
Investigation of ‘Head-to-Tail’-Connected Oligoaryl N,O-Ligands as Recognition Motifs for Cancer-Relevant G-Quadruplexes


Abstract
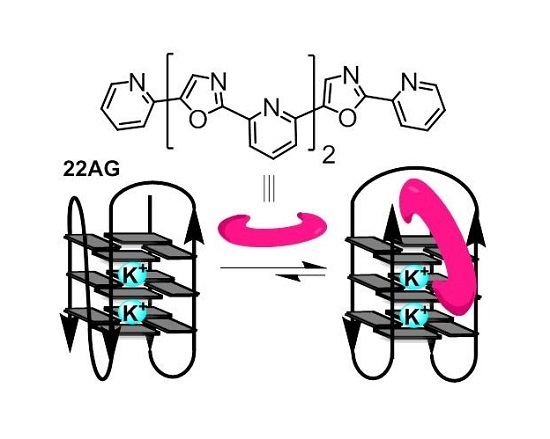
:
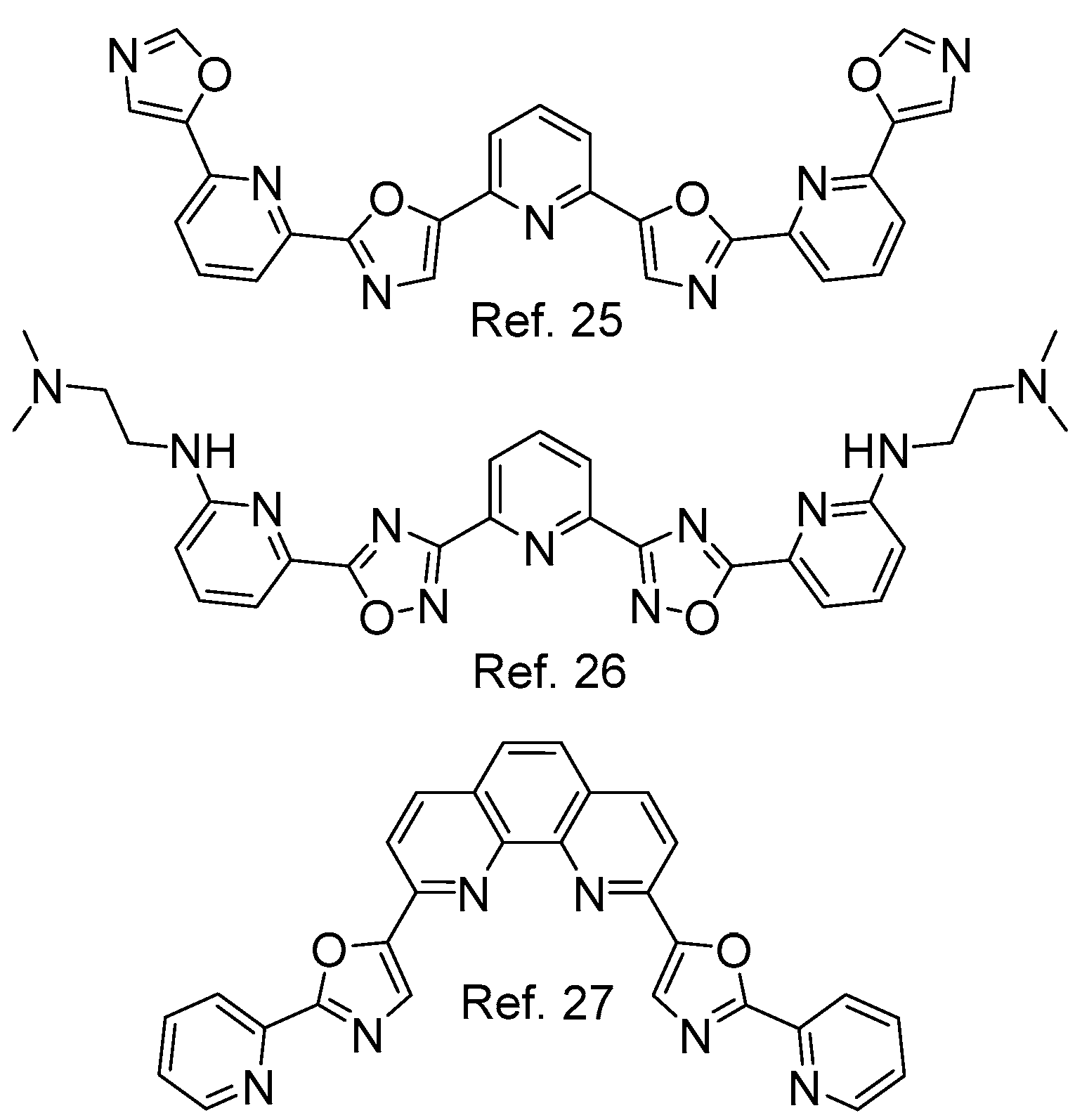
1. Introduction
2. Results
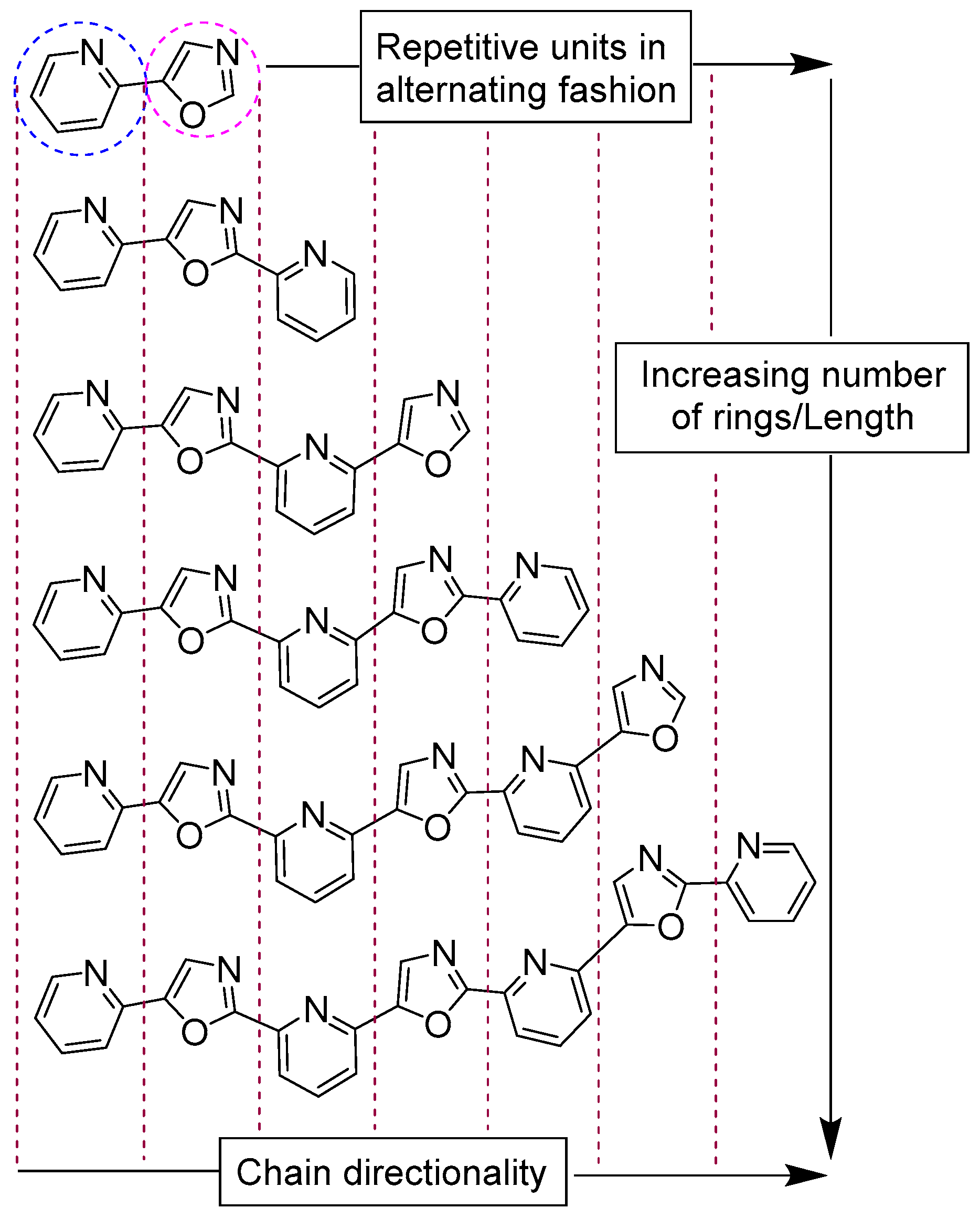
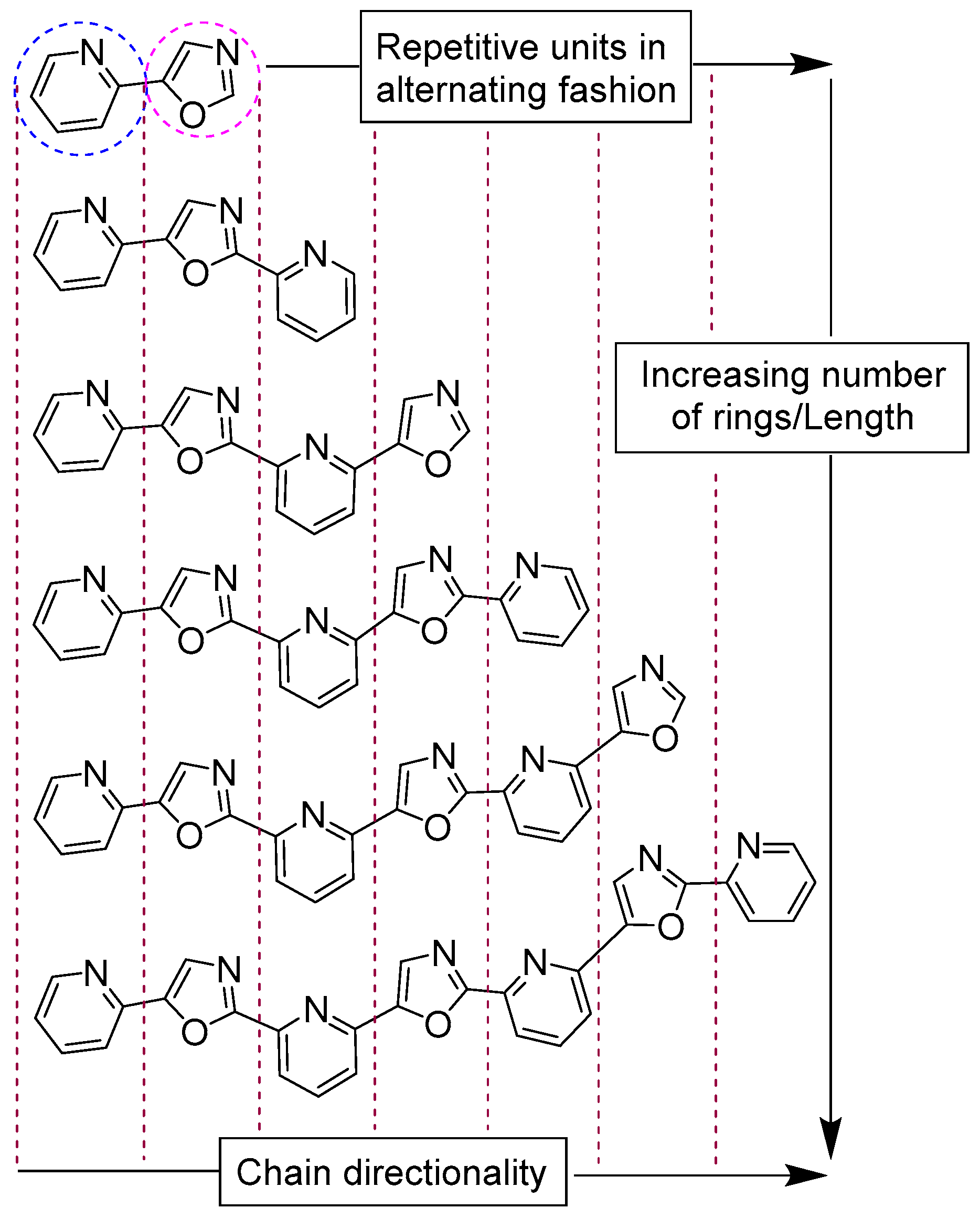
2.1. Synthesis and Purification of a Library of Variable-Length Asymmetric Pyridyl-Oxazole Ligands
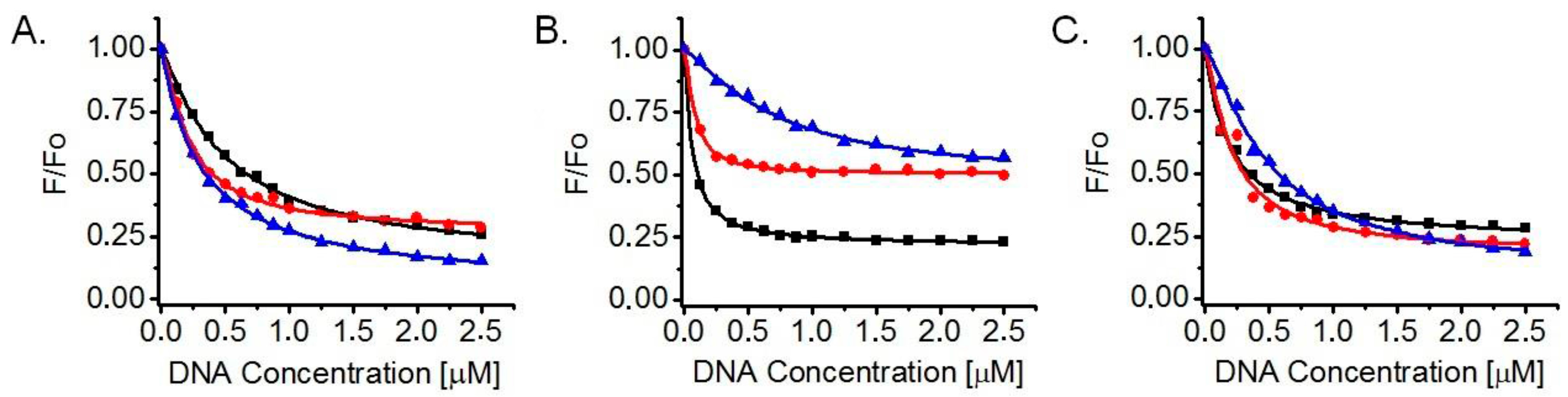
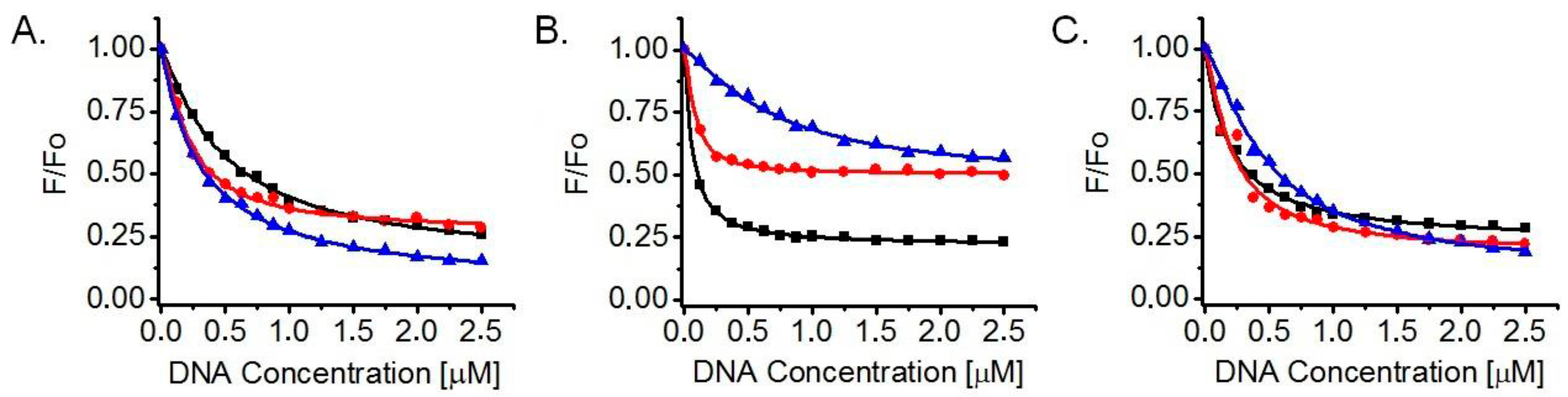
2.2. Determination of Dissociation Constants (KD) and Number of Binding Sites (n) for Ligand Interaction with G-Quadruplexes in Fluorescence Titrations
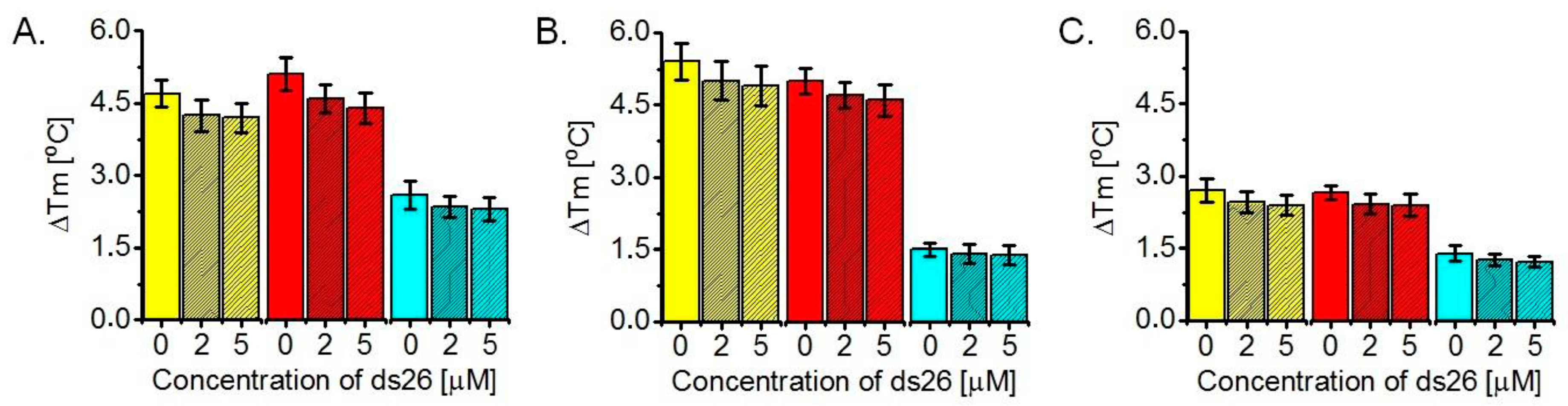
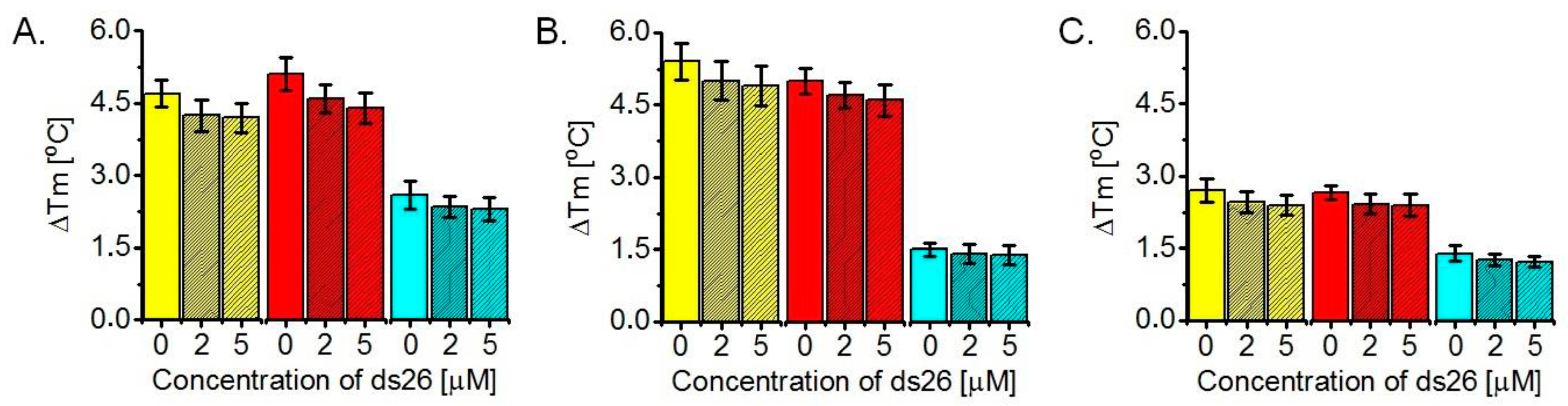
2.3. Determination of ΔTm Values of G-Quadruplexes Upon Ligand Binding, Based on FRET Melting Assays
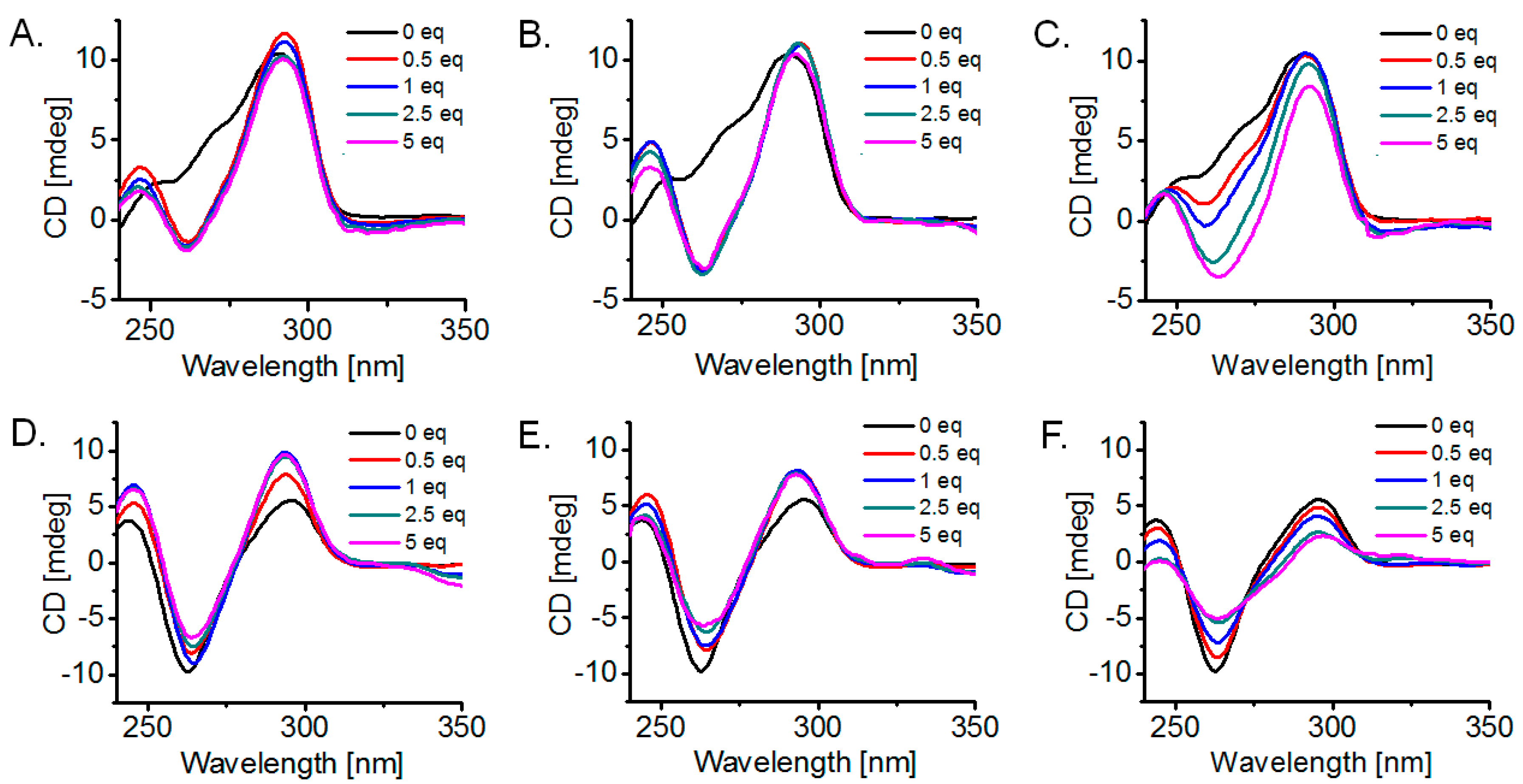
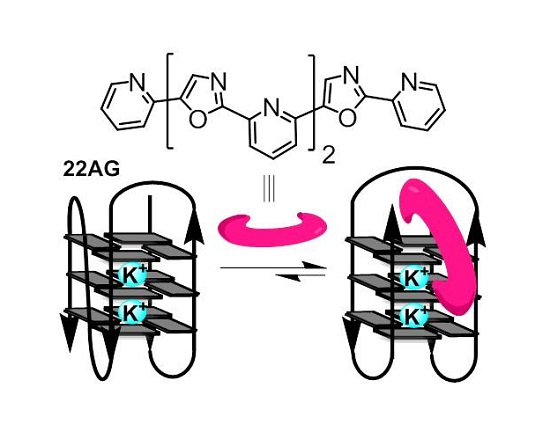
2.4. Detection of G-Quadruplex Conformational Changes upon Ligand Interaction, via Circular Dichroism (CD) Titrations
3. Discussion
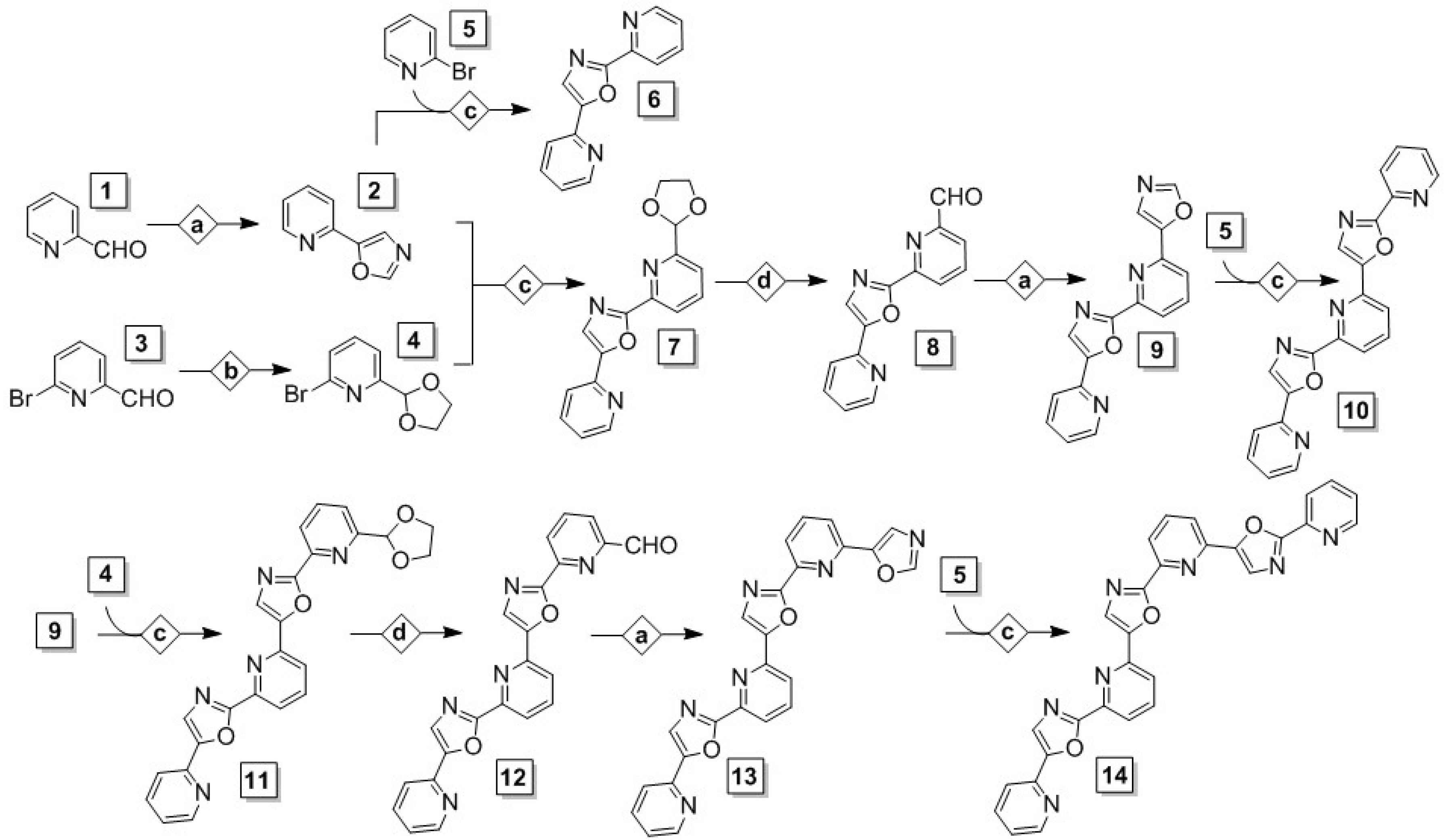
3.1. Synthesis
3.2. Ligand Affinity for DNA G-Quadruplexes and Binding Stoichiometry, Based on Fluorescence Titrations
3.3. Ligand-Induced Thermodynamic Stabilization of Prefolded G-Quadruplexes, Based on FRET Melting Assays
3.4. G-Quadruplex Conformational Changes Promoted by the Ligands, Based on Circular Dichroism (CD) Study
4. Materials and Methods
4.1. General
4.2. Synthetic Methods
4.2.1. General Method for Conversion of Aldehyde to Oxazole (Condition Set a)
4.2.2. General Method for Conversion of Aldehyde to 1,3-Dioxolane (Condition Set b)
