Nucleobase–Guanidiniocarbonyl-Pyrrole Conjugates as Novel Fluorimetric Sensors for Single Stranded RNA




Abstract
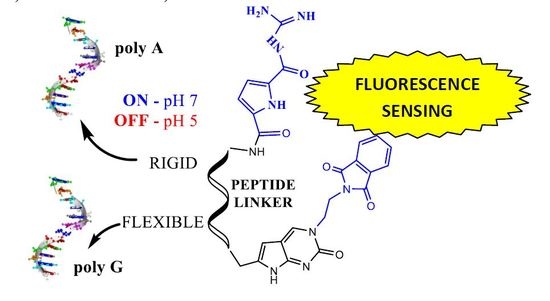
:
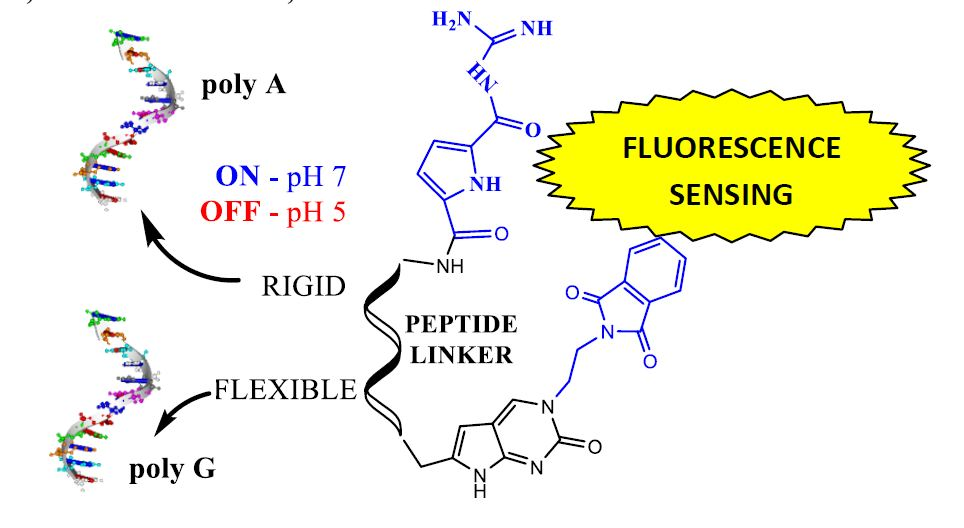
1. Introduction
2. Results and Discussion
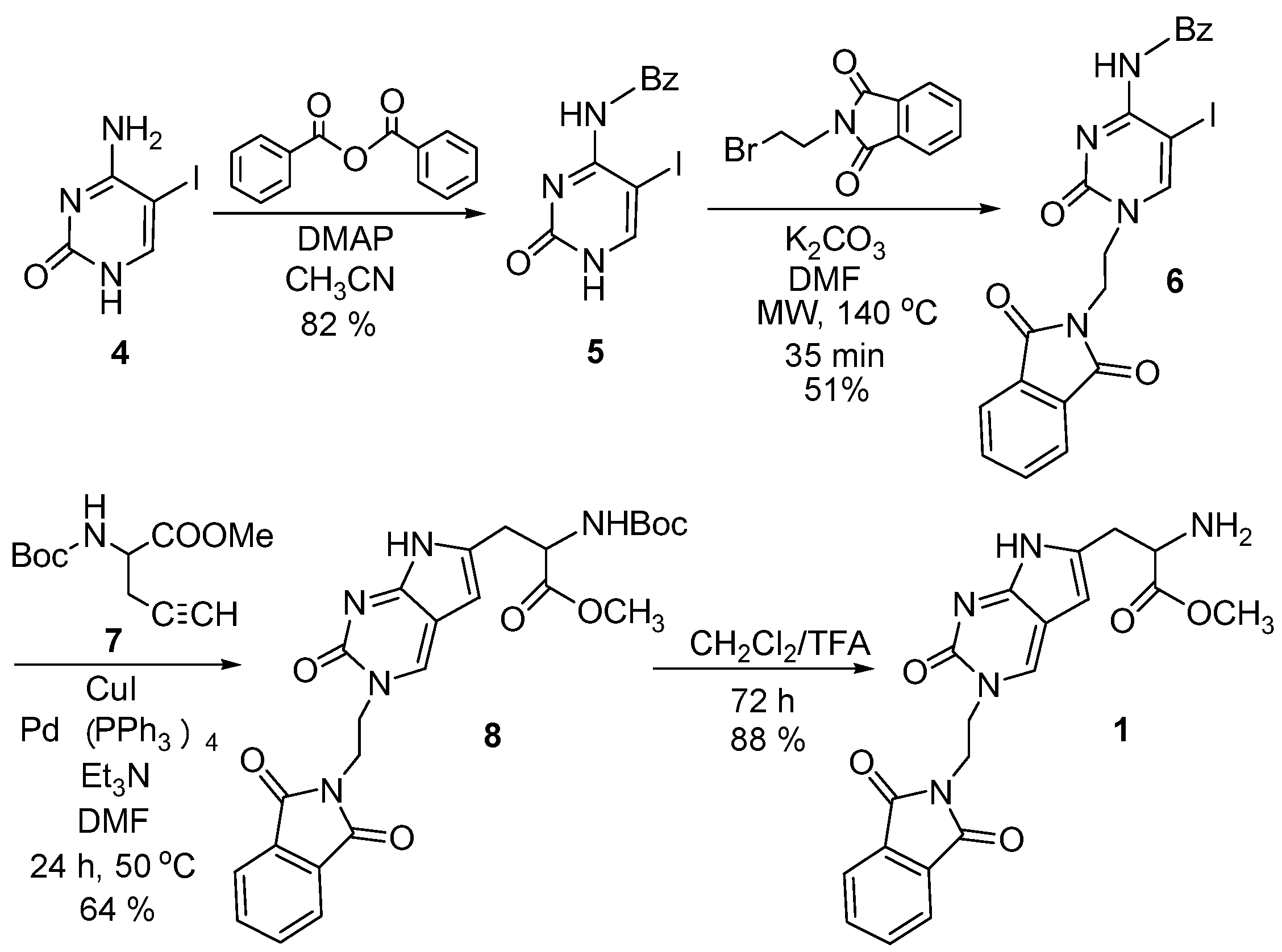
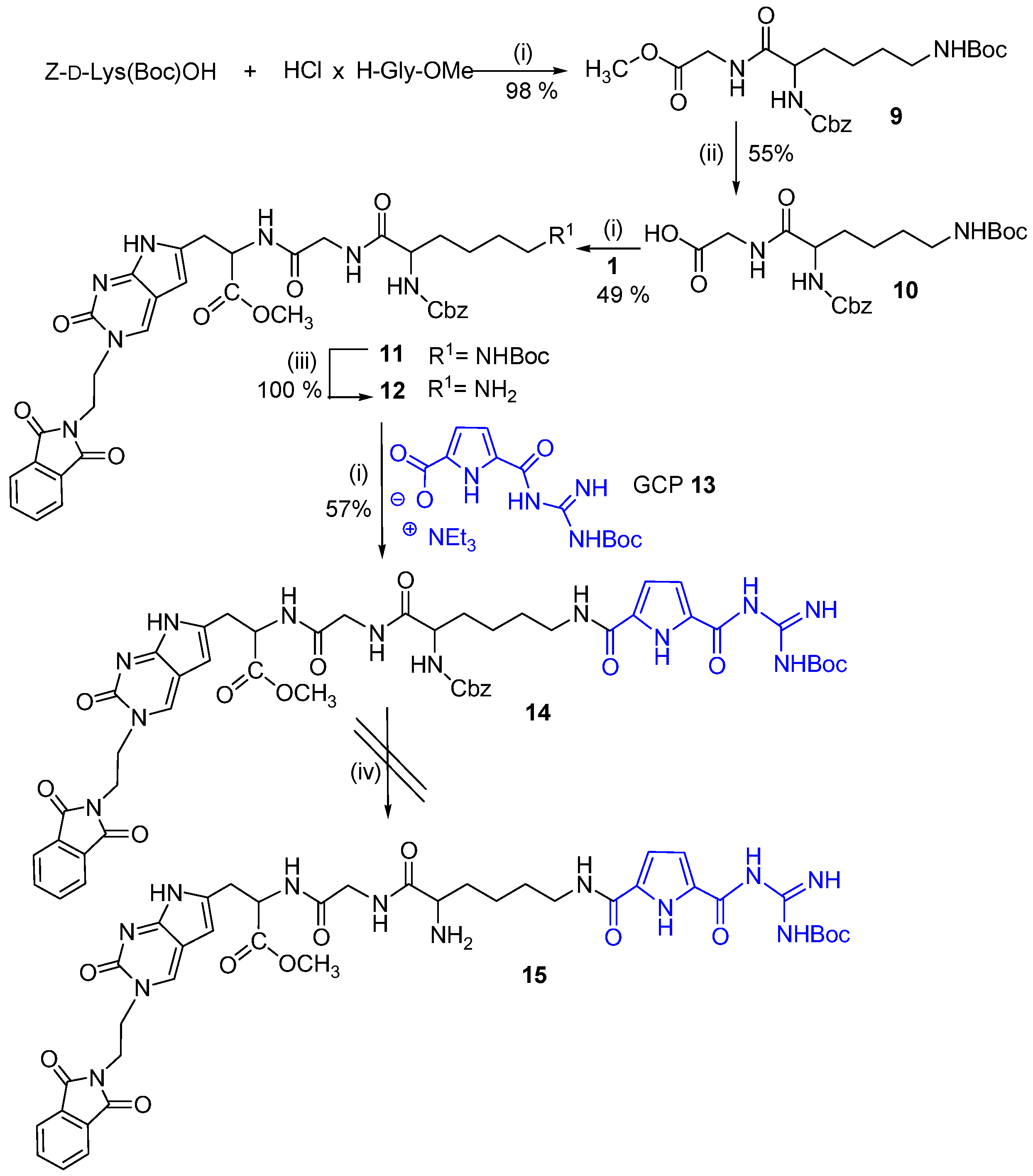
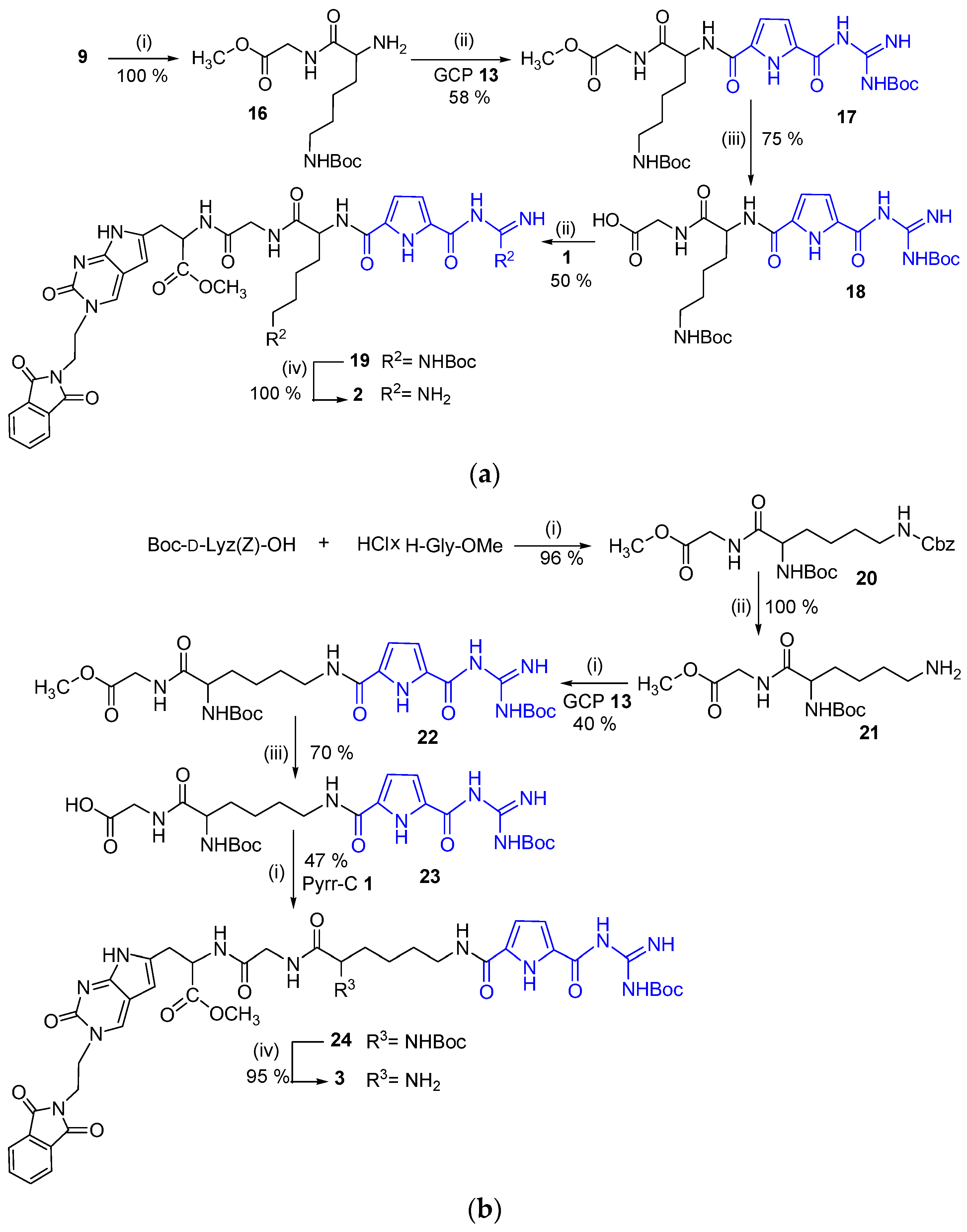
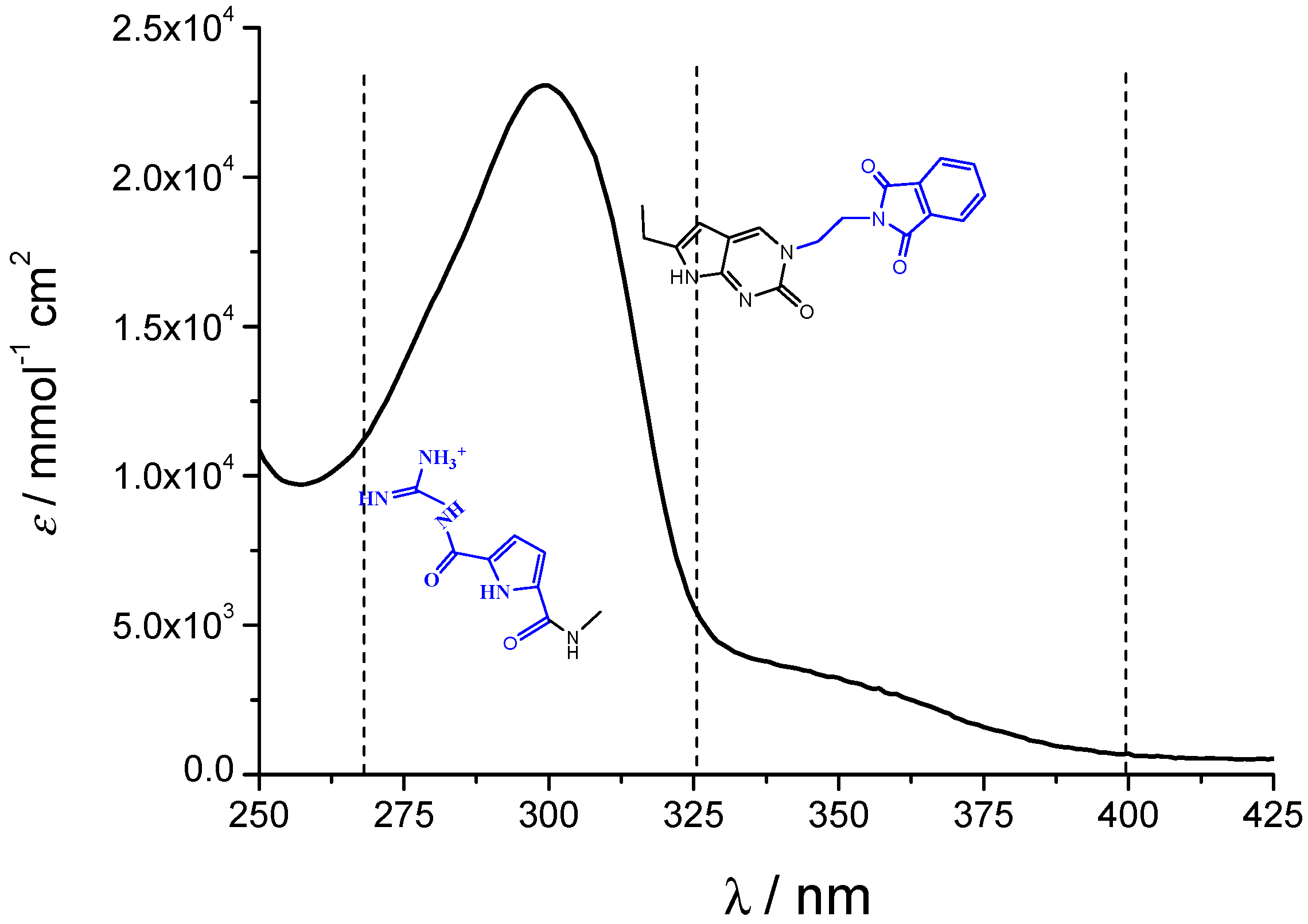
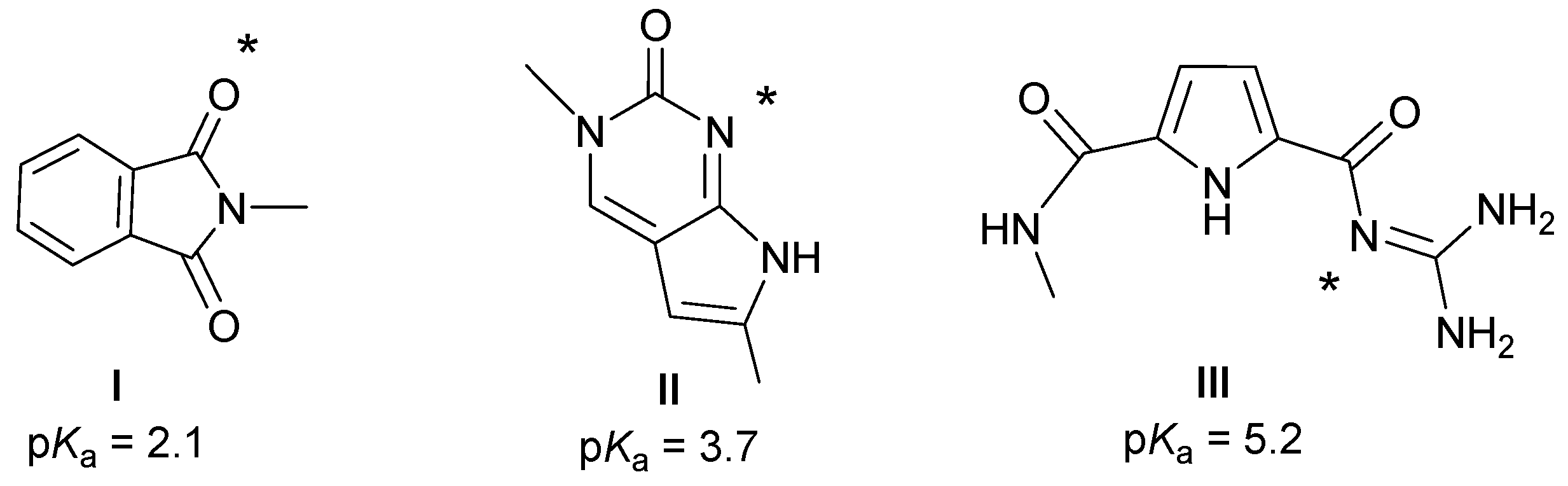
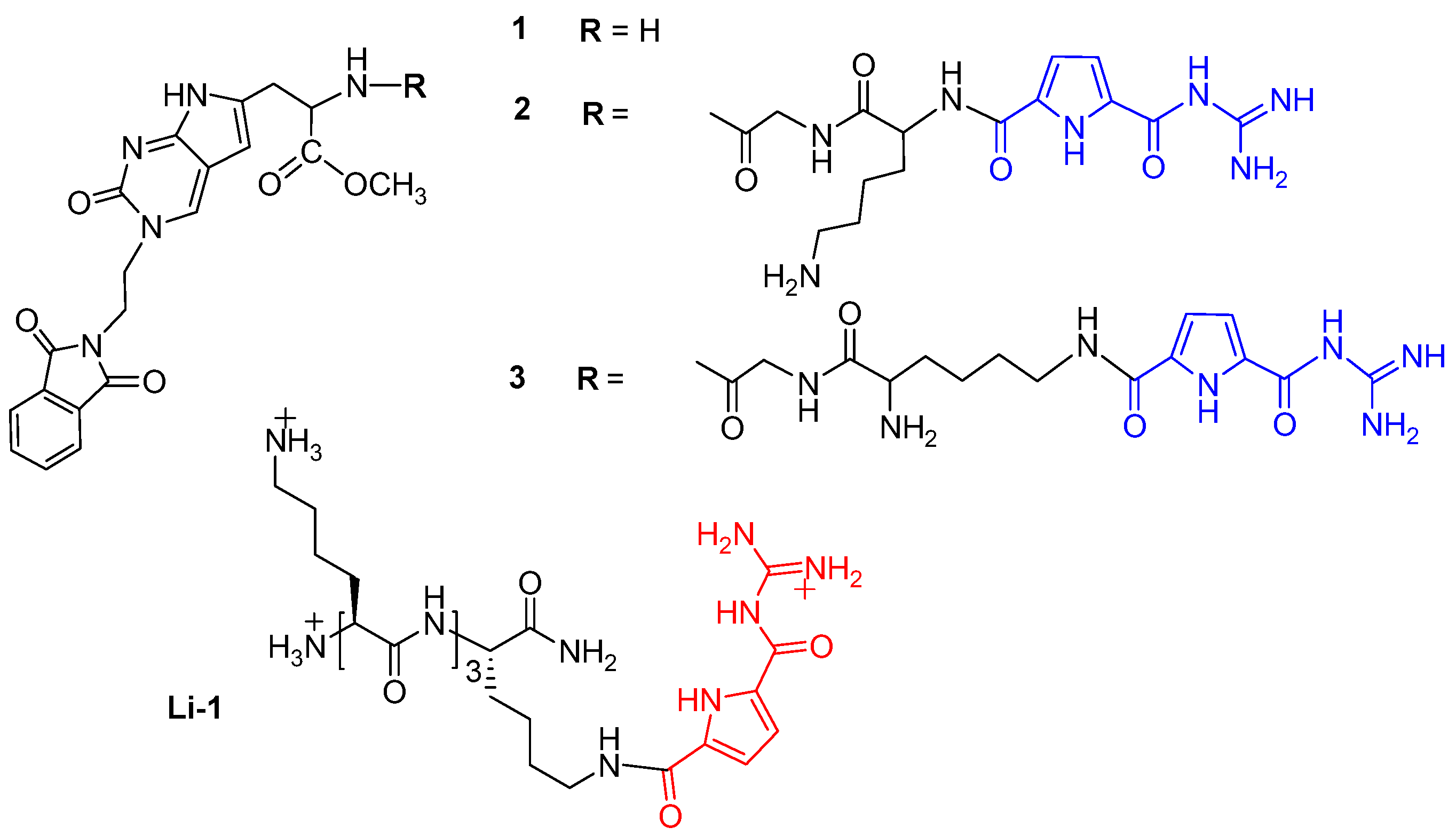
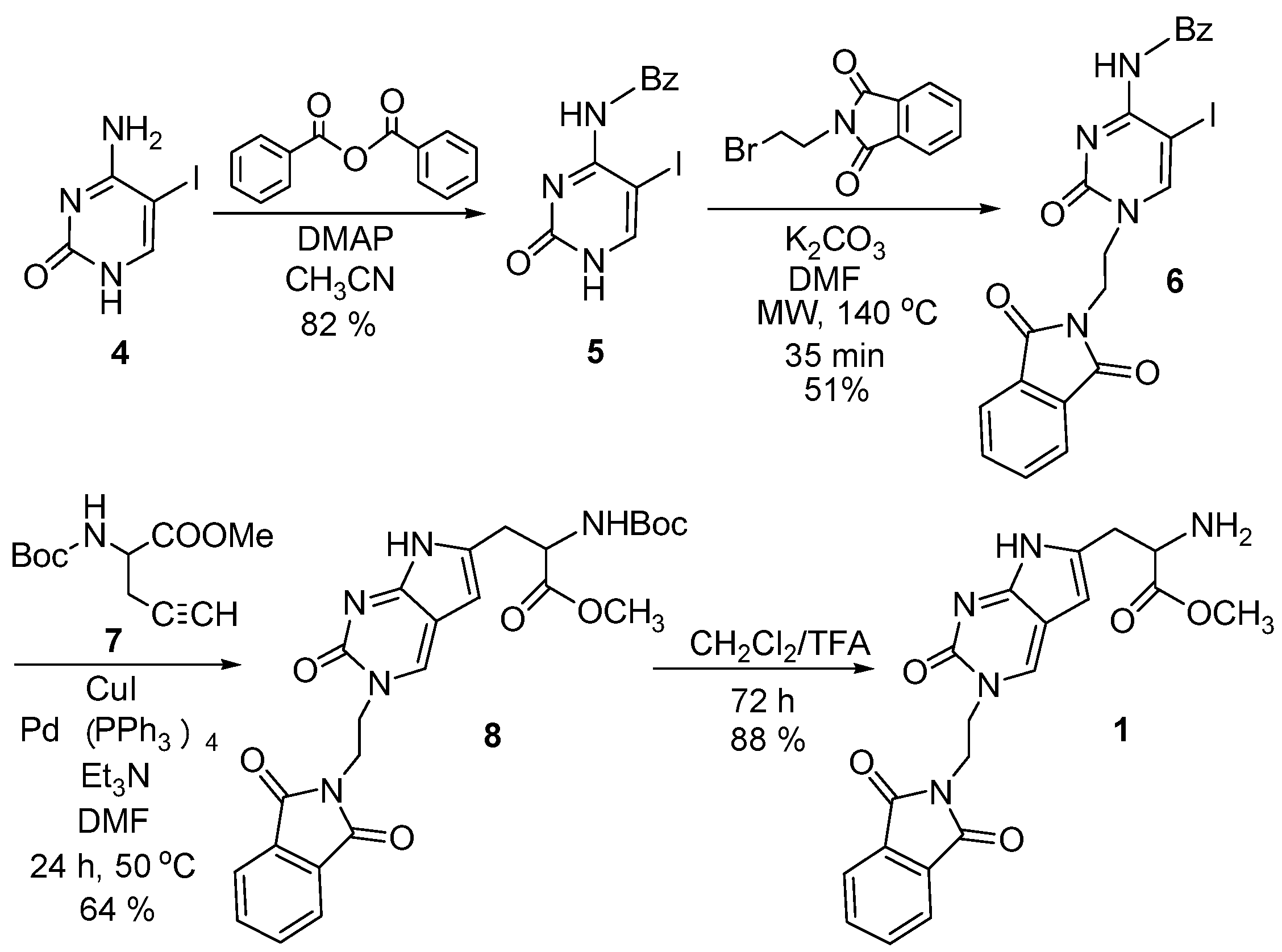
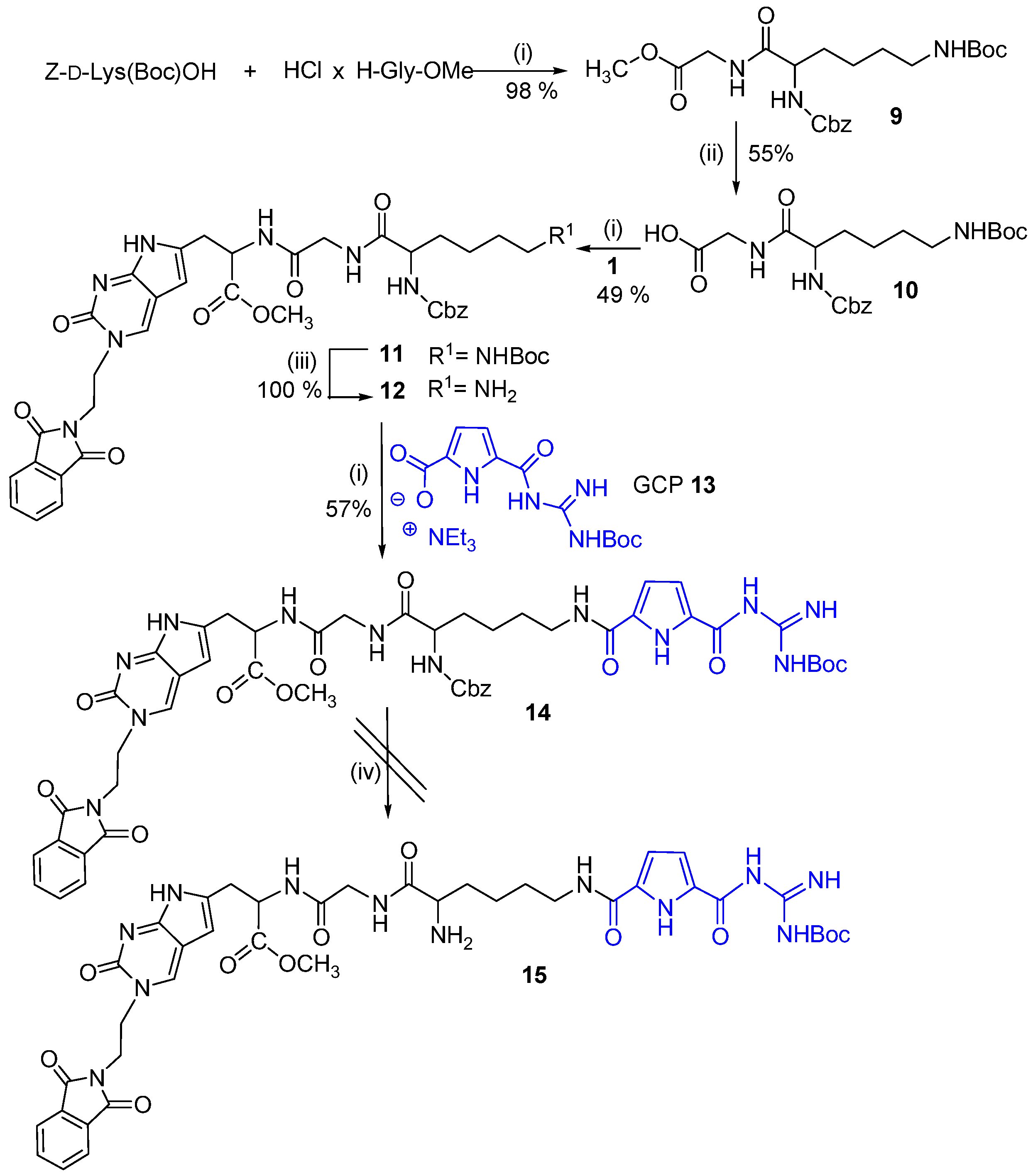
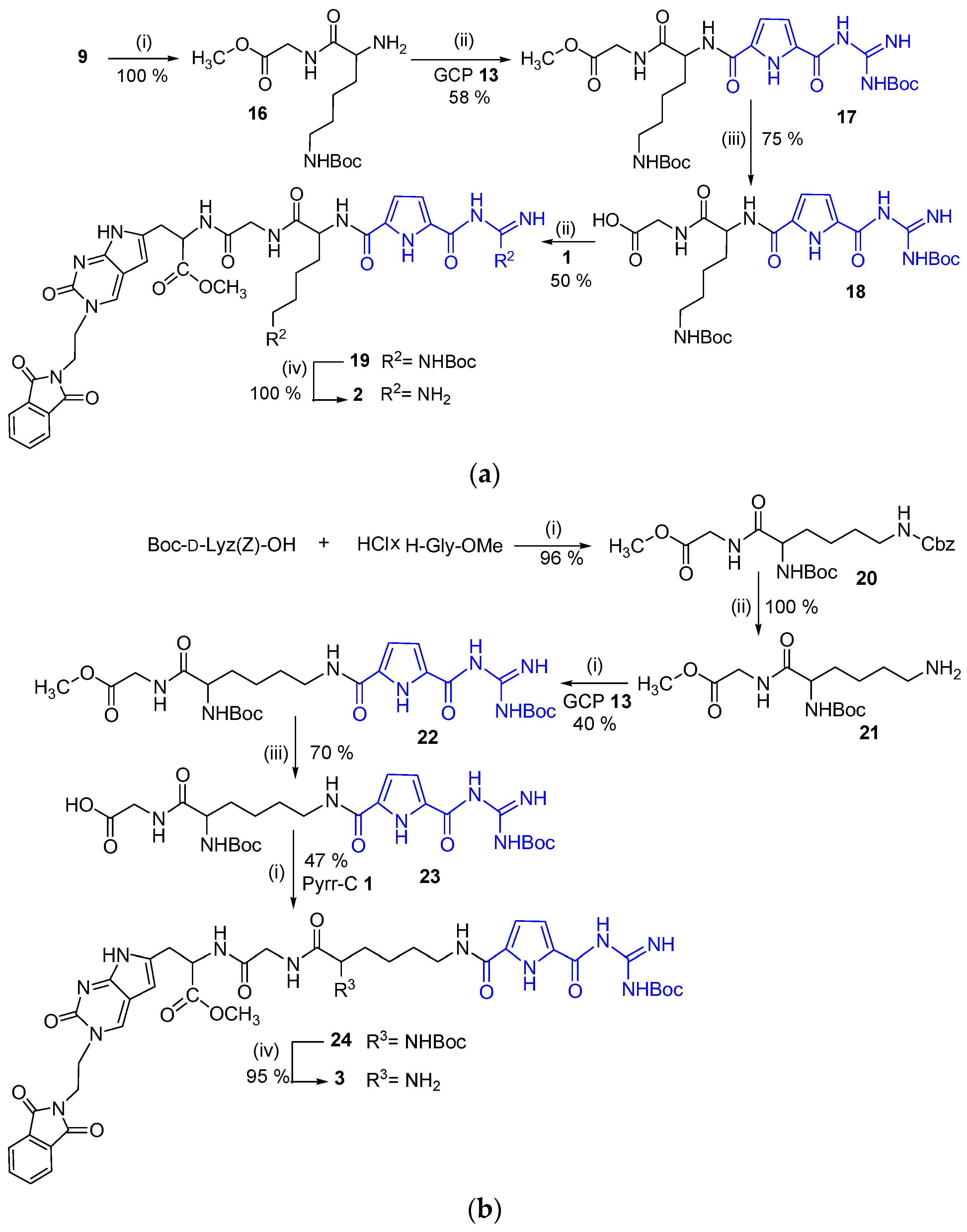
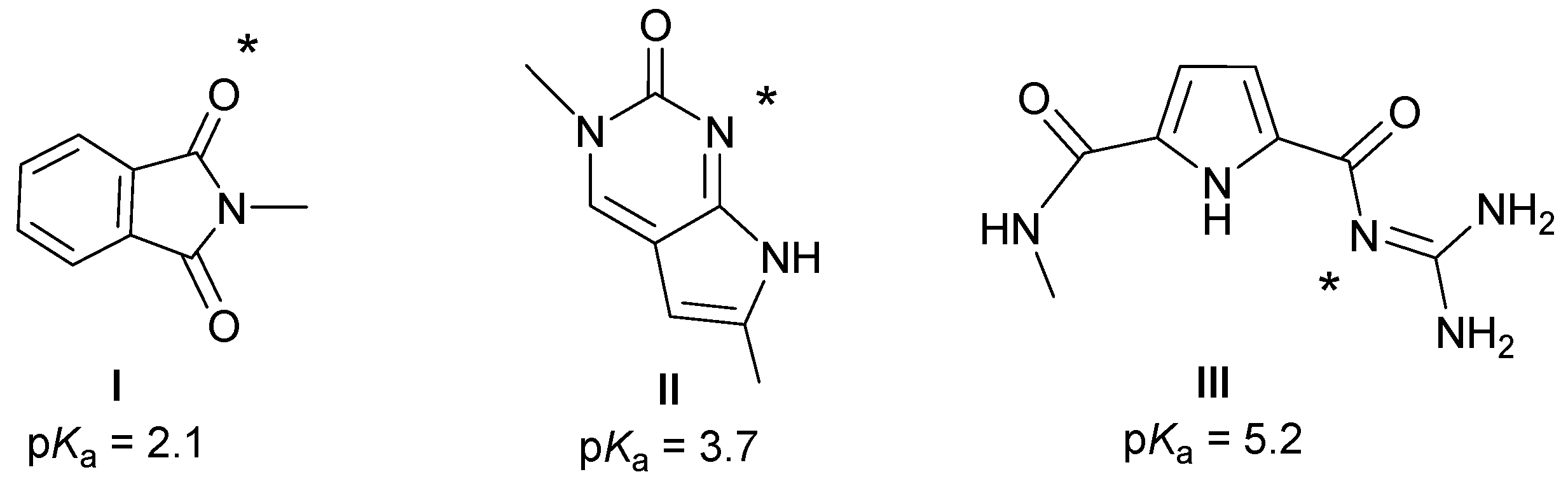
2.1. Chemistry
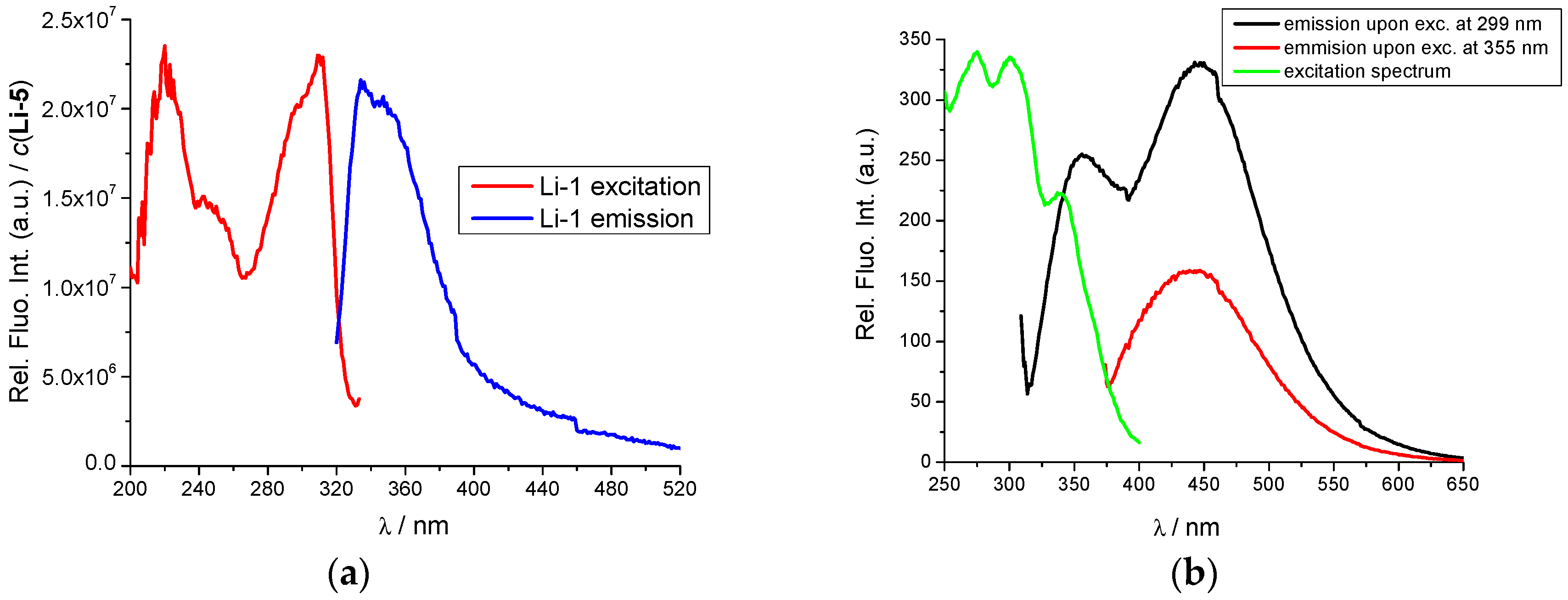
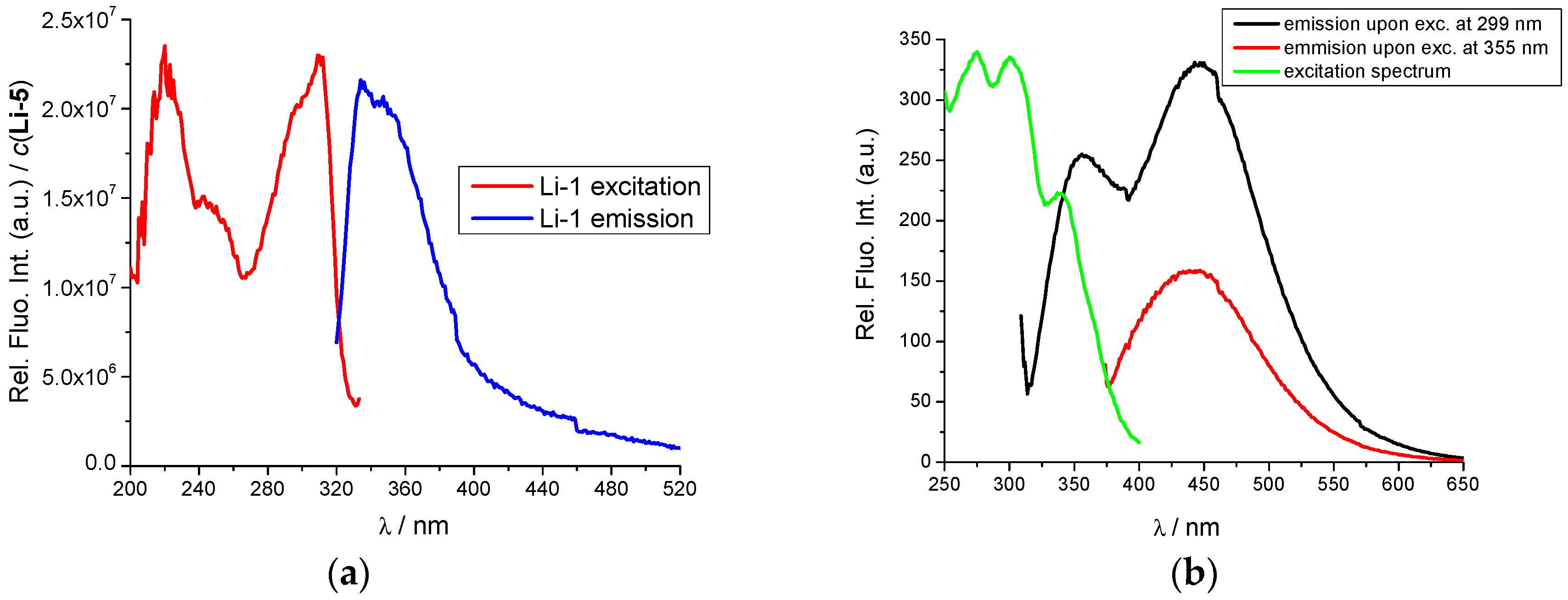
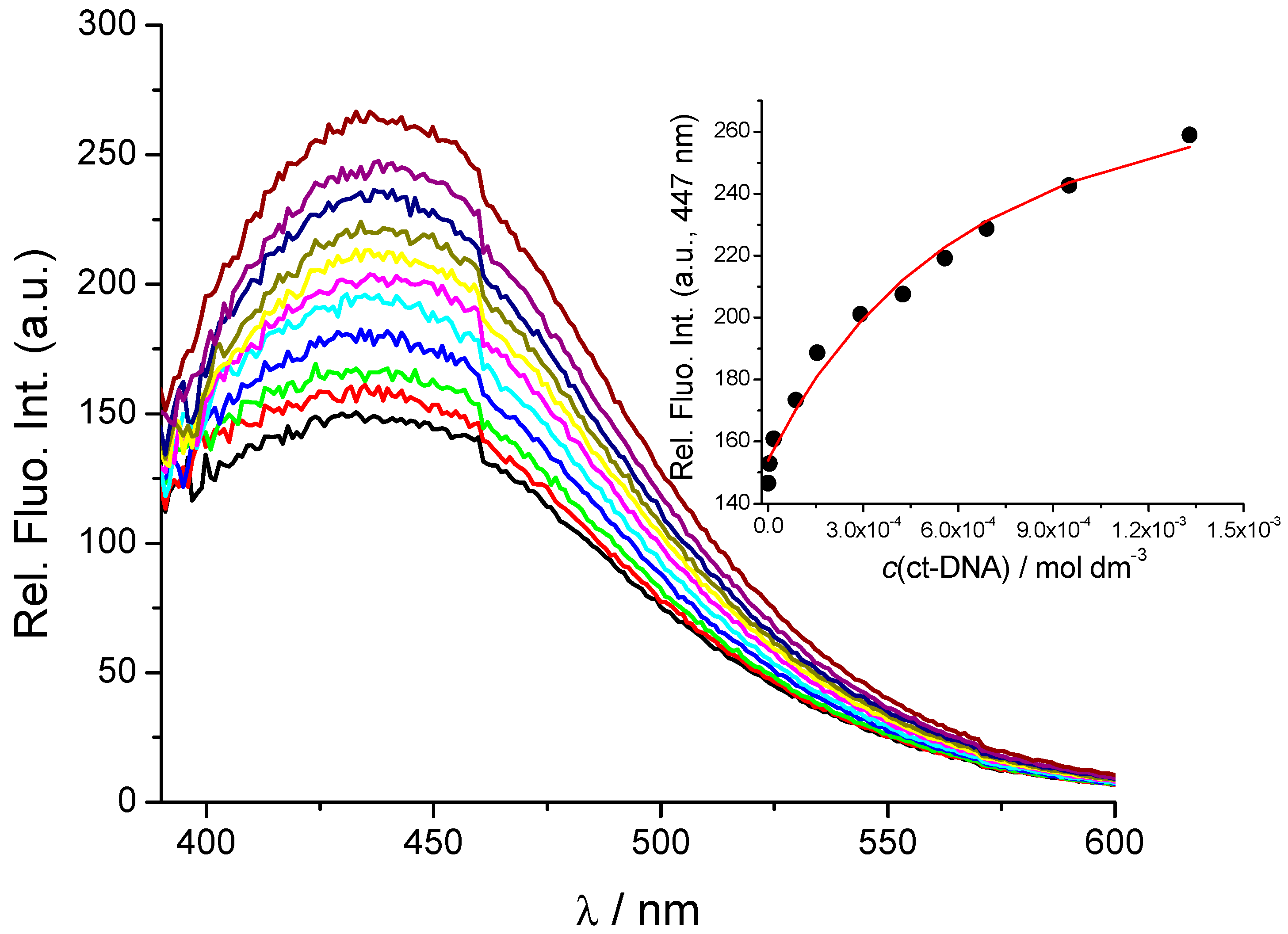
2.2. Interactions with DNA and RNA
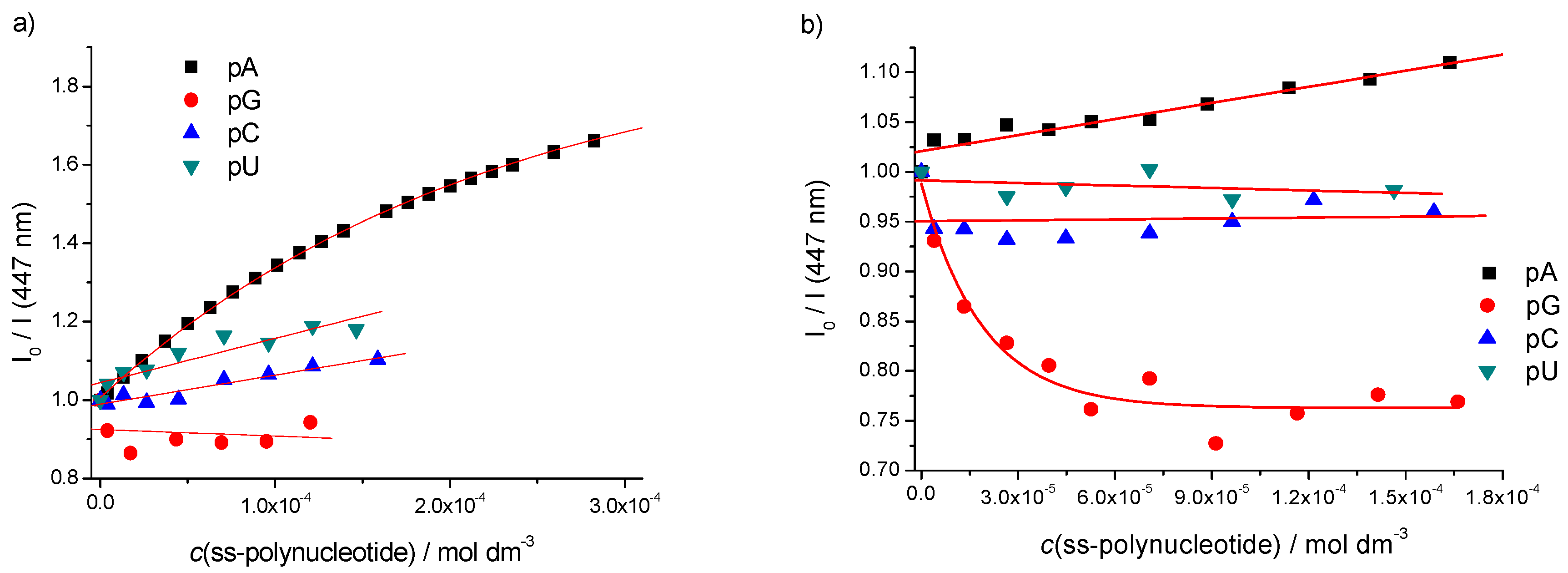
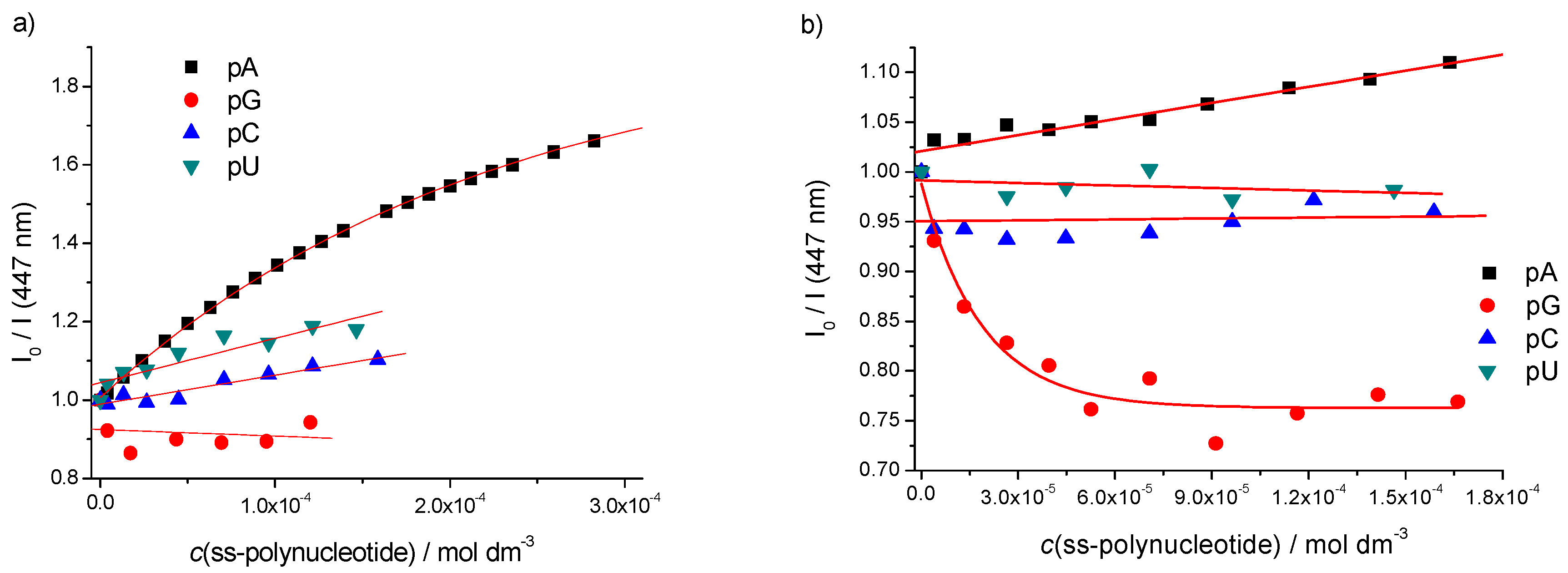
2.3. DNA/RNA Binding Studies
Single Stranded (ss)-Polynucleotides
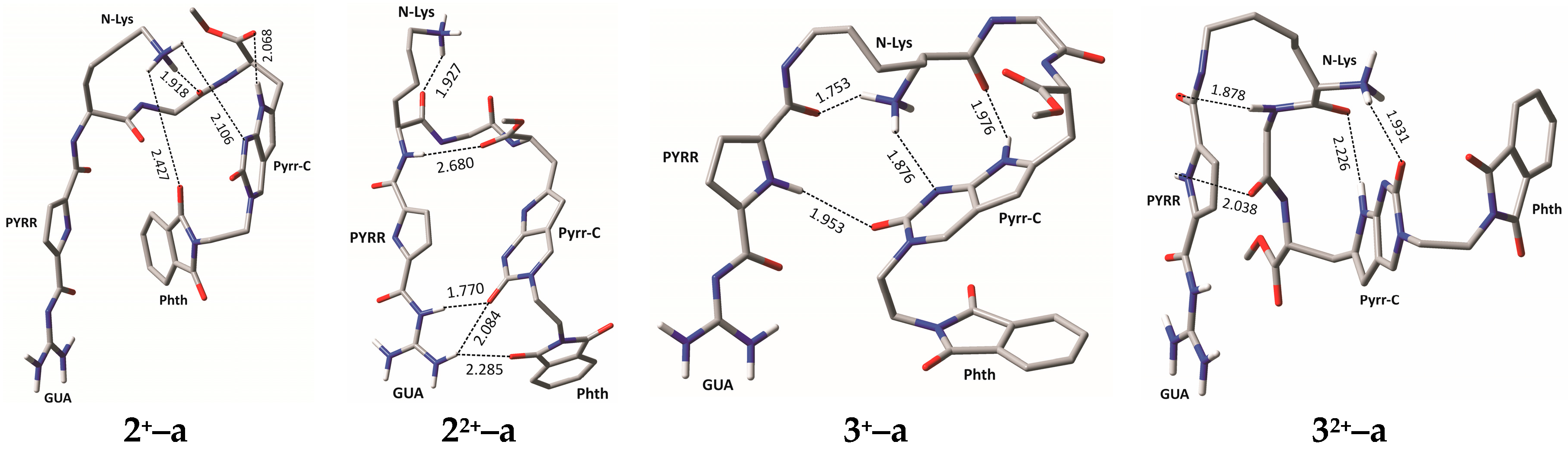
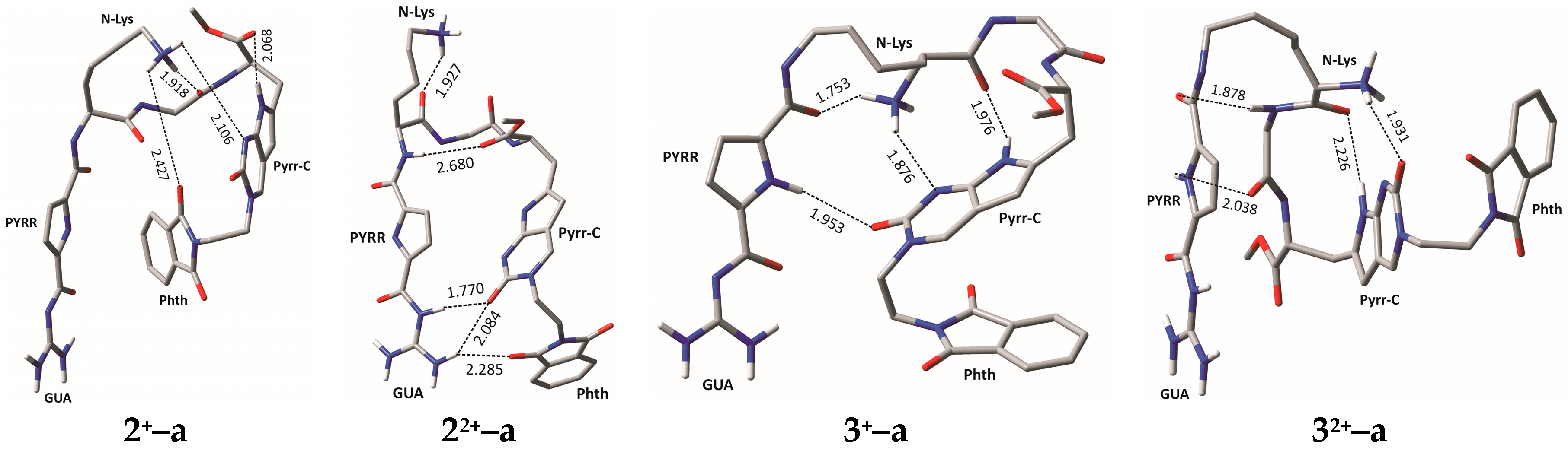
2.4. Molecular Modelling
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Silverman, R.B. The Organic Chemistry of Drug Design and Drug Action; Elsevier Academic Press: New York, NY, USA, 2004. [Google Scholar]
- Demeunynck, M.; Bailly, C.; Wilson, W.D. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004. [Google Scholar]
- Svitkin, Y.V.; Sonenberg, N. An Efficient System for Cap- and Poly(A)-Dependent Translation In Vitro. Methods Mol. Biol. 2004, 257, 155–170. [Google Scholar] [CrossRef]
- Colgan, D.F.; Murthy, K.G.; Prives, C.; Manley, J.L. Cell-cycle related regulation of poly(A) polymerase by phosphorylation. Nature 1996, 384, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.; Castello, A.; Menendez-Arias, L.; Carrasco, L. HIV protease cleaves poly(A)-binding protein. Biochem. J. 2006, 396, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.L.; Gueron, M.; Mergny, J.L.; Helene, C. Intramolecular