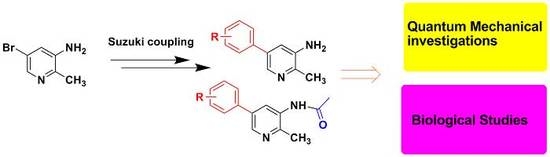
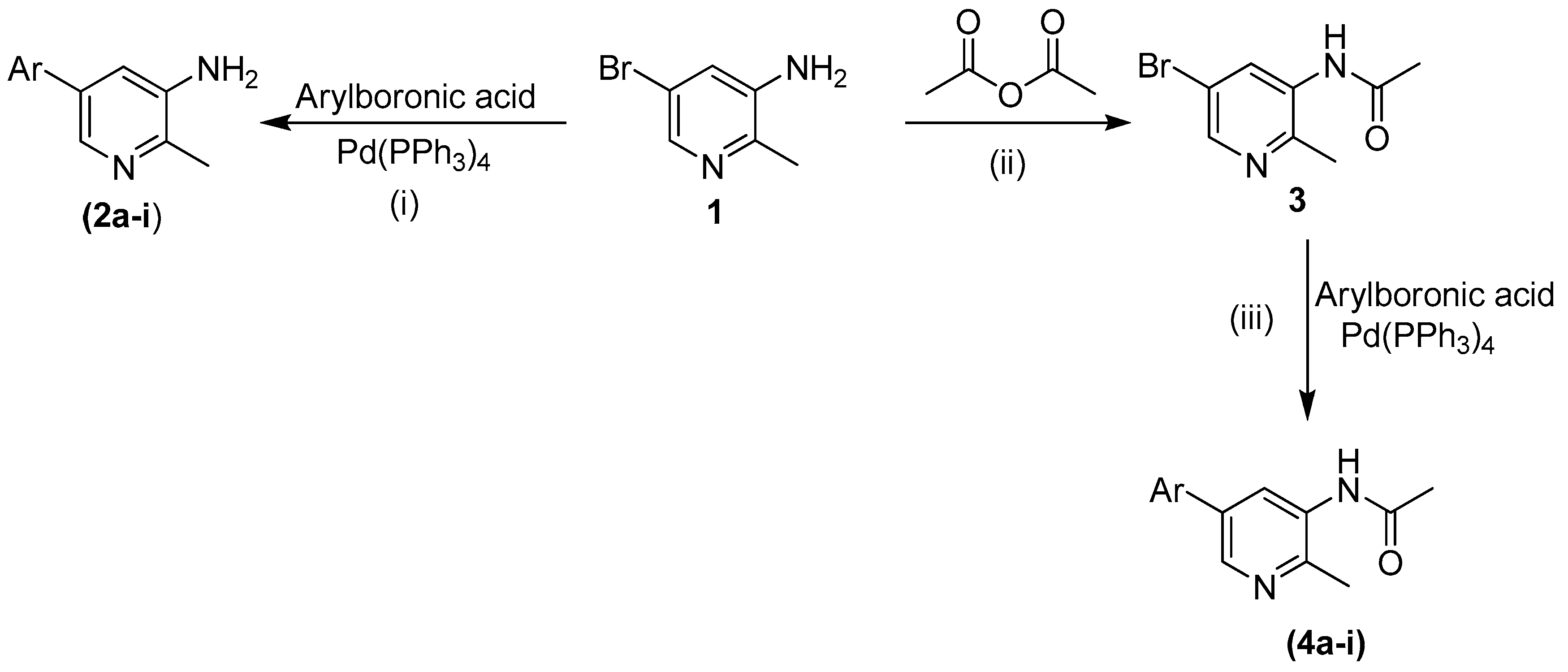
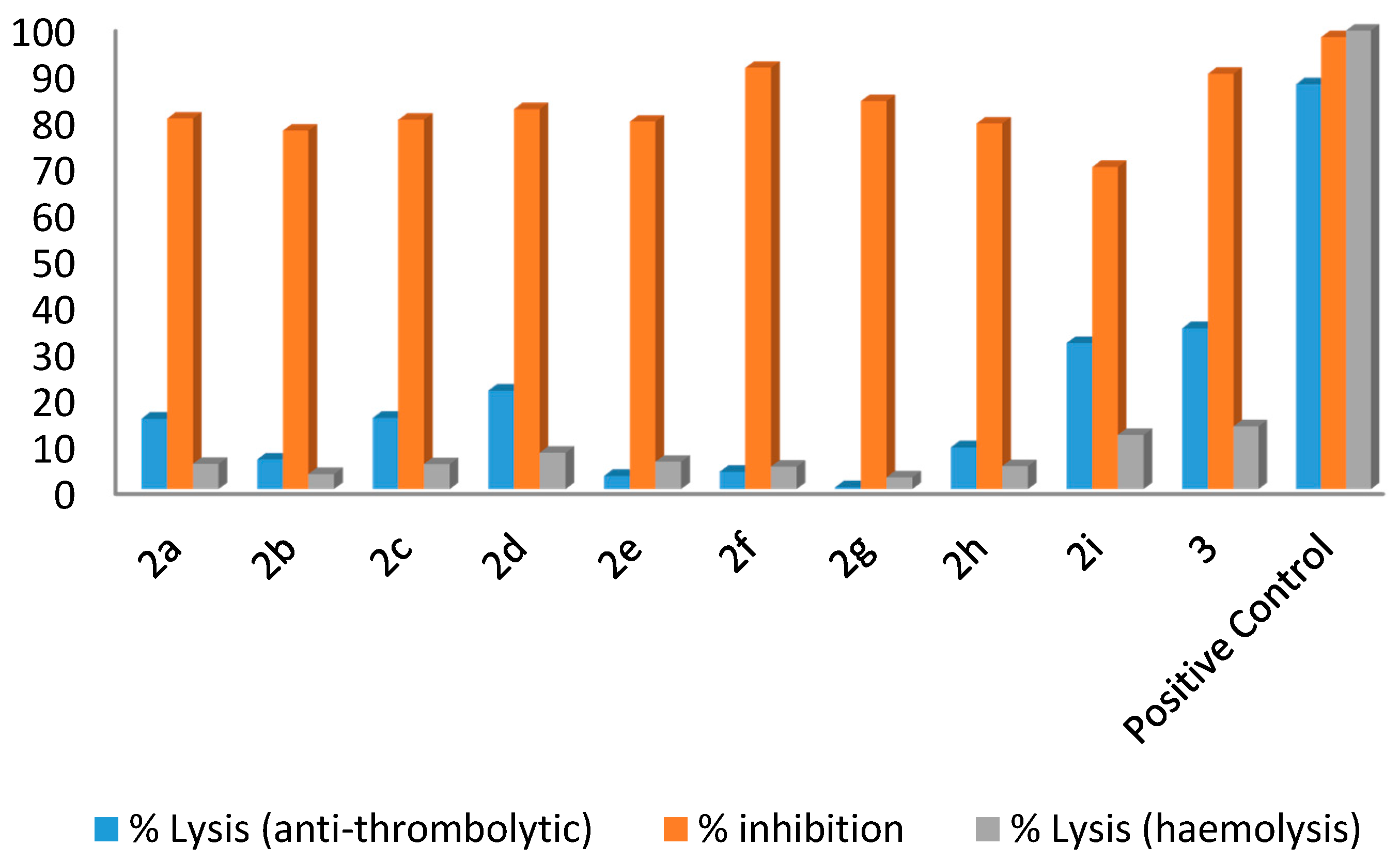
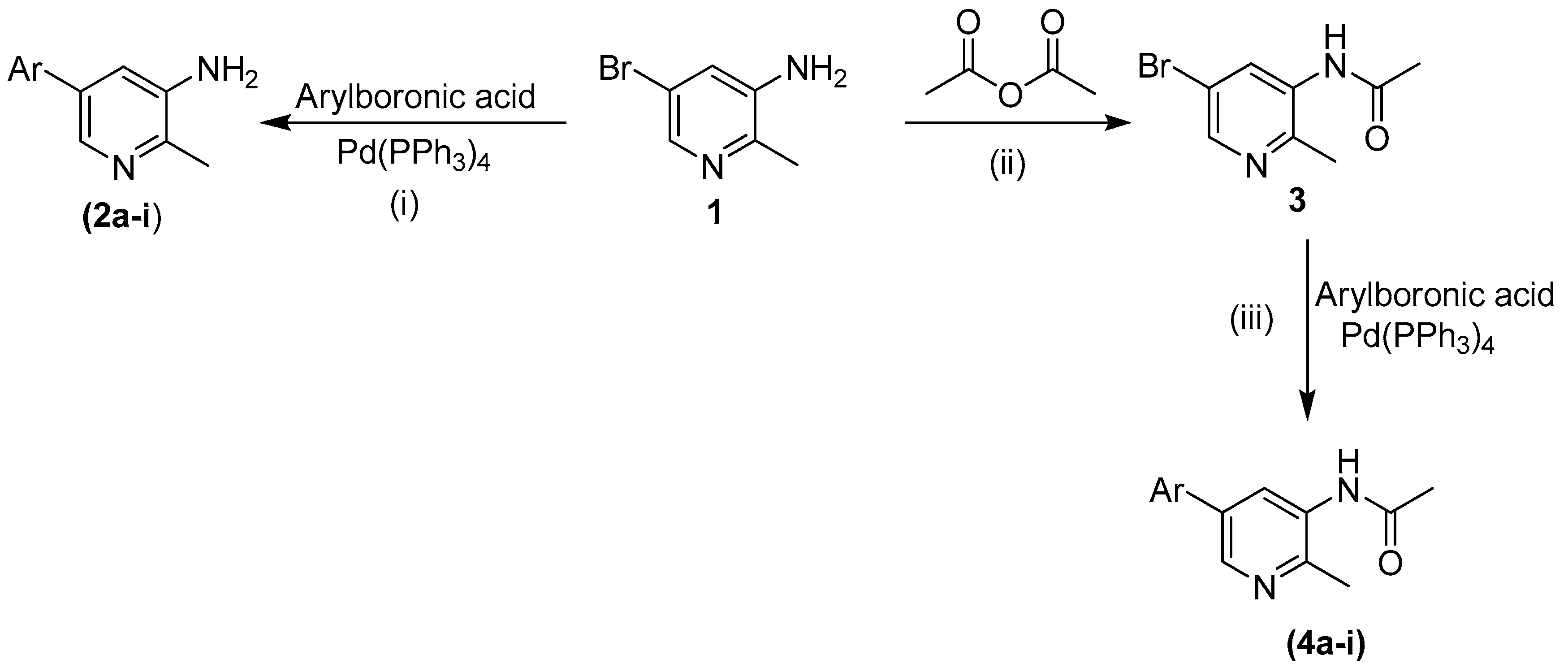
3.2. General Procedure for the Synthesis of Compounds 2a–2i
All compounds were synthesized following the previously reported method [
28]. Briefly 5-bromo-2-methylpyridin-3-amine (
1, 0.2 g with tetrakis(triphenylphosphine)palladium (5 mol %) and 1,4-dioxane (2 mL) were mixed in a Schlenk flask. The reaction mixture was stirred at room temperature for 30 min and then the arylboronic acid (1.1759 mmol), potassium phosphate (2.318 mmol) and water (0.5 mL) were added and the mixture again stirred at 85–95 °C for more than 15 h. After lowering the temperature, the mixture was filtered and diluted with ethyl acetate (50 mL). The excess solvent in the solution was evaporated by rotary evaporator in order to obtain a concentrated solution. Column chromatography was applied to get the desired pure product (silica gel, n-hexane and ethyl acetate). The final products were dried, recrystallized and then characterized using different spectroscopic techniques.