4.2.3. General Method for Oxazole C–H Activation/C–C Cross-Coupling to Bromopyridine (Condition Set c)
4.2.4. General Method for Conversion of 1,3-Dioxolane to Aldehyde (Condition Set d)
4.3. Fluorescence Titrations
4.4. FRET Melting Assays
4.5. Circular Dichroism (CD) Titrations
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sundquist, W.I.; Klug, A. Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 1989, 342, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Patel, D.J. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure 1993, 1, 263–282. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Lee, M.P.H.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Cogoi, S.; Xodo, L.E. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006, 34, 2536–2549. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.T.; Varnai, P.; Bugaut, A.; Reszka, A.P.; Neidle, S.; Balasubramanian, S. A G-rich sequence within the c-kit oncogene promoter forms a parallel G-quadruplex having asymmetric G-tetrad dynamics. J. Am. Chem. Soc. 2009, 131, 13399–13409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Suntharalingam, K.; Brand, N.J.; Barton, P.J.R.; Vilar, R.; Ying, L. Possible regulatory roles of promoter G-quadruplexes in cardiac function-related genes–Human TnIc as a model. PLoS ONE 2013, 8, e53137. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Sommers, J.A.; Zhou, J.; Kaplan, D.L.; Spelbrink, J.N.; Mergny, J.-L.; Brosh, R.M., Jr. DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative twinkle helicase. J. Biol. Chem. 2014, 289, 29975–29993. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Tarsounas, M.; Tijsterman, M. Genomes and G-quadruplexes: For better or for worse. J. Mol. Biol. 2013, 425, 4782–4789. [Google Scholar] [CrossRef] [PubMed]
