Activation of Recombinantly Expressed l-Amino Acid Oxidase from Rhizoctonia solani by Sodium Dodecyl Sulfate

 ,
,  and
and
Abstract
:1. Introduction
2. Results
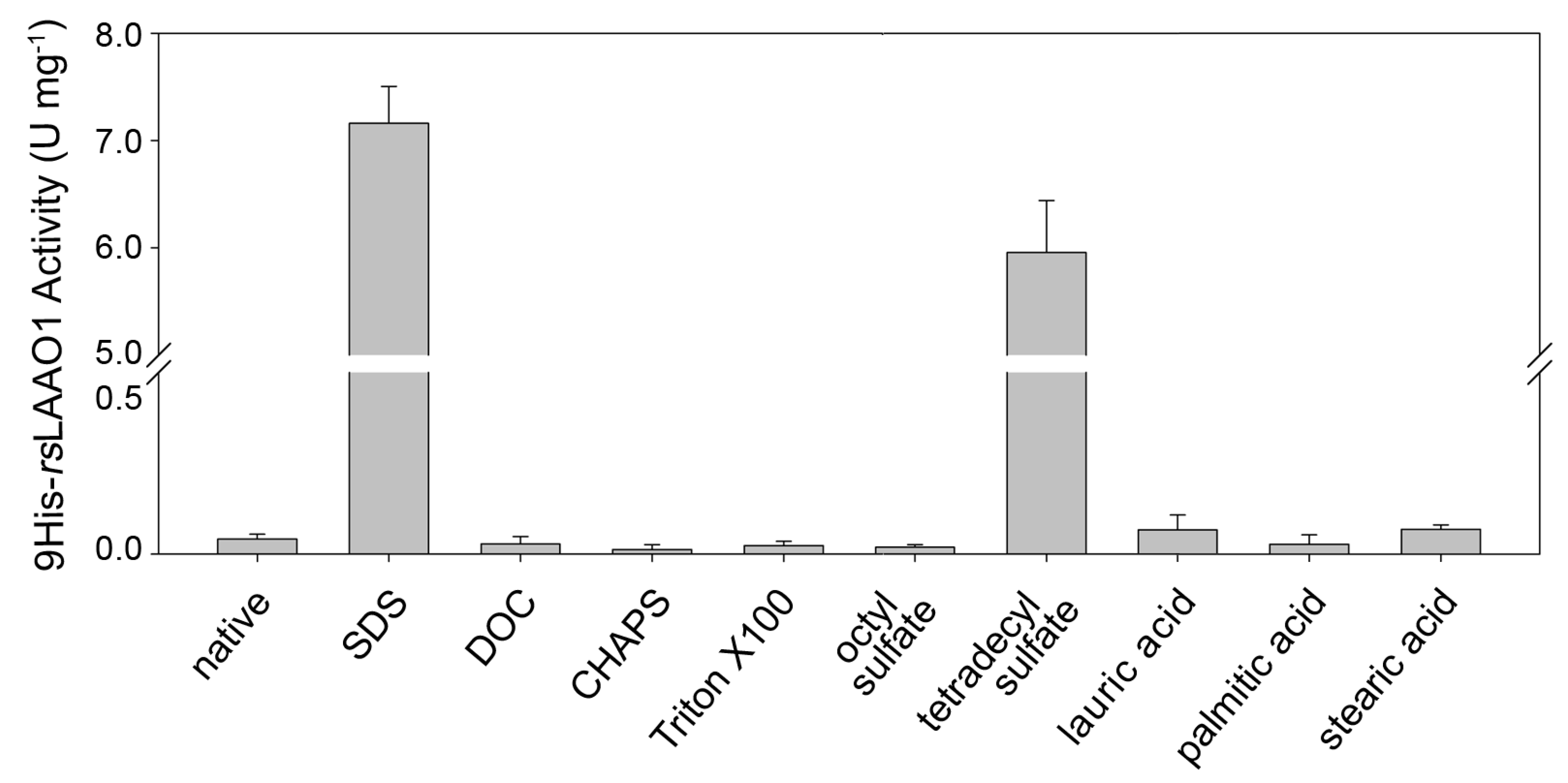
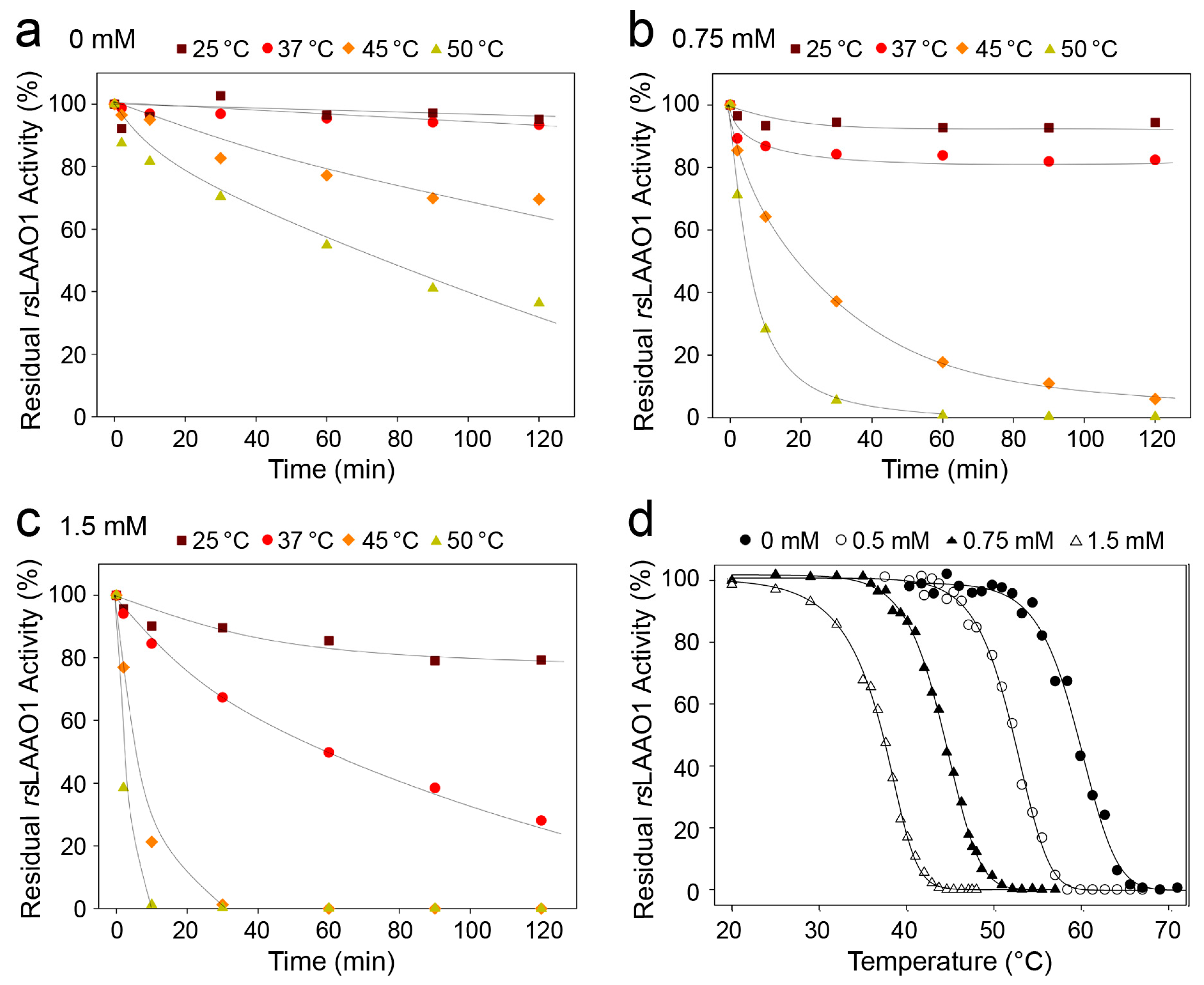
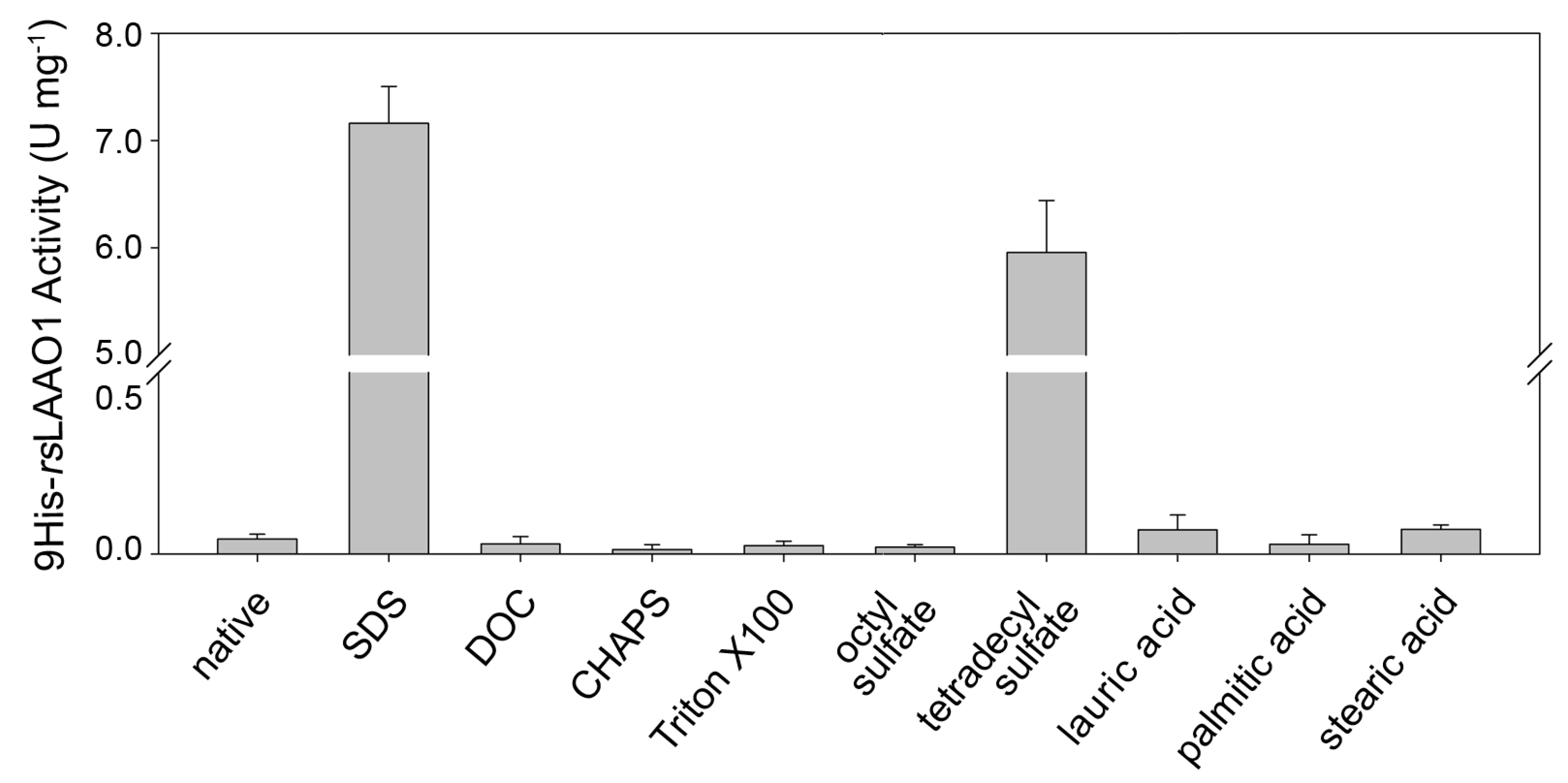
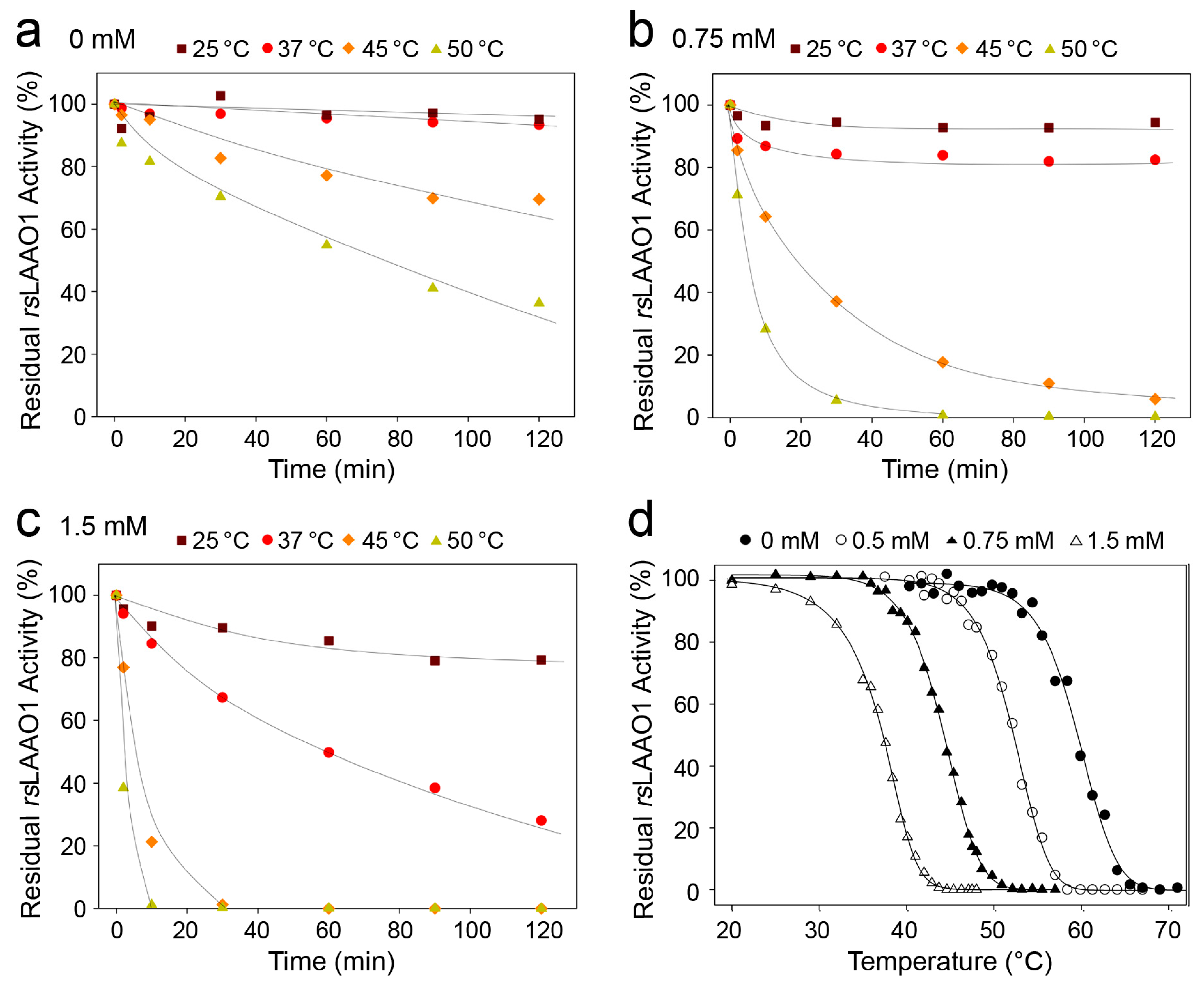
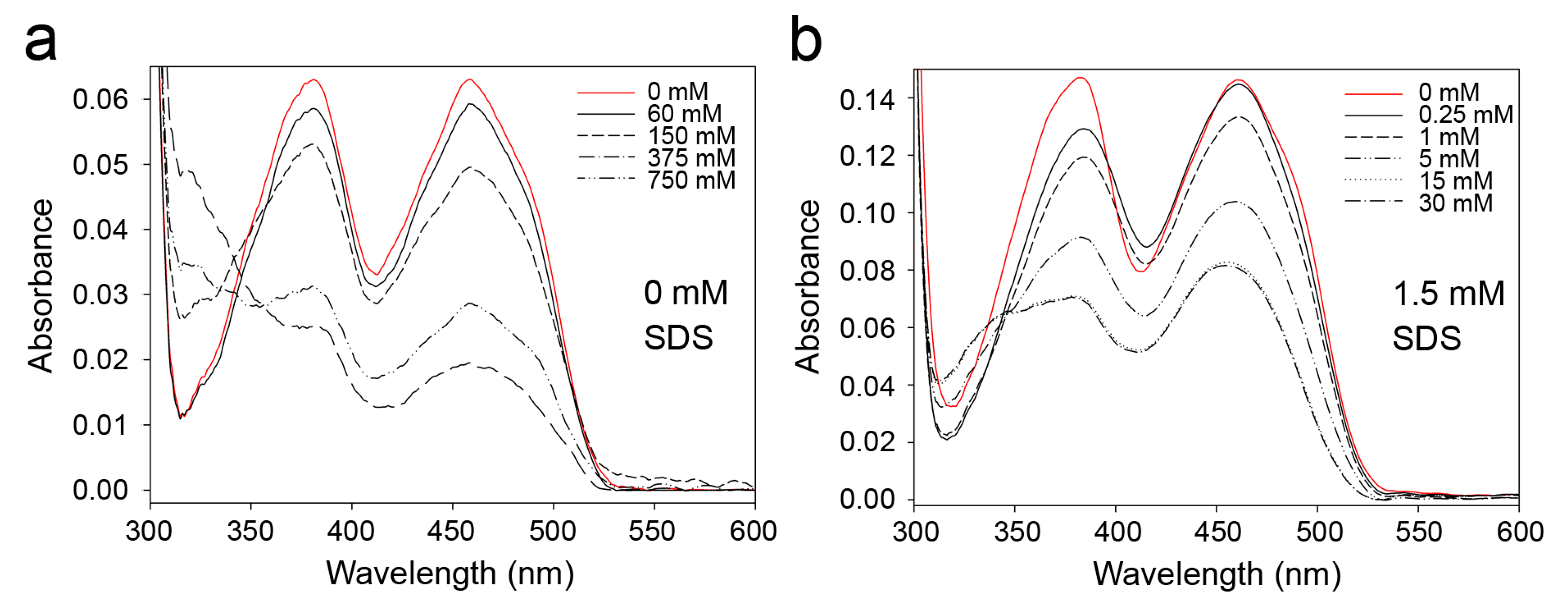
2.1. Effects of Detergents and Fatty Acids on 9His-rsLAAO1 Activity
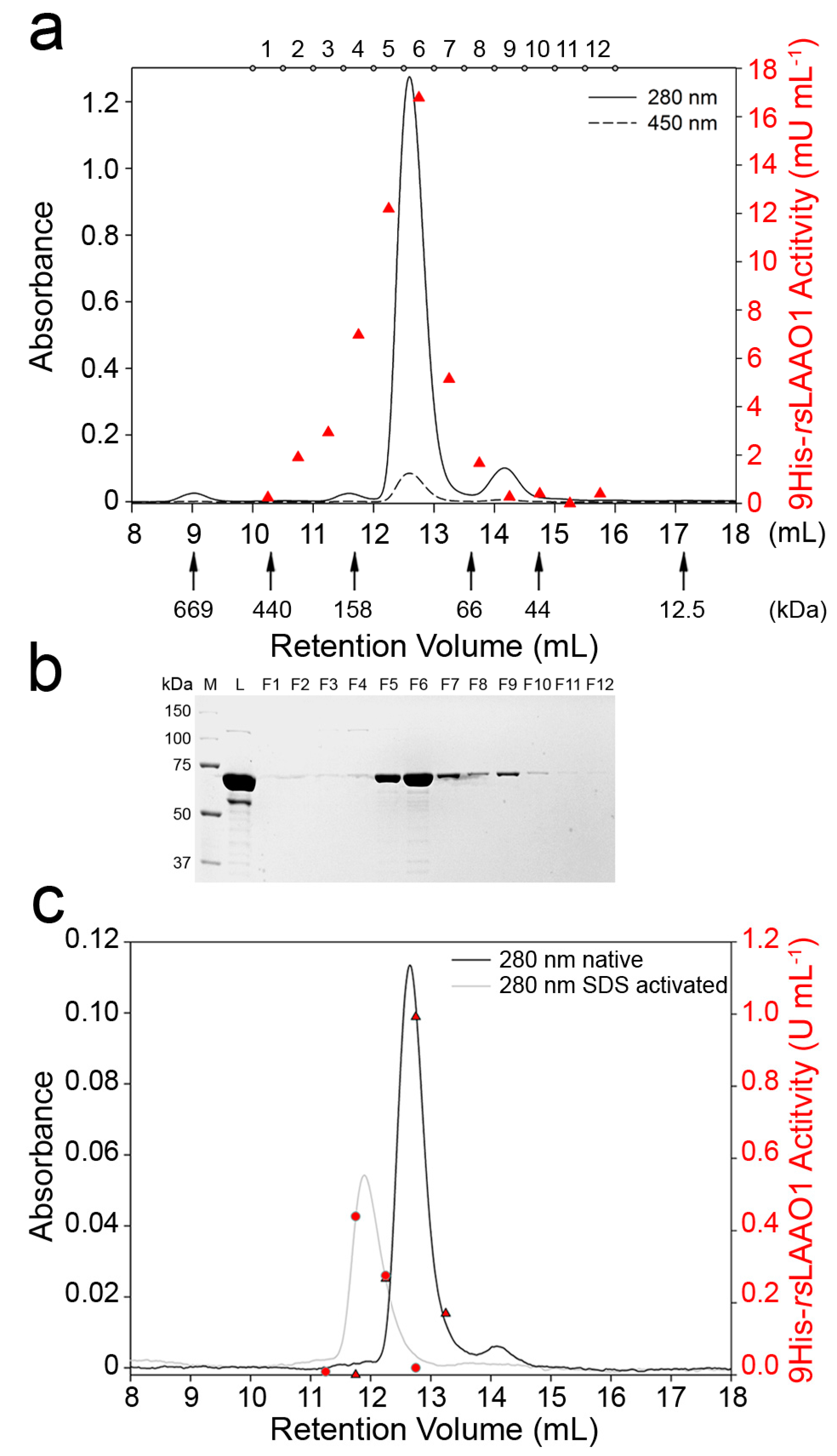
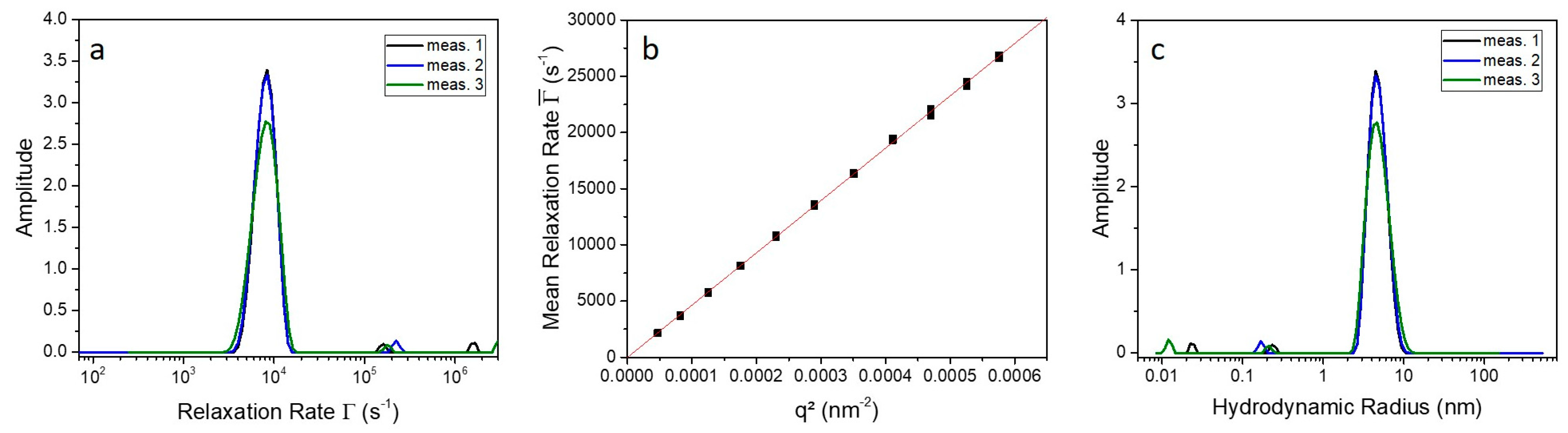
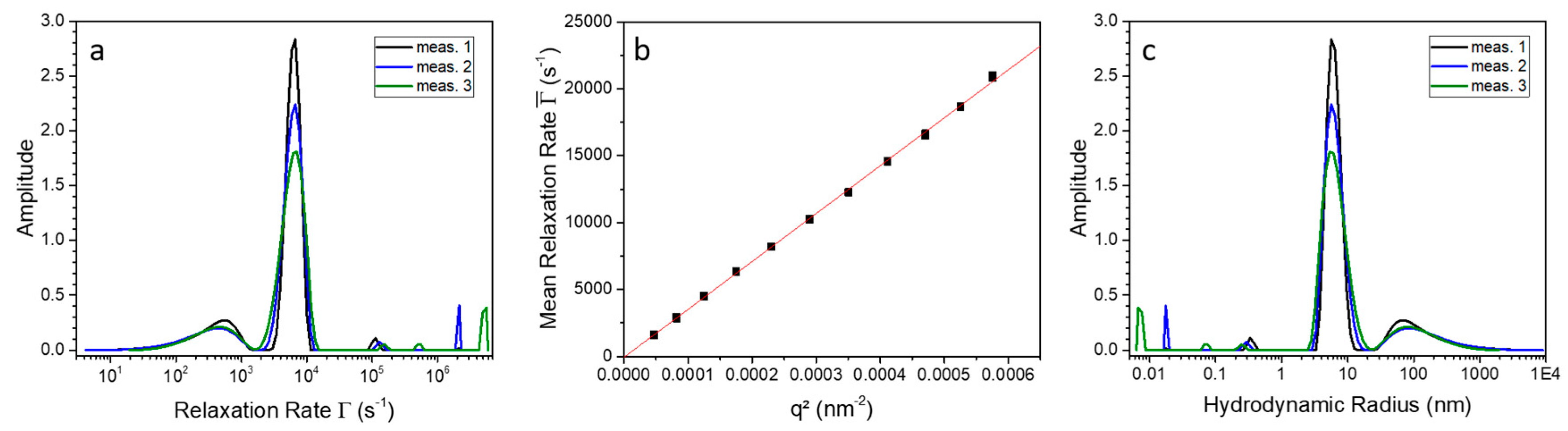
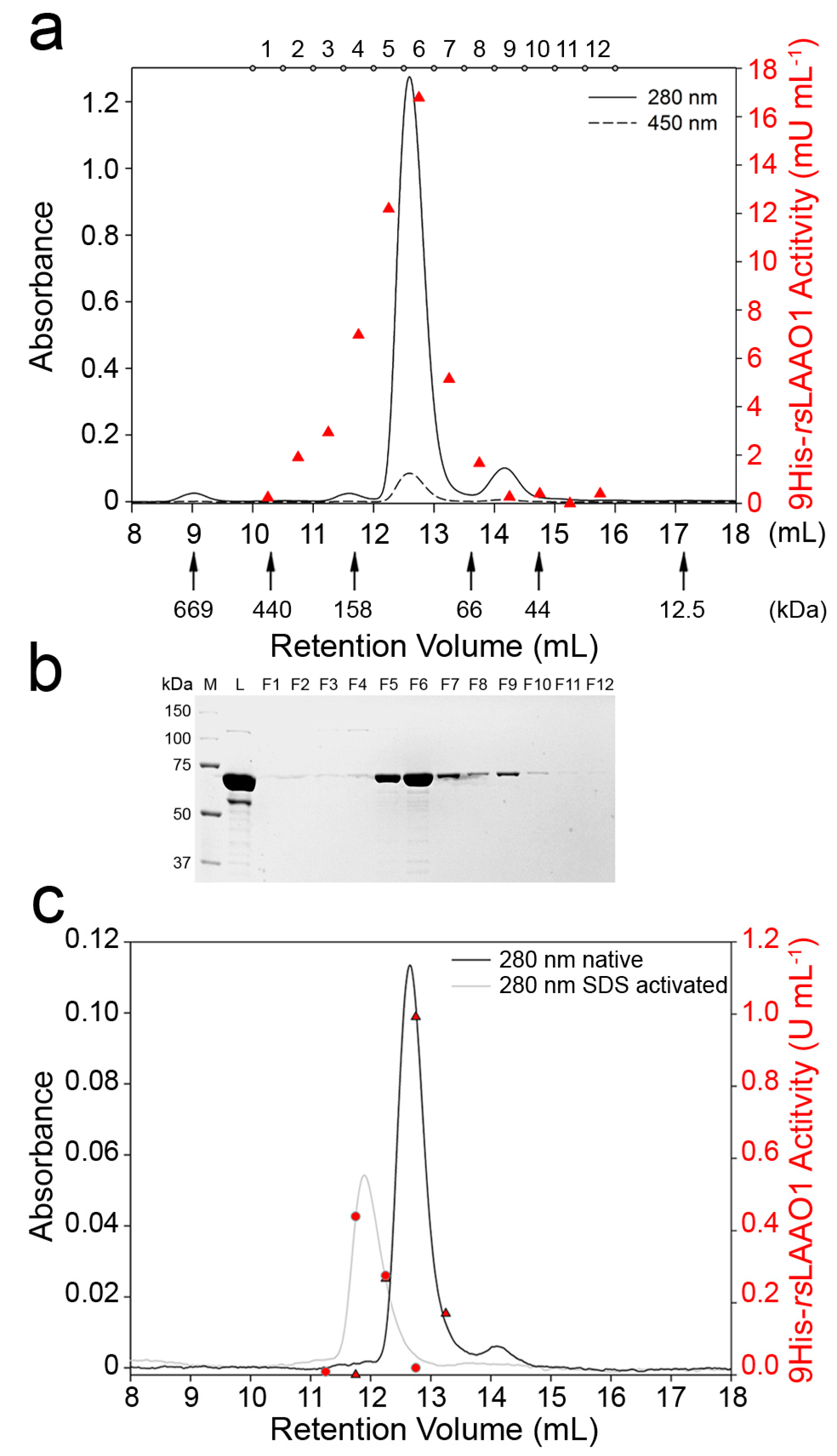
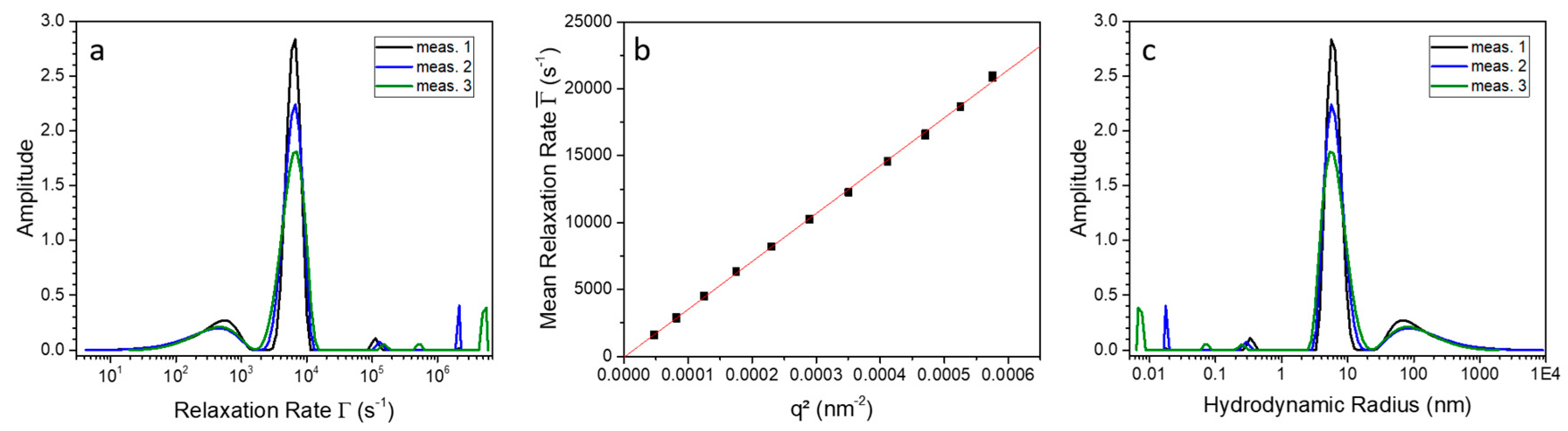
2.2. 9His-rsLAAO1 Behaved as a Dimer in the Native and SDS-Activated State
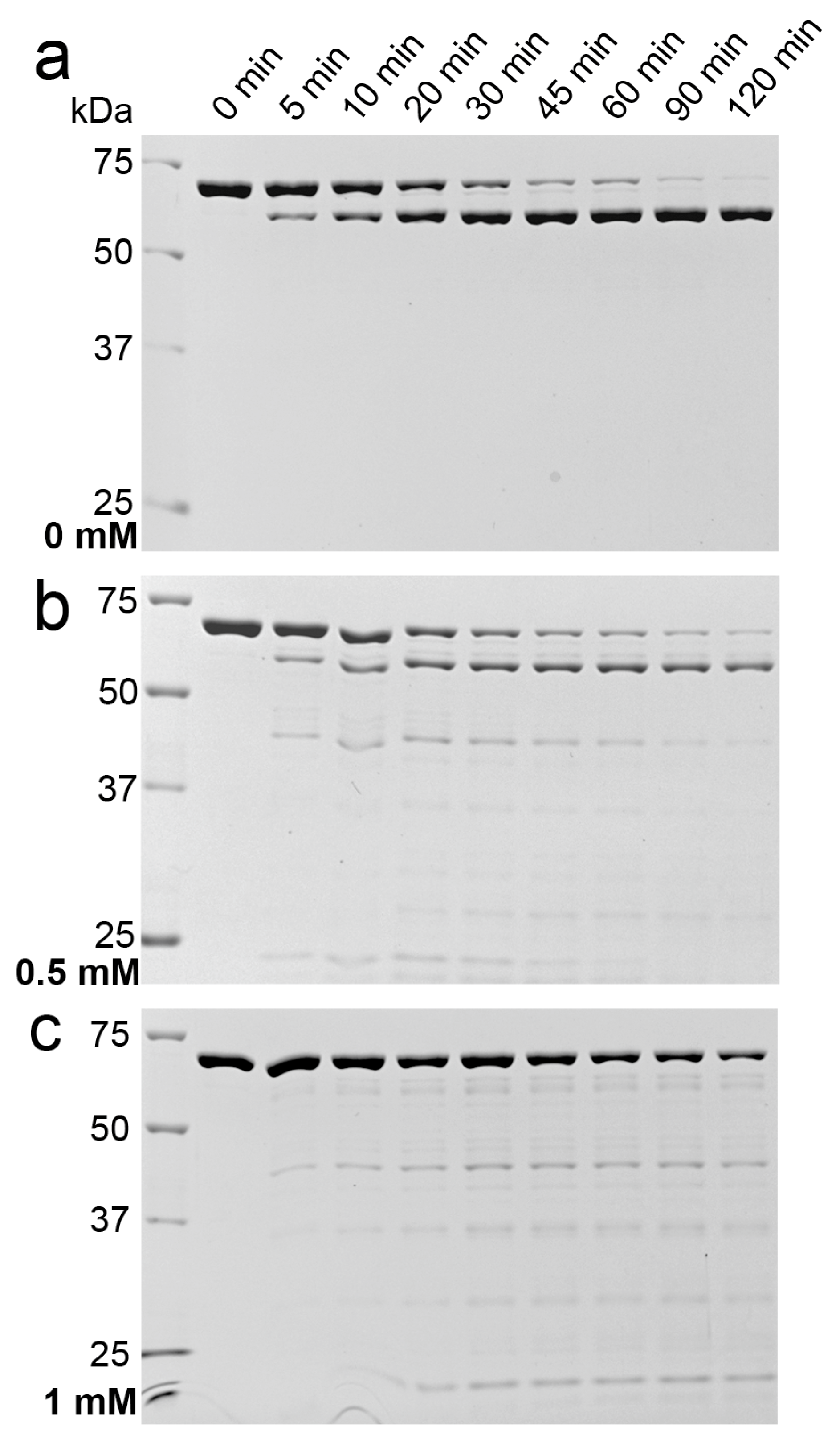
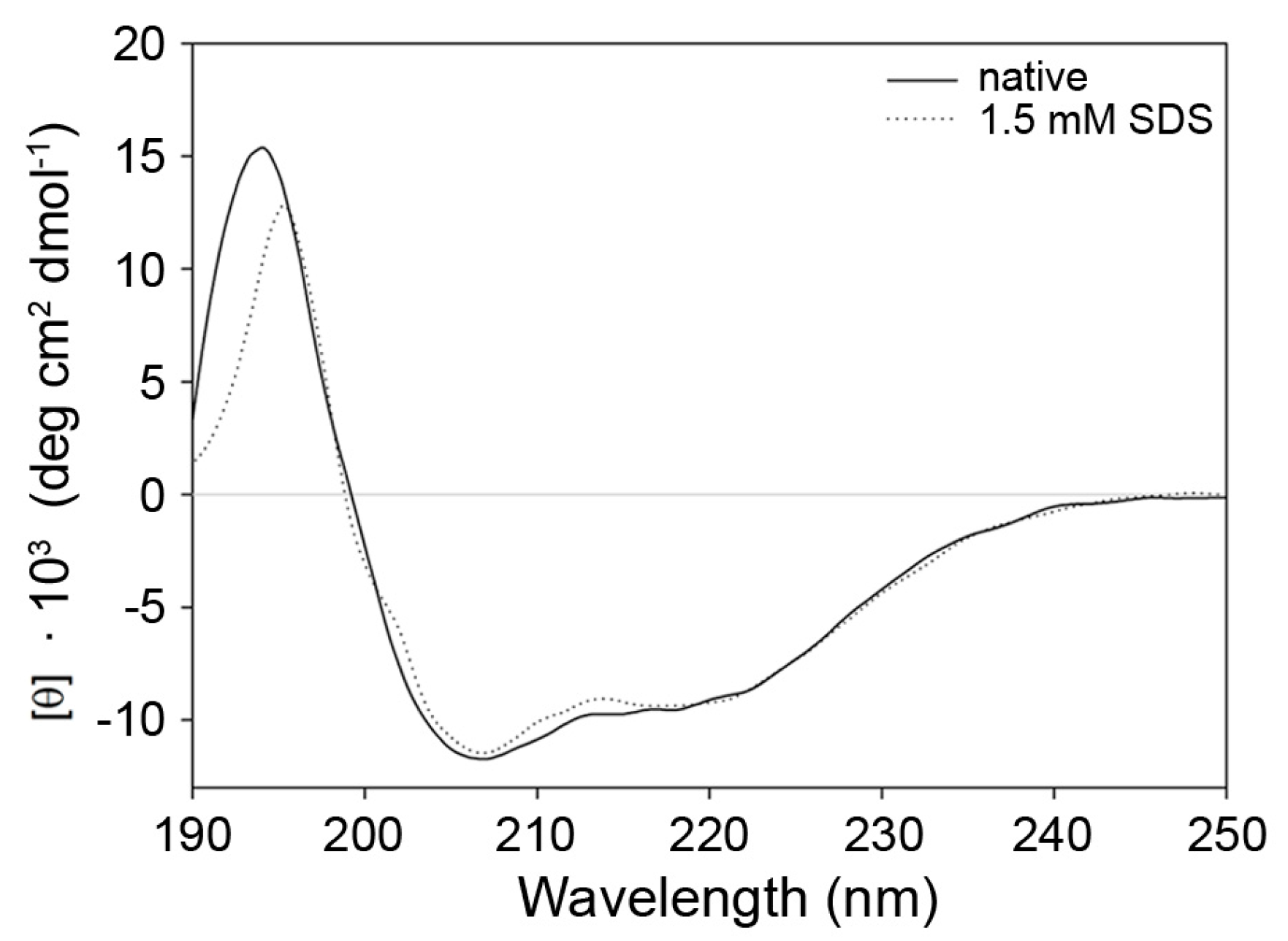
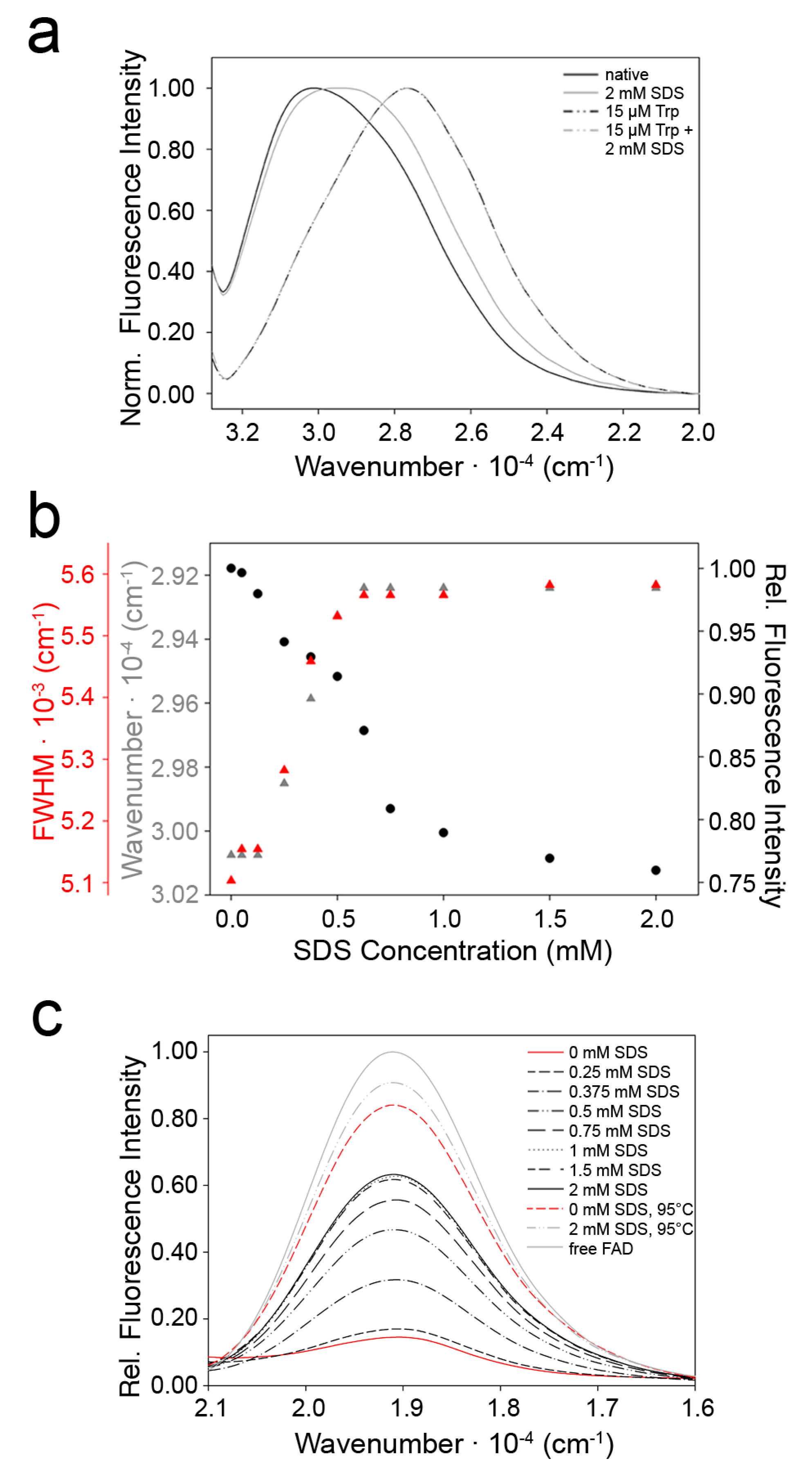
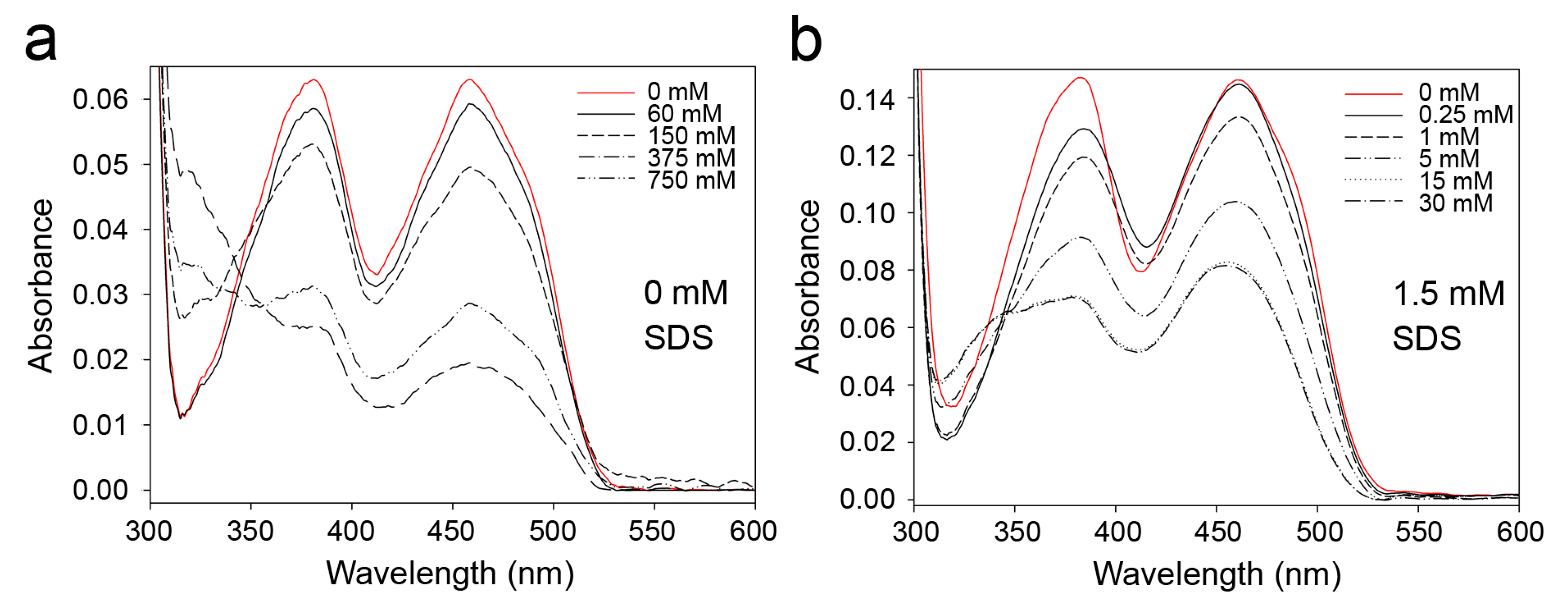
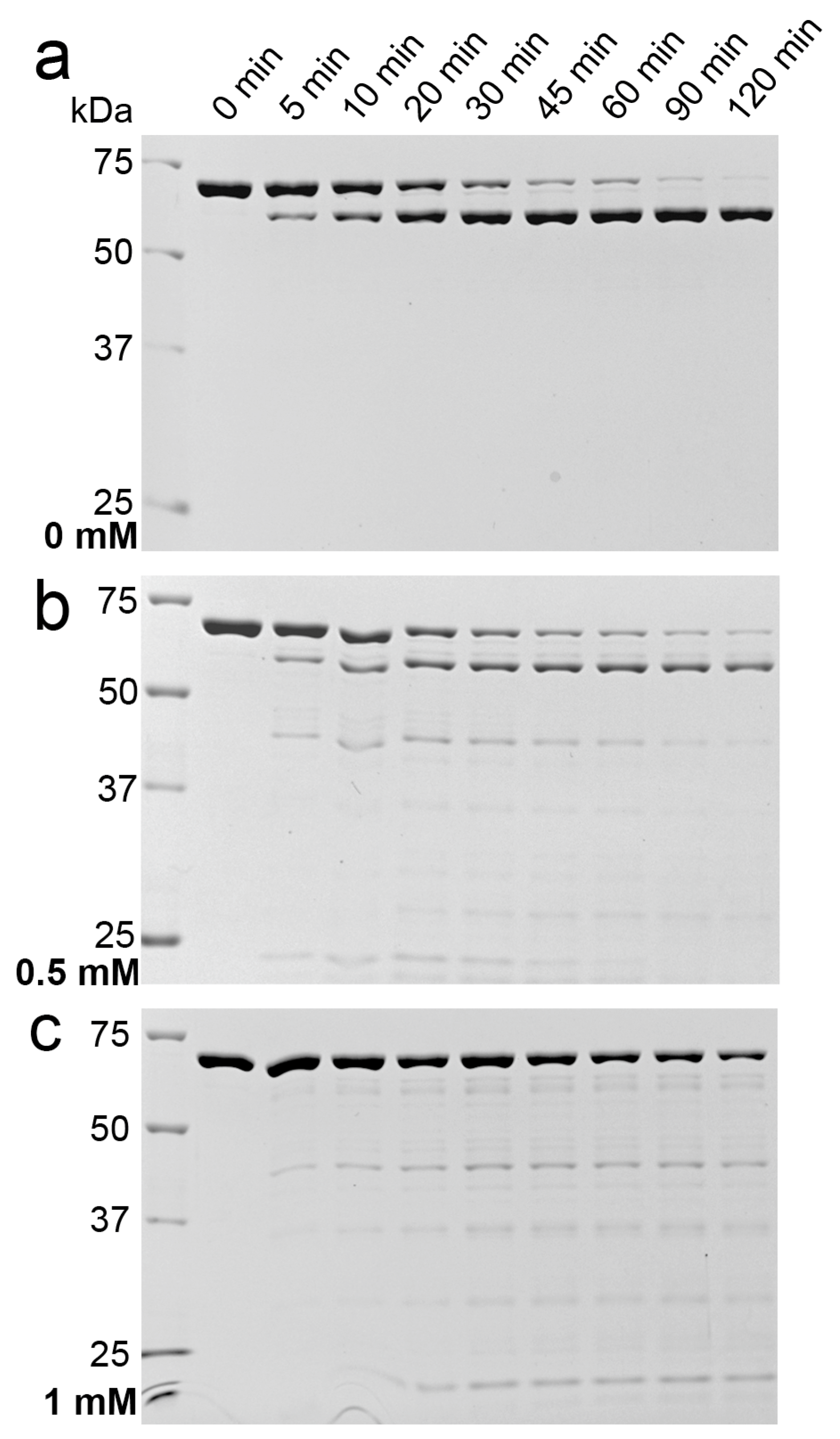
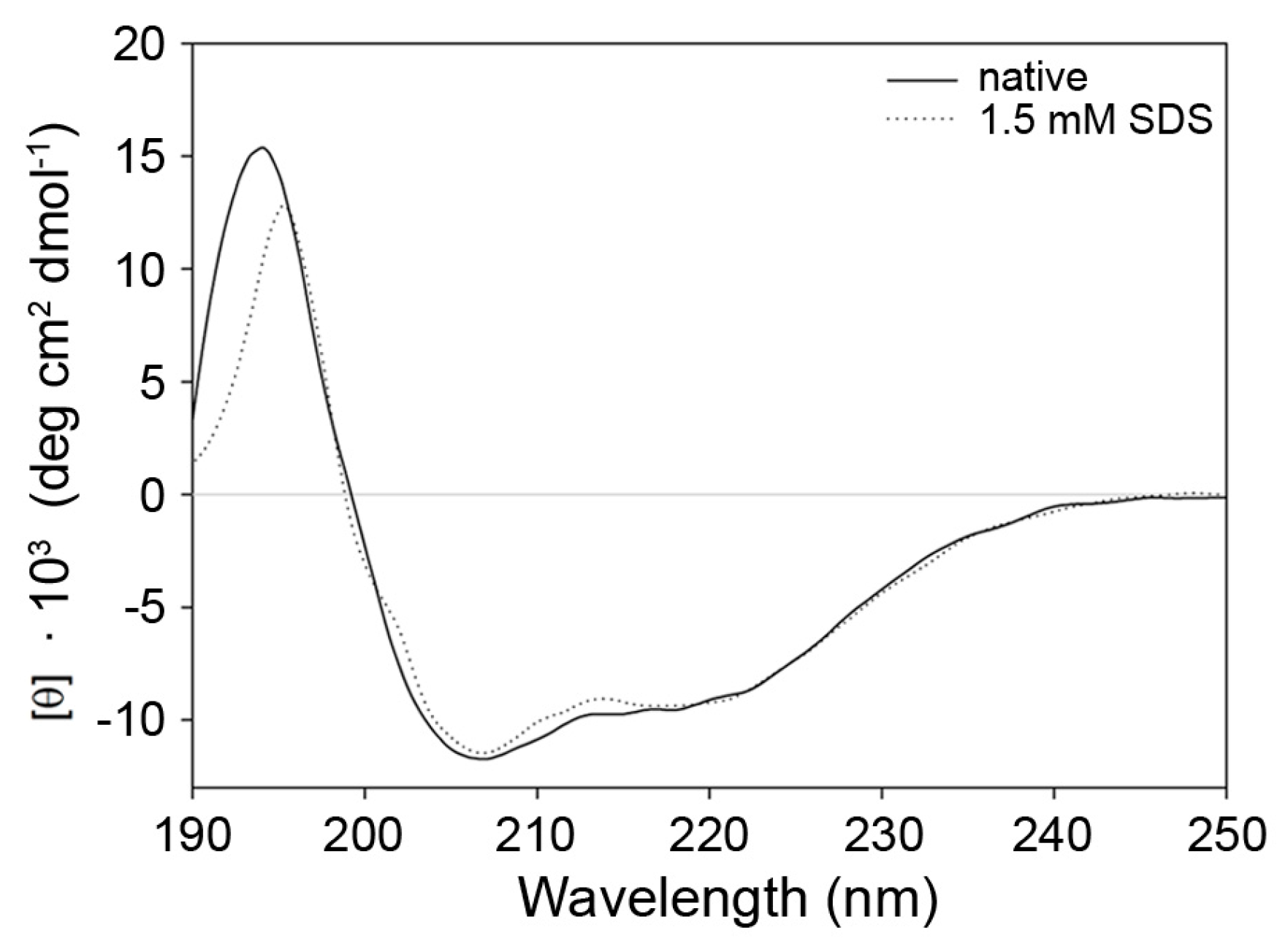
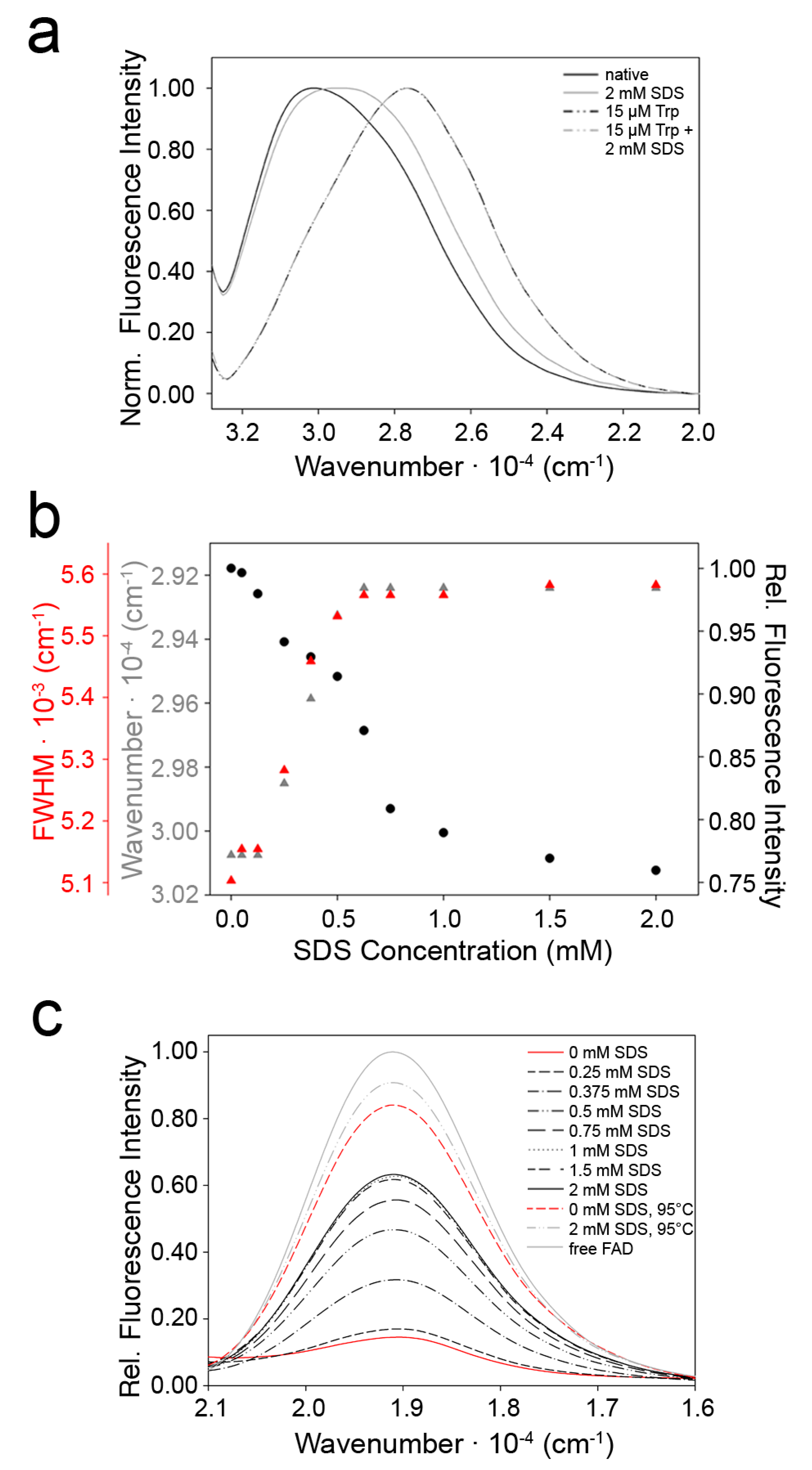
2.3. Indications for Conformational Changes in SDS-Activated 9His-rsLAAO1
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of 9His-rsLAAO1
4.2. Enzyme Assay
4.3. Molecular Mass Determination by Size-Exclusion Chromatography
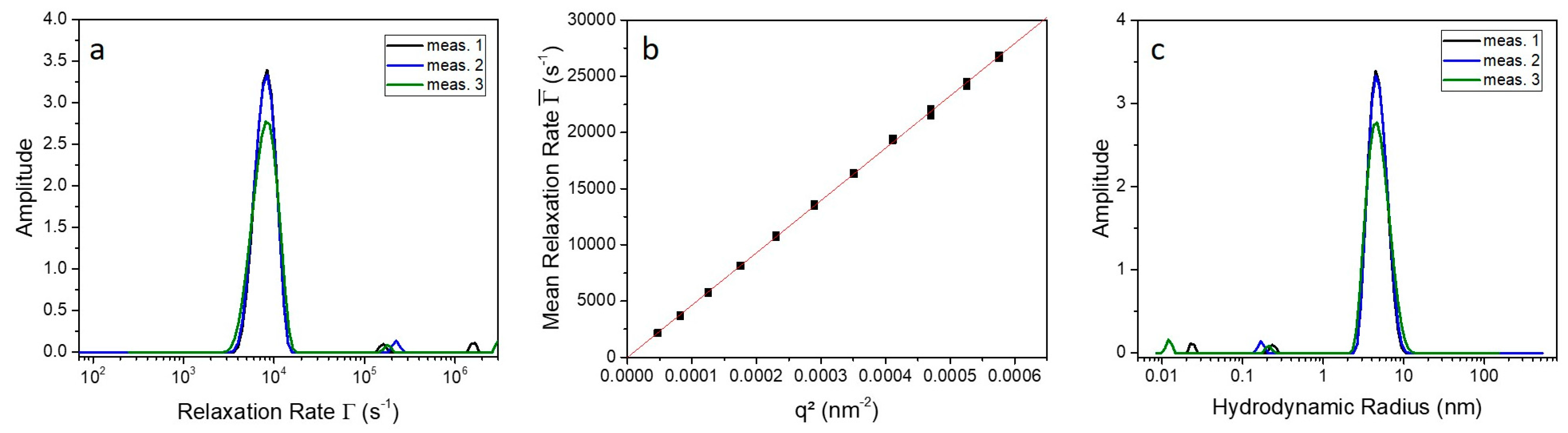
4.4. Photon Correlation Spectroscopy (PCS)
4.5. Circular Dichroism Spectroscopy
