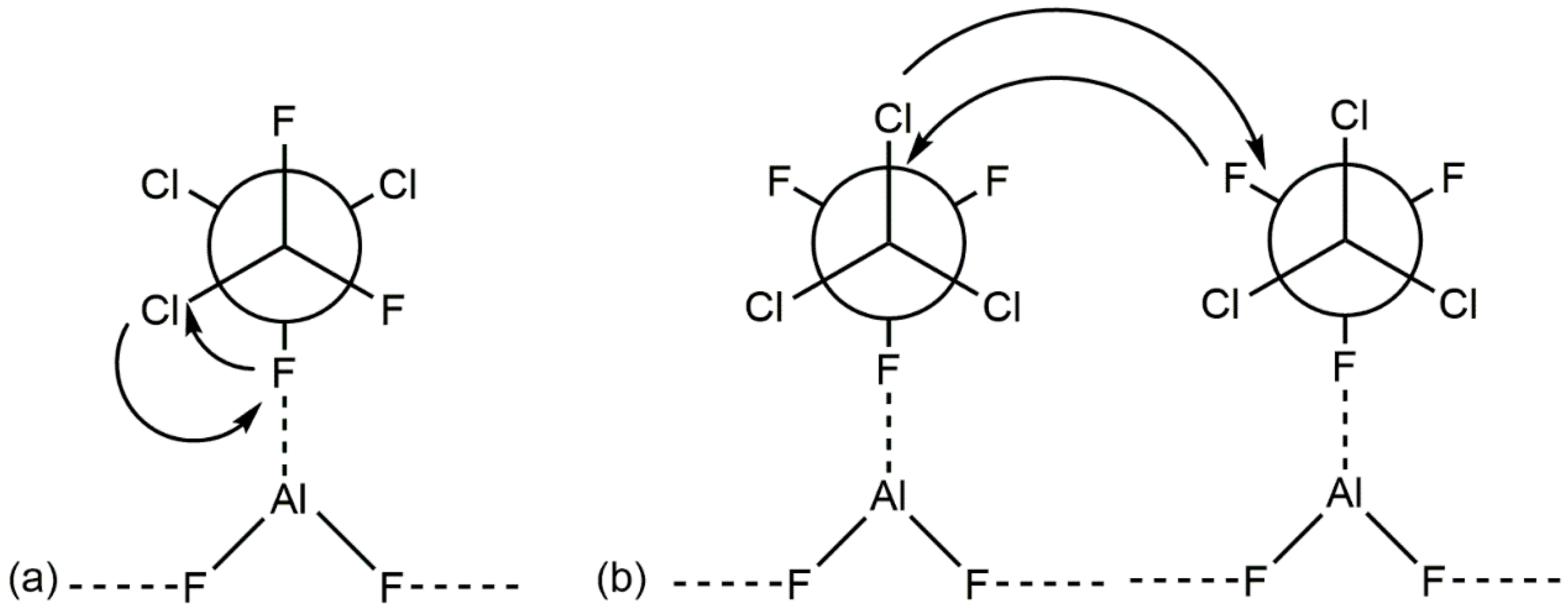
2. Aluminum Fluorides and Halogenated Aluminas
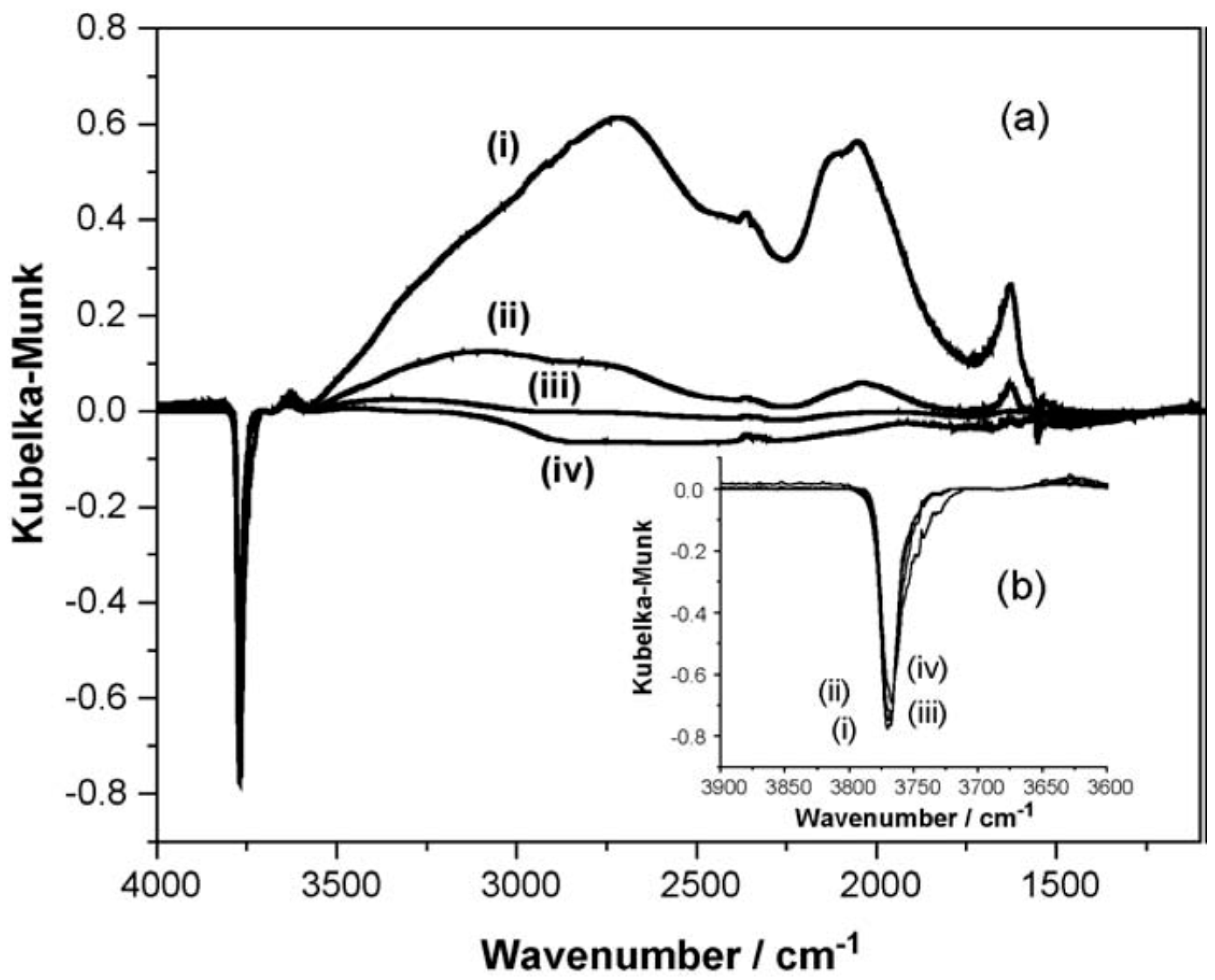
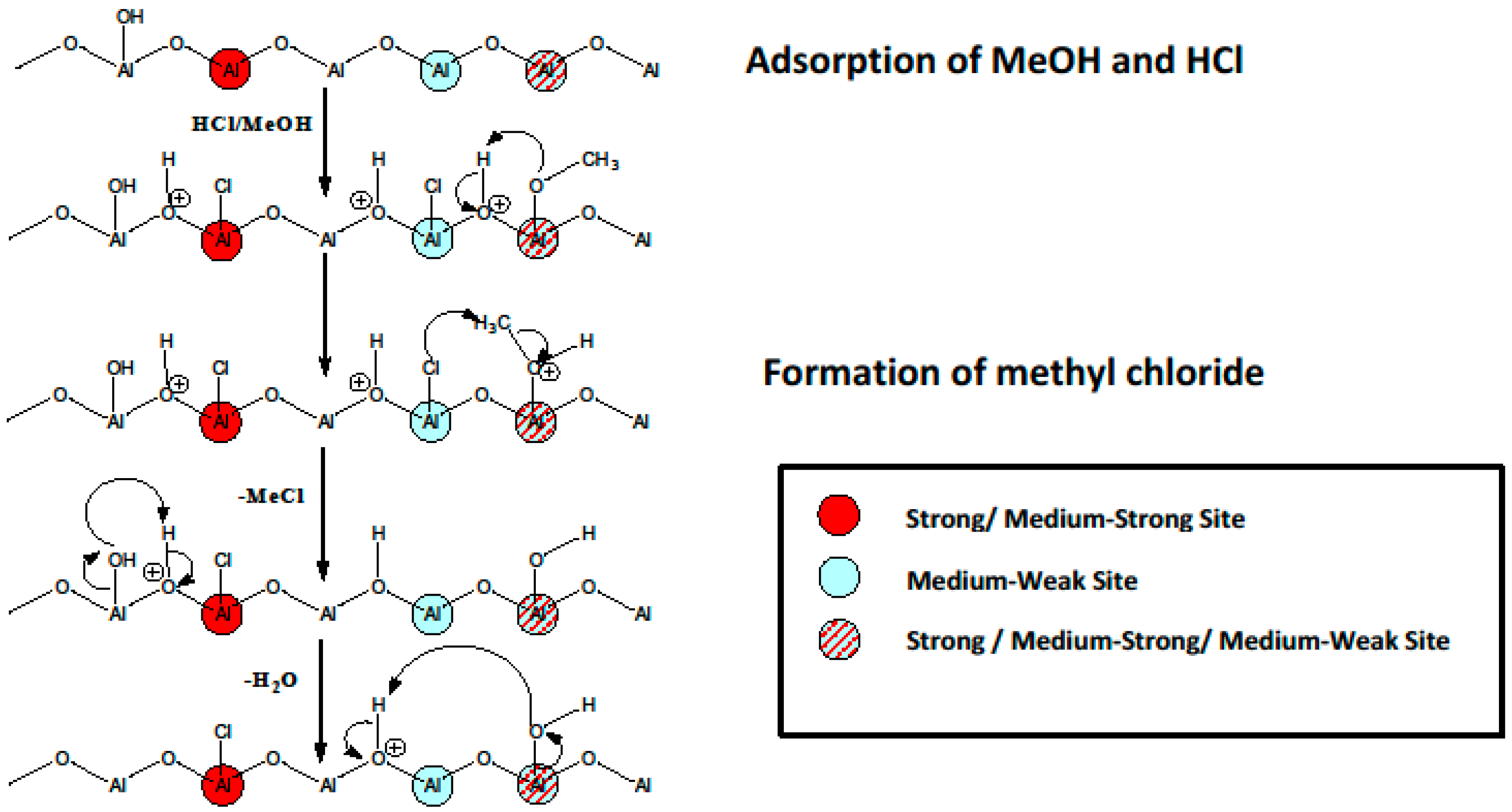
3. Anhydrous Hydrogen Chloride as a Surface Probe
4. Iron(III) and Copper(II) Chlorides as Chlorination Catalysts: Redox and Lewis Acid Possibilities
5. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Tressaud, A. Functionalised Inorganic Fluorides; Wiley: Chichester, UK, 2010. [Google Scholar]
- Lennon, D.; Winfield, J.M. Heterogeneous Catalysts used for Large-Scale Syntheses of Selected Catalytic Applications; Parvulescu, V.I., Kemnitz, E., Eds.; Elsevier: Amsterdam, The Netherlands; Oxford, UK; Boston, MA, USA, 2016; Chapter 7; pp. 193–217. [Google Scholar]
- Le Bail, A.; Jacaboni, C.; Leblanc, M.; De Pape, R.; Duroy, H.; Fourquet, J.L. Crystal Structure of the Metastable form of Aluminium Trifluoride βAlF3 and the Gallium and Indium Homologues. J. Solid State Chem. 1988, 77, 96–101. [Google Scholar]
- Hess, A.; Kemnitz, E. Characterisation of Catalytically Active Sites on Aluminium Oxides, Hydroxyfluorides and Fluorides in Correlation with their Catalytic Behaviour. J. Catal. 1994, 149, 449–457, and references therein. [Google Scholar]
- Kemnitz, E.; Winfield, J.M. Fluoride Catalysts: Their Application to Heterogeneous Catalytic Fluorination and Related Processes. In Advanced Inorganic Fluorides: Synthesis, Characterization and Applications; Nakajima, T., Žemva, B., Tressaud, A., Eds.; Elsevier Science S.A.: Lausanne, Switzerland, 2000; Chapter 12; pp. 367–401. [Google Scholar]
- Krespan, C.G.; Petrov, V.A. The Chemistry of Highly Fluorinated Carbocations. Chem. Rev. 1996, 96, 3269–3301, and references therein. [Google Scholar]
