
Synthesis, Modelling, and Anticonvulsant Studies of New Quinazolines Showing Three Highly Active Compounds with Low Toxicity and High Affinity to the GABA-A Receptor



Abstract
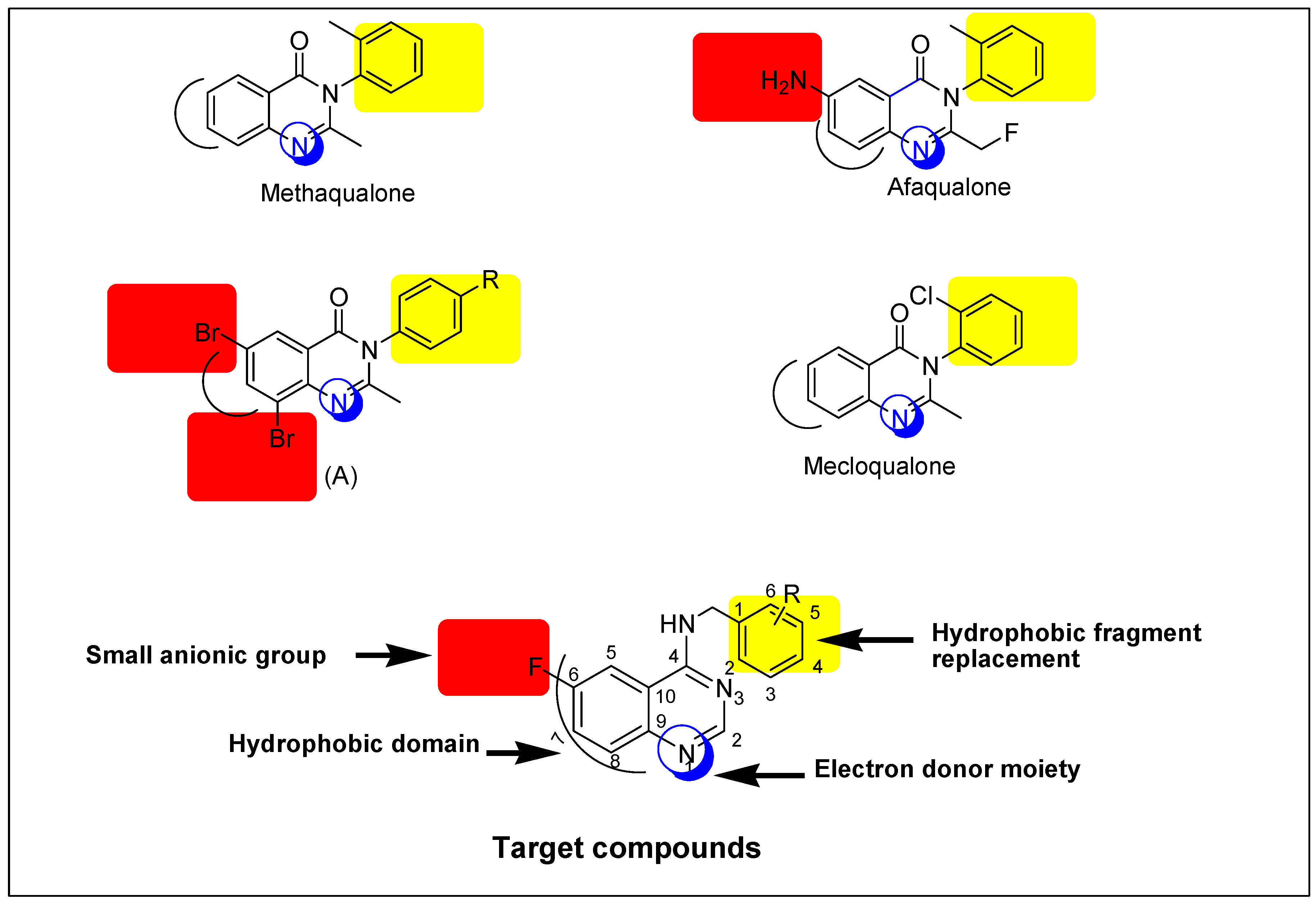
:1. Introduction
2. Results and Discussion
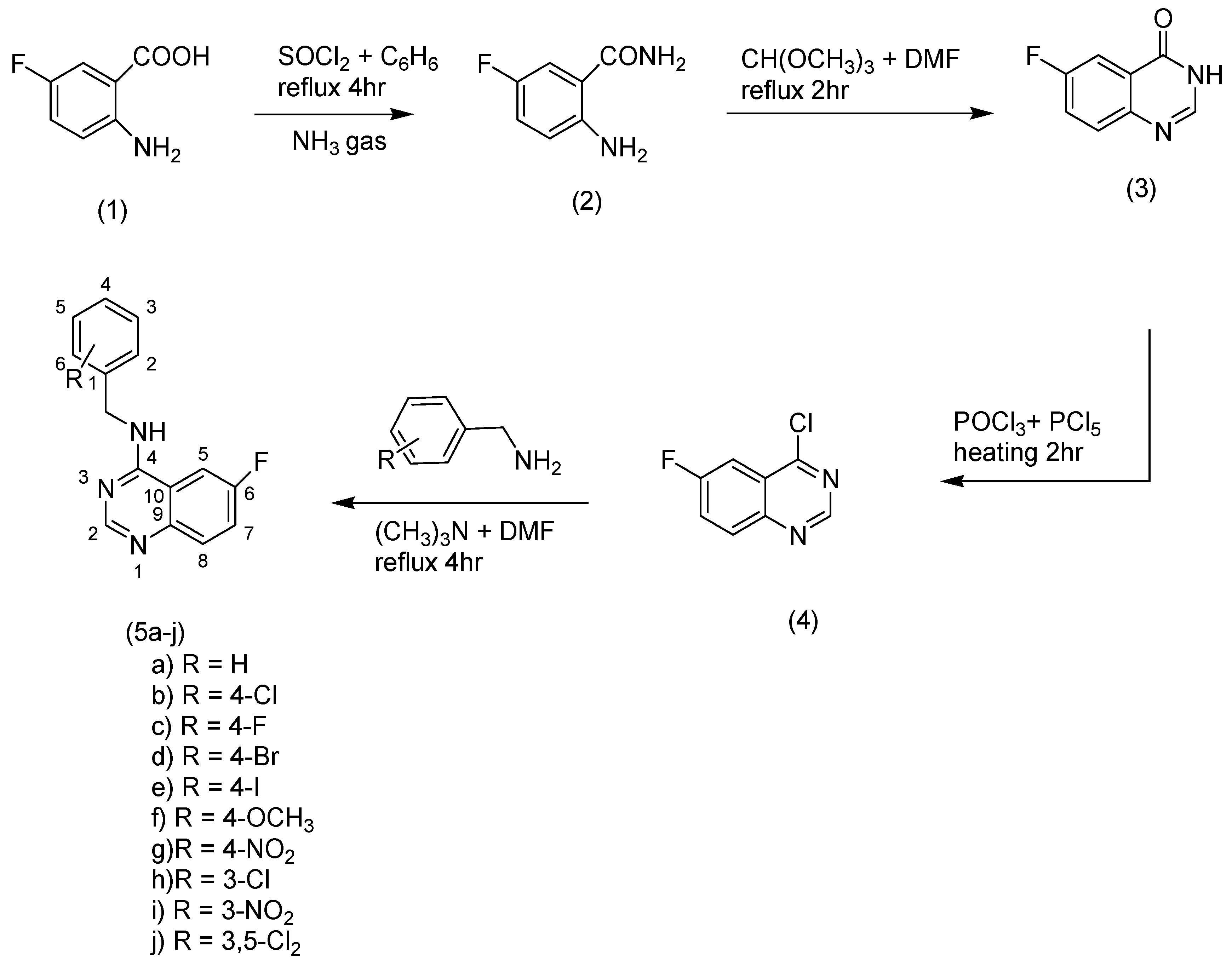
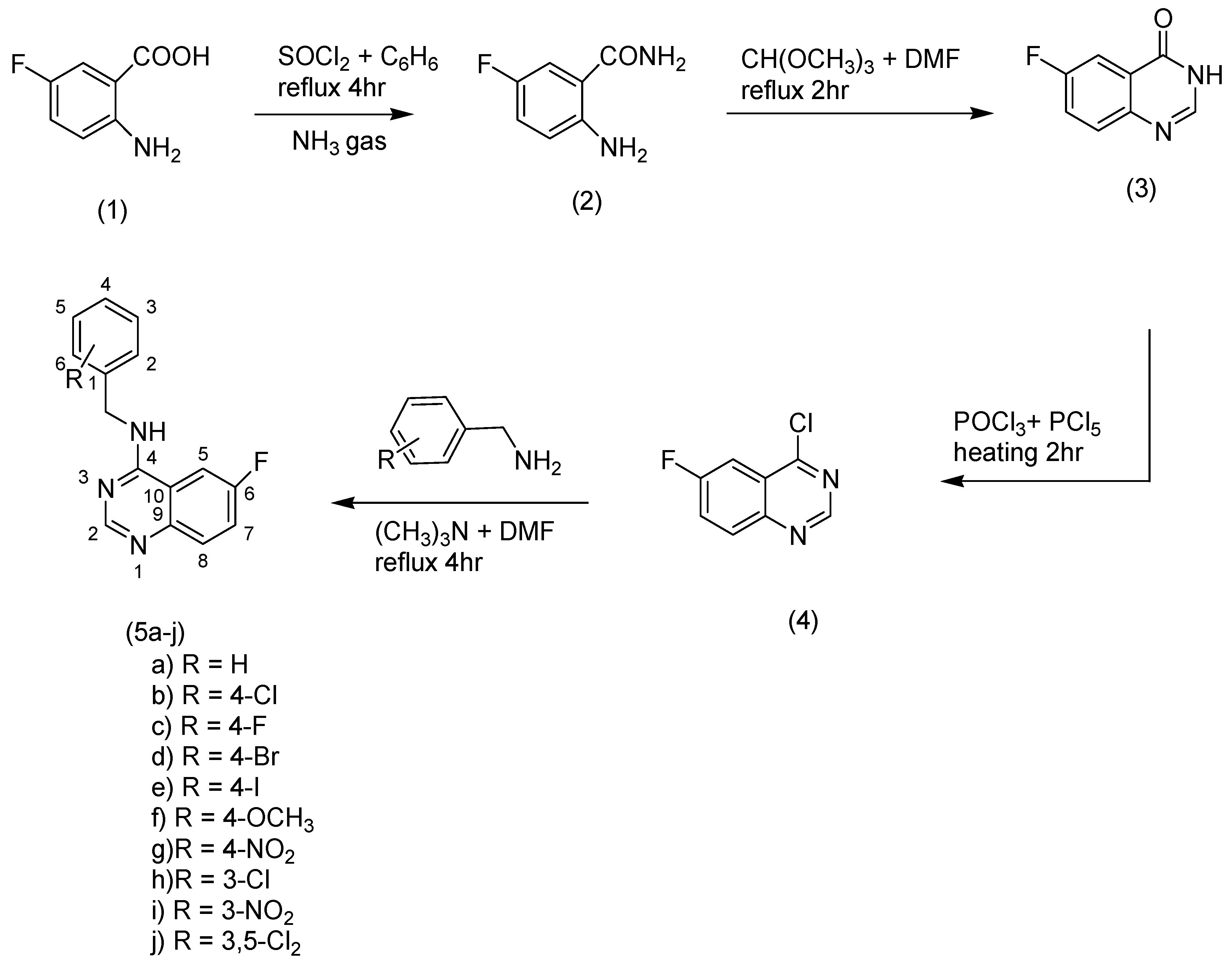
2.1. Chemistry
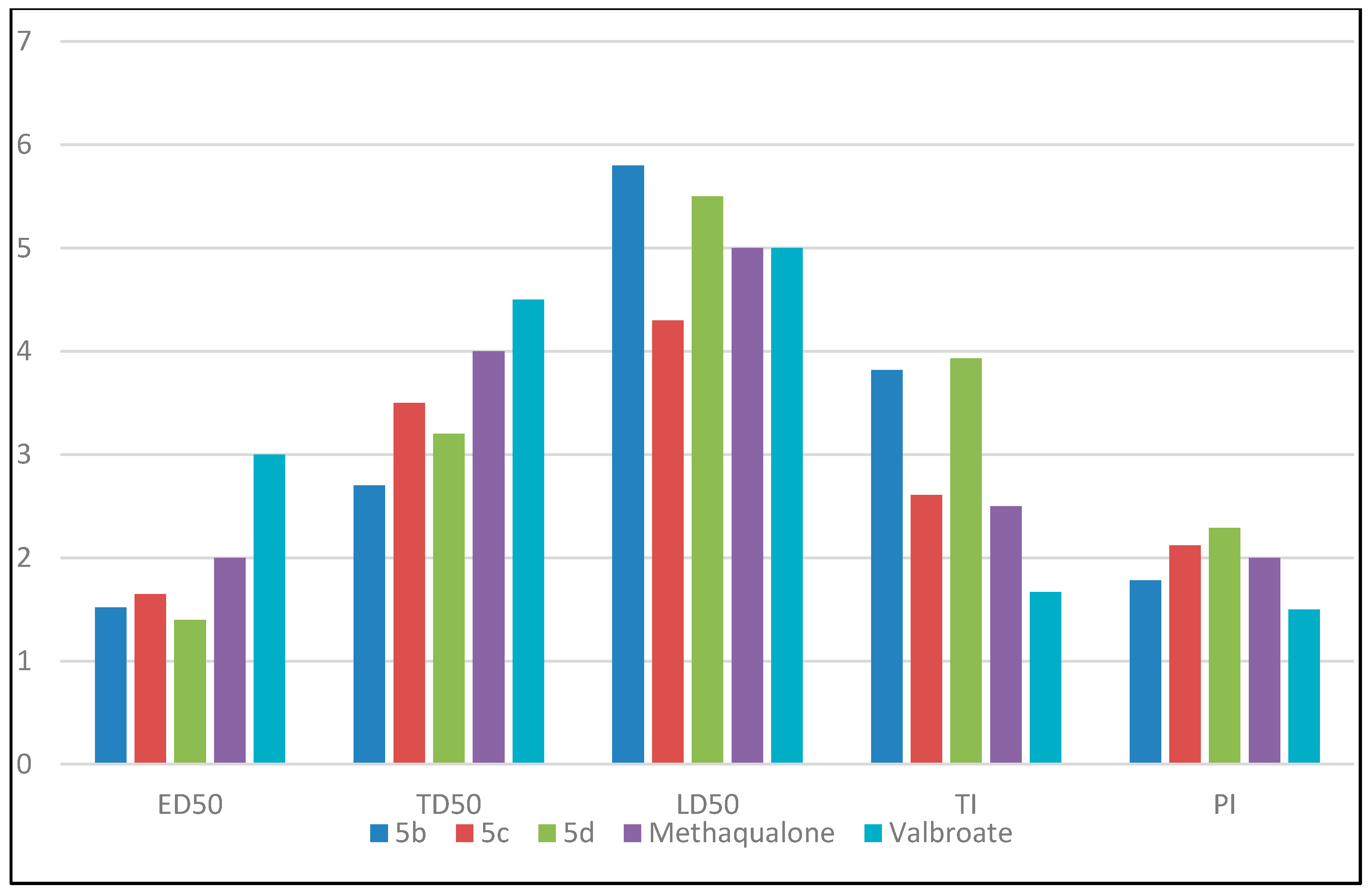
2.2. Anticonvulsant Screening
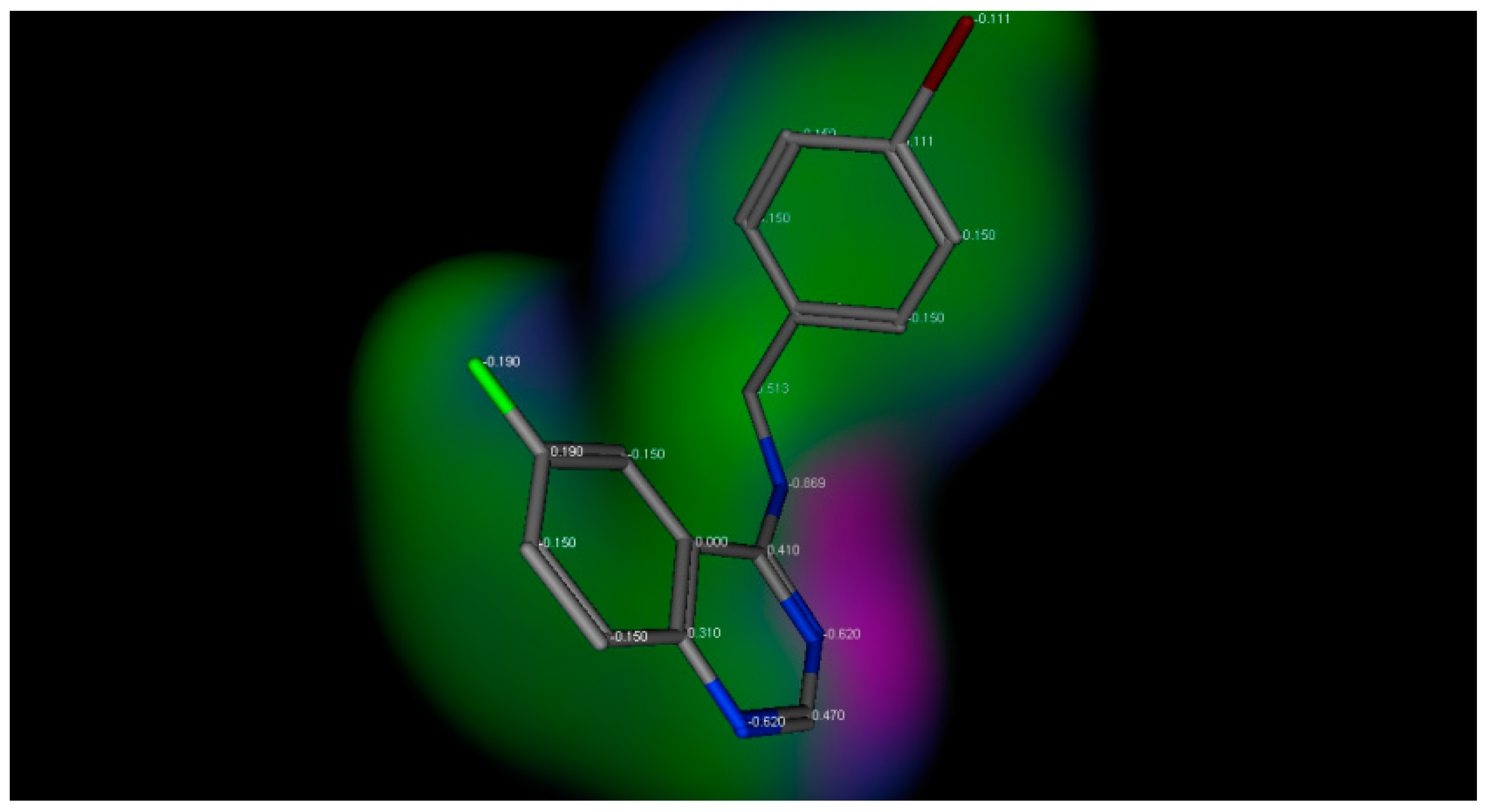
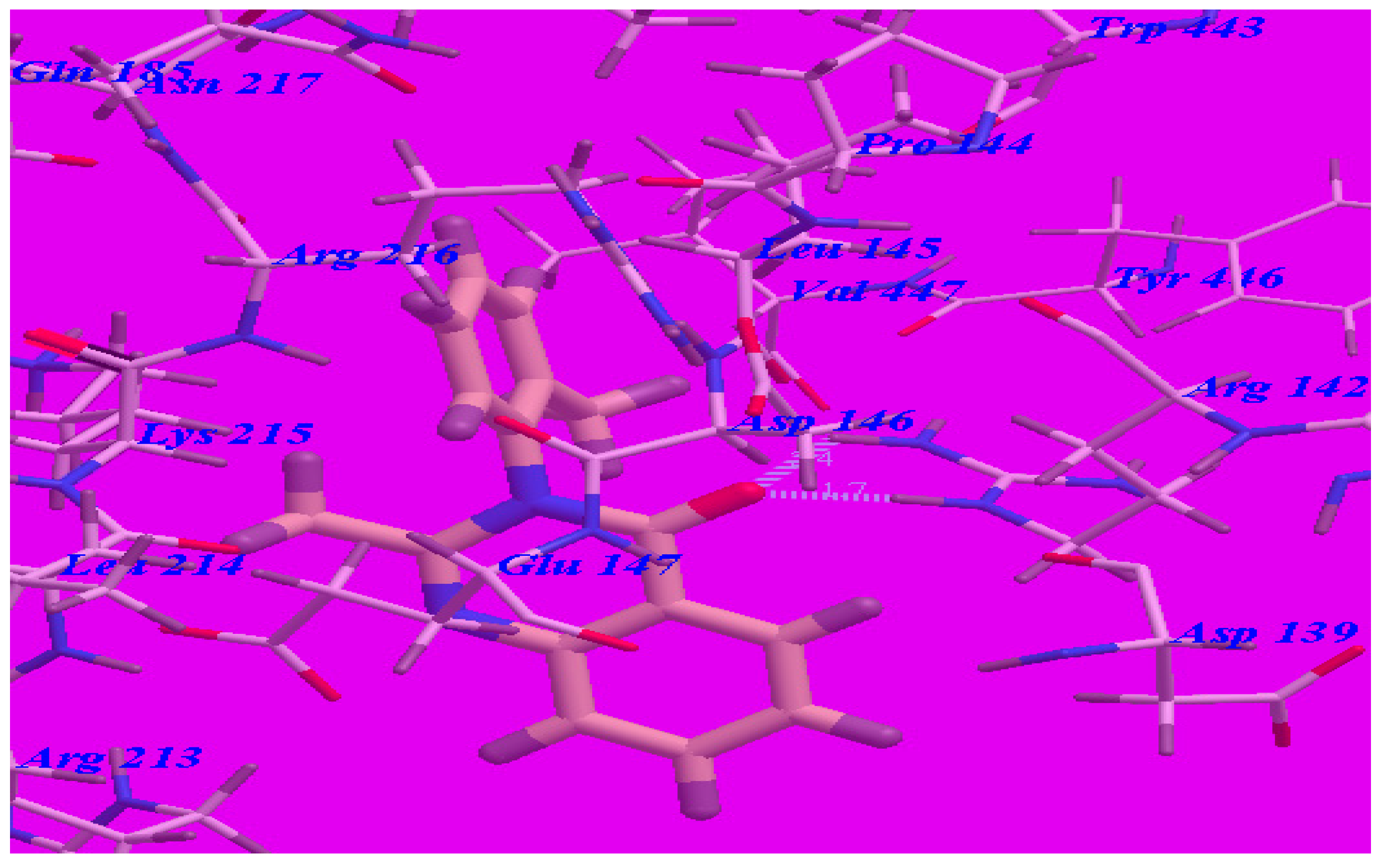
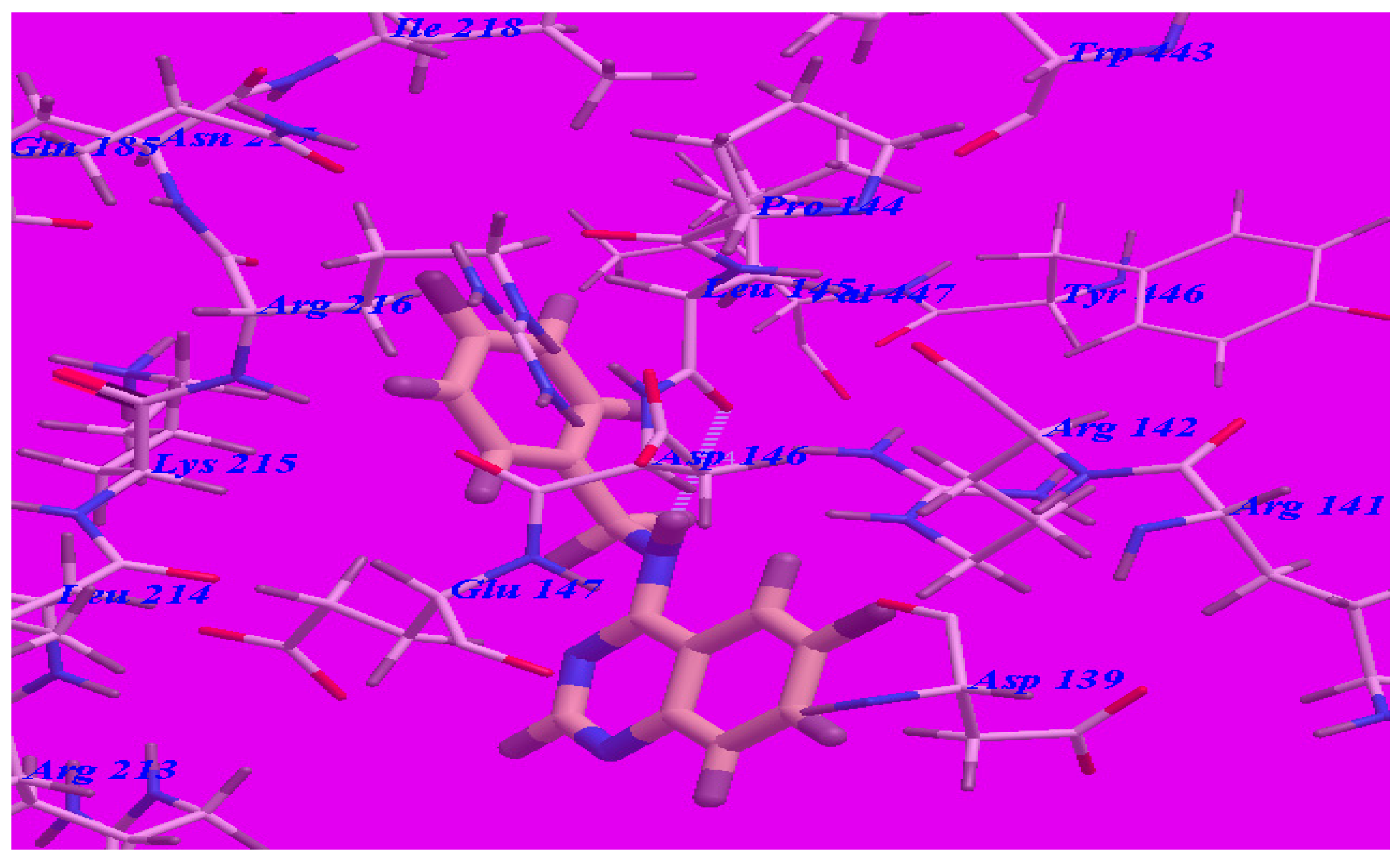
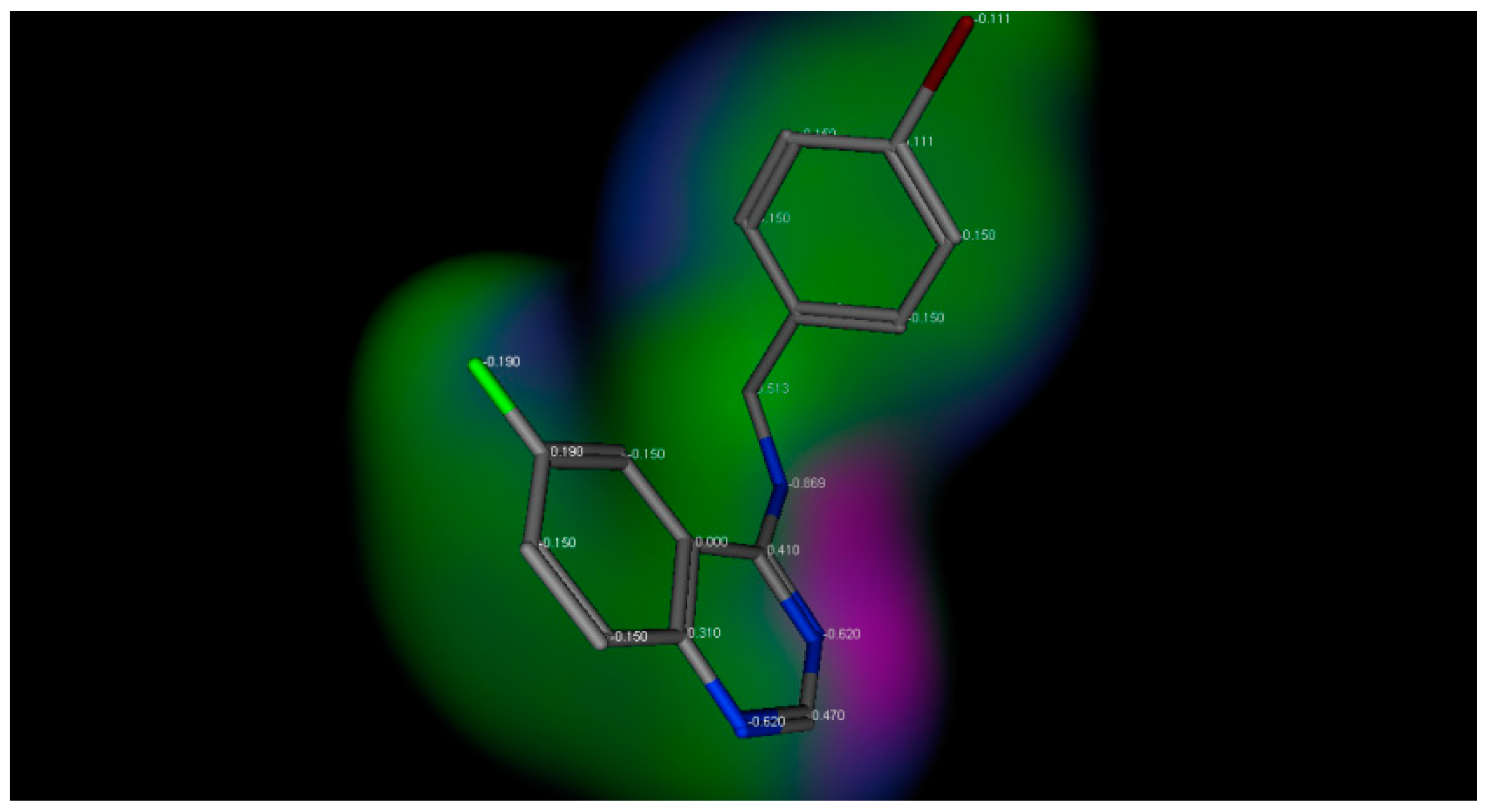
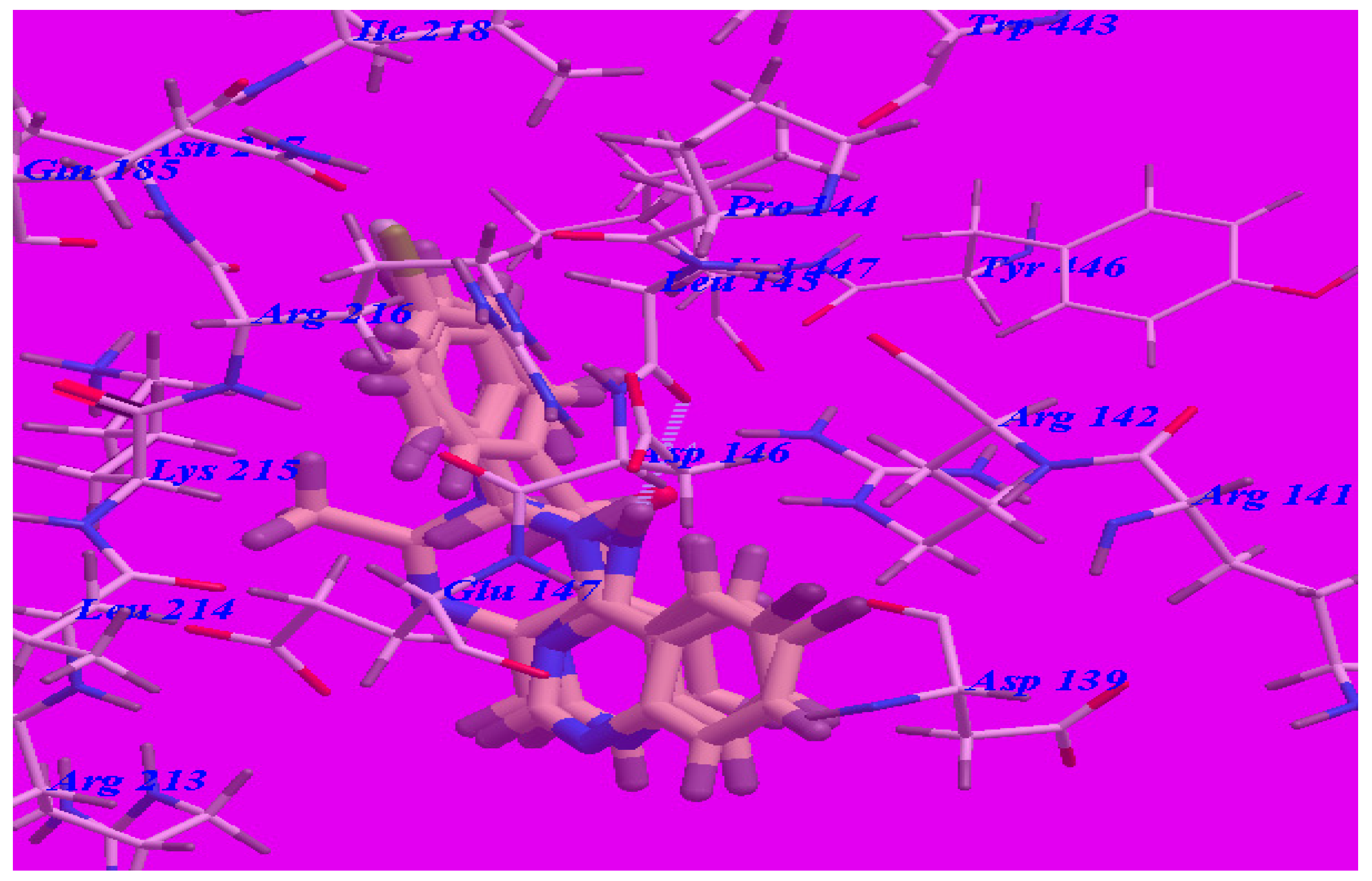
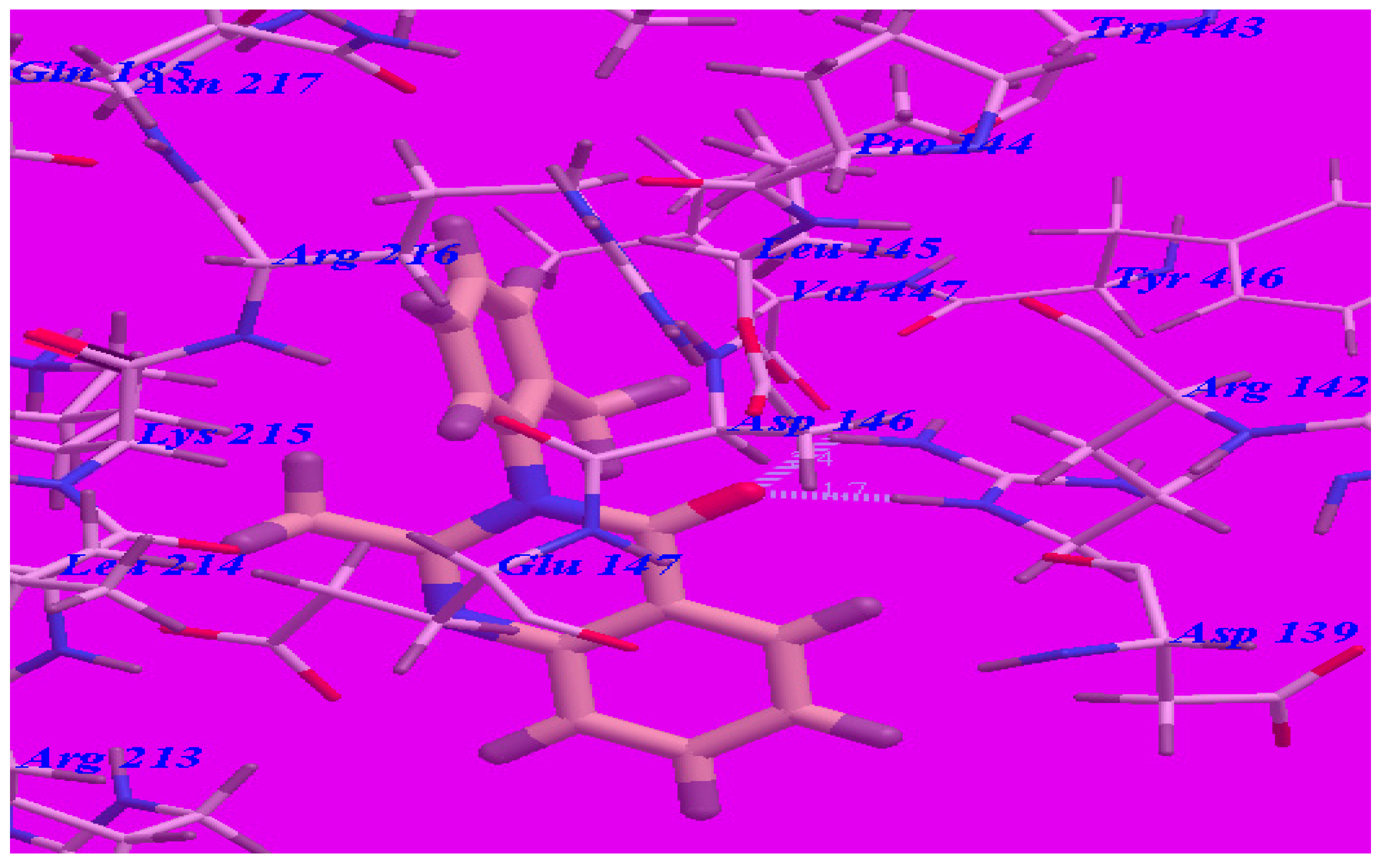
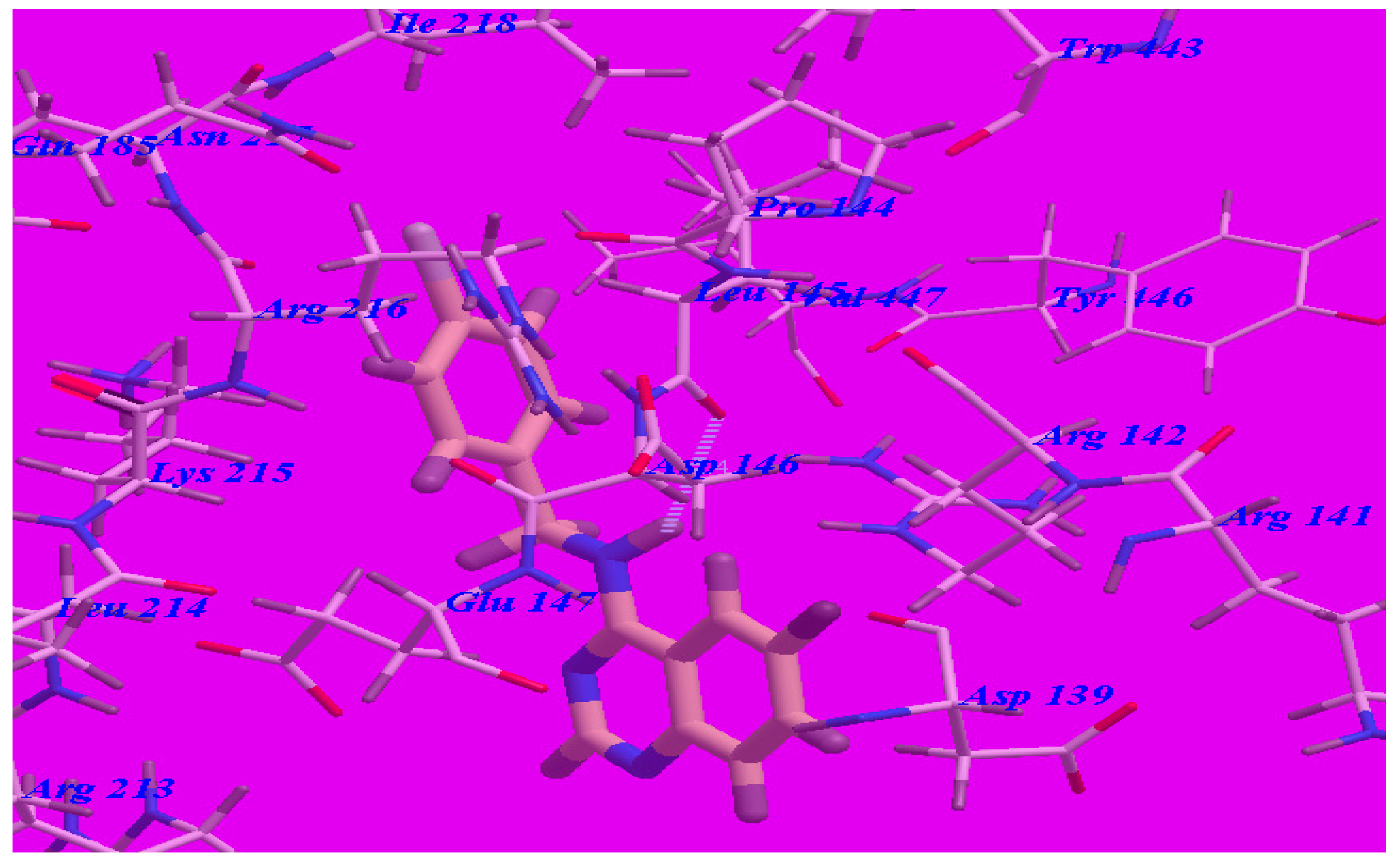
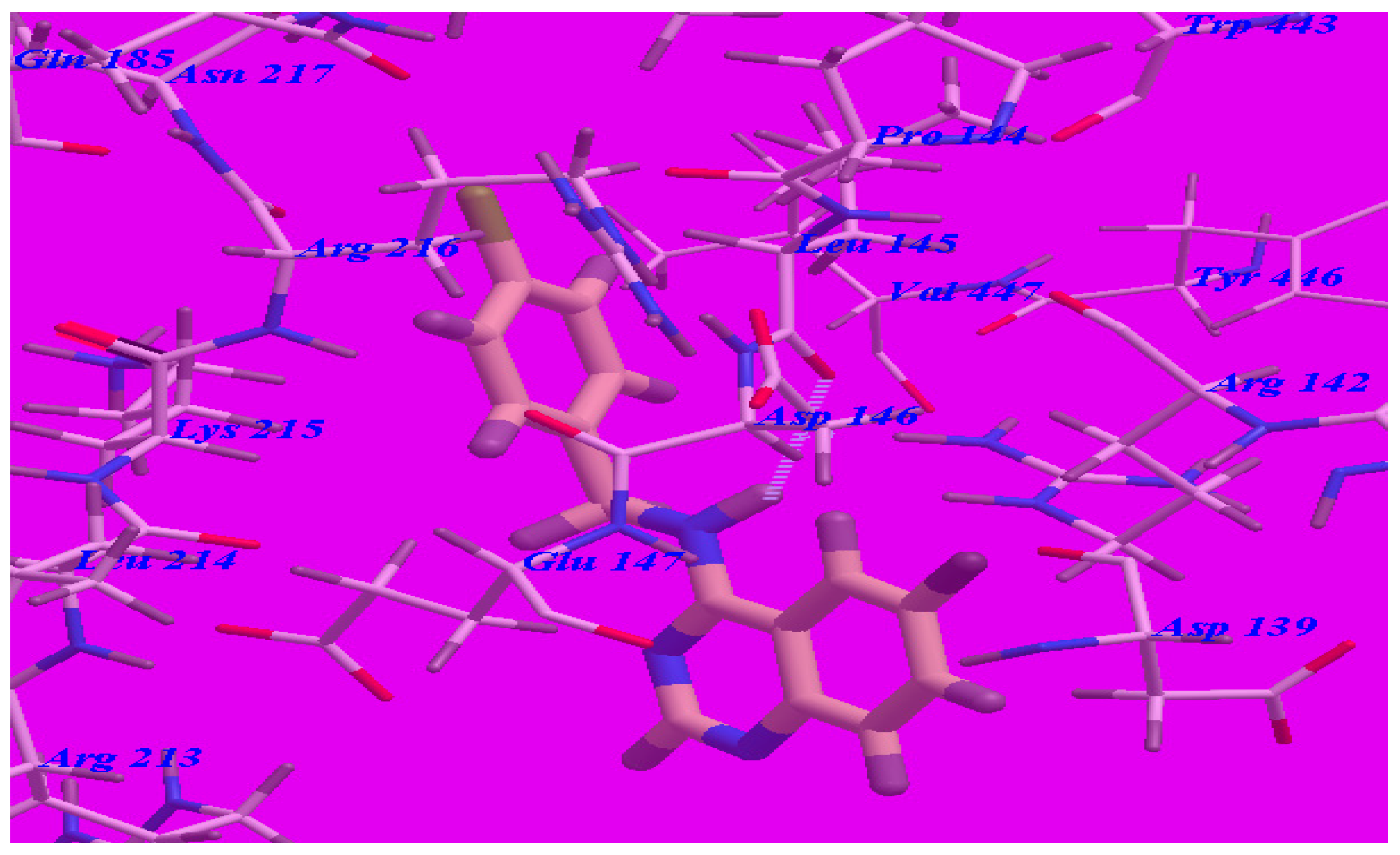
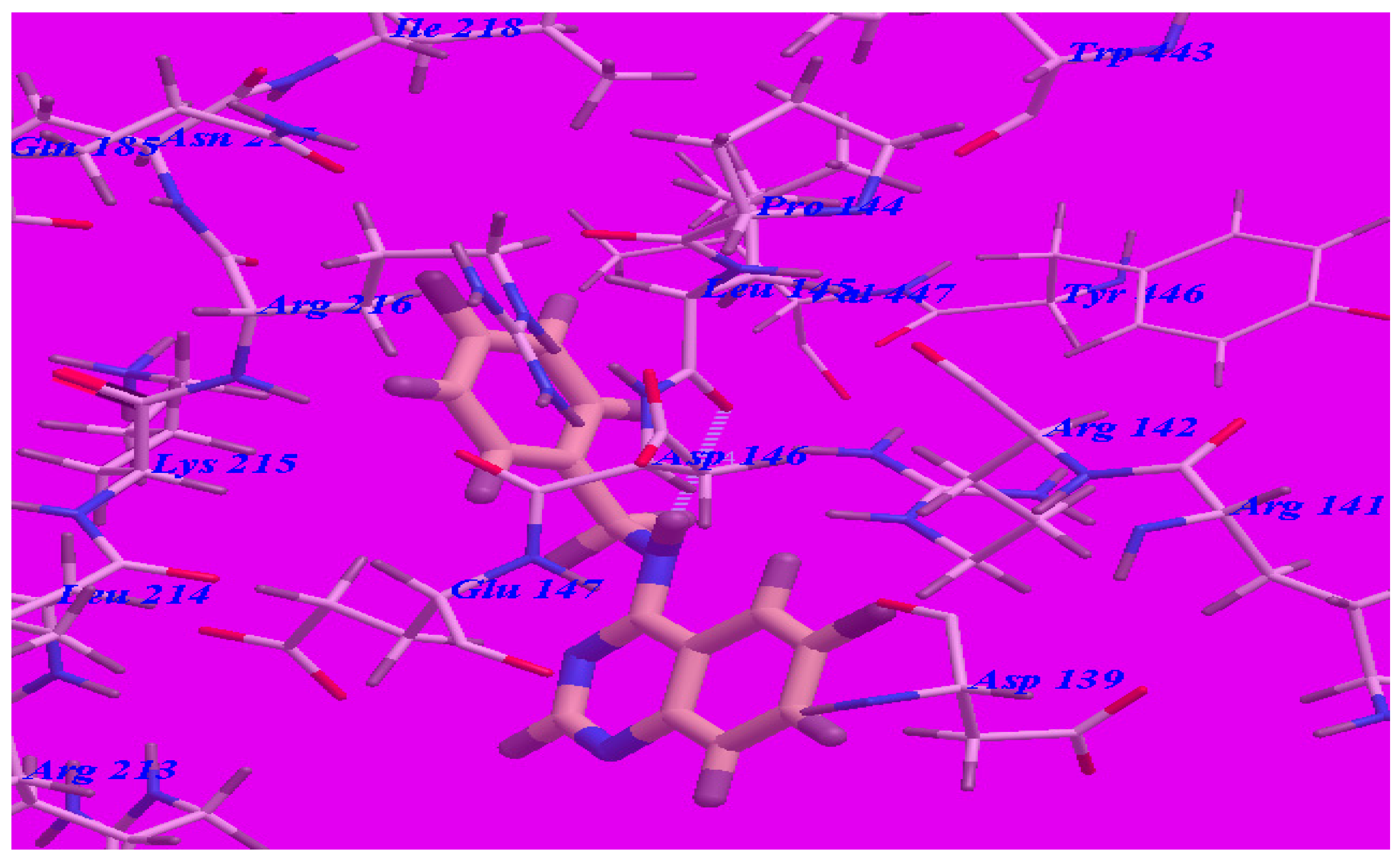
2.3. Docking Studies
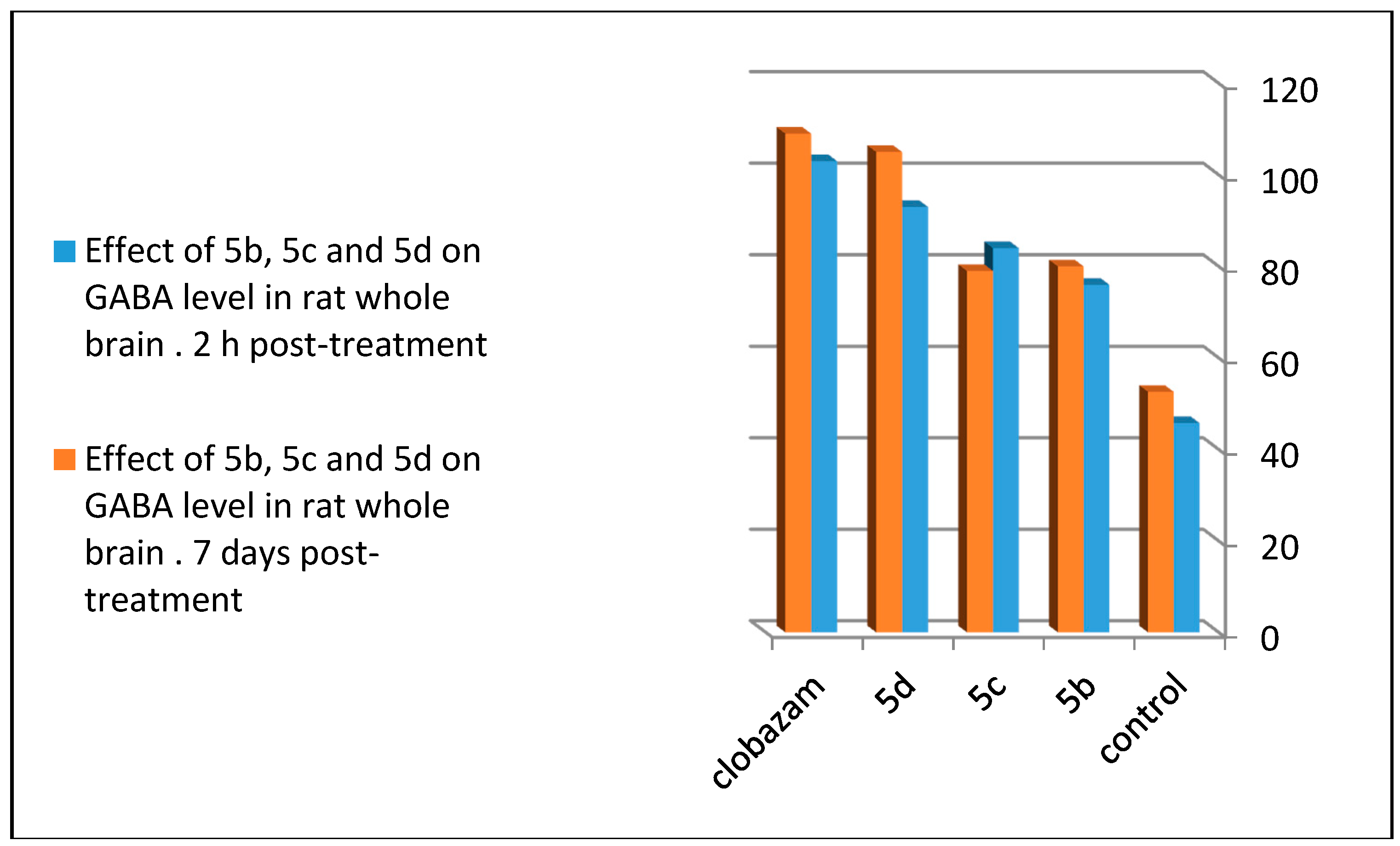
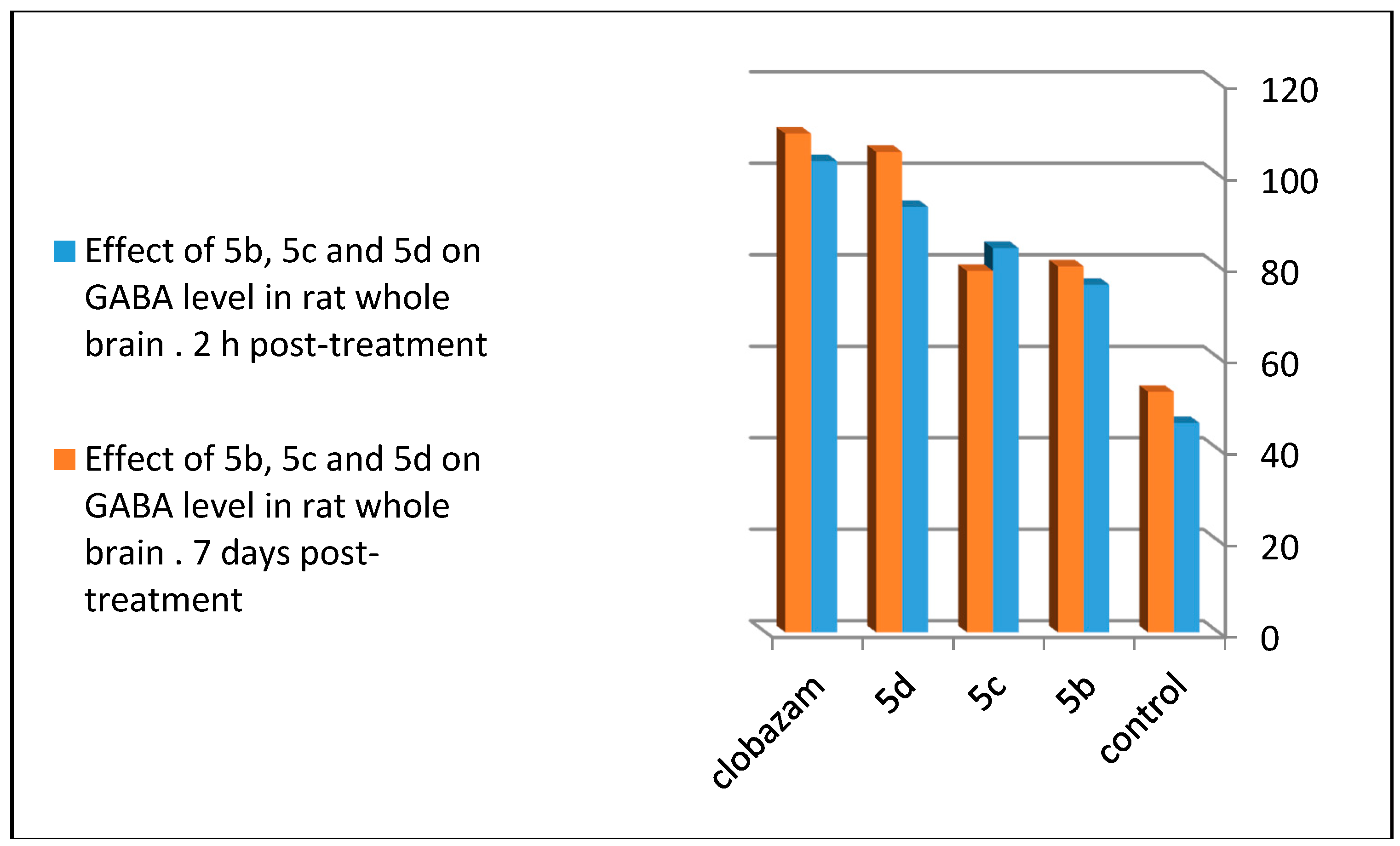
2.4. GABA Enzymatic Assay
3. Materials and Methods
3.1. General
3.2. General Method of the Synthesis of 2-Amino-5-Fluorobenzamide (2)