4.6. Fluorescence Spectroscopy
4.7. Reactivity of FAD with Sulfite
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bender, A.E.; Krebs, H.A. The oxidation of various synthetic α-amino-acids by mammalian d-amino-acid oxidase, l-amino-acid oxidase of cobra venom and the l-amino-acid and d-amino-acid oxidases of Neurospora crassa. Biochem. J. 1950, 46, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Geueke, B.; Hummel, W. A new bacterial l-amino acid oxidase with a broad substrate specificity: Purification and characterization. Enzyme Microb. Technol. 2002, 31, 77–87. [Google Scholar] [CrossRef]
- Nuutinen, J.T.; Timonen, S. Identification of nitrogen mineralization enzymes, l-amino acid oxidases, from the ectomycorrhizal fungi Hebeloma spp. and Laccaria bicolor. Mycol. Res. 2008, 112, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Pollegioni, L.; Motta, P.; Molla, G. l-Amino acid oxidase as biocatalyst: A dream too far? Appl. Microbiol. Biotechnol. 2013, 97, 9323–9341. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.A.; Cheng, C.H.; Lo, C.T.; Liu, S.Y.; Lee, J.W.; Peng, K.C. A Novel l-Amino Acid Oxidase from Trichoderma harzianum ETS 323 Associated with Antagonism of Rhizoctonia solani. J. Agric. Food Chem. 2011, 59, 4519–4526. [Google Scholar] [CrossRef] [PubMed]