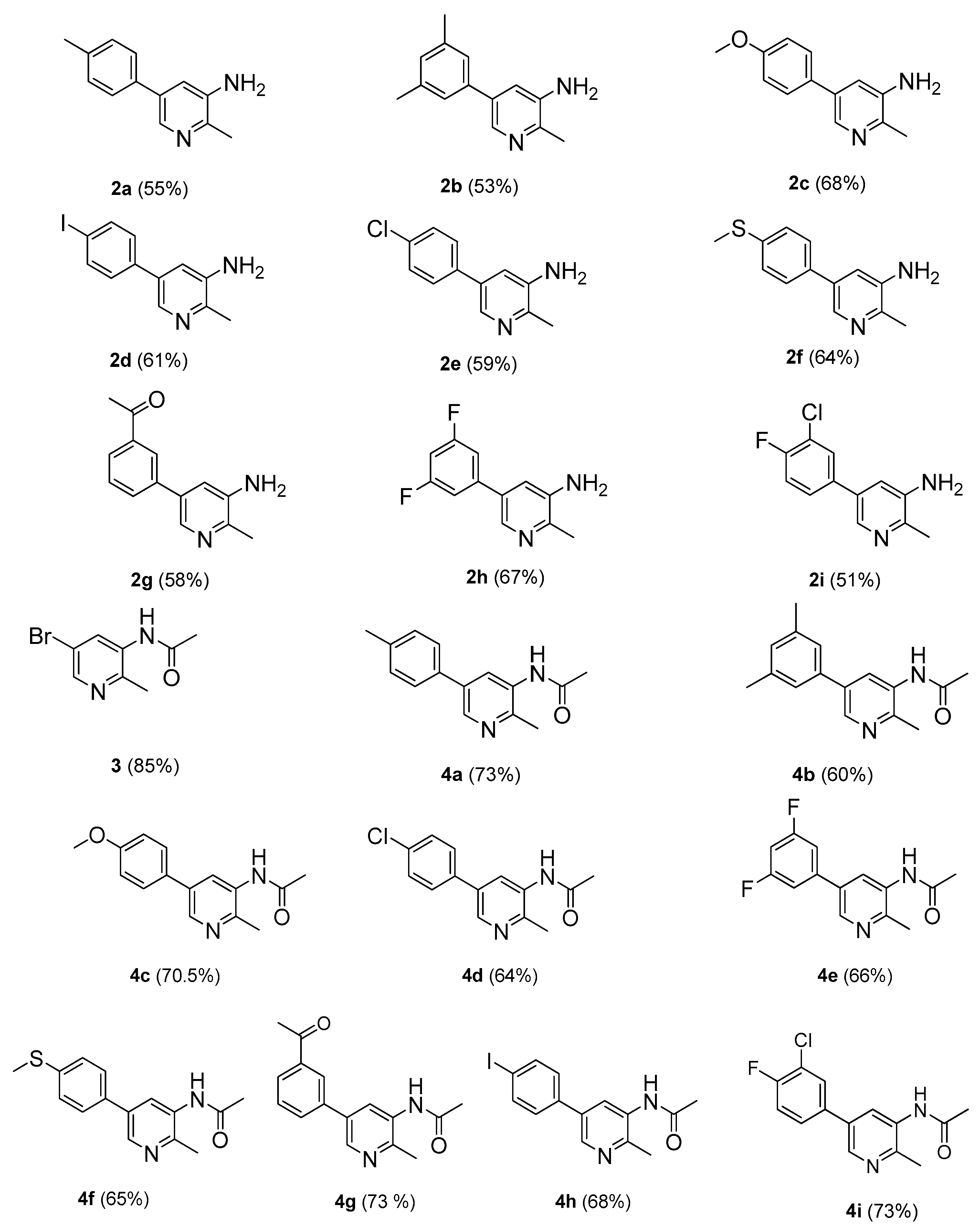
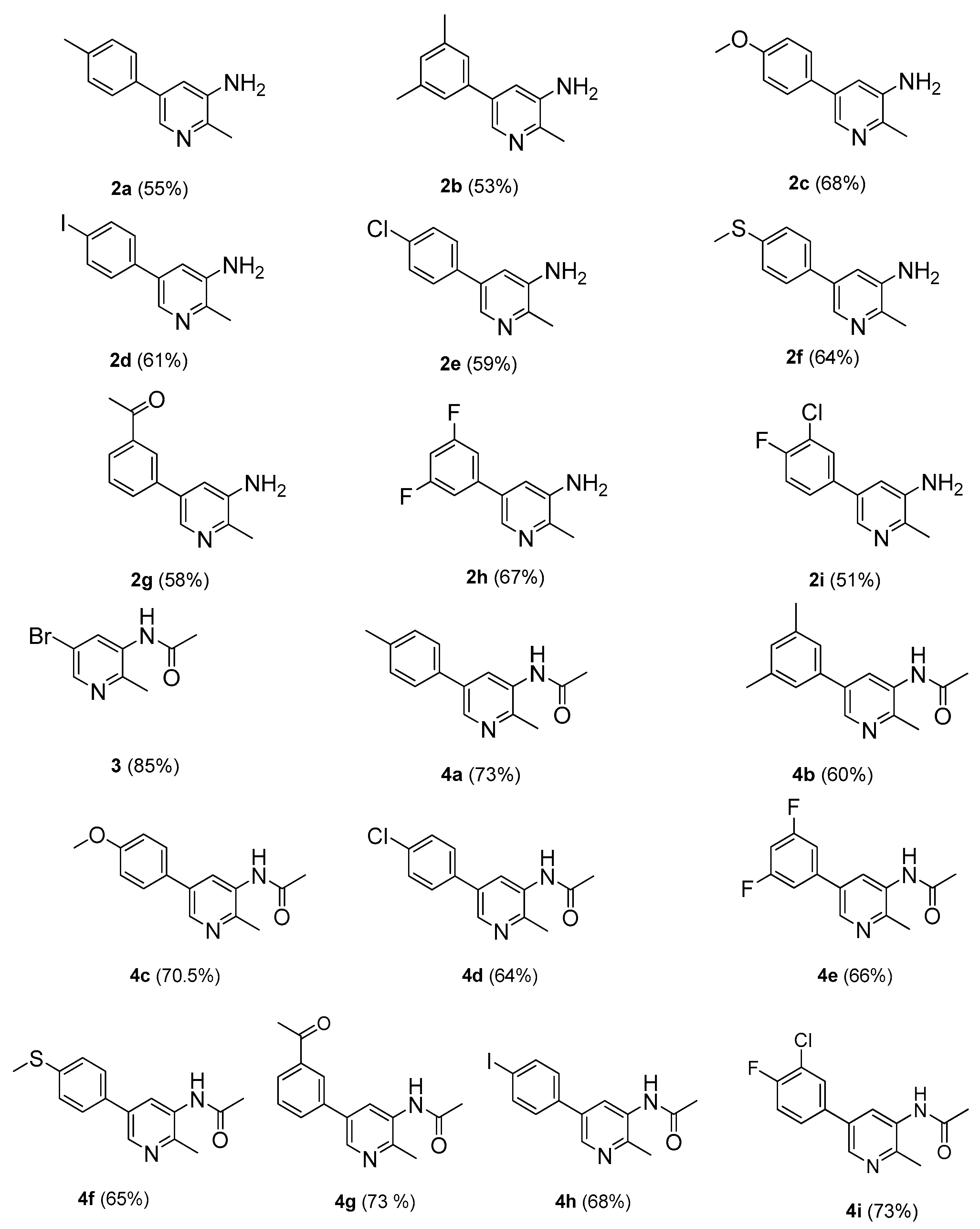
2-Methyl-5(4-methylphenyl)pyridin-3-amine (2a). m.p.: 208–209 °C. 1H-NMR (CDCl3) δ 7.99–7.64 (m, 2H-pyridine), 7.41 (m, 2H-Ar), 7.21 (m, 2H-Ar), 2.50 (s, 3H-methyl), 2.41 (s, 3H-methyl); 13C-NMR (CDCl3 + CD3OD) δ 16.7, 21.3, 120.5, 126.8, 128.8, 132.5, 133.8, 134.9, 144.2, 145.8. EI-MS m/z (+ion mode): 198, [M − NH2]+ = 183, [M − NH2 and CH3]+ = 170, [M − NH2 and 2CH3]+ = 155. Anal. calcd for C13H14N2 (198.26): C, 78.74; H, 7.11. Found: C, 78.69; H, 7.02%.
5-(3,5-Dimethylphenyl)-2-methylpyridin-3-amine (2b). m.p.: 232–233 °C. 1H-NMR (CDCl3) δ 8.2 (s, 1H-pyridine), 7.6 (m, 2H-Ar), 7.3–7.02 (m, 1H-pyridine, 1H-Ar), 2.51 (s, 3H-methyl), 2.41 (s, 3H-methyl); 13C-NMR (CDCl3 + CD3OD) δ 15.5, 21.5, 120.5, 125.3, 130.6, 132.0, 134.1, 136.4, 138.3, 144.3, 145.8. EI-MS m/z (+ion mode): 212, [M − NH2]+ = 197, [M − NH2 and CH3]+ = 183, [M − NH2 and 3CH3]+ = 140. Anal. calcd for C14H16N2 (212.25): C, 79.19; H, 7.59. Found: C, 78.17; H, 7.56%.
5-(4-Methoxyphenyl)-2-methylpyridin-3-amine (2c). m.p.: 232 °C. 1H-NMR (CDCl3) δ 8.12 (s, 1H-pyridine), 7.55 (m, 2H-Ar), 7.31 (m, 1H-pyridine), 7.03 (m, 2H-Ar), 3.9 (s, 3H-methyl), 2.45 (s, 3H-methyl); 13C-NMR (CDCl3 + CD3OD) δ 17.1, 55.6, 114.7, 120.5, 128.4, 129.8, 132.3, 134.3, 144.3, 145.8, 160.3. EI-MS m/z (+ion mode): 214, [M − NH2]+ = 199, [M − NH2 and CH3]+ = 185, [M − OCH3]+ = 184. Anal. calcd for C13H14N2O (214.21): C, 72.79; H, 6.55. Found: C, 72.75; H, 6.53%.