- Krahl, T.; Kemnitz, E. The Very Strong Lewis Acids Aluminium Chlorofluoride (ACF) and Bromofluoride (ABF)—Synthesis, Structure and Lewis Acidity. J. Fluor. Chem. 2006, 127, 663–678, and references therein. [Google Scholar]
- Kemnitz, E.; Skapin, T.; Winfield, J.M. Preparation of Fluorinated γ-Alumina. In Efficient Preparations of Fluorine Compounds, 1st ed.; Roesky, H.W., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Chapter 67; pp. 442–446. [Google Scholar]
- Klapötke, T.M.; McMonagle, F.; Spence, R.R.; Winfield, J.M. γ-Alumina-Supported Boron Trifluoride: Catalysis, Radiotracer Studies and Computations. J. Fluor. Chem. 2006, 127, 1446–1453. [Google Scholar]
- Vimont, A.; Daturi, M.; Winfield, J.M. Investigation of Surface Acidity using a Range of Probe Molecules. In Functionalised Inorganic Fluorides; Tressaud, A., Ed.; Wiley: Chichester, UK, 2010; Chapter 4; pp. 101–139. [Google Scholar]
- Winfield, J.M. Investigating Acidity of Metal Fluoride Surfaces by Spectroscopic and Chemical Methods. J. Fluor. Chem. 2009, 130, 1069–1079. [Google Scholar]
- Nickkho-Amiry, M.; Winfield, J.M. Investigation of Fluorinated Surfaces by means of Radio-Labelled Probe Molecules. J. Fluor. Chem. 2007, 128, 344–352. [Google Scholar]
- Nickkho-Amiry, M.; Eltanany, G.; Wüttke, S.; Rüdiger, S.; Kemnitz, E.; Winfield, J.M. A Comparative Study of Surface Activity in the Amorphous, High Surface Area Solids, Aluminium Fluoride, Magnesium fluoride and Magnesium Fluoride containing Iron(III) or Aluminium(III) Fluoride. J. Fluor. Chem. 2008, 129, 366–375. [Google Scholar]
- McInroy, A.R.; Lundie, D.T.; Winfield, J.M.; Dudman, C.C.; Jones, P.; Parker, S.F.; Lennon, D. The Interaction of Alumina with HCl: An Infrared Spectroscopy, Temperature-Programmed Desorption and Inelastic Neutron Scattering Study. Catal. Today 2006, 114, 403–411. [Google Scholar]