- Oganesian, L.; Karlseder, J. Telomeric armor: The layers of end protection. J. Cell Sci. 2009, 122, 4013–4025. [Google Scholar] [CrossRef] [PubMed]
- Murat, P.; Balasubramanian, S. Existence and consequences of G-quadruplex structures in DNA. Curr. Opin. Genet. Dev. 2014, 25, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S. Quadruplex Nucleic Acids as Novel Therapeutic Targets. J. Med. Chem. 2016, 59, 5989–6011. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Sabò, A.; Guccione, E. Targeting MYC in cancer therapy: RNA processing offers new opportunities. Bioessays 2016, 38, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Laguerre, A.; Stefan, L.; Larrouy, M.; Genest, D.; Novotna, J.; Pirrotta, M.; Monchaud, D. A twice-as-smart synthetic G-quartet: PyroTASQ is both a smart quadruplex ligand and a smart fluorescent probe. J. Am. Chem. Soc. 2014, 136, 12406–12414. [Google Scholar] [CrossRef] [PubMed]
- Shivalingam, A.; Izquierdo, M.A.; Le Marois, A.; Vyšniauskas, A.; Suhling, K.; Kuimova, M.K.; Vilar, R. The interactions between a small molecule and G-quadruplexes are visualized by fluorescence lifetime imaging microscopy. Nat. Commun. 2015, 6, 8178. [Google Scholar] [CrossRef] [PubMed]
- Monchaud, D.; Teulade-Fichou, M.-P. A hitchhiker’s guide to G-quadruplex ligands. Org. Biomol. Chem. 2008, 6, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Monchaud, D.; Granzhan, A.; Saettel, N.; Guédin, A.; Mergny, J.-L.; Teulade-Fichou, M.-P. “One ring to bind them all”—Part I: The efficiency of the macrocyclic scaffold for G-quadruplex DNA recognition. J. Nucleic Acids 2010, 2010, 525862. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.C.; Ulven, T. Macrocyclic G-Quadruplex Ligands. Curr. Med. Chem. 2010, 17, 3438–3448. [Google Scholar] [CrossRef] [PubMed]