Folding of a Fragment of the Cytosine-Rich Strand of Telomeric DNA into an I-Motif. Nucleic Acids Res. 1994, 22, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Manzini, G.; Yathindra, N.; Xodo, L.E. Evidence for intramolecularly folded I-DNA structures in biologically relevant CCC-repeat sequences. Nucleic Acids Res. 1994, 22, 4634–4640. [Google Scholar] [CrossRef] [PubMed]
- Giri, P.; Hossain, M.; Kumar, G.S. RNA specific molecules: Cytotoxic plant alkaloid palmatine binds strongly to poly(A). Bioorg. Med. Chem. Lett. 2006, 16, 2364–2368. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Song, G.; Ren, J.; Chaires, J.B.; Qu, X. Molecular recognition of nucleic acids: Coralyne binds strongly to poly(A). FEBS Lett. 2005, 579, 5035–5039. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.J.; Gray, D.; Kumar, S.; Arya, D.P. Molecular recognition of single-stranded RNA: Neomycin binding to poly(A). FEBS Lett. 2009, 583, 2269–2275. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Constant, J.F.; Demeunynck, M.; Lhomme, J.; Dumy, P. Design of site specific DNA damaging agents for generation of multiply damaged sites. Tetrahedron 2002, 58, 4291–4298. [Google Scholar] [CrossRef]
- Lhomme, J.; Constant, J.F.; Demeunynck, M. Abasic DNA structure, reactivity, and recognition. Biopolymers 1999, 52, 65–83. [Google Scholar] [CrossRef]
- Tumir, L.-M.; Piantanida, I.; Novak, P.; Žinić, M. Interactions of novel phenanthridinium-nucleobase conjugates with complementary and non-complementary nucleotides in aqueous media. J. Phys. Org. Chem. 2002, 15, 599–607. [Google Scholar] [CrossRef]
- Tumir, L.M.; Piantanida, I.; Juranović Cindrić, I.; Hrenar, T.; Meić, Z.; Žinić, M. New permanently charged phenanthridinium-nucleobase conjugates. Interactions with nucleotides and polynucleotides and recognition of ds-polyAH(+). J. Phys. Org. Chem. 2003, 16, 891–899. [Google Scholar] [CrossRef]