- Pollegioni, L.; Molla, G. New biotech applications from evolved d-amino acid oxidases. Trends Biotechnol. 2011, 29, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Rodriguez, S.; Martinez-Gomez, A.I.; Rodriguez-Vico, F.; Clemente-Jimenez, J.M.; Heras-Vazquez, F.J.L. Natural Occurrence and Industrial Applications of d-Amino Acids: An Overview. Chem. Biodivers. 2010, 7, 1531–1548. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, Y.; Rodriguez, G.M.; Atsumi, S. 2-Keto acids based biosynthesis pathways for renewable fuels and chemicals. J. Ind. Microbiol. Biotechnol. 2015, 42, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Hossain, G.S.; Li, J.; Shin, H.-D.; Du, G.; Liu, L.; Chen, J. l-Amino acid oxidases from microbial sources: Types, properties, functions, and applications. Appl. Microbiol. Biotechnol. 2014, 98, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Geueke, B.; Hummel, W. Heterologous expression of Rhodococcus opacus l-amino acid oxidase in Streptomyces lividans. Protein Expr. Purif. 2003, 28, 303–309. [Google Scholar] [CrossRef]
- Tong, H.C.; Chen, W.; Shi, W.Y.; Qi, F.X.; Dong, X.Z. SO-LAAO, a novel l-amino acid oxidase that enables Streptococcus oligofermentans to outcompete Streptococcus mutans by generating H2O2 from peptone. J. Bacteriol. 2008, 190, 4716–4721. [Google Scholar] [CrossRef] [PubMed]