- McInroy, A.L.; Winfield, J.M.; Dudman, C.C.; Jones, P.; Lennon, D. The Development of a New Generation of Methyl Chloride Synthesis Catalysts. Faraday Discuss. 2016, 188, 467–499. [Google Scholar]
- Lundie, D.T.; McInroy, A.R.; Marshall, R.; Winfield, J.M.; Jones, P.; Dudman, C.C.; Parker, S.F.; Mitchell, C.; Lennon, D. Improved Description of the Surface Acidity of η-Alumina. J. Phys. Chem. B 2005, 109, 11592–11601. [Google Scholar]
- Zhang, X.; Fan, X.; Niu, H.; Wang, J. An Ionic Liquid As a Recyclable Medium for the Green Preparation of α,α′-Bis(substituted benzylidine)cycloalkanones Catalysed by FeCl3·6H2O. Green Chem. 2003, 5, 267–269. [Google Scholar]
- Kabalka, G.W.; Yao, M.-L.; Borella, S.; Goins, L.K. Iron Trichloride Mediated Allylation of Lithium Alkoxides through an Unusual Carbon-Oxygen Bond Cleavage. Organometallics 2007, 26, 4112–4114. [Google Scholar]
- Xa, W.-W.; Lin, M.; Huang, Y.; Chen, L.; Zhan, Z.-P. Iron Trichloride Catalysed One-Pot Synthesis of Substituted Oxazoles from Propargylic Acetates and Amides under Microwave Irradiation. Lett. Org. Chem. 2009, 6, 664–665. [Google Scholar]
- Cantagrei, G.; De Canné-Carnavalet, B.; Meyer, C.; Cossy, J. IronTrichloride Promoted Cyclisation of O-Alkynylaryl Isocyanates. Synthesis of 3-(Chloromethylene)Oxindoles. Org. Lett. 2009, 11, 8584–8586. [Google Scholar]
- Schröder, K.; Enthaler, S.; Bitterlich, B.; Schulz, T.; Spannenberg, A.; Tse, M.K.; Junge, K.; Beller, M. Design of and Mechanistic Studies on a Biomimetic Iron-Imidazole Catalyst System for Epoxidation of Olefins with Hydrogen Peroxide. Chem. Eur. J. 2009, 15, 5471–5481. [Google Scholar]