- Tumir, L.-M.; Piantanida, I.; Žinić, M.; Juranović Cindrić, I.; Meić, Z.; Kralj, M.; Tomić, S. Synthesis of phenanthridinium-bis-nucleobase conjugates, interactions with poly U, nucleotides and in vitro antitumour activity of mono- and bis-nucleobase conjugates. Eur. J. Med. Chem. 2006, 41, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Juranović, I.; Meić, Z.; Piantanida, I.; Tumir, L.-M.; Žinić, M. Interactions of phenanthridinium-nucleobase conjugates with polynucleotides in aqueous media. Recognition of poly U. Chem. Commun. 2002, 1432–1433. [Google Scholar] [CrossRef]
- Tumir, L.-M.; Piantanida, I.; Juranović Cindrić, I.; Meić, Z.; Tomić, S.; Žinić, M. Recognition of homo-polynucleotides containing adenine by a phenanthridinium bis-uracil conjugate in aqueous media. Chem. Commun. 2005, 2561–2563. [Google Scholar] [CrossRef] [PubMed]
- Tumir, L.-M.; Grabar, M.; Tomić, S.; Piantanida, I. The interactions of bis-phenanthridinium-nucleobase conjugates with nucleotides: Adenine-conjugate recognizes UMP in aqueous medium. Tetrahedron 2010, 6, 2501–2513. [Google Scholar] [CrossRef]
- Grabar Branilović, M.; Tomić, S.; Tumir, L.-M.; Piantanida, I. The bis-phenanthridinium system flexibility and position of covalently bound uracil finely tunes the interaction with polynucleotides. Mol. BioSyst. 2013, 9, 2051–2062. [Google Scholar] [CrossRef] [PubMed]
- Radić Stojković, M.; Skugor, M.; Tomić, S.; Grabar, M.; Smrečki, V.; Dudek, L.; Grolik, J.; Eilmes, J.; Piantanida, I. Dibenzotetraaza[14]annulene-adenine conjugate recognizes complementary poly dT among ss-DNA/ss-RNA sequences. Org. Biomol. Chem. 2013, 11, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zou, Y.; Li, C.; Sicking, W.; Piantanida, I.; Yi, T.; Schmuck, C. A Molecular Peptide Beacon for the Ratiometric Sensing of Nucleic Acids. J. Am. Chem. Soc. 2012, 134, 1958–1961. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Folgado, L.; Baretić, D.; Piantanida, I.; Marjanović, M.; Kralj, M.; Rehm, T.; Schmuck, C. Guanidiniocarbonylpyrrole-Aryl Derivatives: Structure Tuning for Spectrophotometric Recognition of Specific DNA and RNA Sequences and for Antiproliferative Activity. Chem. Eur. J. 2010, 16, 3036–3056. [Google Scholar] [CrossRef] [PubMed]