- Nuutinen, J.T.; Marttinen, E.; Soliymani, R.; Hilden, K.; Timonen, S. l-Amino acid oxidase of the fungus Hebeloma cylindrosporum displays substrate preference towards glutamate. Microbiology 2012, 158, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Hahn, K.; Neumeister, K.; Mix, A.; Kottke, T.; Groger, H.; Von Mollard, G. Recombinant expression and characterization of a l-amino acid oxidase from the fungus Rhizoctonia solani. Appl. Microbiol. Biotechnol. 2017, 101, 2853–2864. [Google Scholar] [CrossRef] [PubMed]
- Berne, B.J.; Pecora, R. Dynamic Light Scattering; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1976. [Google Scholar]
- Provencher, S.W. A constrained regularization method for inverting data represented by linear algebraic or integral-equations. Comput. Phys. Commun. 1982, 27, 213–227. [Google Scholar] [CrossRef]
- Provencher, S.W. CONTIN—A general-purpose constrained regularization program for inverting noisy linear algebraic and integral-equations. Comput. Phys. Commun. 1982, 27, 229–242. [Google Scholar] [CrossRef]
- Hellweg, T.; Hinssen, H.; Eimer, W. The Ca2+-induced conformational change of gelsolin is located in the carboxyl-terminal half of the molecule. Biophys. J. 1993, 65, 799–805. [Google Scholar] [CrossRef]
- Dubin, S.B.; Clark, N.A.; Benedek, G.B. Measurement of rotational diffusion coefficient of lysozyme by depolarized light scattering—Configuration of lysozyme in solution. J. Chem. Phys. 1971, 54, 5158–5164. [Google Scholar] [CrossRef]
