Dynamic Docking: A Paradigm Shift in Computational Drug Discovery

 and
and 
Abstract
1. Introduction
2. Benefits and Limitations of Static Molecular Docking
2.1. Posing
2.2. Scoring
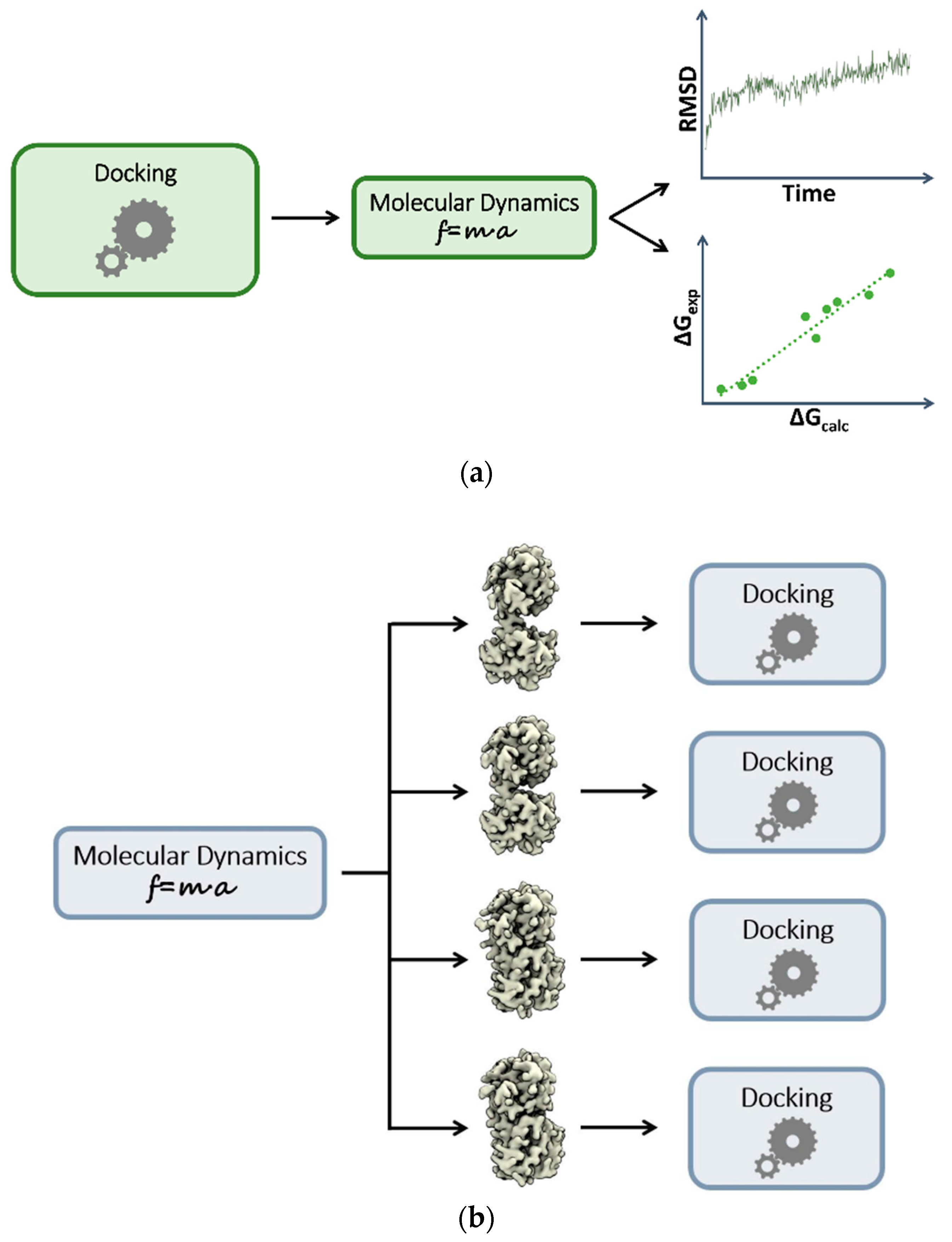
3. Plugging MD into Static Modeling Frameworks
3.1. Combining Docking and Molecular Dynamics Simulations
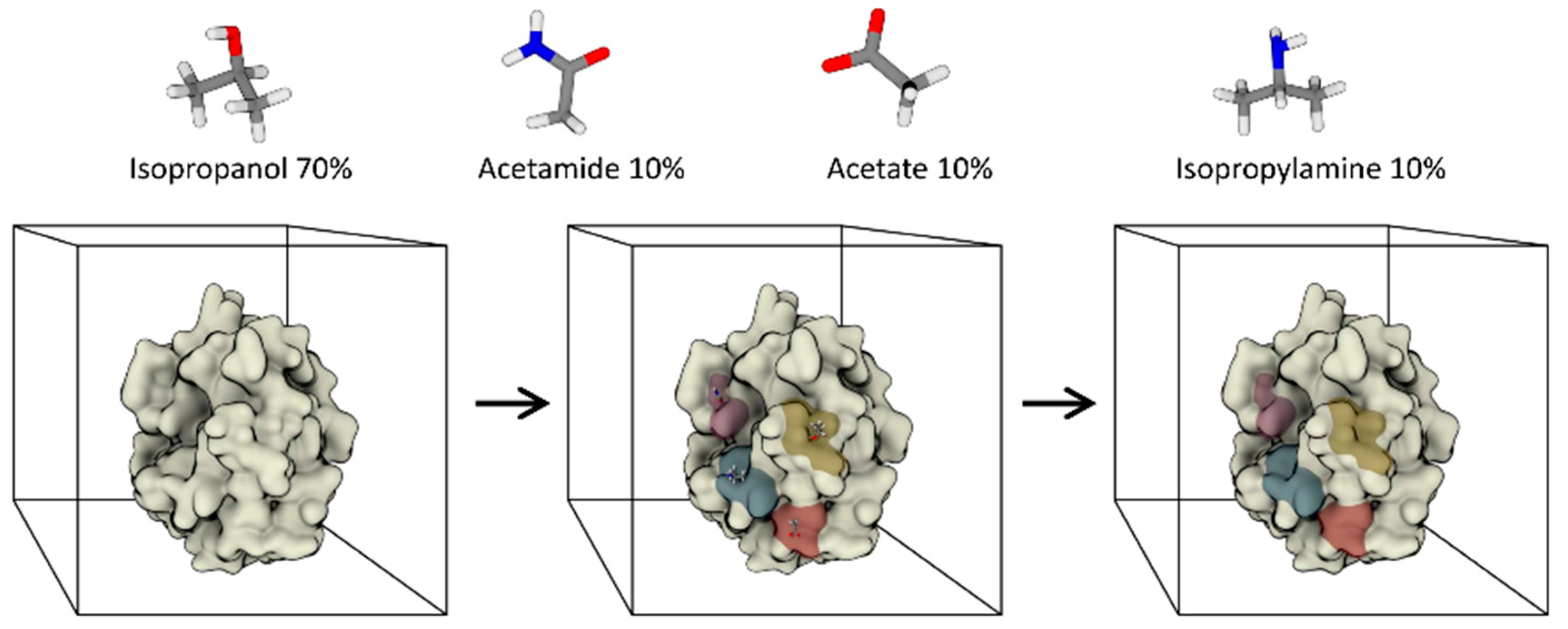
3.2. Fully Dynamic Solvent Mapping
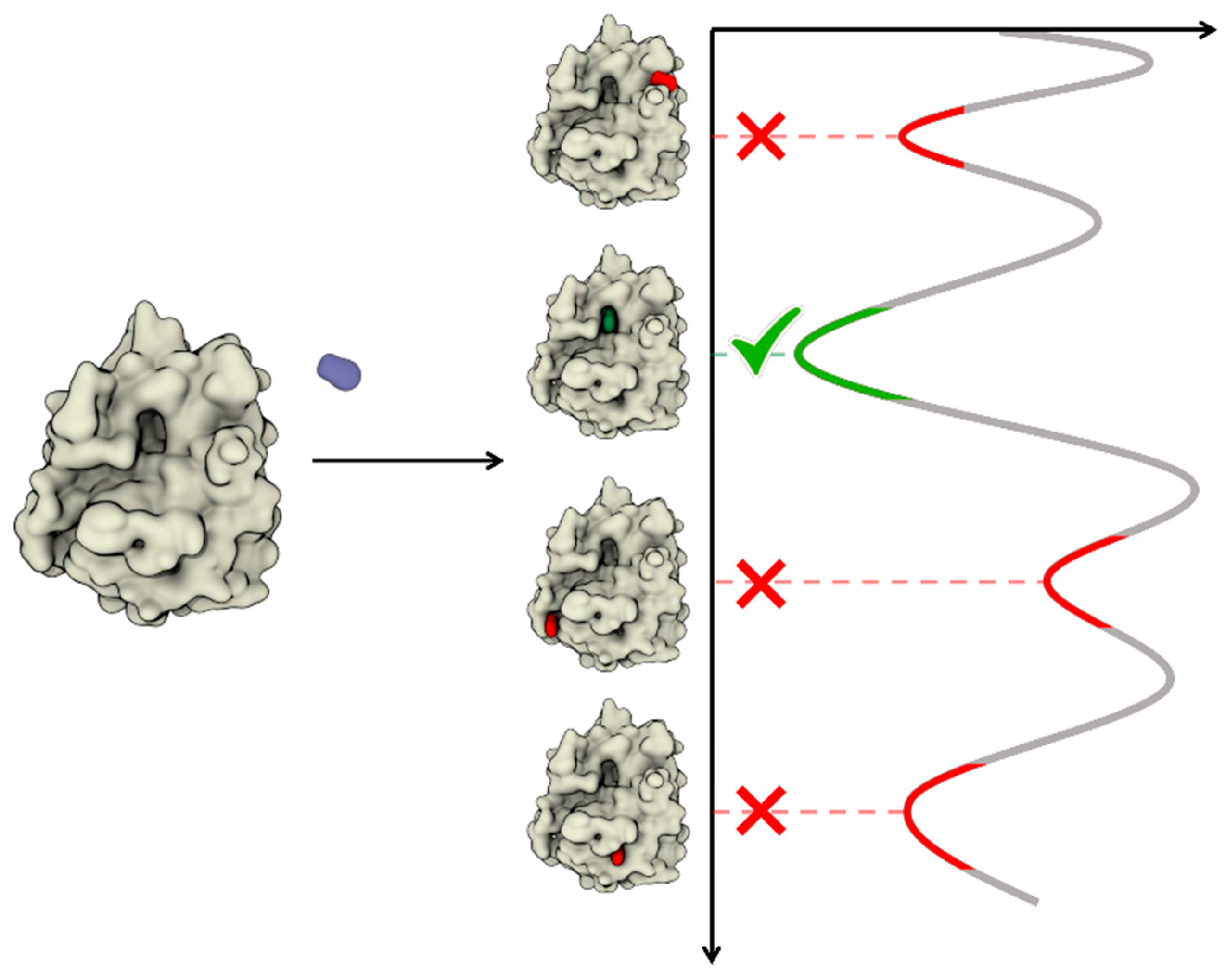
4. Dynamic Docking
4.1. Sampling Strategies
4.1.1. Biased MD Approaches
CV Biasing Methods
Tempering Methods
4.1.2. Unbiased MD Approaches
Brute-Force MD
Discontinuous Approaches
4.2. Estimation of Experimentally Accessible Observables
4.3. Current Challenges and Future Directions
5. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Matter, H.; Sotriffer, C. Applications and Success Stories in Virtual Screening; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2011; ISBN 9783527633326. [Google Scholar]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Carlson, H.A. Protein flexibility and drug design: How to hit a moving target. Curr. Opin. Chem. Biol. 2002, 6, 447–452. [Google Scholar] [CrossRef]
- Copeland, R.A. The drug–target residence time model: A 10-year retrospective. Nat. Rev. Drug Discov. 2015, 15, 87–95. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Cavalli, A. Recent advances in dynamic docking for drug discovery. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1320. [Google Scholar] [CrossRef]
- Lane, T.J.; Shukla, D.; Beauchamp, K.A.; Pande, V.S. To milliseconds and beyond: Challenges in the simulation of protein folding. Curr. Opin. Struct. Biol. 2013, 23, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Abrams, C.; Bussi, G. Enhanced Sampling in Molecular Dynamics Using Metadynamics, Replica-Exchange, and Temperature-Acceleration. Entropy 2013, 16, 163–199. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.M.; Coimbra, J.T.S.; Neves, R.P.P.; Martins, S.A.; Moorthy, N.S.H.N.; Fernandes, P.A.; Ramos, M.J. Protein-Ligand Docking in the New Millennium—A Retrospective of 10 Years in the Field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Lill, M.A. Efficient incorporation of protein flexibility and dynamics into molecular docking simulations. Biochemistry 2011, 50, 6157–6169. [Google Scholar] [CrossRef] [PubMed]
- Feixas, F.; Lindert, S.; Sinko, W.; McCammon, J.A. Exploring the role of receptor flexibility in structure-based drug discovery. Biophys. Chem. 2014, 186, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G. Protein-ligand docking. Front. Biosci. 2011, 16, 2289–2306. [Google Scholar] [CrossRef]
