Structure-Based Design of Potent and Selective Ligands at the Four Adenosine Receptors

 and
and 
Abstract
:1. Introduction
2. Results
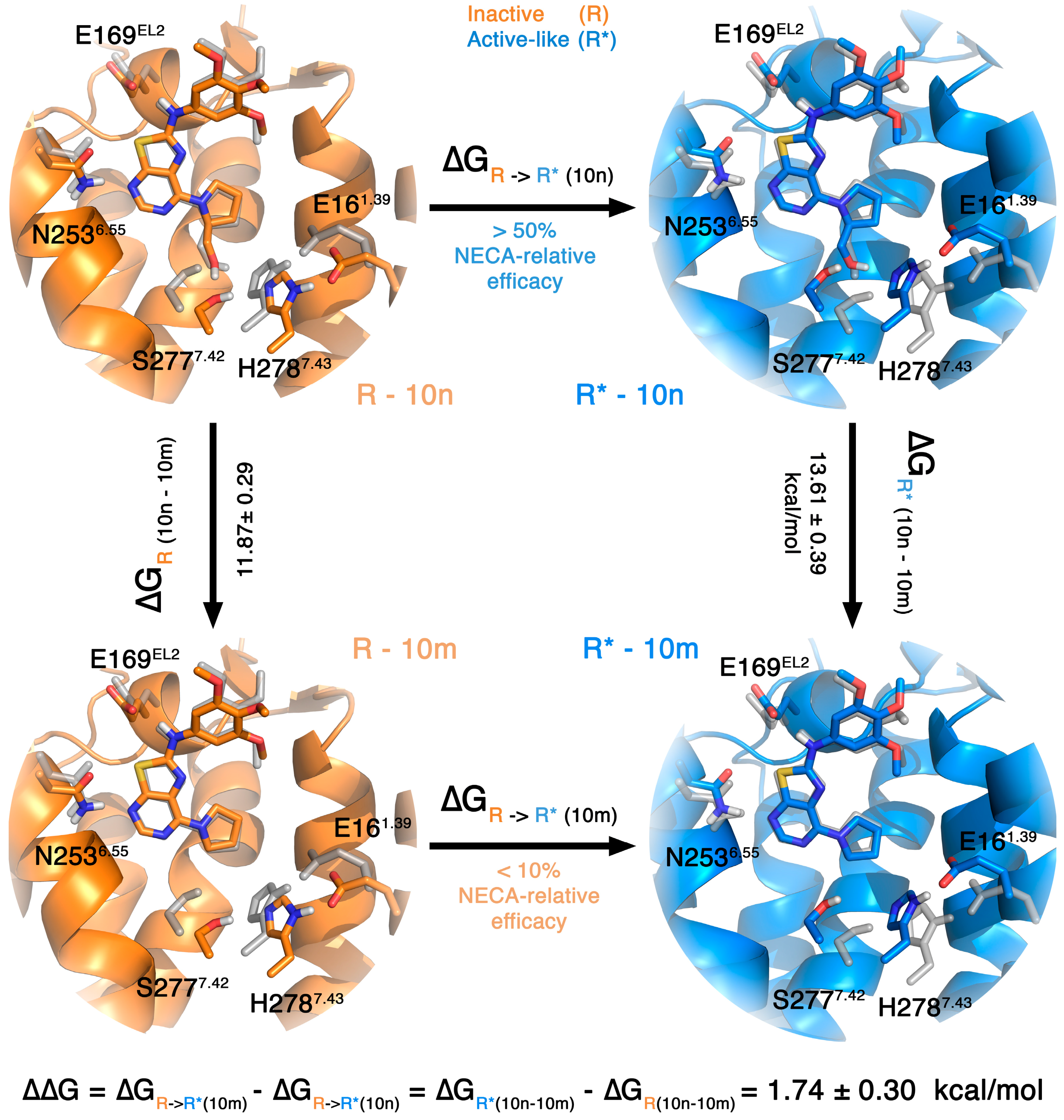
2.1. A2AAR
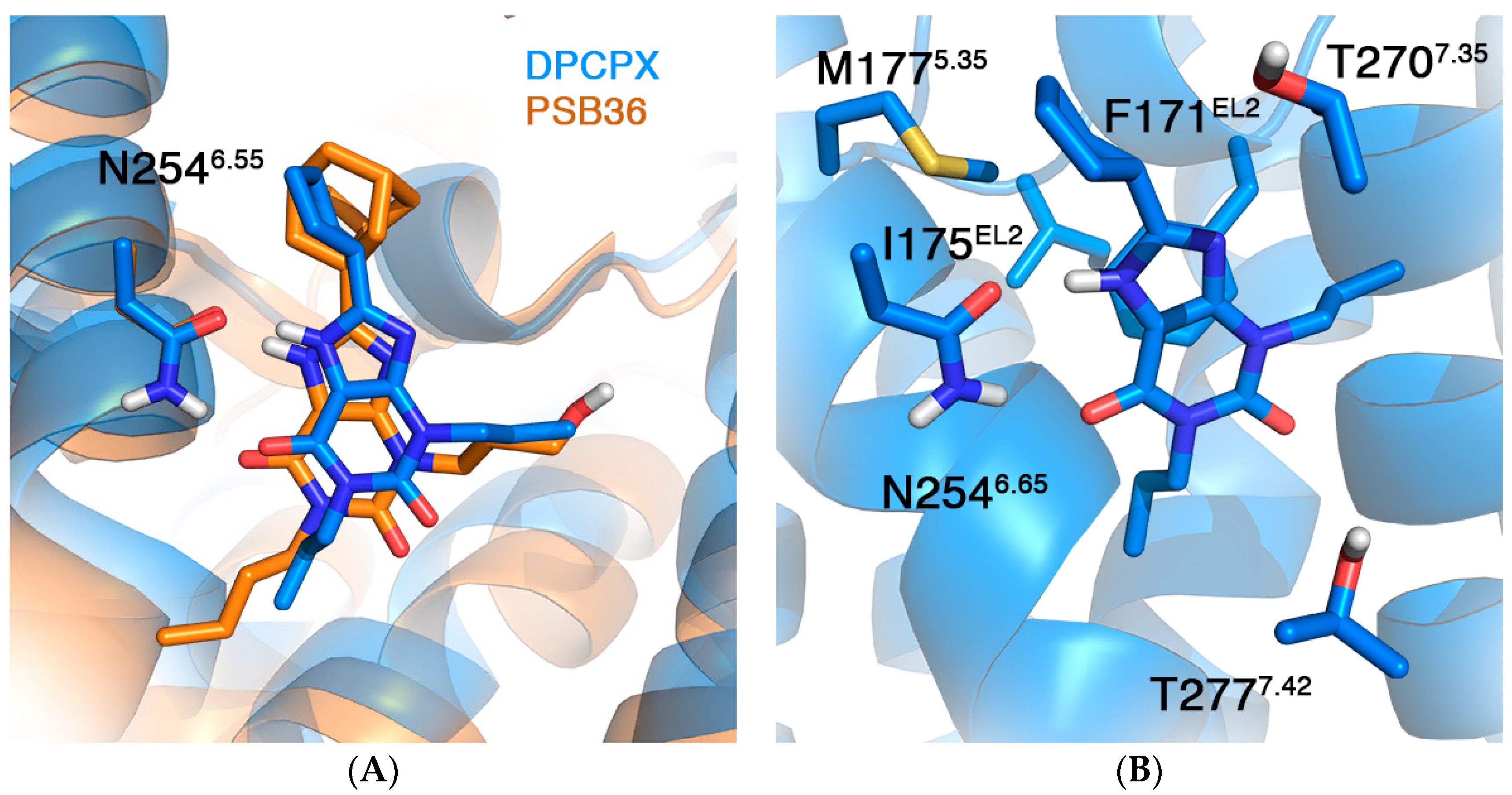
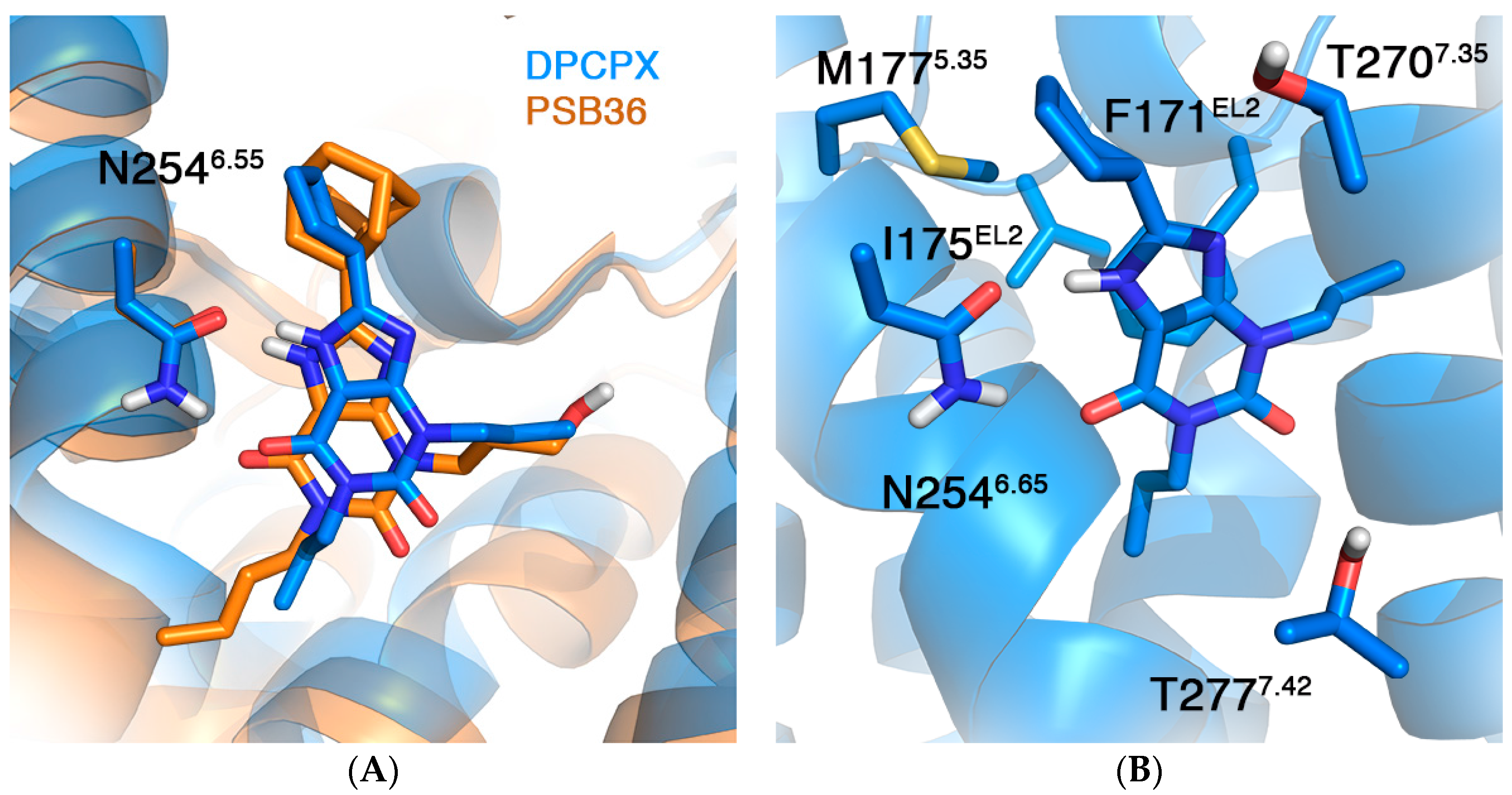
2.2. A1AR
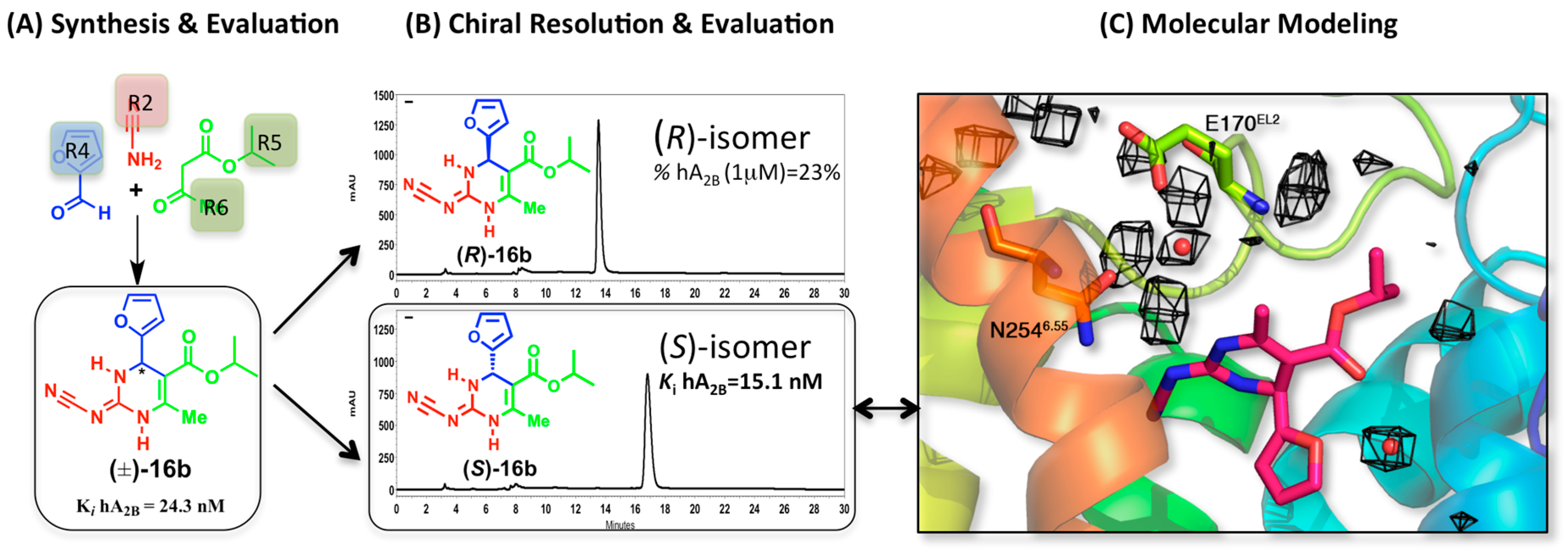
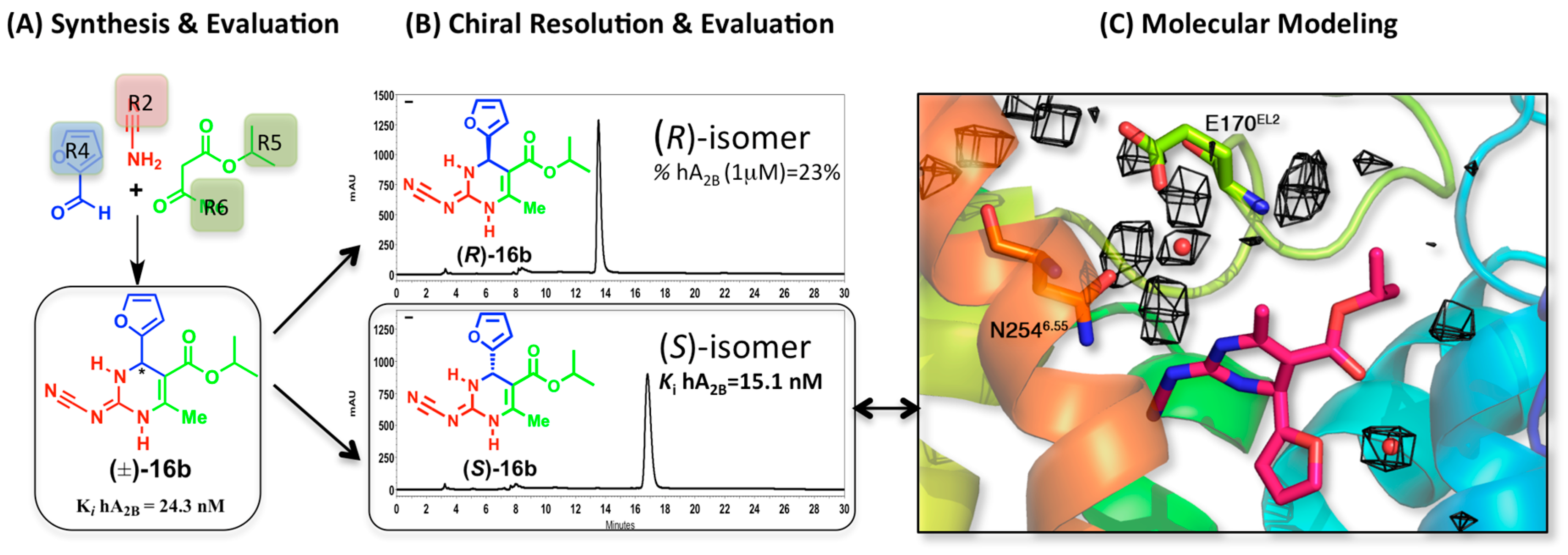
2.3. A2BAR
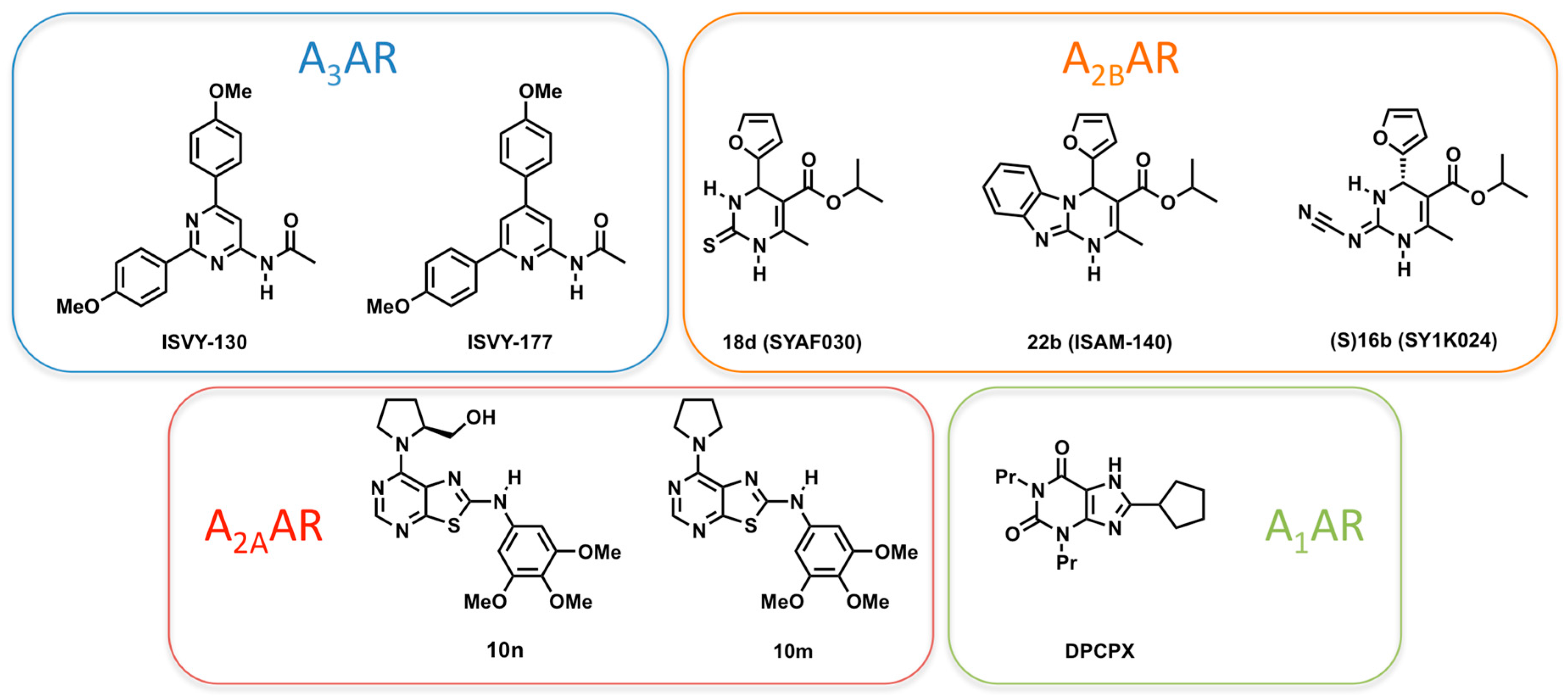
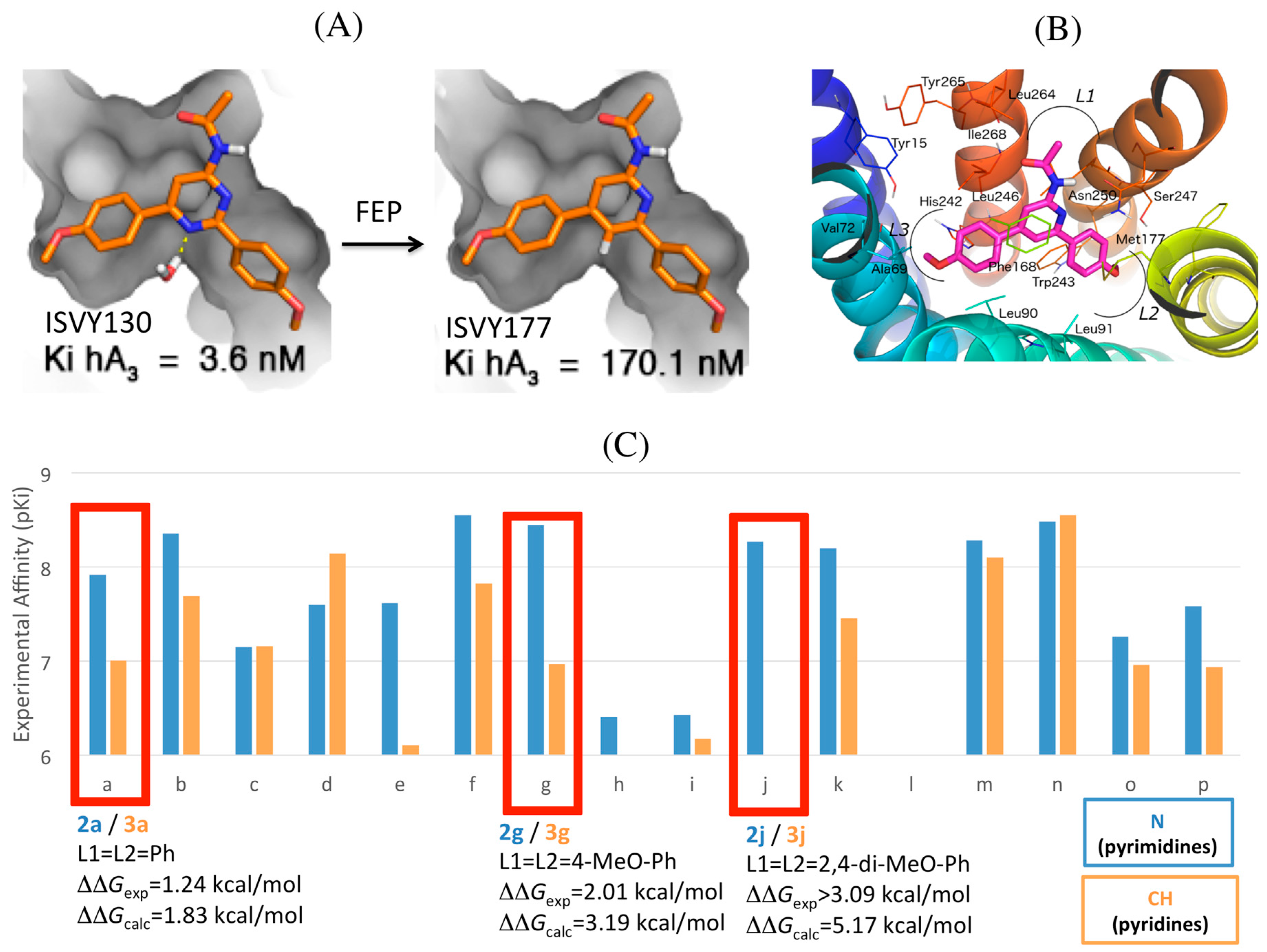
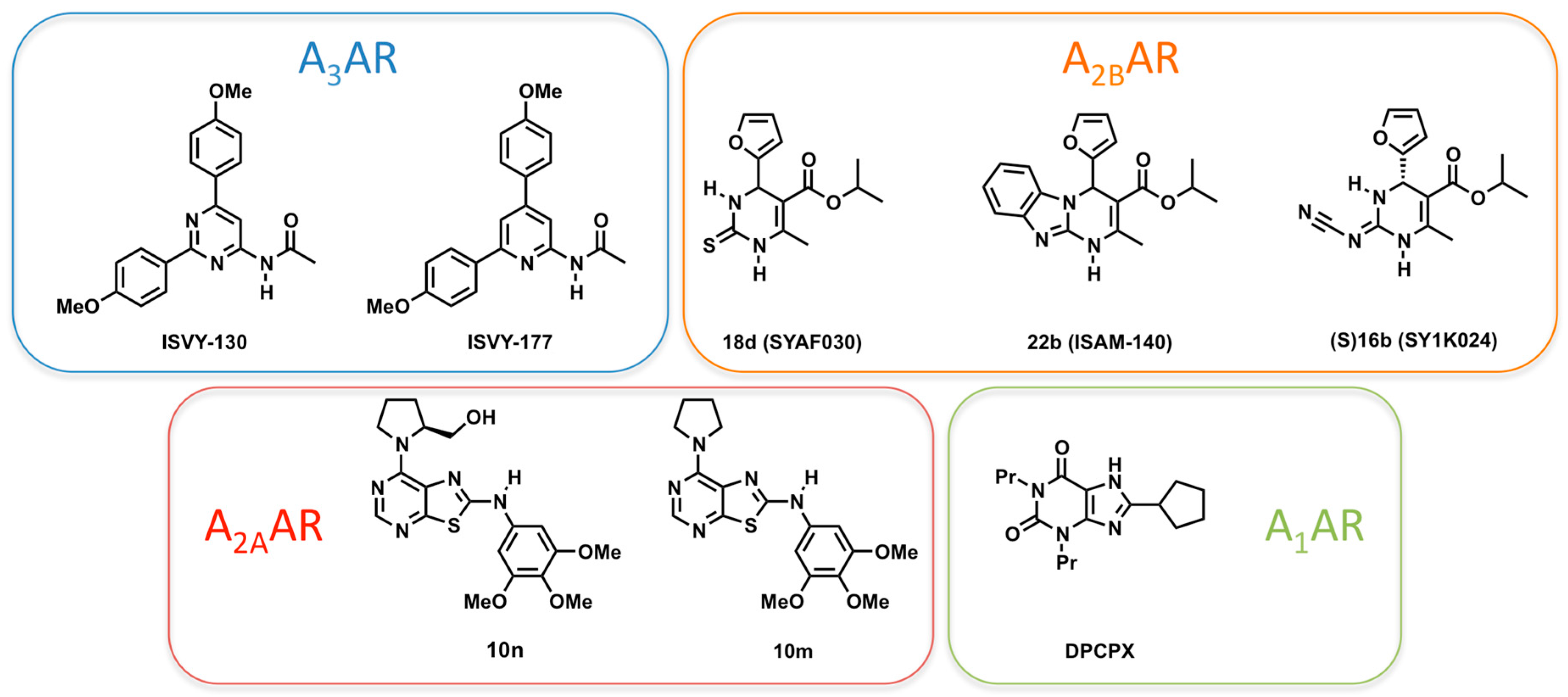
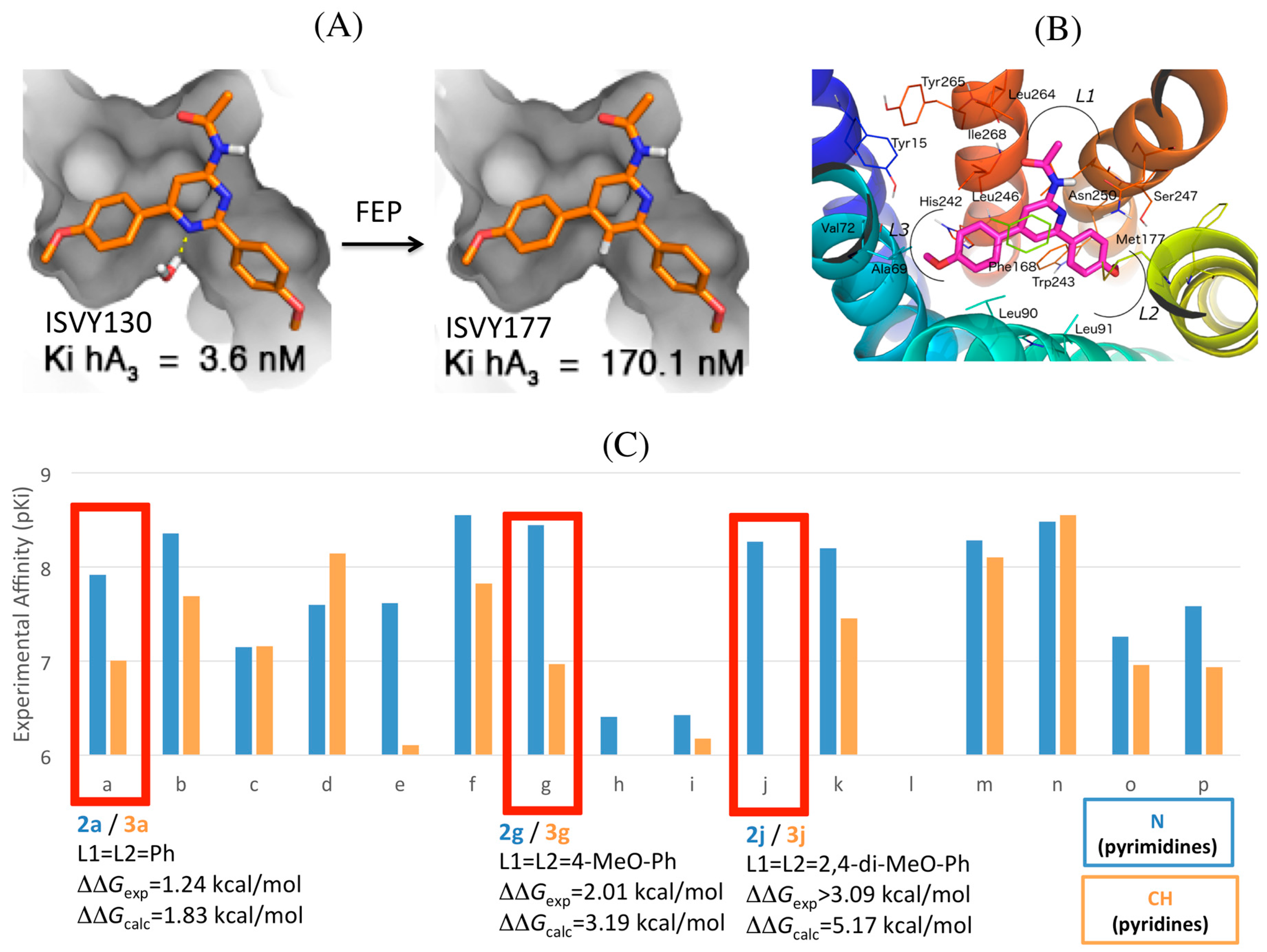
2.4. A3AR
3. Discussion
4. Materials and Methods
4.1. Preparation of Starting Receptor Structures
4.2. Ligand Docking
4.3. Membrane Insertion, Molecular Dynamics Equilibration and Water Sampling
4.4. Free Energy Perturbation Simulations
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2016, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Isberg, V.; Mordalski, S.; Munk, C.; Rataj, K.; Harpsoe, K.; Hauser, A.S.; Vroling, B.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E.; et al. GPCRdb: An information system for G protein-coupled receptors. Nucleic Acids Res. 2016, 44, D356–D364. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Gao, Z.-G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Van Westen, G.J.P.; van den Hoven, O.O.; van der Pijl, R.; Mulder-Krieger, T.; De Vries, H.; Wegner, J.K.; Ijzerman, A.P.; Van Vlijmen, H.W.T.; Bender, A.; Rg, J.; et al. Identifying novel adenosine receptor ligands by simultaneous proteochemometric modeling of rat and human bioactivity data. J. Med. Chem. 2012, 55, 7010–7020. [Google Scholar] [CrossRef] [PubMed]
- Sirci, F.; Goracci, L.; Rodríguez, D.; van Muijlwijk-Koezen, J.; Gutiérrez-de-Terán, H.; Mannhold, R. Ligand-, structure- and pharmacophore-based molecular fingerprints: A case study on adenosine A1, A2A, A2B, and A3 receptor antagonists. J. Comput. Aided. Mol. Des. 2012, 26, 1247–1266. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Phan, K.; Gao, Z.G.; Marko, A.C.; Sali, A.; Jacobson, K.A. Limits of ligand selectivity from docking to models: In silico screening for A1 adenosine receptor antagonists. PLoS ONE 2012, 7, e49910. [Google Scholar] [CrossRef] [PubMed]