- Eimer, W.; Niermann, M.; Eppe, M.A.; Jockusch, B.M. Molecular shape of vinculin in aqueous-solution. J. Mol. Biol. 1993, 229, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, P.; Mysels, K.J. Critical Micelle Concentration of Aqueous Surfactant Systems; NSRDS-NBS 36; U.S. Department of Commerce National Bureau of Standards: Washington, DC, USA, 1971.
- Sammalkorpi, M.; Karttunen, M.; Haataja, M. Ionic Surfactant Aggregates in Saline Solutions: Sodium Dodecyl Sulfate (SDS) in the Presence of Excess Sodium Chloride (NaCl) or Calcium Chloride (CaCl2). J. Phys. Chem. B 2009, 113, 5863–5870. [Google Scholar] [CrossRef] [PubMed]
- Deleage, G.; Geourjon, C. An interactive graphic program for calculating the secondary structure-content of proteins from circular-dichroism spctrum. Comput. Appl. Biosci. 1993, 9, 197–199. [Google Scholar] [PubMed]
- Holmgren, A. Tryptophan fluorescence study of conformational transitions of oxidized and reduced form of thioredoxin. J. Biol. Chem. 1972, 247, 1992–1998. [Google Scholar] [PubMed]
- Massey, V.; Curti, B. A new method of preparation of d-amino acid oxidase apoprotein and a conformational change after its combination with flavin adenine dinucleotide. J. Biol. Chem. 1966, 241, 3417–3423. [Google Scholar] [PubMed]