- Georgiades, S.N.; Abd Karim, N.H.; Suntharalingam, K.; Vilar, R. Interaction of metal complexes with G-quadruplex DNA. Angew. Chem. Int. Ed. 2010, 49, 4020–4034. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.-N.; Zhao, S.-J.; Wu, B.; Li, X.-Z.; Kong, D.-M. A new cationic porphyrin derivative (TMPipEOPP) with large side arm substituents: A highly selective G-quadruplex optical probe. PLoS ONE 2012, 7, e35586. [Google Scholar] [CrossRef] [PubMed]
- Alzeer, J.; Vummidi, B.R.; Roth, P.J.; Luedtke, N.W. Guanidinium-modified phthalocyanines as high-affinity G-quadruplex fluorescent probes and transcriptional regulators. Angew. Chem. Int. Ed. 2009, 48, 9362–9365. [Google Scholar] [CrossRef] [PubMed]
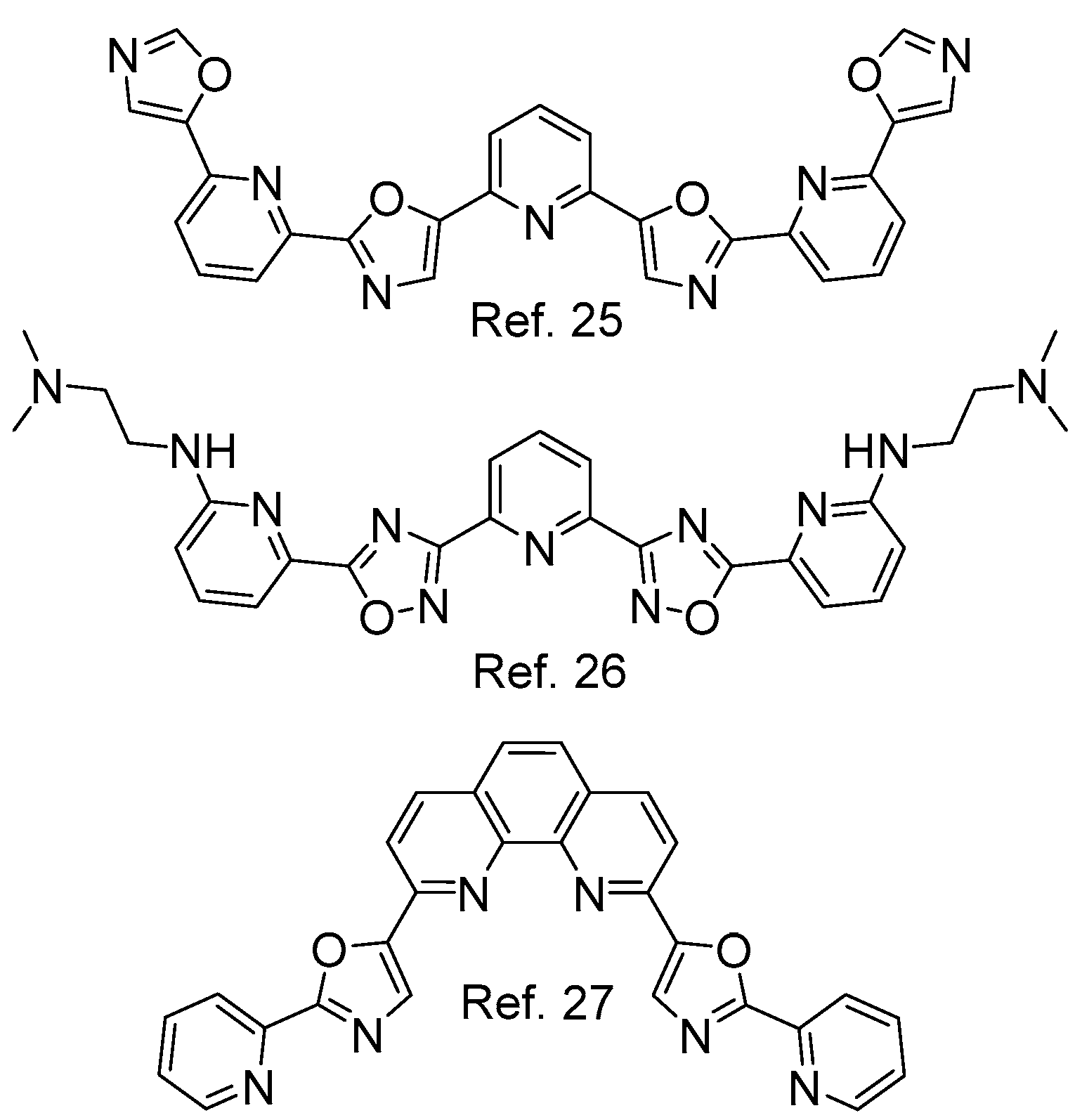
- Hamon, F.; Largy, E.; Guédin-Beaurepaire, A.; Rouchon-Dagois, M.; Sidibe, A.; Monchaud, D.; Mergny, J.-L.; Riou, J.-F.; Nguyen, C.-H.; Teulade-Fichou, M.-P. An acyclic oligoheteroaryle that discriminates strongly between diverse G-quadruplex topologies. Angew. Chem. Int. Ed. 2011, 50, 8745–8749. [Google Scholar] [CrossRef] [PubMed]
- Petenzi, M.; Verga, D.; Largy, E.; Hamon, F.; Doria, F.; Teulade-Fichou, M.-P.; Guédin, A.; Mergny, J.-L.; Mella, M.; Freccero, M. Cationic pentaheteroaryls as selective G-quadruplex ligands by solvent-free microwave-assisted synthesis. Chem. Eur. J. 2012, 18, 14487–14496. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-Silva, J.; Guédin, A.; Salgado, G.F.; Mergny, J.