- Lenselink, E.B.; Jespers, W.; Van Vlijmen, H.W.T.; IJzerman, A.P.; van Westen, G.J.P. Interacting with GPCRs; on the use of interaction fingerprints for virtual screening. J. Chem. Inf. Model. 2016, 56, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Andrews, S.P.; Marshall, F.H. Structurally enabled discovery of adenosine A2A receptor antagonists. Chem. Rev. 2017, 117, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-de-Terán, H.; Sallander, J.; Sotelo, E. Structure-based rational design of adenosine receptor ligands. Curr. Top. Med. Chem. 2017, 17, 40–58. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; Nehmé, R.; Warne, T.; Leslie, A.G.W.; Tate, C.G.; Nehmé, R.; Warne, T.; Leslie, A.G.W.; Tate, C.G. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature 2016, 536, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Glukhova, A.; Thal, D.M.; Nguyen, A.T.; Vecchio, E.A.; Jörg, M.; Scammells, P.J.; May, L.T.; Sexton, P.M.; Christopoulos, A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877.e13. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.K.Y.; Segala, E.; Robertson, N.; Deflorian, F.; Doré, A.S.; Errey, J.C.; Fiez-Vandal, C.; Marshall, F.H.; Cooke, R.M. Structures of human A1 and A2A adenosine receptors with xanthines reveal determinants of selectivity. Structure 2017, 25, 1275–1285.e4. [Google Scholar] [CrossRef] [PubMed]
- Abel, R.; Young, T.; Farid, R.; Berne, B.J.; Friesner, R. A role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. [Google Scholar] [CrossRef] [PubMed]
- Bortolato, A.; Tehan, B.G.; Bodnarchuk, M.S.; Essex, J.W.; Mason, J.S. Water network perturbation in ligand binding: Adenosine A2A antagonists as a case study. J. Chem. Inf. Model. 2013, 53, 1700–1713. [Google Scholar] [CrossRef] [PubMed]