- Buonfiglio, R.; Recanatini, M.; Masetti, M. Protein Flexibility in Drug Discovery: From Theory to Computation. ChemMedChem 2015, 10, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.; dos Santos, R.; Oliva, G.; Andricopulo, A. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Olson, A.J. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J. Comput. Aided Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A Fast Flexible Docking Method using an Incremental Construction Algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E., Jr. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Monod, J.; Wyman, J.; Changeux, J.P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Craig, I.R.; Essex, J.W.; Spiegel, K. Ensemble Docking into Multiple Crystallographically Derived Protein Structures: An Evaluation Based on the Statistical Analysis of Enrichments. J. Chem. Inf. Model. 2010, 50, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Rueda, M.; Bottegoni, G.; Abagyan, R. Recipes for the Selection of Experimental Protein Conformations for Virtual Screening. J. Chem. Inf. Model. 2010, 50, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Rocchia, W.; Rueda, M.; Abagyan, R.; Cavalli, A. Systematic Exploitation of Multiple Receptor Conformations for Virtual Ligand Screening. PLoS ONE 2011, 6, e18845. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Given, J.A.; Bush, B.L.; McCammon, J.A. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 1997, 72, 1047–1069. [Google Scholar] [CrossRef]
- Liu, J.; Wang, R. Classification of Current Scoring Functions. J. Chem. Inf. Model. 2015, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor–ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bottegoni, G.; Raco, C.; De Vivo, M.; Recanatini, M. A computational study of the binding of propidium to the peripheral anionic site of human acetylcholinesterase. J. Med. Chem. 2004, 47, 3991–3999. [Google Scholar] [CrossRef] [PubMed]
- Perdih, A.; Hrast, M.; Pureber, K.; Barreteau, H.; Grdadolnik, S.G.; Kocjan, D.; Gobec, S.; Solmajer, T.; Wolber, G. Furan-based benzene mono- and dicarboxylic acid derivatives as multiple inhibitors of the bacterial Mur ligases (MurC–MurF): Experimental and computational characterization. J. Comput. Aided Mol. Des. 2015, 29, 541–560. [Google Scholar] [CrossRef] [PubMed]
- Sakano, T.; Mahamood, M.I.; Yamashita, T.; Fujitani, H. Molecular dynamics analysis to evaluate docking pose prediction. Biophys. Physicobiol. 2016, 13, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef] [PubMed]
- Decherchi, S.; Masetti, M.; Vyalov, I.; Rocchia, W. Implicit solvent methods for free energy estimation. Eur. J. Med. Chem. 2015, 91, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Zwanzig, R.W. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys. 1954, 22, 1420–1426. [Google Scholar] [CrossRef]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Masetti, M.; Cavalli, A.; Recanatini, M.; Gervasio, F.L. Exploring complex protein-ligand recognition mechanisms with coarse metadynamics. J. Phys. Chem. B 2009, 113, 4807–4816. [Google Scholar] [CrossRef] [PubMed]