3.3. General Method of the Synthesis of 6-Fluroquinazolin-4(3H)-one (3)
3.4. General Method of the Synthesis of 4-Chloro-6-Fluoro-Quinazoline (4)
3.5. General Method of the Synthesis of Quinazoline Derivatives (5a–j)
3.6. Docking Studies
3.7. Pharmacological Tests
3.8. Anticonvulsant Screening
3.9. Neurotoxicity Screening
3.10. GABA Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zayed, M.F.; Ayyad, R.R. Some novel anticonvulsant agents derived from Phthalazinedione. Arzneimittelforschung 2012, 62, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Jatav, V.; Mishra, P.; Kashaw, S.; Stables, J.P. CNS depressant and anticonvulsant activities of some novel 3-[5-substituted1,3,4-thiadiazole-2-yl]-2-styryl quinazoline-4(3H)-ones. Eur. J. Med. Chem. 2008, 43, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- El-Naser Ossman, A.R.; El-Sayed Barakat, S. Synthesis and Anticonvulsant Activity of Some new 3-(p-Sulfamoylphenyl)-4(3H)-Quinazolinones. Arzneimittelforsch 1994, 44, 915–919. [Google Scholar] [PubMed]
- Jin, H.G.; Sun, X.Y.; Chai, K.Y.; Piao, H.R.; Quan, Z.S. Anticonvulsant and toxicity evaluation of some 7-alkoxy-4,5-dihydro-[1,2,4]triazolo[4,3-a]quinoline-1(2H)-ones. Bioorg. Med. Chem. 2006, 14, 6868–6873. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F.; Ahmed, E.A.; Omar, A.M.; Abdelrahim, A.S.; El-Adl, K. Design, synthesis and biological evaluation studies of novel quinazolinone derivatives as anticonvulsant agents. Med. Chem. Res. 2013, 22, 5823–5831. [Google Scholar] [CrossRef]
- Zayed, M.F. New fluorinated quinazolinone derivatives as anticonvulsant agents. J. Taibah Univ. Med. Sci. 2014, 9, 104–109. [Google Scholar] [CrossRef]
- Elhelby, A.A.; Ayyad, R.R.; Zayed, M.F. Synthesis and biological evalution of some novel quinoxaline derivatives as anticonvulsant agents. Arzneimittelforschung 2011, 61, 379–381. [Google Scholar] [PubMed]