- Lenselink, E.B.; Beuming, T.; Sherman, W.; van Vlijmen, H.W.T.; IJzerman, A.P. Selecting an optimal number of binding site waters to improve virtual screening enrichments against the adenosine A2A receptor. J. Chem. Inf. Model. 2014, 54, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Burger, S.K.; Ayers, P.W.; Vöhringer-Martinez, E. Computational study of the binding modes of caffeine to the adenosine A2A receptor. J. Phys. Chem. B 2011, 115, 13880–13890. [Google Scholar] [CrossRef] [PubMed]
- Doré, A.S.; Robertson, N.; Errey, J.C.; Ng, I.; Hollenstein, K.; Tehan, B.; Hurrell, E.; Bennett, K.; Congreve, M.; Magnani, F.; et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 2011, 19, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Boukharta, L.; Gutiérrez-de-Terán, H.; Åqvist, J. Computational prediction of alanine scanning and ligand binding energetics in G-protein coupled receptors. PLoS Comput. Biol. 2014, 10, e1003585. [Google Scholar] [CrossRef] [PubMed]
- Keränen, H.; Åqvist, J.; Gutiérrez-de-Terán, H. Free energy calculations of A2A adenosine receptor mutation effects on agonist binding. Chem. Commun. 2015, 51, 3522–3525. [Google Scholar] [CrossRef] [PubMed]
- Keränen, H.; Gutiérrez-de-Terán, H.; Åqvist, J. Structural and energetic effects of A2A adenosine receptor mutations on agonist and antagonist binding. PLoS ONE 2014, 9, e108492. [Google Scholar] [CrossRef] [PubMed]
- Nøhr, A.C.; Jespers, W.; Shehata, M.A.; Floryan, L.; Isberg, V.; Andersen, K.B.; Åqvist, J.; Gutiérrez-de-Terán, H.; Bräuner-Osborne, H.; Gloriam, D.E. The GPR139 reference agonists 1a and 7c, and tryptophan and phenylalanine share a common binding site. Sci. Rep. 2017, 7, 1128. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-de-Terán, H.; Keränen, H.; Azuaje, J.; Rodríguez, D.; Åqvist, J.; Sotelo, E. Computer-aided design of GPCR ligands. Methods Mol. Biol. 2015, 1272, 271–291. [Google Scholar] [CrossRef] [PubMed]