- Colizzi, F.; Perozzo, R.; Scapozza, L.; Recanatini, M.; Cavalli, A. Single-Molecule Pulling Simulations Can Discern Active from Inactive Enzyme Inhibitors. J. Am. Chem. Soc. 2010, 132, 7361–7371. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.-P.P. A steered molecular dynamics mediated hit discovery for histone deacetylases. Phys. Chem. Chem. Phys. 2014, 16, 3777. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Carmona, S.; Schmidtke, P.; Luque, F.J.; Baker, L.; Matassova, N.; Davis, B.; Roughley, S.; Murray, J.; Hubbard, R.; Barril, X. Dynamic undocking and the quasi-bound state as tools for drug discovery. Nat. Chem. 2016, 9, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Baron, R.; McCammon, J.A. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput. Aided Mol. Des. 2008, 22, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Perryman, A.L.; Schames, J.R.; McCammon, J.A. Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J. Am. Chem. Soc. 2002, 124, 5632–5633. [Google Scholar] [CrossRef] [PubMed]
- Masetti, M.; Cavalli, A.; Recanatini, M. Modeling the hERG potassium channel in a phospholipid bilayer: Molecular dynamics and drug docking studies. J. Comput. Chem. 2008, 29, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Buonfiglio, R.; Ferraro, M.; Falchi, F.; Cavalli, A.; Masetti, M.; Recanatini, M. Collecting and Assessing Human Lactate Dehydrogenase-A Conformations for Structure-Based Virtual Screening. J. Chem. Inf. Model. 2013, 53, 2792–2797. [Google Scholar] [CrossRef] [PubMed]
- Loving, K.; Alberts, I.; Sherman, W. Computational Approaches for Fragment-Based and De Novo Design. Curr. Top. Med. Chem. 2010, 10, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Miranker, A.; Karplus, M. Functionality maps of binding sites: A multiple copy simultaneous search method. Proteins Struct. Funct. Genet. 1991, 11, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Carlson, H.A.; Masukawa, K.M.; Rubins, K.; Bushman, F.D.; Jorgensen, W.L.; Lins, R.D.; Briggs, J.M.; McCammon, J.A. Developing a Dynamic Pharmacophore Model for HIV-1 Integrase. J. Med. Chem. 2000, 43, 2100–2114. [Google Scholar] [CrossRef] [PubMed]
- Lexa, K.W.; Carlson, H.A. Protein flexibility in docking and surface mapping. Q. Rev. Biophys. 2012, 45, 301–343. [Google Scholar] [CrossRef] [PubMed]
- Ghanakota, P.; Carlson, H.A. Driving Structure-Based Drug Discovery through Cosolvent Molecular Dynamics. J. Med. Chem. 2016, 59, 10383–10399. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.N.; Bellamacina, C.R.; Ding, X.; Jeffery, C.J.; Mattos, C.; Petsko, G.A.; Ringe, D. An Experimental Approach to Mapping the Binding Surfaces of Crystalline Proteins. J. Phys. Chem. 1996, 100, 2605–2611. [Google Scholar] [CrossRef]
- Bakan, A.; Nevins, N.; Lakdawala, A.S.; Bahar, I. Druggability Assessment of Allosteric Proteins by Dynamics Simulations in the Presence of Probe Molecules. J. Chem. Theory Comput. 2012, 8, 2435–2447. [Google Scholar] [CrossRef] [PubMed]