5-(4-Iodophenyl)-2-methylpyridin-3-amine (2d). m.p.: 258 °C. 1H-NMR (CDCl3) δ 8.09 (s, 1H-pyridine), 7.71–7.49 (m, 4H-Ar), 7.15 (s, 1H-pyridine), 2.5 (s, 3H-methyl); 13C-NMR (CDCl3 + CD3OD) δ 15.8, 94.1, 120.1, 130, 132.1, 134.5, 135.7, 138.4, 144.2, 145.5. EI-MS m/z (+ion mode): 310, [M − NH2]+ = 295, [M − NH2 and CH3]+ = 281, [M − I]+ = 184. Anal. calcd for C12H11IN2 (310.13): C, 46.47; H, 3.58. Found: C, 46.41; H, 3.52%.
5-(4-Chlorophenyl)-2-methylpyridin-3-amine (2e). m.p.: 228 °C. 1H-NMR (CDCl3) δ 8.2 (s, 1H-pyridine), 7.71–7.41 (m, 4H-Ar), 7.11 (s, 1H-pyridine), 2.5 (s, 3H, methyl); 13C-NMR (CDCl3 + CD3OD) δ 15.7, 120.4, 128.3, 129.8, 132, 134.3, 135.3, 144.2, 145.6. EI-MS m/z (+ion mode): 310, [M − NH2]+ = 295, [M − NH2 and CH3]+ = 281, [M − I]+ = 184. Anal. calcd for C12H11ClN2 (218.68): C, 66.01; H, 5.07. Found: C, 66.0; H, 5.03%.
2-Methyl-5-[4-(methylsulfonyl)phenyl]pyridin-3-amine (2f). m.p.: 240 °C. 1H-NMR (CDCl3) δ 8.23 (s, 1H-pyridine), 7.69–7.11 (m, 4H-Ar, 1H-pyridine), 2.59 (s, 3H, methyl), 2.5 (s, 3H, methyl). 13C-NMR (CDCl3 + CD3OD) δ 14.6, 17, 120.1, 127.2, 128.3, 132.2, 133.4, 135, 139.5, 144, 146.1. EI-MS m/z (+ion mode): 230, [M − NH2]+ = 215, [M − NH2 and CH3]+ = 201, [M − SCH3]+ = 184. Anal. calcd for C13H14N2S (230.33): C, 66.98; H, 5.99. Found: C, 66.95; H, 5.95%.
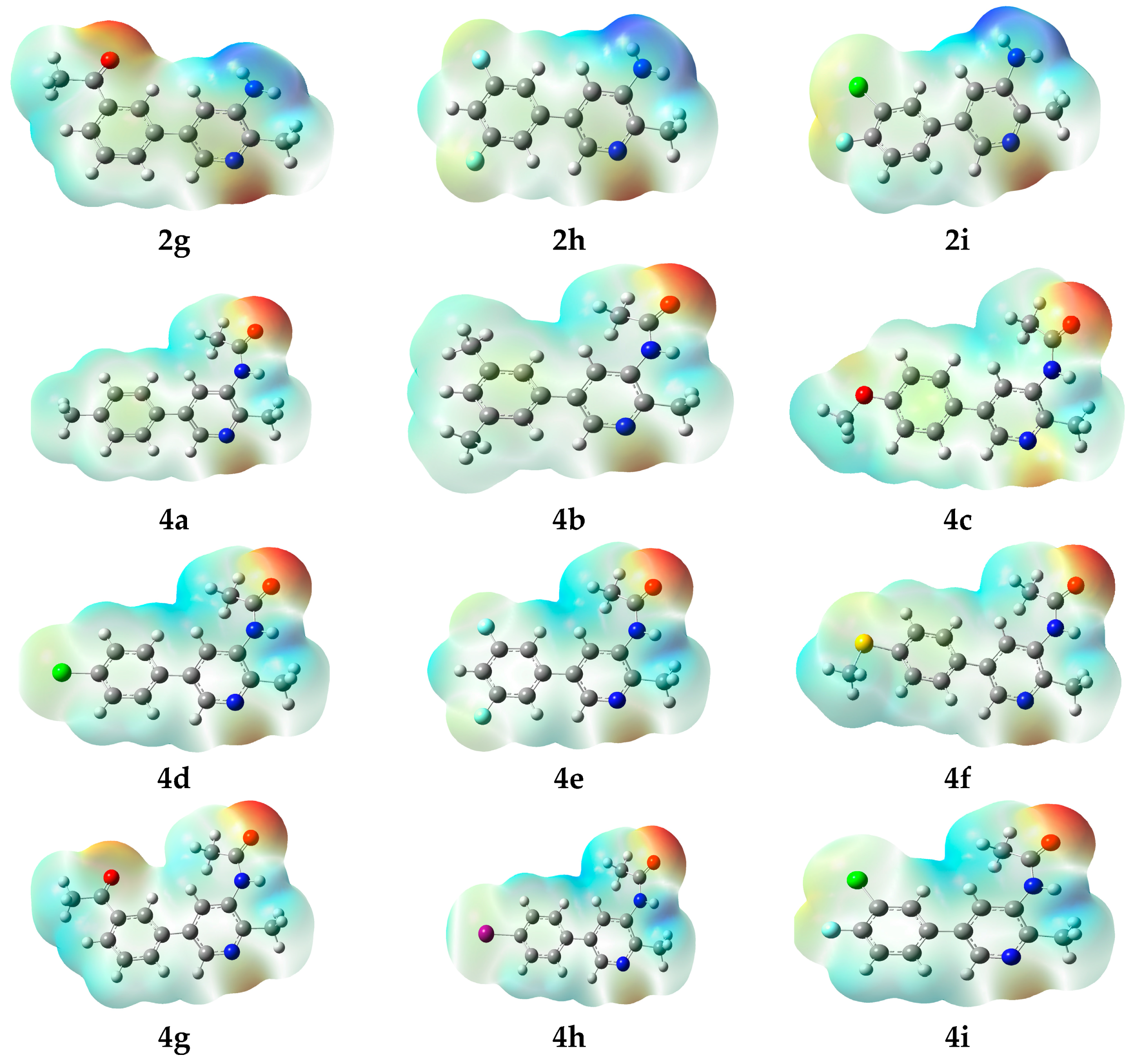
1-[3-(5-Amino-6-methylpyridin-3-yl)phenyl]ethanone (2g). m.p.: 270 °C. 1H-NMR (CDCl3) δ 8.24 (d, J = 3.4 Hz, 1H-Ar), 8.17 (s, 1H-pyridine), 7.96–7.49 (m, 3H-Ar), 7.25 (s, 1H-pyridine), 2.7 (s, 3H-COCH3), 2.5 (s, 3H-methyl). 13C-NMR (CDCl3 + CD3OD) δ 16.3, 26.4, 120.5, 126.5, 128.2, 129.7.131.2, 132.8, 134.8, 136.6, 138, 144.5, 146.1, 195. EI-MS m/z (+ion mode): 226 [M + H]+: [M − NH2]+ = 211: [M − NH2 and CH3]+ = 197: [M − OCH3]+ = 184. Anal. calcd for C14H14N2O (226.19): C, 74.27; H, 6.18. Found: C, 74.26; H, 6.17%.