- Buchwald, S.L.; Bolm, C. On the Role of Metal Contaminants in Catalyses with FeCl3. Angew. Chem. Int. Ed. 2009, 48, 5586–5587. [Google Scholar]
- Schröder, K.; Junge, K.; Spannenberg, A.; Beller, M. Design of a Bio-Inspired Imidazole-Based Iron Catalyst for Epoxidation of Olefins: Mechanistic Insights. Catal. Today 2010, 157, 364–370. [Google Scholar]
- Su, L.; Lei, C.-Y.; Fan, W.-Y.; Liu, L.-X. FeCl3-Mediated Reaction of Alkynols with Iodine: An Efficient and Convenient Route to Vinyl Iodides. Synth. Commun. 2011, 41, 1200–1207. [Google Scholar]
- Blevins, D.W.; Yao, M.-L.; Yong, L. Iron Trichloride Promoted Hydrolysis of Potassium Organotrifluoroborates. Tetrahedron Lett. 2011, 52, 6534–6536. [Google Scholar]
- Yeh, M.-C.P.; Fang, C.-W.; Lin, H.-H. Facile Synthesis of Azaspirocycles via Iron Trichloride Promoted Cyclisation/Chlorination of Cyclic 8-Aryl-5-aza-5-tosyl-2-en-7-yn-1-ols. Org. Lett. 2012, 14, 1830–1833. [Google Scholar]
- Della Casa, C.; Bertinelli, F.; Costa Bizzarri, P.; Salatelli, E. Properties of a Hydroxydecyl-Functionalized Polythiophene Synthesized by the Iron Trichloride Route. Adv. Mater. 1995, 7, 1005–1007. [Google Scholar]
- Tanaka, K.; Mihara, T.; Koide, N. Synthesis and Physical Properties of Regioregular Poly(3-Alkoxy-4-Methylthiophene)s. Polym. J. 2004, 36, 628–663. [Google Scholar]
- Svoboda, J.; Bláha, M.; Sedláček, J.; Vohlídal, H.; Balcar, H.; Mav-Golež, I.; Žigon, M. New Approaches to the Synthesis of Pure Conjugated Polymers. Acta Chim. Slov. 2006, 53, 407–416. [Google Scholar]
- Cloutet, E.; Mumtaz, M.; Cramail, H. Synthesis of PEDOT Latexes by Dispersion Polymerization in Aqueous Media. Mater. Sci. Eng. C 2009, 29, 377–382. [Google Scholar]