- Seco, J.; Luque, F.J.; Barril, X. Binding site detection and druggability index from first principles. J. Med. Chem. 2009, 52, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, D.; Barril, X. Molecular simulations with solvent competition quantify water displaceability and provide accurate interaction maps of protein binding sites. J. Med. Chem. 2014, 57, 8530–8539. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, D.; Barril, X. Relationship between Protein Flexibility and Binding: Lessons for Structure-Based Drug Design. J. Chem. Theory Comput. 2014, 10, 2608. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; MacKerell, A.D. Computational Fragment-Based Binding Site Identification by Ligand Competitive Saturation. PLoS Comput. Biol. 2009, 5, e1000435. [Google Scholar] [CrossRef] [PubMed]
- Raman, E.P.; Yu, W.; Lakkaraju, S.K.; MacKerell, A.D. Inclusion of Multiple Fragment Types in the Site Identification by Ligand Competitive Saturation (SILCS) Approach. J. Chem. Inf. Model. 2013, 53, 3384–3398. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, S.K.; Raman, E.P.; Yu, W.; MacKerell, A.D. Sampling of Organic Solutes in Aqueous and Heterogeneous Environments Using Oscillating Excess Chemical Potentials in Grand Canonical-like Monte Carlo-Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 2281–2290. [Google Scholar] [CrossRef] [PubMed]
- Lexa, K.W.; Carlson, H.A. Full protein flexibility is essential for proper hot-spot mapping. J. Am. Chem. Soc. 2011, 133, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Ghanakota, P.; Carlson, H.A. Moving beyond Active-Site Detection: MixMD Applied to Allosteric Systems. J. Phys. Chem. B 2016, 120, 8685–8695. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.R.; Hu, H.P.; Ruvinsky, A.M.; Sherman, W.; Favia, A.D. Deciphering Cryptic Binding Sites on Proteins by Mixed-Solvent Molecular Dynamics. J. Chem. Inf. Model. 2017, 57, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.; Masetti, M.; Recanatini, M.; Cavalli, A.; Bottegoni, G. Mapping cholesterol interaction sites on serotonin transporter through coarse-grained molecular dynamics. PLoS ONE 2016, 11, e0166196. [Google Scholar] [CrossRef] [PubMed]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Grubmüller, H.; Heymann, B.; Tavan, P. Ligand binding: Molecular mechanics calculation of the streptavidin-biotin rupture force. Science 1996, 271, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [PubMed]
- Mark, A.E.; van Gunsteren, W.F.; Berendsen, H.J.C. Calculation of relative free energy via indirect pathways. J. Chem. Phys. 1991, 94, 3808–3816. [Google Scholar] [CrossRef]
- Nakajima, N.; Nakamura, H.; Kidera, A. Multicanonical Ensemble Generated by Molecular Dynamics Simulation for Enhanced Conformational Sampling of Peptides. J. Phys. Chem. B 1997, 101, 817–824. [Google Scholar] [CrossRef]
- Gervasio, F.L.; Laio, A.; Parrinello, M. Flexible docking in solution using metadynamics. J. Am. Chem. Soc. 2005, 127, 2600–2607. [Google Scholar] [CrossRef] [PubMed]
- Provasi, D.; Bortolato, A.; Filizola, M. Exploring molecular mechanisms of ligand recognition by opioid receptors with metadynamics. Biochemistry 2009, 48, 10020–10029. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, V.; Bonomi, M.; Parrinello, M. Funnel metadynamics as accurate binding free-energy method. Proc. Natl. Acad. Sci. USA 2013, 110, 6358–6363. [Google Scholar] [CrossRef] [PubMed]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Piana, S.; Laio, A. A Bias-Exchange Approach to Protein Folding. J. Phys. Chem. B 2007, 111, 4553–4559. [Google Scholar] [CrossRef] [PubMed]