5-(3,5-Difluorophenyl)-2-methylpyridin-3-amine (2h). m.p.: 208 °C. 1H-NMR (CDCl3) δ 7.9 (s, 1H-pyridine), 7.57 (m, 1H-pyridine), 7.55–6.82 (m, 3H-Ar), 2.5 (s, 3H, methyl). 13C-NMR (CDCl3 + CD3OD) δ 16.3, 104.8, 111.2, 120, 132.4, 134.5, 139.3, 144.3, 145.8. EI-MS m/z (+ion mode): 220 [M + H]+: [M − NH2]+ = 205: [M − NH2 and CH3]+ = 191: [M − 2F]+ = 184. Anal. calcd for C12H10F2N2 (220.22): C, 65.45; H, 4.56. Found: C, 65.41; H, 4.54%.
5-(3-Chloro-4-fluorophenyl)-2-methylpyridin-3-amine (2i). m.p.: 240 °C. 1H-NMR (CDCl3) δ 8.22 (s, 1H-pyridine), 7.72–7.08 (m, 3H-Ar, 1H-pyridine), 2.5 (s, 3H, methyl). 13C-NMR (CDCl3 + CD3OD) δ 16.6, 117.1, 120.4, 121.1, 128, 129.4, 132.2, 133.1, 134.6, 143.9, 145.2. EI-MS m/z (+ion mode): 236 [M + H]+: [M − NH2]+ = 221: [M − NH2 and CH3]+ = 207: [M − F]+ = 218: [M − Cl]+ = 202: [M − F and Cl]+ = 184. Anal. calcd for C12H10ClFN2 (236.67): C, 60.90; H, 4.27. Found: C, 60.85; H, 4.22%.
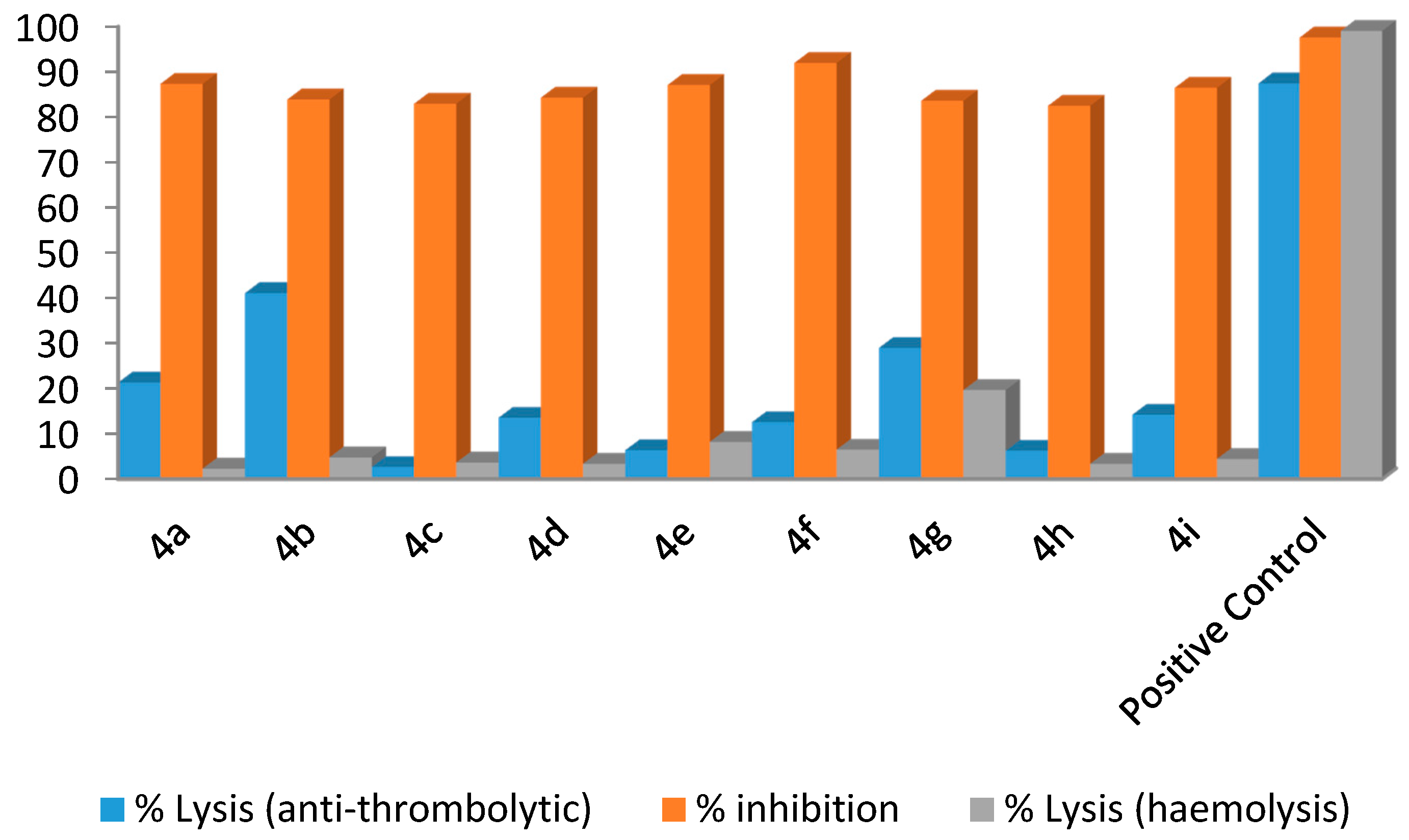
3.4. General Procedure for the Synthesis of Compounds 4a–4i
All compounds were synthesized by reported method [
28].
N-[5-bromo-2-methylpyridine-3-yl]acetamide (
3, 0.1 g), tetrakis(triphenylphosphine)-palladium (5 mol %) and 1,4-dioxane (2 mL) were placed in the Schlenk flask at room temperature and the mixture was stirred for 30 min . Then the appropriate arylboronic acid (1.1 mmol), potassium phosphate (1.5 mmol) and H
2O (0.5 mL) were added to the mixture, which was stirred and kept at 85–95 °C for more than 15 h. After reaching room temperature, the mixture was filtered and then diluted with ethyl acetate (50 mL). The excess solvent was evaporated by rotary evaporator in order to obtain a concentrated solution. Column chromatography (silica gel,