- Yaziji, V.; Rodríguez, D.; Gutiérrez-de-Terán, H.; Coelho, A.; Caamaño, O.; García-Mera, X.; Brea, J.; Loza, M.I.; Cadavid, M.I.; Sotelo, E. Pyrimidine derivatives as potent and selective A3 adenosine receptor antagonists. J. Med. Chem. 2011, 54, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Yaziji, V.; Rodríguez, D.; Coelho, A.; García-Mera, X.; El Maatougui, A.; Brea, J.; Loza, M.I.; Cadavid, M.I.; Gutiérrez-De-Terán, H.; Sotelo, E. Selective and potent adenosine A3 receptor antagonists by methoxyaryl substitution on the N-(2,6-diarylpyrimidin-4-yl)acetamide scaffold. Eur. J. Med. Chem. 2013, 59, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Azuaje, J.; Carbajales, C.; González-Gómez, M.; Coelho, A.; Caamaño, O.; Gutiérrez-de-Terán, H.; Sotelo, E. Pyrazin-2(1 H )-ones as a novel class of selective A3 adenosine receptor antagonists. Future Med. Chem. 2015, 7, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Crespo, A.; El Maatougui, A.; Azuaje, J.; Escalante, L.; Majellaro, M.; Loza, M.I.; Brea, J.; Cadavid, M.I.; Gutiérrez-de-Terán, H.; Sotelo, E. Exploring the influence of the substituent at position 4 in a series of 3,4-dihydropyrimidin-2(1H)-one A2B adenosine receptor antagonists. Chem. Heterocycl. Compd. 2017, 53, 316–321. [Google Scholar] [CrossRef]
- Bharate, S.B.S.S.; Singh, B.; Kachler, S.; Oliveira, A.; Kumar, V.; Bharate, S.B.S.S.; Vishwakarma, R.A.; Klotz, K.-N.; Gutiérrez de Terán, H. Discovery of 7-(prolinol-N-yl)-2-phenylamino-thiazolo[5,4-d]pyrimidines as novel non-nucleoside partial agonists for the A2A adenosine receptor: Prediction from molecular modeling. J. Med. Chem. 2016, 59, 5922–5928. [Google Scholar] [CrossRef] [PubMed]