- Pietrucci, F.; Marinelli, F.; Carloni, P.; Laio, A. Substrate binding mechanism of HIV-1 protease from explicit-solvent atomistic simulations. J. Am. Chem. Soc. 2009, 131, 11811–11818. [Google Scholar] [CrossRef] [PubMed]
- Soderhjelm, P.; Tribello, G.A.; Parrinello, M. Locating binding poses in protein-ligand systems using reconnaissance metadynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 5170–5175. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Yamaguchi, T.; Okamoto, Y. Replica-exchange molecular dynamics simulation of small peptide in water and in ethanol. Chem. Phys. Lett. 2005, 412, 280–284. [Google Scholar] [CrossRef]
- Ostermeir, K.; Zacharias, M. Advanced replica-exchange sampling to study the flexibility and plasticity of peptides and proteins. Biochim. Biophys. Acta 2013, 1834, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Luitz, M.P.; Zacharias, M. Protein-ligand docking using Hamiltonian replica exchange simulations with soft core potentials. J. Chem. Inf. Model. 2014, 54, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Kappel, K.; Miao, Y.; McCammon, J.A. Accelerated molecular dynamics simulations of ligand binding to a muscarinic G-protein-coupled receptor. Q. Rev. Biophys. 2015, 48, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, N.; Yonezawa, Y.; Nakamura, H.; Higo, J. Protein-inhibitor flexible docking by a multicanonical sampling: Native complex structure with the lowest free energy and a free-energy barrier distinguishing the native complex from the others. Proteins Struct. Funct. Bioinform. 2007, 70, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Bekker, G.-J.; Kamiya, N.; Araki, M.; Fukuda, I.; Okuno, Y.; Nakamura, H. Accurate Prediction of Complex Structure and Affinity for a Flexible Protein Receptor and Its Inhibitor. J. Chem. Theory Comput. 2017, 13, 2389–2399. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tajkhorshid, E. Electrostatic funneling of substrate in mitochondrial inner membrane carriers. Proc. Natl. Acad. Sci. USA 2008, 105, 9598–9603. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Kim, E.T.; Eastwood, M.P.; Dror, R.O.; Seeliger, M.A.; Shaw, D.E. How Does a Drug Molecule Find Its Target Binding Site? J. Am. Chem. Soc. 2011, 133, 9181–9183. [Google Scholar] [CrossRef] [PubMed]
- Buch, I.; Giorgino, T.; De Fabritiis, G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 10184–10189. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Pan, A.C.; Arlow, D.H.; Borhani, D.W.; Maragakis, P.; Shan, Y.; Xu, H.; Shaw, D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 13118–13123. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O.; et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Decherchi, S.; Berteotti, A.; Bottegoni, G.; Rocchia, W.; Cavalli, A. The ligand binding mechanism to purine nucleoside phosphorylase elucidated via molecular dynamics and machine learning. Nat. Commun. 2015, 6, 6155. [Google Scholar] [CrossRef] [PubMed]