- Lanzi, M.; Paganin, L.; Errani, F. Synthesis, Characterization and Photovoltaic Properties of a New Thiophene-Based Double-Cable Polymer with Pendent Fullerene Group. Polymer 2012, 53, 2134–2145. [Google Scholar]
- Reed, D.J. Chlorocarbons and Chlorohydrocarbons. In Kirk-Othmer Encyclopaedia of Chemical Technology, 4th ed.; Kroschwitz, J.I., Howe-Grant, M., Eds.; John Wiley & Sons: New York, NY, USA, 1993; Volume 5, pp. 1017–1028. [Google Scholar]
- Weissermel, K.; Arpe, H.-J. Industrial Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2003; pp. 449–450. [Google Scholar]
- Delaude, L.; Lazlo, P. Aromatic Chlorination of Toluene and of Anisole Using Clay-Supported FeCl3 and m-Chloroperbenzoic Acid. A Biomimetic Approach. Catal. Lett. 1990, 5, 35–44. [Google Scholar]
- Kawaguchi, T.; Suzuki, Y.; Saito, R. Trichloroethylene and Perchloroethylene Coproduction by Ethylene Process. Ind. Eng. Chem. 1970, 62, 36–41. [Google Scholar]
- Sutherland, I.W.; Hamilton, N.G.; Dudman, C.C.; Jones, P.; Lennon, D.; Winfield, J.M. Chlorination and Dehydrochlorination Reactions Relevant to the Manufacture of Trichloroethene and Tetrachloroethene. Part 1. Reaction Pathways. Appl. Catal. A Gen. 2011, 399, 1–11. [Google Scholar]
- Sutherland, I.W.; Hamilton, N.G.; Dudman, C.C.; Jones, P.; Lennon, D.; Winfield, J.M. Chlorination Reactions Relevant to the Manufacture of Trichloroethene and Tetrachloroethene. Part 2. Effects of Chlorine Supply. Appl. Catal. A Gen. 2014, 471, 149–156. [Google Scholar]
- Taylor, D.S.C. Catalysis in the System 1,2-Dichloroethane, Iron(III) Chloride. Ph.D. Thesis, University of Glasgow, Glasgow, UK, 2002. [Google Scholar]
- Fontana, C.M.; Gorin, E.; Kidder, G.A.; Meredith, C.S. Ternary System, Cuprous Chloride-Cupric Chloride-Potassium Chloride and its Equilibrium Chlorine Pressures. Ind. Eng. Chem. 1952, 44, 363–368. [Google Scholar]