- Massey, V.; Muller, F.; Feldberg, R.; Schuman, M.; Sullivan, P.A.; Howell, L.G.; Mayhew, S.G.; Matthews, R.G.; Foust, G.P. Reactivity of flavoproteins with sulfite—Possible relevance to problem of oxygen reactivity. J. Biol. Chem. 1969, 244, 3999–4006. [Google Scholar] [PubMed]
- Massey, V.; Hemmerich, P. Active-site probes of flavoproteins. Biochem. Soc. Trans. 1980, 8, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Saeidian, S.; Keyhani, E.; Keyhani, J. Effect of ionic detergents, nonionic detergents, and chaotropic agents on polyphenol oxidase activity from dormant saffron (Crocus sativus L.) corms. J. Agric. Food Chem. 2007, 55, 3713–3719. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Nellaiappan, K. On the latency and nature of phenoloxidase present in the left colleterial gland of the cockroach Periplaneta americana. Arch. Insect Biochem. Physiol. 1990, 15, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Nellaiappan, K.; Sugumaran, M. On the presence of prophenoloxidase in the hemolymph of the horseshoe crab, Limulus. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 1996, 113, 163–168. [Google Scholar] [CrossRef]
- Wittenberg, C.; Triplett, E.L. A detergent-activated tyrosinase from Xenopus-laevis 2. Detergent activation and binding. J. Biol. Chem. 1985, 260, 2542–2546. [Google Scholar]