- Scharfman, H. Upregulation of Multidrug Resistance Transporters in the Epileptic Brain. Epilepsy Curr. 2002, 2, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.K.; El-Adl, K.; Zayed, M.F.; Mahdy, H.A. Design, synthesis, docking and biological evaluation of some novel 5-chloro-2-substituted sulfanylbenzoxazole derivatives as anticonvulsant agents. Med. Chem. Res. 2014, 24, 99–114. [Google Scholar] [CrossRef]
- Krall, R.L.; Penry, J.K.; White, B.G.; Kupferberg, H.J.; Swinyard, E.A. Antiepileptic Drug Development: II. Anticonvulsant Drug Screening. Epilepsia 1968, 19, 409–428. [Google Scholar] [CrossRef]
- Topliss, J.G. A manual method for applying the Hansch approach to drug design. J. Med. Chem. 1977, 20, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Englert, L.; Biela, A.; Zayed, M.F.; Heine, A.; Hangauer, D.; Klebe, G. Displacement of disordered water molecules from hydrophobic pocket creates enthalpic signature: Binding of phosphonamidate to the S1’-pocket of thermolysin. Biochim. Biophys. Acta 2010, 1800, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Kirk, L.K. Fluorine in medicinal chemistry: Recent therapeutic applications of fluorinated small molecules. J. Fluorine. Chem. 2006, 127, 1013–1029. [Google Scholar] [CrossRef]