- Arganbright, R.P.; Yates, W.F. Chlorinations with Cupric Chloride. J. Org. Chem. 1962, 27, 1205–1208. [Google Scholar]
- Cavani, F. Catalytic Selective Oxidation: The Forefront in the Challenge for a more Sustainable Chemical Industry. Catal. Today 2010, 157, 8–15. [Google Scholar]
- Leofanti, G.; Padovan, M.; Garilli, M.; Carmello, D.; Zecchina, A.; Spoto, G.; Bordiga, S.; Turnes Palomino, G.; Lamberti, C. Alumina-Supported Copper Chloride 1. Characterization of freshly Prepared Catalyst. J. Catal. 2000, 189, 91–104. [Google Scholar]
- Leofanti, G.; Padovan, M.; Garilli, M.; Carmello, D.; Marra, G.L.; Zecchina, A.; Spoto, G.; Bordiga, S.; Lamberti, C. Alumina-Supported Copper Chloride 2. Effect of Aging and Thermal Treatments. J. Catal. 2000, 189, 105–116. [Google Scholar]
- Leofanti, G.; Marsella, A.; Cremaschi, B.; Garilli, M.; Zecchina, A.; Spoto, G.; Bordiga, S.; Fisicaro, P.; Berlier, G.; Prestipino, C.; et al. Alumina-Supported Copper Chloride 3. Effect of Exposure to Ethylene. J. Catal. 2001, 202, 279–295. [Google Scholar]
- Leofanti, G.; Marsella, A.; Cremaschi, B.; Garilli, M.; Zecchina, A.; Spoto, G.; Bordiga, S.; Fisicaro, P.; Prestipino, C.; Villain, F.; et al. Alumina-Supported Copper Chloride 4. Effect of Exposure to O2 and HCl. J. Catal. 2002, 205, 375–381. [Google Scholar]
- Lamberti, C.; Prestipino, C.; Bonino, F.; Capello, L.; Bordiga, S.; Spoto, G.; Zecchina, A.; Diaz Moreno, S.; Cremaschi, B.; Garilli, M.; et al. The Chemistry of the Oxychlorination Catalyst: An In Situ, Time-Resolved XANES Study. Angew. Chem. Int. Ed. 2002, 41, 2341–2344. [Google Scholar]