- Gröger, K.; Baretić, D.; Piantanida, I.; Marjanović, M.; Kralj, M.; Grabar, M.; Tomić, S.; Schmuck, C. Guanidiniocarbonyl-pyrrole-aryl conjugates as nucleic acid sensors: Switch of binding mode and spectroscopic responses by introducing additional binding sites into the linker. Org. Biomol. Chem. 2011, 9, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Klemm, K.; Radić Stojković, M.; Horvat, G.; Tomišić, V.; Piantanida, I.; Schmuck, C. Interactions of Multicationic Bis(guanidiniocarbonylpyrrole) Receptors with Double-Stranded Nucleic Acids: Syntheses, Binding Studies, and Atomic Force Microscopy Imaging. Chem. Eur. J. 2012, 18, 1352–1363. [Google Scholar] [CrossRef] [PubMed]
- Radić Stojković, M.; Piotrowski, P.; Schmuck, C.; Piantanida, I. A short, rigid linker between pyrene and guanidiniocarbonyl-pyrrole induced a new set of spectroscopic responses to the ds-DNA secondary structure. Org. Biomol. Chem. 2015, 13, 1629–1633. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Matković, M.; Piantanida, I.; Schmuck, C. Incorporation of arginine mimetic residue into peptides for recognition of double stranded nucleic acid structure: Binding and aggregation studies. Bioorg. Med. Chem. 2017, 25, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.H.E.; Dambenieks, A.K.; Viirre, R.D. Fluorescent 7-deazapurine derivatives from 5-iodocytosine via a tandem cross-coupling-annulation reaction with terminal alkynes. Synlett 2004, 13, 2400–2402. [Google Scholar] [CrossRef]