- Crespo, A.; El Maatougui, A.; Biagini, P.; Azuaje, J.; Coelho, A.; Brea, J.; Loza, M.I.; Cadavid, M.I.; García-Mera, X.; Gutiérrez-de-Terán, H.; et al. Discovery of 3,4-dihydropyrimidin-2(1H)-ones as a novel class of potent and selective A2B adenosine receptor antagonists. ACS Med. Chem. Lett. 2013, 4, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Azuaje, J.; Jespers, W.; Yaziji, V.; Mallo, A.; Majellaro, M.; Caamaño, O.; Loza, M.I.; Cadavid, M.I.; Brea, J.; Åqvist, J.; et al. Effect of nitrogen atom substitution in A3 adenosine receptor binding: N-(4,6-diarylpyridin-2-yl)acetamides as potent and selective antagonists. J. Med. Chem. 2017, 60, 7502–7511. [Google Scholar] [CrossRef] [PubMed]
- Carbajales, C.; Azuaje, J.; Oliveira, A.; Loza, M.I.; Brea, J.; Cadavid, M.I.; Masaguer, C.F.; García-Mera, X.; Gutiérrez-de-Terán, H.; Sotelo, E. Enantiospecific recognition at the A2B adenosine receptor by alkyl 2-cyanoimino-4-substituted-6-methyl-1,2,3,4-tetrahydropyrimidine-5-carboxylates. J. Med. Chem. 2017, 60, 3372–3382. [Google Scholar] [CrossRef] [PubMed]
- El Maatougui, A.; Azuaje, J.; González-Gómez, M.; Miguez, G.; Crespo, A.; Carbajales, C.; Escalante, L.; García-Mera, X.; Gutiérrez-de-Terán, H.; Sotelo, E. Discovery of potent and highly selective A2B adenosine receptor antagonist chemotypes. J. Med. Chem. 2016, 59, 1967–1983. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Andrews, S.P.; Doré, A.S.; Hollenstein, K.; Hurrell, E.; Langmead, C.J.; Mason, J.S.; Ng, I.W.; Tehan, B.; Zhukov, A.; et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J. Med. Chem. 2012, 55, 1898–1903. [Google Scholar] [CrossRef] [PubMed]
- Al Jaroudi, W.; Iskandrian, A.E. Regadenoson: A new myocardial stress agent. J. Am. Coll. Cardiol. 2009, 54, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.N.; Singh, U.C.; Bash, P.A.; Kollman, P.A. Free energy perturbation calculations on binding and catalysis after mutating Asn 155 in subtilisin. Nature 1987, 328, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar] [CrossRef]