-L.; Queiroz, J.A.; Cabrita, E.J.; Cruz, C. Phenanthroline-bis-oxazole ligands for binding and stabilization of G-quadruplexes. Biochim. Biophys. Acta 2017, 1861, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Shin-ya, K.; Wierzba, K.; Matsuo, K.-L.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-Y.; Vankayalapati, H.; Shin-ya, K.; Wierzba, K.; Hurley, L.H. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. J. Am. Chem. Soc. 2002, 124, 2098–2099. [Google Scholar] [CrossRef] [PubMed]
- Rzuczek, S.G.; Pilch, D.S.; Liu, A.; Liu, L.; LaVoie, E.J.; Rice, J.E. Macrocyclic pyridyl polyoxazoles: Selective RNA and DNA G-quadruplex ligands as antitumor agents. J. Med. Chem. 2010, 53, 3632–3644. [Google Scholar] [CrossRef] [PubMed]
- Besselievre, F.; Mahuteau-Betzer, F.; Grierson, D.S.; Piquel, S. Ligandless Microwave-Assisted Pd/Cu-Catalyzed Direct Arylation of Oxazoles. J. Org. Chem. 2008, 73, 3278–3280. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Satoh, T.; Hirano, K.; Matsuo, D.; Orita, A.; Otera, J.; Miura, M. Synthesis of 2,5-diaryloxazoles through van Leusen reaction and copper-mediated direct arylation. Tetrahedron Lett. 2009, 50, 3273–3276. [Google Scholar] [CrossRef]
- Georgiades, S.N.; Rizeq, N. Synthesis of a ‘propeller-like’ oligoheteroaryl with alternating pyridine and oxazole motifs. Synlett 2015, 26, 489–493. [Google Scholar] [CrossRef]
- Rizeq, N.; Georgiades, S.N. Linear and branched pyridyl-oxazole oligomers: Synthesis and circular dichroism detectable effect on c-myc G-quadruplex helicity. Eur. J. Org. Chem. 2016, 2016, 122–131. [Google Scholar] [CrossRef]