- Woo, J.S.; Meyer, R.B.; Gamper, H.B. G/C-modified oligodeoxynucleotides with selective complementarity: Synthesis and hybridization properties. Nucleic Acids Res. 1996, 24, 2470–2475. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Martin, C.T. Fluorescence characterization of the transcription bubble in elongation complexes of T7 RNA polymerase. J. Mol. Biol. 2001, 308, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.H.E.; Viirre, R.D.; McCourt, N.; Tse, J. Nucleobase modified peptide nucleic acid. Nucleosides Nucleotides Nucleic Acids 2003, 22, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Larock, R.C. Palladium-catalyzed annulation. J. Organomet. Chem. 1999, 576, 111–124. [Google Scholar] [CrossRef]
- Chang, P.K. 2′-Deoxy-5-iodocytidine 5′-triphosphatetriphosphate and T-deoxy-5-iodouridine 5′-triphosphate. In Nucleic Acid Chemistry: Improved and New Synthetic Procedures, Methods and Techniques; Townsend, L.B., Tipson, R.S., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1978; Volume 2, pp. 779–782. [Google Scholar]
- Watanabe, K.A.; Su, T.-L.; Klein, R.S.; Chu, C.K.; Matsuda, A.; Chun, M.W.; Lopez, C.; Fox, J.J. Nucleosides. 123. Synthesis of Antiviral Nucleosides-5-Substituted 1-(2-Deoxy-2-Halogeno-Beta-d-Arabinofuranosyl)Cytosines and -Uracils. Some Structure Activity Relationships. J. Med. Chem. 1983, 26, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Holý, A.; Rosenberg, I.; Dvořáková, H. Synthesis of N-(2-Phosphonylmethoxyethyl) Derivatives of Heterocyclic Bases. Collect. Czechoslov. Chem. Commun. 1989, 54, 2190–2210. [Google Scholar] [CrossRef]
- Hudson, R.H.E.; Dambenieks, A.K. Synthesis of N1-unsubstitued 5-alkynylcytosine and derivatives thereof. Heterocycles 2006, 68, 1325–1328. [Google Scholar] [CrossRef]
- Dondoni, A.; Perrone, D. Synthesis of 1,1-Dimethylethyl(S)-4-formyl-2,2-dimethyl-3-oxazolidinecarboxylate by oxidation of the alcohol. Org. Synth. 2000, 77, 64–77. [Google Scholar] [CrossRef]
- Schmuck, C.; Bickert, V.; Merschky, M.; Geiger, L.; Rupprecht, D.; Dudaczek, J.; Wich, P.; Rehm, T.; Machon, U. A facile and efficient multi-gram synthesis of N-protected 5-(Guanidinocarbonyl)-1H-pyrrole-2-carboxylic acids. Eur. J. Org. Chem. 2008, 2, 324–329. [Google Scholar] [CrossRef]