- Lane, J.R.; Klein Herenbrink, C.; van Westen, G.J.P.; Spoorendonk, J.A.; Hoffmann, C.; IJzerman, A.P. A novel nonribose agonist, LUF5834, engages residues that are distinct from those of adenosine-like ligands to activate the adenosine A2A receptor. Mol. Pharmacol. 2012, 81, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.N.; Baltos, J.A.; Thomas, T.; Nguyen, T.D.; Munoz, L.L.; Gregory, K.J.; White, P.J.; Sexton, P.M.; Christopoulos, A.; May, L.T. Extracellular loop 2 of the adenosine A1 receptor has a key role in orthosteric ligand affinity and agonist efficacy. Mol. Pharmacol. 2016, 90, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Townsend-Nicholson, A.; Beukers, M.W.; Schofield, P.R.; Ijzerman, A.P. Thermodynamics of full agonist, partial agonist, and antagonist binding to wild-type and mutant adenosine A1 receptors. Biochem. Pharmacol. 1998, 56, 1437–1445. [Google Scholar] [CrossRef]
- Palaniappan, K.K.; Gao, Z.G.; Ivanov, A.A.; Greaves, R.; Adachi, H.; Besada, P.; Hea, O.K.; Ae, Y.K.; Seung, A.C.; Lak, S.J.; et al. Probing the binding site of the A1 adenosine receptor reengineered for orthogonal recognition by tailored nucleosides. Biochemistry 2007, 46, 7437–7448. [Google Scholar] [CrossRef] [PubMed]
- Townsend-Nicholson, A.; Schofield, P.R.A. Threonine residue in the seventh transmembrane domain of the human A1 adenosine receptor mediates specific agonist binding. J. Biol. Chem. 1994, 269, 2373–2376. [Google Scholar] [PubMed]
- Gutiérrez-de-Terán, H.; Bello, X.; Rodríguez, D. Characterization of the dynamic events of GPCRs by automated computational simulations. Biochem. Soc. Trans. 2013, 41, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Esguerra, M.; Siretskiy, A.; Bello, X.; Sallander, J.; Gutiérrez-de-Terán, H. GPCR-ModSim: A comprehensive web based solution for modeling G-protein coupled receptors. Nucleic Acids Res. 2016, 44, W455–W462. [Google Scholar] [CrossRef] [PubMed]