- Prestipino, C.; Bordiga, S.; Lamberti, C.; Vidotto, S.; Garilli, M.; Cremaschi, B.; Marsella, A.; Leofanti, G.; Fisicaro, P.; Spoto, G.; et al. Structural Determination of Copper Species on the Alumina-Supported Copper Chloride Catalyst: A Detailed EXAFS Study. J. Phys. Chem. B 2003, 107, 5022–5030. [Google Scholar]
- Muddada, N.B.; Olsbye, U.; Caccialupi, L.; Cavani, F.; Leofanti, G.; Gianolio, D.; Bordiga, S.; Lamberti, C. Influence of Additives in Defining the Active Phase of the Ethylene Oxychlorination Catalyst. Phys. Chem. Chem. Phys. 2010, 12, 5605–5618. [Google Scholar]
- Muddada, N.B.; Olsbye, U.; Leofanti, G.; Gianolio, D.; Bonino, F.; Bordiga, S.; Fuglerud, T.; Vidotto, S.; Marsella, A.; Lamberti, C. Quantification of Copper Phases, their Reducibility and Dispersion in Doped-CuCl2/Al2O3 Catalysts for Ethylene Oxychlorination. Dalton Trans. 2010, 39, 8437–8449. [Google Scholar]
- Zhang, T.; Troll, C.; Rieger, B.; Kintrup, J.; Schlüter, O.F.-K.; Weber, R. Reaction Kinetics of Oxychlorination of Carbon Monoxide to Phosgene based on Copper(II) Chloride. Appl. Catal. A Gen. 2009, 357, 51–57. [Google Scholar]
- Zhang, T.; Troll, C.; Rieger, B.; Kintrup, J.; Schlüter, O.F.-K.; Weber, R. Composition Optimisation of Silica-Supported Copper(II) Chloride Substance for Phosgene Production. Appl. Catal. A Gen. 2009, 365, 20–27. [Google Scholar]
- Zhang, T.; Troll, C.; Rieger, B.; Kintrup, J.; Schlüter, O.F.-K.; Weber, R. Oxychlorination of CO to Phosgene in a Three-Step Reaction Cycle and Corresponding Catalytic Mechanism. J. Catal. 2010, 270, 76–85. [Google Scholar]

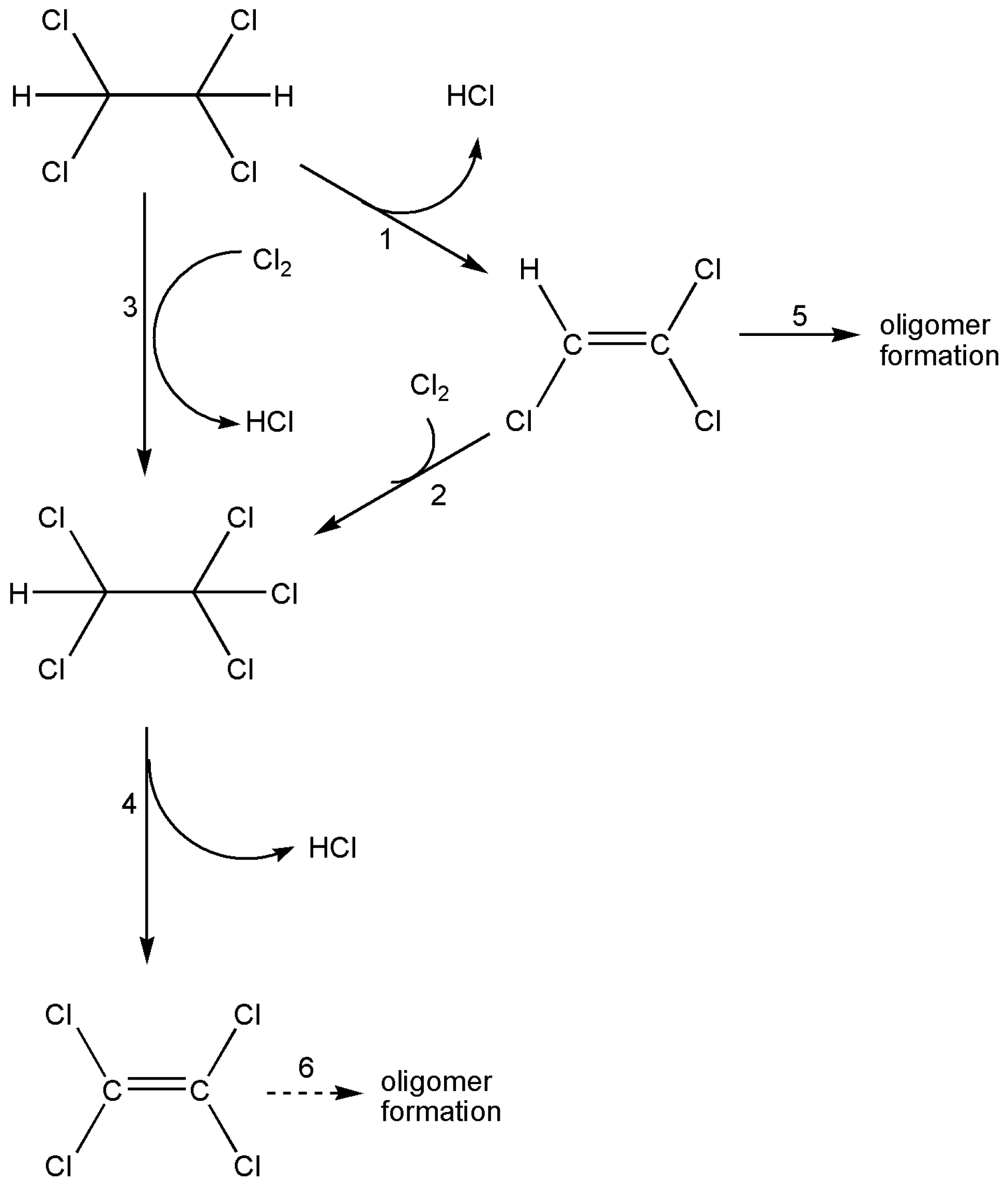

{kind=link}
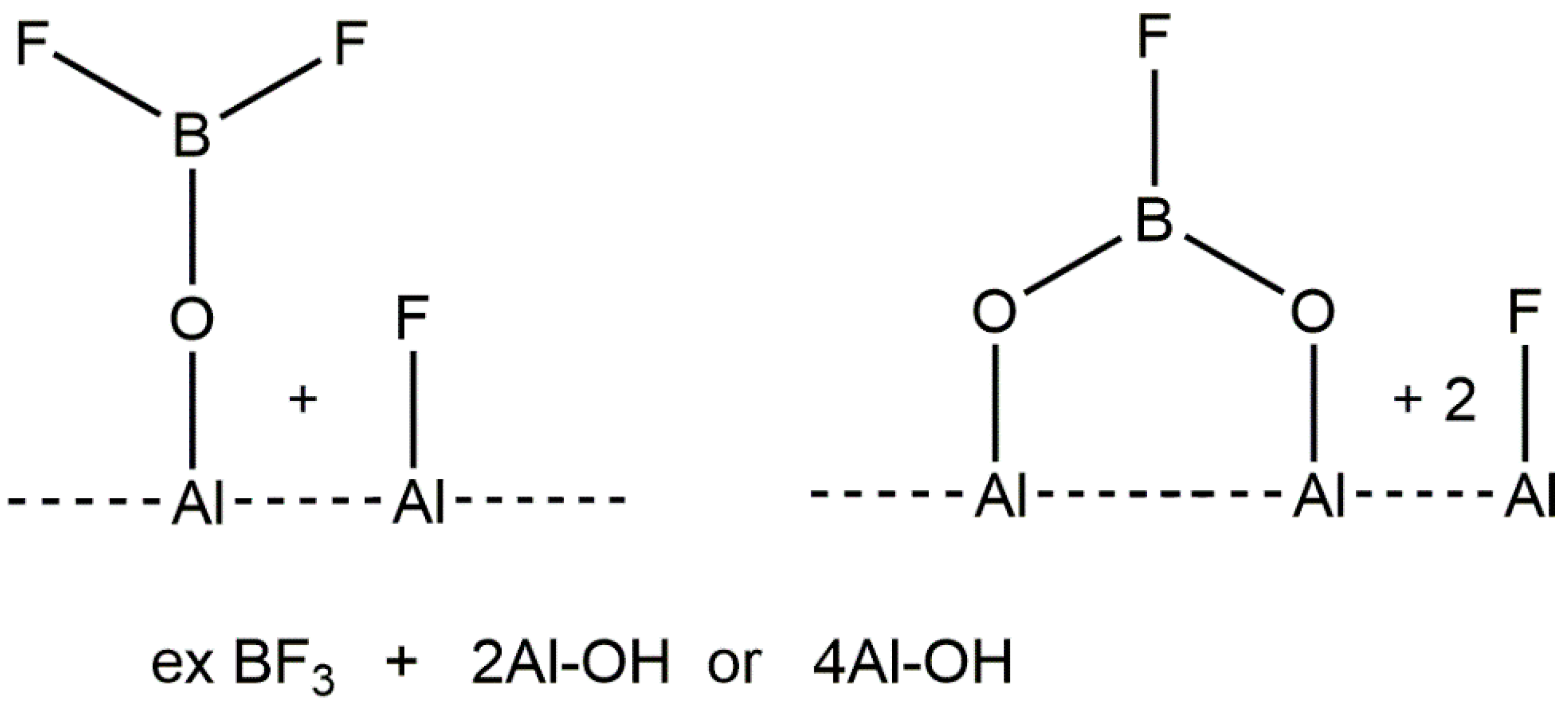
{kind=link}
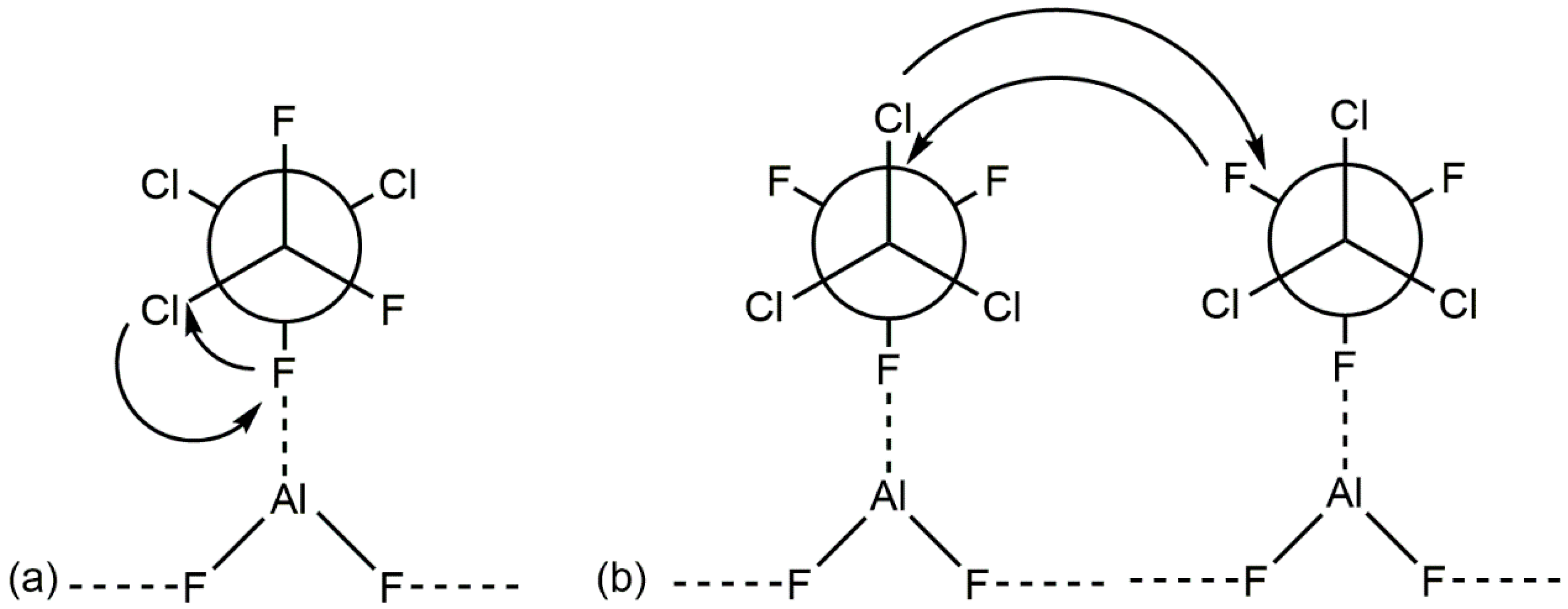
{kind=link}
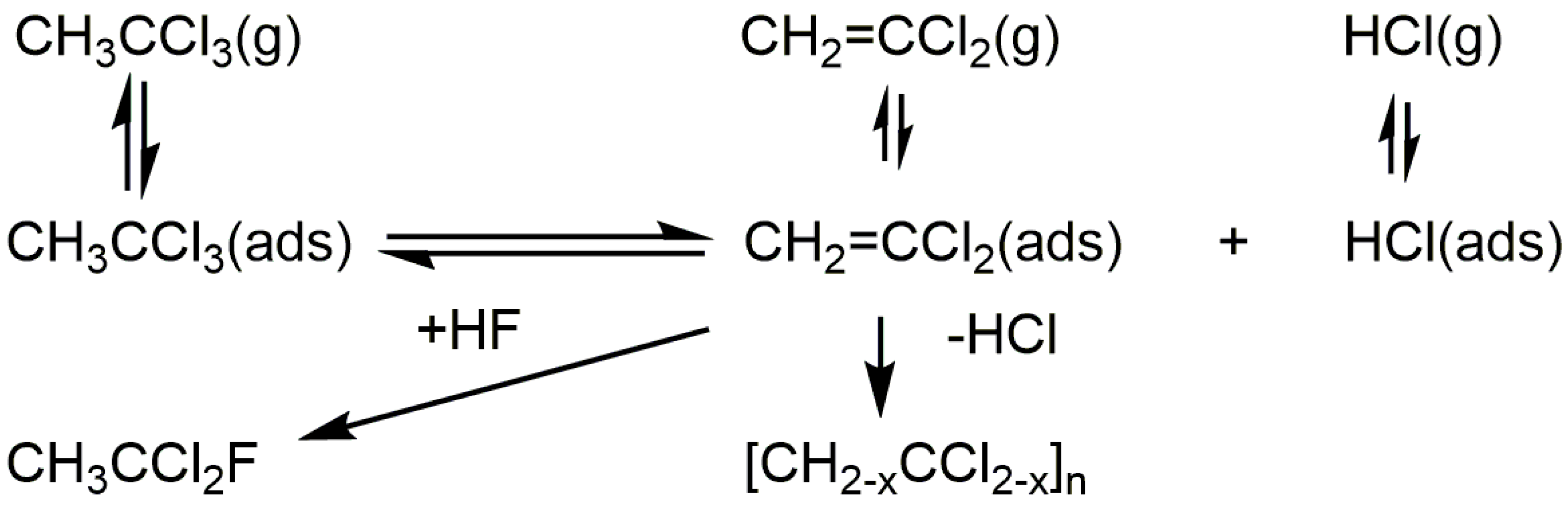