- Golbeck, J.H.; Cammarata, K.V. Spinach thylakoid polyphenol oxidase—Isolation, activation, and properties of the native chloroplast enzyme. Plant Physiol. 1981, 67, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Kanade, S.R.; Paul, B.; Rao, A.G.A.; Gowda, L.R. The conformational state of polyphenol oxidase from field bean (Dolichlos lablah) upon SDS and acid-pH activation. Biochem. J. 2006, 395, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Espin, J.C.; Wichers, H.J. Activation of a latent mushroom (Agaricus bisporus) tyrosinase isoform by sodium dodecyl sulfate (SDS). kinetic properties of the SDS-activated isoform. J. Agric. Food Chem. 1999, 47, 3518–3525. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. Rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Koppel, D.E. Analysis of macromolecular polydispersity in intensity correlation spectroscopy—Method of cumulants. J. Chem. Phys. 1972, 57, 4814–4820. [Google Scholar] [CrossRef]
- Kelly, S.M.; Price, N.C. The Use of Circular Dichroism in the Investigation of Protein Structure and Function. Curr. Protein Pept. Sci. 2000, 1, 349–384. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the expression plasmid encoding MBP-P-9His-rsLAAO1 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calculated | SEC (without SDS) | SEC with SDS | |
|---|---|---|---|
| molecular weight | 150 kDa (dimer) | 120 kDa (dimer) | 168 kDa (dimer) |