n-hexane and ethyl acetate?) was applied to obtain the desired pure products. The final product was dried and recrystallized and further analyzed using different spectroscopic techniques.
5-(3-Chloro-4-fluorophenyl)-2-methylpyridin-3-amine (4a). m.p.: 310 °C. 1H-NMR (CDCl3) δ 8.77 (s, 1H-pyridine), 8.7 (d, J =2.5 Hz, 1H-pyridine), 7.6 (m, 2H-Ar), 7.4 (m, 2H-Ar), 2.65 (s, 3H, methyl), 2.5 (s, 3H, methyl), 2.4 (s, 3H, COCH3). 13C-NMR (CDCl3 + CD3OD) δ 16.4, 21.5, 24.2, 126.1, 127.6, 129.2, 132.1, 133.2, 134, 136.8, 149.2, 168.2. EI-MS m/z (+ion mode): 240 [M + H]+: [M − CH3]+ = 226: [M − 2CH3]+ = 212. Anal. calcd for C15H16N2O (240.3): C, 74.89; H, 6.69. Found: C, 74.87; H, 6.65%.
N-[5-(3,5-Dimethylphenyl)-2-methylpyridine-3-yl]acetamide (4b). m.p.: 338 °C. 1H-NMR (CDCl3) δ 8.6 (s, 1H-pyridine), 8.5 (s, 1H-pyridine), 7.49 (d, 2H-Ar), 7.35 (s, 1H-Ar), 2.49 (s, 3H, methyl), 2.32 (s, 6H, methyl), 2.05 (s, 3H, COCH3). 13C-NMR (CDCl3 + CD3OD) δ 16.5, 21.7, 24, 125.2, 126.6, 130.1, 133.5, 136.4, 138.6, 149.3, 167.9. EI-MS m/z (+ion mode): 254 [M + H]+: [M − CH3]+ = 240: [M − 3CH3]+ = 212. Anal. calcd for C16H18N2O (254.33): C, 75.48; H, 7.09. Found: C, 75.45; H, 7.07%.
N-[5-(4-Methoxyphenyl)-2-methylpyridine-3-yl]acetamide (4c). m.p.: 337 °C. 1H-NMR (CDCl3) δ 8.27 (s, 1H-pyridine), 7.7 (d, J = 2.3 Hz, 1H-pyridine), 7.15–7.05 (m, 4H-Ar), 3.95 (s, 3H, methoxy), 2.6 (s, 3H, methyl), 1.75 (s, 3H, COCH3). 13C-NMR (CDCl3 + CD3OD) δ 16.6, 24, 55.6, 114.7, 126.2, 128.3, 129.9, 133.3, 136.8, 149.4, 160.5, 168.3. EI-MS m/z (+ion mode): 256 [M + H]+: [M − CH3]+ = 242: [M − 2CH3]+ = 228. Anal. calcd for C15H16N2O2 (256.30): C, 70.29; H, 6.26. Found: C, 70.27; H, 6.24%.