- Ferruz, N.; Harvey, M.J.; Mestres, J.; De Fabritiis, G. Insights from Fragment Hit Binding Assays by Molecular Simulations. J. Chem. Inf. Model. 2015, 55, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Bisignano, P.; Doerr, S.; Harvey, M.J.; Favia, A.D.; Cavalli, A.; De Fabritiis, G. Kinetic characterization of fragment binding in AmpC β-lactamase by high-throughput molecular simulations. J. Chem. Inf. Model. 2014, 54, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Green, H.F.; Valant, C.; Borhani, D.W.; Valcourt, J.R.; Pan, A.C.; Arlow, D.H.; Canals, M.; Lane, J.R.; Rahmani, R.; et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 2013, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Pande, V.S.; Beauchamp, K.; Bowman, G.R. Everything you wanted to know about Markov State Models but were afraid to ask. Methods 2010, 52, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Noé, F. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014, 25, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Plattner, N.; Noé, F. Protein conformational plasticity and complex ligand-binding kinetics explored by atomistic simulations and Markov models. Nat. Commun. 2015, 6, 7653. [Google Scholar] [CrossRef] [PubMed]
- Doerr, S.; De Fabritiis, G. On-the-Fly Learning and Sampling of Ligand Binding by High-Throughput Molecular Simulations. J. Chem. Theory Comput. 2014, 10, 2064–2069. [Google Scholar] [CrossRef] [PubMed]
- Doerr, S.; Harvey, M.J.; Noé, F.; De Fabritiis, G. HTMD: High-Throughput Molecular Dynamics for Molecular Discovery. J. Chem. Theory Comput. 2016, 12, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Ferruz, N.; Tresadern, G.; Pineda-Lucena, A.; De Fabritiis, G. Multibody cofactor and substrate molecular recognition in the myo-inositol monophosphatase enzyme. Sci. Rep. 2016, 6, 30275. [Google Scholar] [CrossRef] [PubMed]
- Stanley, N.; Pardo, L.; Fabritiis, G. De The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci. Rep. 2016, 6, 22639. [Google Scholar] [CrossRef] [PubMed]
- Sabbadin, D.; Moro, S. Supervised Molecular Dynamics (SuMD) as a Helpful Tool To Depict GPCR–Ligand Recognition Pathway in a Nanosecond Time Scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Sabbadin, D.; Ciancetta, A.; Deganutti, G.; Cuzzolin, A.; Moro, S. Exploring the recognition pathway at the human A2A adenosine receptor of the endogenous agonist adenosine using supervised molecular dynamics simulations. Med. Chem. Commun. 2015, 6, 1081–1085. [Google Scholar] [CrossRef]
- Zeller, F.; Luitz, M.P.; Bomblies, R.; Zacharias, M. Multiscale Simulation of Receptor–Drug Association Kinetics: Application to Neuraminidase Inhibitors. J. Chem. Theory Comput. 2017, 13, 5097–5105. [Google Scholar] [CrossRef] [PubMed]
- Ermak, D.L.; McCammon, J.A. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 1978, 69, 1352–1360. [Google Scholar] [CrossRef]