- Dunham, N.W.; Miya, T.S. A note on a simple apparatus for detecting neurological deficit in rats and mice. J. Am. Pharm. Assoc. 1957, 46, 208–209. [Google Scholar] [CrossRef]
- Crivori, P.; Cruciani, G.; Carrupt, P.A.; Testa, B. Predicting blood-brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 2000, 11, 2204–2216. [Google Scholar] [CrossRef]
- Yogeeswari, P.; Sriram, D.; Thirumurugan, R.; Raghavendran, J.V.; Sudhan, K.; Pavana, R.K.; Stables, J. Discovery of N-(2,6-dimethylphenyl)-substituted semicarbazones as anticonvulsants: hybrid pharmacophore-based design. J. Med. Chem. 2005, 48, 6202–6211. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.F.; Rathman, T.L.; Sleevi, M.C.; Campbell, J.A.; Greenwood, T.D.J. Synthesis and anticonvulsant activity of some new 2-substituted 3-aryl-4(3H)-quinazolinone. J. Med. Chem. 1990, 33, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Swinyard, E.A.; Brown, W.C.; Goodman, L.S. Comparative Assays of Antiepileptic Drugs in Mice and Rats. J. Pharmacol. Exp. Ther. 1952, 106, 319–330. [Google Scholar] [PubMed]
- Chen, L.; Sun, Y.X.; Chai, Y.K.; Lee, J.S.; Song, M.S.; Quan, Z.S. Synthesis and anticonvulsant evaluation of 4-(4-alkoxylphenyl)-3-ethyl-4H-1,2,4-triazoles as open-chain analogues of 7-alkoxyl-4,5-dihydro[1,2,4]triazolo[4,3-a]quinolines. Bioorg. Med. Chem. 2007, 15, 6775–6781. [Google Scholar] [CrossRef] [PubMed]
- Zappalà, M.; Grasso, S.; Micale, N.; Zuccalà, G.; Menniti, F.S.; Ferreri, G.; Desarro, G.; Micheli, C. 1-Aryl-6,7-methylenedioxy-3H-quinazolin-4-ones as Anticonvulsant Agents. Bioorg. Med. Chem. Lett. 2003, 13, 4427–4430. [Google Scholar] [CrossRef] [PubMed]
- Ajeet, K.A. Designing of hybrid form of benzothiazole-quinazoline as GABA-A inhibitor with anticonvulsant profile: An in-silico approach. Am. J. Pharm. Sci. 2013, 1, 116–120. [Google Scholar] [CrossRef]