N-[5-(4-Chlorophenyl)-2-methylpyridine-3-yl]acetamide (4d). m.p.: 316 °C. 1H-NMR (CDCl3) δ 8.48 (s, 1H-pyridine), 8.22 (s, 1H-pyridine), 7.55 (m, 2H-Ar), 7.45 (m, 2H-Ar), 2.5 (s, 3H, methyl), 2.22 (s, 3H, methyl). 13C-NMR (CDCl3 + CD3OD) δ 16.1, 24.2, 126.6, 128.7, 129.9, 133.4, 134.7, 136.5, 149.2, 169.1. EI-MS m/z (+ion mode): 260 [M + H]+: [M − CH3]+ = 247: [M − Cl]+ = 226: [M − Cl and CH3]+ = 212. Anal. calcd for C14H13ClN2O (260.72): C, 64.49; H, 5.08. Found: C, 64.44; H, 5.02%.
N-[5-(3,5-Difluorophenyl)-2-methylpyridine-3-yl]acetamide (4e). m.p.: 310 °C. 1H-NMR (CDCl3) δ 8.73 (s, 1H-pyridine), 8.46 (s, 1H-pyridine) 7.29 (m, 2H-Ar), 6.64 (s, 1H-Ar), 2.53 (s, 3H-methyl), 2.04 (s, 3H-methyl). 13C-NMR (CDCl3 + CD3OD) δ 16.6, 24.3, 104.9, 111.2, 126.7, 133.8, 136.5, 139.3, 149.5, 164.8, 168.4. EI-MS m/z (+ion mode): 262 [M + H]+: [M − CH3]+ = 248: [M − 2F]+ = 226. Anal. calcd for C14H12F2N2O (262.23): C, 64.12; H, 4.61. Found: C, 64.08; H, 4.54%.
N-[2-Methy-5-(4-methylsulfonyl)phenylpyridin-3-yl]acetamide (4f). m.p.: 325 °C. 1H-NMR (CDCl3) δ 7.69–7.39 (m, 4H-Ar, 2H-pyridine), 2.55 (s, 3H-methyl), 1.7 (s, 3H-methyl). 13C-NMR (CDCl3 + CD3OD) δ 14.7, 16.8, 24.2, 127.1, 128.2, 132.8, 133.8, 136.2, 139.1, 149.6, 168.5. EI-MS m/z (+ion mode): 272 [M + H]+: [M − CH3]+ = 258: [M − 2CH3]+ = 244. Anal. calcd for C15H16N2OS (272.37): C, 66.15; H, 5.92. Found: C, 66.10; H, 5.91%.
N-[5-(3-Acetylphenyl)-2-methylpyridine-3-yl]acetamide (4g). m.p.: 340 °C. 1H-NMR (CDCl3) δ 8.33 (s, 1H-pyridine), 8.25 (d, J = 1.9 Hz, 1H-pyridine), 8.0 (m, 1H-Ar), 7.80 (m, 1H-Ar), 7.68 (m, 1H-Ar), 7.4 (m, 1H-Ar), 2.71 (s, 3H, methyl), 2.51 (s, 3H, COCH3), 2.09 (s, 3H, methyl). 13C-NMR (CDCl3 + CD3OD) δ 16.5, 24.6, 26.3, 125.8, 126.9, 127.7, 128.6, 129.5, 132, 133.7, 136.2, 137.3, 148.6, 167.8, 196.6. EI-MS m/z (+ion mode): 268 [M + H]+: [M − CH3]+ = 254: [M − OCH3]+ = 226. Anal. calcd for C16H16N2O2 (268.29): C, 71.59; H, 5.99. Found: C, 71.55; H, 5.96%.
N-[5-(4-iodophenyl)-2-methylpyridine-3-yl]acetamide (4h). m.p.: 333 °C. 1H-NMR (CDCl3) δ 8.73 (s, 1H-pyridine), 8.46 (d, J = 2.5 Hz, 1H-pyridine), 7.99 (m, 2H-Ar), 7.56 (m, 2H-Ar), 2.53 (s, 3H, methyl), 2.04 (s, 3H, COCH3). 13C-NMR (CDCl3 + CD3OD) δ 16.7, 23.9, 94.5, 126.3, 128.2, 130.5, 133.6, 135.7, 136.9, 138.4, 149.1, 168.8. EI-MS m/z (+ion mode): 352 [M + H]+: [M − CH3]+ = 338: [M − F]+ = 226: [M − F and CH3]+ = 212. Anal. calcd for C14H13IN2O (352.17): C, 47.75; H, 3.72. Found: C, 47.71; H, 3.67%.
N-[5-(3-chloro-4-fluorophenyl)-2-methylpyridine-3-yl]acetamide (4i). m.p.: 325 °C. 1H-NMR (CDCl3) δ 8.2 (s, 1H-pyridine) 7.71 (m, 1H-pyridine), 7.5 (m, 1H-Ar), 7.35 (d, J = 8.2 Hz, 1H-Ar), 7.09 (d, J = 8.1, 1H-Ar), 2.45 (s, 3H, methyl), 1.7 (s, 3H, COCH3). 13C-NMR (CDCl3 + CD3OD) δ 15.9, 24.4, 117.8, 126.3, 127.9, 129.5, 132.9, 135.5, 136.9, 150, 158.1, 167.5. EI-MS m/z (+ion mode): 279 [M + H]+: [M − CH3]+ = 265: [M − F]+ = 261: [M − Cl]+ = 244: [M − Cl and F]+ = 226. Anal. calcd for C14H12ClFN2O (278.71): C, 60.33; H, 4.34. Found: C, 60.27; H, 4.29%.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}