- Van Leusen, A.M.; Hoogenboom, B.E.; Siderius, H. A novel and efficient synthesis of oxazoles from tosylmethylisocyanide and carbonyl compounds. Tetrahedron Lett. 1972, 23, 2369–2372. [Google Scholar] [CrossRef]
- Nimitz, J.S. Synthesis of some new symmetrically substituted 2,2’-dipyridyl sulphides. Synth. Commun. 1981, 11, 273–280. [Google Scholar] [CrossRef]
- Mandal, P.K.; Dutta, P.; Roy, S.C. A mild and efficient method for selective cleavage of ketals and acetals using lithium chloride in water-dimethyl sulfoxide. Tetrahedron Lett. 1997, 38, 7271–7274. [Google Scholar] [CrossRef]
- Kerkour, A.; Mergny, J.-L.; Salgado, G.F. NMR based model of human telomeric repeat G-quadruplex in complex with 2,4,6-triarylpyridine family ligand. Biochim. Biophys. Acta 2017, 1861, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Carver, M.; Yang, D. Polymorphism of human telomeric quadruplex structures. Biochimie 2008, 90, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Noer, S.L.; Preus, S.; Gudnason, D.; Aznauryan, M.; Mergny, J.-L.; Birkedal, V. Folding dynamics and conformational heterogeneity of human telomeric G-quadruplex structures in Na+ solutions by single molecule FRET microscopy. Nucleic Acids Res. 2016, 44, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Mathad, R.I.; Hatzakis, E.; Dai, J.; Yang, D. c-MYC promoter G-quadruplex formed at the 5’-end of NHE III1 element: Insights into biological relevance and parallel-stranded G-quadruplex stability. Nucleic Acids Res. 2011, 39, 9023–9033. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Wu, J.; Shao, F.; Yan, J. Stability and Kinetics of c-MYC Promoter G-Quadruplexes Studied by Single-Molecule Manipulation. J. Am. Chem. Soc. 2015, 137, 2424–2427. [Google Scholar] [CrossRef] [PubMed]