- Dourtoglou, V.; Gross, B.; Lambropoulou, V.; Zioudrou, C. O-Benzotriazolyl-N,N,N′,N′-Tetramethyluronium Hexafluorophosphate as Coupling Reagent for the Synthesis of Peptides of Biological Interest. Synthesis 1984, 7, 572–574. [Google Scholar] [CrossRef]
- Cantor, C.R.; Scimmel, P.R. Biophysical Chemistry; WH Freeman and Co.: San Francisco, CA, USA, 1980; Volume 3, pp. 1109–1181. [Google Scholar]
- Scatchard, G. The attractions of proteins for small molecules and ions. Ann. N.Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- Mc Ghee, J.D.; Hippel, P.H. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Rodger, A.; Norden, B. Circular Dichroism and Linear Dichroism; Oxford University Press: New York, NY, USA, 1997; Chapter 2. [Google Scholar]
- Eriksson, M.; Nordén, B. Linear and Circular Dichroism of Drug-Nucleic Acid Complexes. Methods Enzymol. 2001, 340, 68–98. [Google Scholar] [CrossRef] [PubMed]
- Borštnar, R.; Repič, M.; Kamerlin, S.C.L.; Vianello, R.; Mavri, J. Computational study of the pKa values of potential catalytic residues in the active site of monoamine oxidase B. J. Chem. Theory Comput. 2012, 8, 3864–3870. [Google Scholar] [CrossRef] [PubMed]
- Schwamm, R.J.; Vianello, R.; Maršavelski, A.; García, M.A.; Claramunt, R.M.; Alkorta, I.; Saame, J.; Leito, I.; Fitchett, C.M.; Edwards, A.J.; et al. 15N NMR spectroscopy, X-ray and neutron diffraction, quantum-chemical calculations, and UV/Vis-spectrophotometric titrations as complementary techniques for the analysis of pyridine-supported bicyclic guanidine superbases. J. Org. Chem. 2016, 81, 7612–7625. [Google Scholar] [CrossRef] [PubMed]
- Coles, M.P.; Aragon-Saez, P.J.; Oakley, S.H.; Hitchcock, P.B.; Davidson, M.G.; Maksić, Z.B.; Vianello, R.; Leito, I.; Kaljurand, I.; Apperley, D.C. Superbasicity of a bis-guanidino compound with a flexible linker: A theoretical and experimental study. J. Am. Chem. Soc. 2009, 131, 16858–16868. [Google Scholar] [CrossRef] [PubMed]