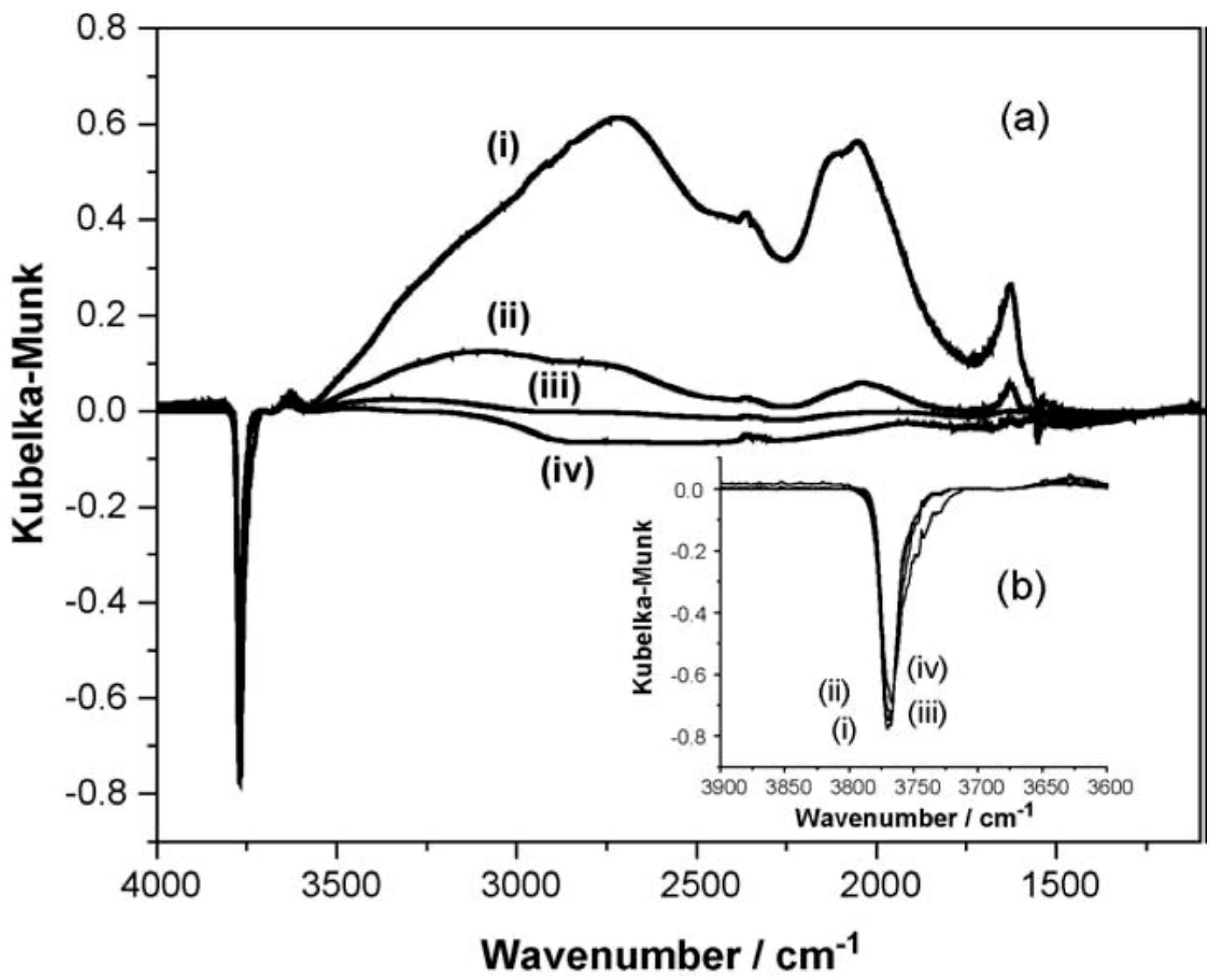{kind=link}
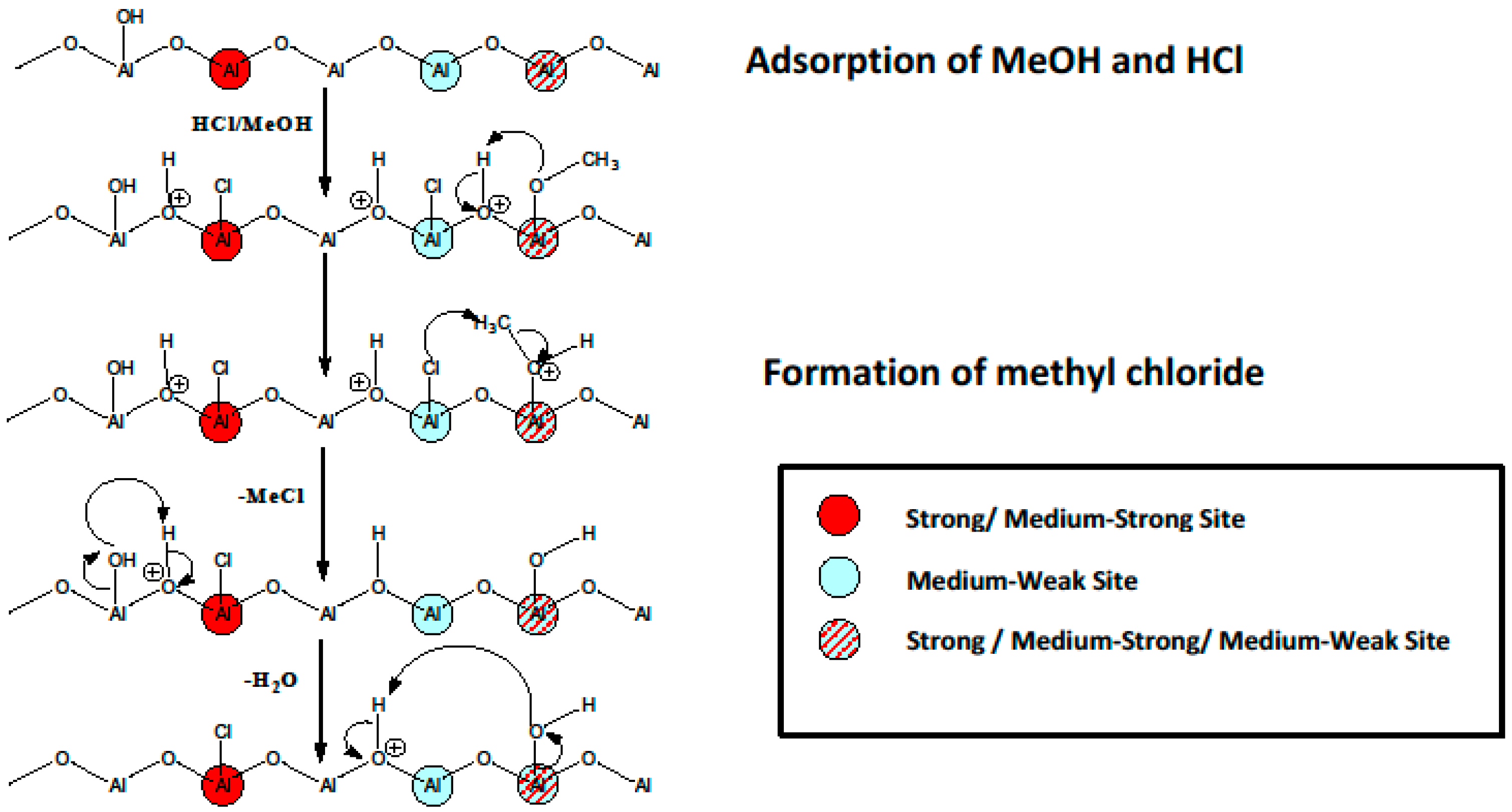{kind=link}
{kind=link}
| Fluorination Conditions a | BET Area (m2·g−1) | Fluorine Content (%) | Other Properties |
|---|---|---|---|
| SF4 static conditions, nominally room temperature, 2 h, procedure repeated ×2 | 80–90 b | ca. 22 c | Some Brønsted sites also |
| SF4 20% in N2, flow conditions, 523 K, 2 h | 67 | 47.1 | γ-alumina present; possibly an amorphous phase also |
| CHF3 20% in N2, then 100%, flow conditions, 623 K, total time 5 h | 34 d | 58.4 | α- and β-AlF3 present; possibly an amorphous phase |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lennon, D.; Winfield, J.M. Metal Fluorides, Metal Chlorides and Halogenated Metal Oxides as Lewis Acidic Heterogeneous Catalysts. Providing Some Context for Nanostructured Metal Fluorides. Molecules 2017, 22, 201. https://doi.org/10.3390/molecules22020201
Lennon D, Winfield JM. Metal Fluorides, Metal Chlorides and Halogenated Metal Oxides as Lewis Acidic Heterogeneous Catalysts. Providing Some Context for Nanostructured Metal Fluorides. Molecules. 2017; 22(2):201. https://doi.org/10.3390/molecules22020201
Chicago/Turabian StyleLennon, David, and John M. Winfield. 2017. "Metal Fluorides, Metal Chlorides and Halogenated Metal Oxides as Lewis Acidic Heterogeneous Catalysts. Providing Some Context for Nanostructured Metal Fluorides" Molecules 22, no. 2: 201. https://doi.org/10.3390/molecules22020201
APA StyleLennon, D., & Winfield, J. M. (2017). Metal Fluorides, Metal Chlorides and Halogenated Metal Oxides as Lewis Acidic Heterogeneous Catalysts. Providing Some Context for Nanostructured Metal Fluorides. Molecules, 22(2), 201. https://doi.org/10.3390/molecules22020201