| theoretical radius | = 3.5 nm | = 3.3 nm | = 3.7 nm |
| theoretical hydrodynamic radius | = 3.8 nm | = 3.6 nm | = 4.0 nm |
| 4.6 nm | 4.6 nm | 6.0 nm | |
| 1.2 | 1.3 | 1.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hahn, K.; Hertle, Y.; Bloess, S.; Kottke, T.; Hellweg, T.; Fischer von Mollard, G. Activation of Recombinantly Expressed l-Amino Acid Oxidase from Rhizoctonia solani by Sodium Dodecyl Sulfate. Molecules 2017, 22, 2272. https://doi.org/10.3390/molecules22122272
Hahn K, Hertle Y, Bloess S, Kottke T, Hellweg T, Fischer von Mollard G. Activation of Recombinantly Expressed l-Amino Acid Oxidase from Rhizoctonia solani by Sodium Dodecyl Sulfate. Molecules. 2017; 22(12):2272. https://doi.org/10.3390/molecules22122272
Chicago/Turabian StyleHahn, Katharina, Yvonne Hertle, Svenja Bloess, Tilman Kottke, Thomas Hellweg, and Gabriele Fischer von Mollard. 2017. "Activation of Recombinantly Expressed l-Amino Acid Oxidase from Rhizoctonia solani by Sodium Dodecyl Sulfate" Molecules 22, no. 12: 2272. https://doi.org/10.3390/molecules22122272
APA StyleHahn, K., Hertle, Y., Bloess, S., Kottke, T., Hellweg, T., & Fischer von Mollard, G. (2017). Activation of Recombinantly Expressed l-Amino Acid Oxidase from Rhizoctonia solani by Sodium Dodecyl Sulfate. Molecules, 22(12), 2272. https://doi.org/10.3390/molecules22122272