- Baraldi, P.G.; Preti, D.; Borea, P.A.; Varani, K. Medicinal chemistry of A3 adenosine receptor modulators: Pharmacological activities and therapeutic implications. J. Med. Chem. 2012, 55, 5676–5703. [Google Scholar] [CrossRef] [PubMed]
- Ciancetta, A.; Jacobson, K. Structural probing and molecular modeling of the A3 adenosine receptor: A focus on agonist binding. Molecules 2017, 22, 449. [Google Scholar] [CrossRef] [PubMed]
- Pennington, L.D.; Moustakas, D.T. The necessary nitrogen atom: A versatile high-impact design element for multiparameter optimization. J. Med. Chem. 2017, 60, 3552–3579. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Lebon, G.; Warne, T.; Edwards, P.C.; Bennett, K.; Langmead, C.J.; Leslie, A.G.W.; Tate, C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 2011, 474, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.-P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.T.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Šali, A. MODELLER: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2014-3: Maestro, version 9.9; Schrödinger, LLC: New York, NY, USA, 2014.
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–632. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Marelius, J.; Kolmodin, K.; Feierberg, I.; Åqvist, J.; Aqvist, J. Q: A molecular dynamics program for free energy calculations and empirical valence bond simulations in biomolecular systems. J. Mol. Graph. Model. 1998, 16, 213–225. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 33–38. [Google Scholar] [CrossRef]
- King, G.; Warshel, A. A surface constrained all-atom solvent model for effective simulations of polar solutions. J. Chem. Phys. 1989, 91, 3647. [Google Scholar] [CrossRef]