- Votapka, L.W.; Amaro, R.E. Multiscale Estimation of Binding Kinetics Using Brownian Dynamics, Molecular Dynamics and Milestoning. PLOS Comput. Biol. 2015, 11, e1004381. [Google Scholar] [CrossRef] [PubMed]
- Votapka, L.W.; Jagger, B.R.; Heyneman, A.L.; Amaro, R.E. SEEKR: Simulation Enabled Estimation of Kinetic Rates, A Computational Tool to Estimate Molecular Kinetics and Its Application to Trypsin-Benzamidine Binding. J. Phys. Chem. B 2017, 121, 3597–3606. [Google Scholar] [CrossRef] [PubMed]
- General, I.J. A Note on the Standard State’s Binding Free Energy. J. Chem. Theory Comput. 2010, 6, 2520–2524. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; McCammon, J.A. Molecular Recognition and Ligand Association. Annu. Rev. Phys. Chem. 2013, 64, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Bernetti, M.; Cavalli, A.; Mollica, L. Protein–ligand (un)binding kinetics as a new paradigm for drug discovery at the crossroad between experiments and modelling. Med. Chem. Commun. 2017, 8, 534–550. [Google Scholar] [CrossRef]
- Deganutti, G.; Moro, S. Estimation of kinetic and thermodynamic ligand-binding parameters using computational strategies. Future Med. Chem. 2017, 9, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- BiKi Technologies. Available online: http://www.bikitech.com/ (accessed on 16 November 2017).
- Harvey, M.J.; Giupponi, G.; Fabritiis, G. De ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Mohanty, U.; Noehre, J.; Sawyer, T.K.; Sherman, W.; Krilov, G. Probing the α-Helical Structural Stability of Stapled p53 Peptides: Molecular Dynamics Simulations and Analysis. Chem. Biol. Drug Des. 2010, 75, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Todorov, I.T.; Smith, W.; Trachenko, K.; Dove, M.T. DL_POLY_3: New dimensions in molecular dynamics simulations via massive parallelism. J. Mater. Chem. 2006, 16, 1911. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Procacci, P. Hybrid MPI/OpenMP Implementation of the ORAC Molecular Dynamics Program for Generalized Ensemble and Fast Switching Alchemical Simulations. J. Chem. Inf. Model. 2016, 56, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xia, Z.; Zhang, J.; Best, R.; Wu, C.; Ponder, J.W.; Ren, P. Polarizable Atomic Multipole-Based AMOEBA Force Field for Proteins. J. Chem. Theory Comput. 2013, 9, 4046–4063. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | Searching Algorithm | Native Scoring Function 1 | License |
|---|---|---|---|
| AutoDock [16] | Stochastic | Force-Field based | Free for Academia |
| DOCK [17] | Systematic | Force-Field based | Free for Academia |
| FlexX [18] | Systematic | Empirical | Paid |
| Glide [19] | Systematic | Empirical | Paid |
| GOLD [20] | Stochastic | Force-Field based | Paid |
| ICM [21] | Stochastic | Force-Field based | Paid |
| MOE [22] | Stochastic | Force-Field based | Paid |
| Author (Year) | Complex | Multiple Ligands | No. of Runs | Aggregate Time | Productive Runs 1 | Time to Binding |
|---|---|---|---|---|---|---|
| Brute Force MD | ||||||