- Lõkov, M.; Tshepelevitsh, S.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the basicity of conjugated nitrogen heterocycles in different media. Eur. J. Org. Chem. 2017, 30, 4475–4489. [Google Scholar] [CrossRef]
- Chaires, J.B.; Dattagupta, N.; Crothers, D.M. Studies on interaction of anthracycline antibiotics and deoxyribonucleic acid: Equilibrium binding studies on interaction of daunomycin with deoxyribonucleic acid. Biochemistry 1982, 21, 3933–3940. [Google Scholar] [CrossRef] [PubMed]
- Palm, B.S.; Piantanida, I.; Žinić, M.; Schneider, H.J. The interaction of new 4,9-diazapyrenium compounds with double stranded nucleic acids. J. Chem. Soc. Perkin Trans. 2000, 2, 385–392. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Lacroix, L. Analysis of thermal melting curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Betz, R.M.; Botello-Smith, W.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, UK, 2009. [Google Scholar]
- Jeddi, I.; Saiz, L. Three-dimensional modeling of single stranded DNA hairpins for aptamer-based biosensors. Sci. Rep. 2017, 7, 1178. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.G. Acids and Bases: Solvent Effects on Acid-Base Strength; Oxford University Press: Oxford, UK, 2013; ISBN 978-0199670529. [Google Scholar]
- Horak, E.; Vianello, R.; Hranjec, M.; Krištafor, S.; Karminski Zamola, G.; Murković Steinberg, I. Benzimidazole acrylonitriles as multifunctional push-pull chromophores: Spectral characterisation, protonation equilibria and nanoaggregation in aqueous solution. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2017, 178, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Maršavelski, A.; Vianello, R. What a difference a methyl group makes: The selectivity of monoamine oxidase B towards histamine and N-methylhistamine. Chem. Eur. J. 2017, 23, 2915–2925. [Google Scholar] [CrossRef] [PubMed]