- De Cian, A.; Guittat, L.; Kaiser, M.; Sacca, B.; Amrane, S.; Bourdoncle, A.; Alberti, P.; Teulade-Fichou, M.-P.; Lacroix, L.; Mergny, J.-L. Fluorescence-based melting assays for studying quadruplex ligands. Methods 2007, 42, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, A.; Spada, G.P.; da Silva, M.W. Circular dicroism of quadruplex structures. Top. Curr. Chem. 2013, 330, 67–86. [Google Scholar] [PubMed]
- Vorlíčková, M.; Kejnovská, I.; Saqi, J.; Renčiuk, D.; Bednářová, K.; Motlová, J.; Kypr, J. Circular dichroism and guanine quadruplexes. Methods 2012, 57, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Paramasivan, S.; Rujan, I.; Bolton, P.H. Circular dichroism of quadruplex DNAs: Applications to structure, cation effects and ligand binding. Methods 2007, 43, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Víglaský, V.; Bauer, L.; Tlučková, K. Structural features of intra- and intermolecular G-quadruplexes derived from telomeric repeats. Biochemistry 2010, 49, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.M.; Wen, J.-D.; Gray, C.W.; Repges, R.; Repges, C.; Raabe, G.; Fleischhauer, J. Measured and calculated CD spectra of G-quartets stacked with the same or opposite polarities. Chirality 2008, 20, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, K.; Kumar, G.S. Interaction of berberine, palmatine, coralyne, and sanguinarine to quadruplex DNA: A comparative spectroscopic and calorimetric study. Biochim. Biophys. Acta 2011, 1810, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Allenmark, S. Induced circular dichroism by chiral molecular interaction. Chirality 2003, 15, 409–422. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of all compounds are available from the authors upon request. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | 22AG/K+ | 22AG/Na+ | Myc2345-Pu22/K+ |
|---|---|---|---|
| 10 | KD = (3.2 ± 0.1) × 10−7 | KD = (7.2 ± 0.8) × 10−7 | KD = (4.8 ± 0.3) × 10−7 |
| n = 1.0 ± 0.0 | n = 1.3 ± 0.1 | n = 1.3 ± 0.1 | |
| 13 | KD = (2.2 ± 0.1) × 10−7 | KD = (0.8 ± 0.1) × 10−7 | KD = (1.7 ± 0.1) × 10−7 |
| n = 1.3 ± 0.1 | n = 1.4 ± 0.2 | n = 1.2 ± 0.1 | |
| 14 | KD = (5.0 ± 0.3) × 10−7 | KD = (0.6 ± 0.0) × 10−7 | KD = (1.9 ± 0.1) × 10−7 |
| n = 1.1 ± 0.1 | n = 1.1 ± 0.1 | n = 1.0 ± 0.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizeq, N.; Georgiades, S.N. Investigation of ‘Head-to-Tail’-Connected Oligoaryl N,O-Ligands as Recognition Motifs for Cancer-Relevant G-Quadruplexes. Molecules 2017, 22, 2160. https://doi.org/10.3390/molecules22122160
Rizeq N, Georgiades SN. Investigation of ‘Head-to-Tail’-Connected Oligoaryl N,O-Ligands as Recognition Motifs for Cancer-Relevant G-Quadruplexes. Molecules. 2017; 22(12):2160. https://doi.org/10.3390/molecules22122160
Chicago/Turabian StyleRizeq, Natalia, and Savvas N. Georgiades. 2017. "Investigation of ‘Head-to-Tail’-Connected Oligoaryl N,O-Ligands as Recognition Motifs for Cancer-Relevant G-Quadruplexes" Molecules 22, no. 12: 2160. https://doi.org/10.3390/molecules22122160
APA StyleRizeq, N., & Georgiades, S. N. (2017). Investigation of ‘Head-to-Tail’-Connected Oligoaryl N,O-Ligands as Recognition Motifs for Cancer-Relevant G-Quadruplexes. Molecules, 22(12), 2160. https://doi.org/10.3390/molecules22122160