- Lee, F.S.; Warshel, A. A local reaction field method for fast evaluation of long-range electrostatic interactions in molecular simulations. J. Chem. Phys. 1992, 97, 3100. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.J.; Ciccotti, G.; Berendsen, H.J.H. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Zwanzig, R.W. High-temperature equation of state by a perturbation method. I. nonpolar gases. J. Chem. Phys. 1954, 22, 1420. [Google Scholar] [CrossRef]
Sample Availability: Calculations of the affinity of compounds DPCPX for A1AR, and 10m and 10n on A2AAR are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | ∆∆G (kcal/mol) | |
|---|---|---|
| In Vitro | In Silico | |
| F171AEL2 [40] | 4.32 a | 6.15 ± 0.69 |
| I175AEL2 [40] | 0.98 | 0.26 ± 0.39 |
| M177A5.37 [40] | 1.06 | 1.84 ± 0.46 |
| N254A6.55 | ND b | 4.14 ± 0.67 |
| T270A7.34 [40] | 0.46 | 0.40 ± 0.48 |
| T277A7.41 [41,42,43] | −0.32 ± 0.19 c | −2.77 ± 0.55 −0.42 ± 0.54 d |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jespers, W.; Oliveira, A.; Prieto-Díaz, R.; Majellaro, M.; Åqvist, J.; Sotelo, E.; Gutiérrez-de-Terán, H. Structure-Based Design of Potent and Selective Ligands at the Four Adenosine Receptors. Molecules 2017, 22, 1945. https://doi.org/10.3390/molecules22111945
Jespers W, Oliveira A, Prieto-Díaz R, Majellaro M, Åqvist J, Sotelo E, Gutiérrez-de-Terán H. Structure-Based Design of Potent and Selective Ligands at the Four Adenosine Receptors. Molecules. 2017; 22(11):1945. https://doi.org/10.3390/molecules22111945
Chicago/Turabian StyleJespers, Willem, Ana Oliveira, Rubén Prieto-Díaz, María Majellaro, Johan Åqvist, Eddy Sotelo, and Hugo Gutiérrez-de-Terán. 2017. "Structure-Based Design of Potent and Selective Ligands at the Four Adenosine Receptors" Molecules 22, no. 11: 1945. https://doi.org/10.3390/molecules22111945
APA StyleJespers, W., Oliveira, A., Prieto-Díaz, R., Majellaro, M., Åqvist, J., Sotelo, E., & Gutiérrez-de-Terán, H. (2017). Structure-Based Design of Potent and Selective Ligands at the Four Adenosine Receptors. Molecules, 22(11), 1945. https://doi.org/10.3390/molecules22111945