- Hranjec, M.; Horak, E.; Babić, D.; Plavljanin, S.; Srdović, Z.; Murković Steinberg, I.; Vianello, R.; Perin, N. Fluorescent benzimidazo[1,2-a]quinolines: Synthesis, spectroscopic and computational studies of protonation equilibria and metal ion sensitivity. New J. Chem. 2017, 41, 358–371. [Google Scholar] [CrossRef]
- Despotović, I.; Vianello, R. Engineering exceptionally strong oxygen superbases with 1,8-diazanaphthalene di-N-oxides. Chem. Commun. 2014, 50, 10941–10944. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–3 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polynucleotide | 2 | 3 | ||
|---|---|---|---|---|
| pH 7 | pH 5 | pH 7 | pH 5 | |
| log Ks/ c I/I0 | log Ks/ c I/I0 | log Ks/ c I/I0 | log Ks/ c I/I0 | |
| ct-DNA | 3.7/1.8 | 4.0/1.5 | 3.7/1.6 | 4.4/1.5 |
| poly A–poly U | b | 3.5/1.8 | b | b |
| poly A | 4.1/2.4 | b | b | d 4/1.2 |
| poly G | b | b | d 5/0.8 | d 4/0.9 |
| poly C | b | b | b | b |
| poly U | b | b | b | b |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ban, Ž.; Žinić, B.; Vianello, R.; Schmuck, C.; Piantanida, I. Nucleobase–Guanidiniocarbonyl-Pyrrole Conjugates as Novel Fluorimetric Sensors for Single Stranded RNA. Molecules 2017, 22, 2213. https://doi.org/10.3390/molecules22122213
Ban Ž, Žinić B, Vianello R, Schmuck C, Piantanida I. Nucleobase–Guanidiniocarbonyl-Pyrrole Conjugates as Novel Fluorimetric Sensors for Single Stranded RNA. Molecules. 2017; 22(12):2213. https://doi.org/10.3390/molecules22122213
Chicago/Turabian StyleBan, Željka, Biserka Žinić, Robert Vianello, Carsten Schmuck, and Ivo Piantanida. 2017. "Nucleobase–Guanidiniocarbonyl-Pyrrole Conjugates as Novel Fluorimetric Sensors for Single Stranded RNA" Molecules 22, no. 12: 2213. https://doi.org/10.3390/molecules22122213
APA StyleBan, Ž., Žinić, B., Vianello, R., Schmuck, C., & Piantanida, I. (2017). Nucleobase–Guanidiniocarbonyl-Pyrrole Conjugates as Novel Fluorimetric Sensors for Single Stranded RNA. Molecules, 22(12), 2213. https://doi.org/10.3390/molecules22122213