- Kumar, P.; Shrivastava, B.; Pandeya, S.N.; Stables, J.P. Design, synthesis and potential 6 Hz psychomotor seizure test activity of some novel 2-(substituted)-3-{[substituted]amino}quinazolin-4(3H)-one. Eur. J. Med. Chem. 2011, 46, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.A. GABAA Receptor Pharmacology. Pharmacol. Ther. 1996, 69, 173–198. [Google Scholar] [CrossRef]
- Ibrahim, M.K.; El-Adl, K.; Al-Karmalawy, A.A. Design, synthesis, molecular docking and anticonvulsant evaluation of novel 6-iodo-2-phenyl-3-substituted-quinazolin-4(3H)-ones. Bull. Fac. Pharm. Cairo Univ. 2015, 53, 101–116. [Google Scholar] [CrossRef]
- Kuo, S.; Hour, M.; Huang, L.; Lee, K. Preparation of 2-Phenyl-4-quinazolinones and 2-Phenyl-4-alkoxyquinazolines. U.S. Patent 6,479,499, 12 December 2002. [Google Scholar]
- Protein Data Bank. Crystal Structure of a Human Gamma-Aminobutyric Acid Receptor, the GABA(A)R-beta3 Homopentamer. Available online: http://www.rcsb.org/pdb/explore/explore.do?structureId=4COF (accessed on 20 December 2016).