| Shan et al. (2011) | PP1/Src kinase | y | 7 | 115 µs | 3 | 15.1–1.9–0.6 µs |
| Dasatinib/Src kinase | y | 4 | 35 µs | 1 | 2.3 µs | |
| Buch et al. (2011) | Benzamidine/Trypsine | n | 495 | 49.5 µs | 187 | 15–90 ns |
| Dror et al. (2011) | Dihydroalprenolol/β2AR | y | 40 | 111.8 µs | 5 | NA |
| Alprenolol/β2AR | y | 10 | 14 µs | 1 | NA | |
| Propranolol/β2AR | y | 21 | 35.7 µs | 0 | - | |
| Isoprotenerol/β2AR | y | 1 | 15.0 µs | 0 | - | |
| Dihydroalprenolol/β1AR | y | 10 | 55.5 µs | 2 | NA | |
| Kruse et al. (2012) | ACh/M3 R | y | 1 | 25 µs | 1 | 9.5 µs |
| Tiotropium/M3 R | y | 3 | 18 µs | 0 | - | |
| Tiotropium/M2 R | y | 3 | 16.2 µs | 0 | - | |
| Decherchi et al. (2015) | DADMe-immucilin-H/PNP | y | 14 | 7 µs | 3 | 340 ns |
| Discontinuous Approaches | ||||||
| Sabbadin et al. (2014) | ZM241385/hA2A | n | 3 | - | 1 | 59 ns |
| T4G/hA2A | n | 3 | - | 1 | 62 ns | |
| T4E/hA2A | n | 3 | - | 1 | 105 ns | |
| Caffeine/hA2A | n | 3 | - | 1 | 15.2 ns | |
| Cuzzolin et al. (2016) | Ellagic Acid/CK2 | n | 3 | - | 0 | - |
| SAPS/GSTP1-1 | n | 3 | - | 2 | 27–19 ns | |
| Benzen-1,2-diol/PRDX5 | n | 3 | - | 3 | 17.4–31.2–18 ns | |
| (S)-naproxen/HSA | n | 3 | - | 0 | - | |
| (S)-fluoxetin/LeuT | n | 3 | - | 0 | - | |
| NECA/hA2A | n | 3 | - | 0 | - | |
| Zeller et al. (2017) | Oseltamivir/neuraminidase | n | 676 | 50.0 µs | ~20 | NA |
| Zanamivir/neuraminidase | n | 606 | 35.7 µs | ~20 | NA | |
| Software | GPU Support | Biased MD Support | PLUMED 2.3 Patch Available | License |
|---|---|---|---|---|
| MD Engines | ||||
| ACEMD [114] | x | x | x 1 | Free Serial Version (for Academia) |
| AMBER [115] | x | x | x | Paid |
| CHARMM [116] | x | x | Free Serial Version | |
| Desmond [117] | x | x | Free for Academia | |
| DL_POLY [118] | x | x | x 1 | Free for Academia |
| GROMACS [119] | x | x | x | Free |
| LAMMPS [120] | x | x | x | Free |
| NAMD [121] | x | x | x | Free |
| ORAC [122] | x | Free | ||
| Tinker [123] | x | Free | ||
| Software Interfaces | ||||
| BiKi Life Sciences | - | - | - | Paid |
| HTMD | - | - | - | Free Basic Version (for Academia) |
| SEEKR | - | - | - | Free |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gioia, D.; Bertazzo, M.; Recanatini, M.; Masetti, M.; Cavalli, A. Dynamic Docking: A Paradigm Shift in Computational Drug Discovery. Molecules 2017, 22, 2029. https://doi.org/10.3390/molecules22112029
Gioia D, Bertazzo M, Recanatini M, Masetti M, Cavalli A. Dynamic Docking: A Paradigm Shift in Computational Drug Discovery. Molecules. 2017; 22(11):2029. https://doi.org/10.3390/molecules22112029
Chicago/Turabian StyleGioia, Dario, Martina Bertazzo, Maurizio Recanatini, Matteo Masetti, and Andrea Cavalli. 2017. "Dynamic Docking: A Paradigm Shift in Computational Drug Discovery" Molecules 22, no. 11: 2029. https://doi.org/10.3390/molecules22112029
APA StyleGioia, D., Bertazzo, M., Recanatini, M., Masetti, M., & Cavalli, A. (2017). Dynamic Docking: A Paradigm Shift in Computational Drug Discovery. Molecules, 22(11), 2029. https://doi.org/10.3390/molecules22112029