- Roberts, E. Methods in Enzymology; Academic Press: New York, NY, USA, 1962; Volume VI, p. 612. [Google Scholar]
- Sample Availability: Samples of the compounds are currently not available from the authors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | PTZ (% Protection) | MES (% Protection) |
|---|---|---|
| 5a | 83 | 50 |
| 5b | 100 | 100 |
| 5c | 100 | 100 |
| 5d | 100 | 100 |
| 5e | 100 | 83 |
| 5f | 83 | 66 |
| 5g | 66 | 50 |
| 5h | 66 | 33 |
| 5i | 33 | 33 |
| 5j | 50 | 33 |
| Methaqualone | 100 | 100 |
| Valproate | 100 | 100 |
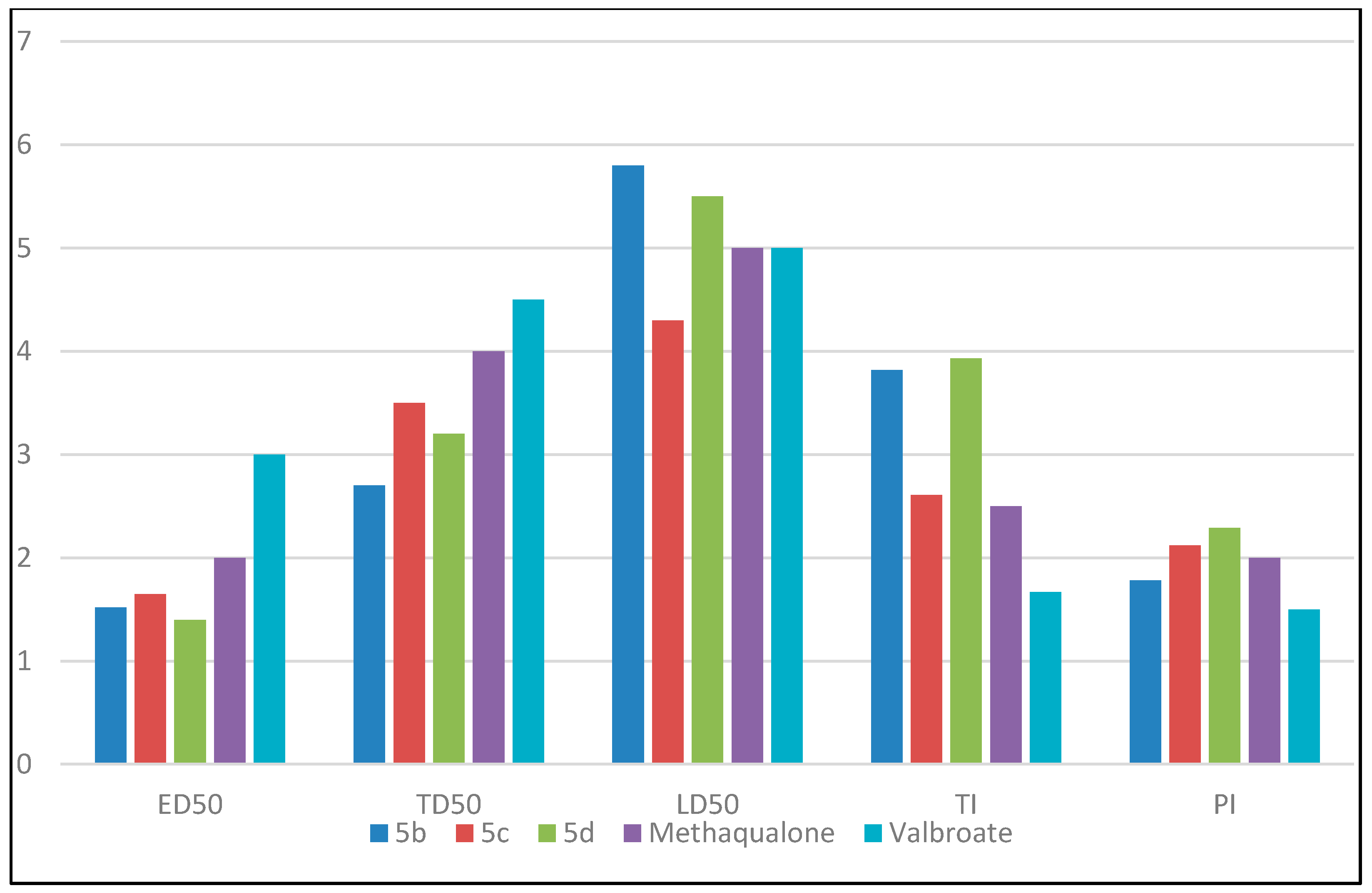
| Compound | ED50 (mg/kg) | TD50 (mg/kg) | LD50 (mg/kg) | Therapeutic Index (TI) | Protective Index (PI) |
|---|---|---|---|---|---|
| 5b | 152 | 270 | 580 | 3.82 | 1.78 |
| 5c | 165 | 350 | 430 | 2.61 | 2.12 |
| 5d | 140 | 320 | 550 | 3.93 | 2.29 |
| Methaqualone | 200 | 400 | 500 | 2.5 | 2 |
| Valproate | 300 | 450 | 500 | 1.67 | 1.5 |
| Compound | ∆G (kcal·mol−1) |
|---|---|
| 5a | –55.74 |
| 5b | –68.97 |
| 5c | –67.50 |
| 5d | –69.68 |
| 5e | –66.84 |
| 5f | –58.81 |
| 5g | –62.65 |
| 5h | –65.68 |
| 5i | –56.04 |
| 5j | –65.76 |
| Methaqualone | –50.69 |
| GABA con. (μgm/100mg of Brain Tissue) | ||
|---|---|---|
| Compound | 2 h Post-Treatment (100 mg/kg) | Seven Days Post-Treatment (30 mg/kg) |
| Control | 45.8 ± 5.7 | 52.6 ± 3.4 |
| 5b | 84 ± 4.42 ** | 79 ± 4.21 *** |
| 5c | 76 ± 5.28 ** | 80 ± 4.26 *** |
| 5d | 93 ± 8.42 * | 105 ± 3.1 |
| Clobazam | 103 ± 5.9 | 109 ± 5.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayed, M.F.; Ihmaid, S.K.; Ahmed, H.E.A.; El-Adl, K.; Asiri, A.M.; Omar, A.M. Synthesis, Modelling, and Anticonvulsant Studies of New Quinazolines Showing Three Highly Active Compounds with Low Toxicity and High Affinity to the GABA-A Receptor. Molecules 2017, 22, 188. https://doi.org/10.3390/molecules22020188
Zayed MF, Ihmaid SK, Ahmed HEA, El-Adl K, Asiri AM, Omar AM. Synthesis, Modelling, and Anticonvulsant Studies of New Quinazolines Showing Three Highly Active Compounds with Low Toxicity and High Affinity to the GABA-A Receptor. Molecules. 2017; 22(2):188. https://doi.org/10.3390/molecules22020188
Chicago/Turabian StyleZayed, Mohamed F., Saleh K. Ihmaid, Hany E. A. Ahmed, Khaled El-Adl, Ahmed M. Asiri, and Abdelsattar M. Omar. 2017. "Synthesis, Modelling, and Anticonvulsant Studies of New Quinazolines Showing Three Highly Active Compounds with Low Toxicity and High Affinity to the GABA-A Receptor" Molecules 22, no. 2: 188. https://doi.org/10.3390/molecules22020188
APA StyleZayed, M. F., Ihmaid, S. K., Ahmed, H. E. A., El-Adl, K., Asiri, A. M., & Omar, A. M. (2017). Synthesis, Modelling, and Anticonvulsant Studies of New Quinazolines Showing Three Highly Active Compounds with Low Toxicity and High Affinity to the GABA-A Receptor. Molecules, 22(2), 188. https://doi.org/10.3390/molecules22020188